

Abstract
Histone variants can replace canonical histones in the nucleosome and modify chromatin structure and gene expression. The histone variant H3.3 preferentially associates with active chromatin and has been implicated in the regulation of a diverse range of developmental processes. However, the mechanisms by which H3.3 may regulate gene activity are unclear and gene duplication has hampered an analysis of H3.3 function in mouse. Here, we report that the specific knockdown of H3.3 in fertilized mouse zygotes leads to developmental arrest at the morula stage. This phenotype can be rescued by exogenous H3.3 but not by canonical H3.1 mRNA. Loss of H3.3 leads to over-condensation and mis-segregation of chromosomes as early as the two-cell stage, with corresponding high levels of aneuploidy, but does not appear to affect zygotic gene activation at the two-cell stage or lineage gene transcription at the morula stage. H3.3-deficient embryos have significantly reduced levels of markers of open chromatin, such as H3K36me2 and H4K16Ac. Importantly, a mutation in H3.3K36 that disrupts H3K36 methylation (H3.3K36R) does not rescue the H3.3 knockdown (KD) phenotype. In addition, H3.3 KD embryos have increased incorporation of linker H1. Knockdown of Mof (Kat8), an acetyltransferase specific for H4K16, similarly leads to excessive H1 incorporation. Remarkably, pan-H1 RNA interference (RNAi) partially rescues the chromosome condensation of H3.3 KD embryos and allows development to the blastocyst stage. These results reveal that H3.3 mediates a balance between open and condensed chromatin that is crucial for the fidelity of chromosome segregation during early mouse development.
Keywords: Epigenetics, Mouse preimplantation development, Histone variant H3.3, Linker histone H1, Chromatin condensation, Chromosome segregation, Histone methylation, Histone acetylation
INTRODUCTION
The fundamental unit of chromatin is the nucleosome, which is composed of two copies each of histones H2A, H2B, H3 and H4. The linker histone H1 lies adjacent to the nucleosomes and facilitates chromosome condensation to establish higher order chromatin structures (Robinson et al., 2008). Chromatin architecture can be altered by distinct mechanisms, including histone markers, chromatin remodeling, and incorporation of variants of the canonical histones. H3.3 is an H3 variant of particular interest owing to its preferential association with active chromatin (Ahmad and Henikoff, 2002). This and other observations have lead to the proposal of an ‘H3 barcode hypothesis’ (Hake and Allis, 2006). According to this hypothesis, H3.3 would mark euchromatic regions of the genome and contribute to maintaining their transcriptional activity during development. However, it should be noted that H3.3 is also found localized to promoters of silent genes and some heterochromatic regions (Goldberg et al., 2010; Eustermann et al., 2011), suggesting that the function of H3.3 might be more complex than to simply maintain gene expression during development.
Much of the difficulty in assessing the developmental function of particular histones arises from the high level of redundancy of duplicated histone genes that code for identical proteins. In mammals, H3.3 is a maternal factor that can arise from both H3f3a and H3f3b genes and is dynamically distributed during oogenesis (Chen et al., 2011) and through fertilization and cleavage stages to the blastocyst stage (Torres-Padilla et al., 2006). Dramatic epigenetic changes occur during this developmental period, and both DNA demethylation (Gu et al., 2011) and the dynamic regulation of histone marks (Erhardt et al., 2003; Puschendorf et al., 2008) are known to be essential for embryogenesis. It has been reported that the exogenous expression of a mutant version of H3.3, H3.3K27R, affects heterochromatin formation and disrupts development to the blastocyst stage (Santenard et al., 2010), suggesting that endogenous H3.3 might play a crucial role during this period. Here, we describe an analysis of H3.3 function during early mouse development. We find that H3.3 maintains a decondensed chromatin state and antagonizes H1 incorporation, and that the balance between H3.3 and H1 is key for proper chromosome segregation and development to the blastocyst stage.
MATERIALS AND METHODS
Collection and culture of preimplantation embryos
All animal treatment was performed in accordance with the guidelines of the University of California, San Francisco (UCSF) Institutional Animal Care and Use Committee (IACUC). Mice were purchased from Simonsen Laboratories. C57BL/6 females were superovulated by administration of 7.5 I.U. of pregnant mare serum gonadotropin [PMSG; obtained from the National Hormone Pituitary Program (NHPP), Harbor-UCLA Medical Center, CA, USA] and 48 hours later with 7.5 I.U. of human chorionic gonadotropin (hCG; Sigma-Aldrich). Females were mated with DBA/2 males, and 18-19 hours later females with copulation plugs were sacrificed and early zygotes were collected. Embryos were cultured in KSOM-AA medium (Millipore) at 37°C in 5% CO2.
Microinjection of morpholinos, siRNAs and mRNAs into early zygotes
Zygotes were microinjected as described previously (Oh et al., 2011). Approximately 2-5 pl of the solution was injected into the cytoplasm. The following morpholino sequences were used: H3.3A: AAGGACACCTCCTTACTTACCCCCC; H3.3B: TTTTTTTCACCCAAAGCCGAGTCTG (GeneTools). Morpholino solutions were injected at a stock concentration of 1 mM. H1 siRNA sense strand sequence: GAGAAGAACAACAGCCGCA. Mof siRNA (M-048962, SMARTpool: siGENOME, Dhamacon) and non-targeting siRNA#1 (D-001700-01, siSTABLE, Dhamacon) were used. siRNA solutions were injected at a stock concentration of 0.5 μM or 2 μM (Okada et al., 2010). Mutagenesis of H3.3K36R was carried out using the QuikChange Lightning Site-directed Mutagenesis Kit (Agilent) using the following primer sequences: H3.3K36R F: CCATCCACCGGCGGAGTGAGGAAGCCACAT; H3.3K36R R: ATGTGGCTTCCTCACTCCGCCGGTGGATGG. mRNAs for microinjection were generated by T3 mMessage mMachine (Ambion) and poly(A) tailing kit (Ambion). mRNAs were purified by RNeasy columns (Qiagen) and diluted in Nuclease-free water (Ambion). To avoid toxicity from overexpression of exogenous histones via mRNA microinjection, we titrated mRNA concentrations and all injections were carried out using a stock concentration of 25 ng/μl (Okada et al., 2010). mRNAs were used for rescue experiments only when normal blastocyst formation rates (>75% of injected embryos developed to blastocysts) were achieved after injection of each single mRNA (see examples in Fig. 4B). For experiments relating to assessing embryonic development, at least three biological replicates were carried out.
Fig. 4.

The H3.3K36 residue is required for development. (A) Mutation of K36 to R abolishes the rescue ability of exogenous H3.3-GFP mRNA in H3.3 MO-injected embryos. Left panel: Brightfield (BF) and GFP fluorescence images of E4.5 embryos injected at the zygote stage with H3.3 MOs, H3.3 MOs plus H3.3-GFP mRNA or H3.3 MOs plus H3.3 K36R-GFP mRNA. Right panel: Quantification of blastocyst formation percentage. Scale bar: 100 μm. (B) Exogenous expression of wild-type H3.3 or H3.3K36R at the concentrations used does not affect development. Left panel: Brightfield (BF) and GFP fluorescence images of E4.5 embryos injected at the zygote stage with H3.3-GFP or H3.3K36R-GFP mRNA. Right panel: Quantification of blastocyst formation percentage. Scale bar: 100 μm. All error bars indicate s.d.
Mitotic chromosome spreads
Microinjected embryos were cultured in KSOM-AA with 0.25 μg/ml nocodazole (Sigma-Aldrich) overnight to synchronize the blastomeres at metaphase. Chromosome spreads were prepared as described (Hodges and Hunt, 2002) with the following modifications: Zona pellucida was first removed by treatment with acidic Tyrode’s solution (Millipore). Embryos were then treated with hypotonic solution (50% fetal bovine serum in water) and transferred into a drop of fixative solution (1% paraformaldehyde containing 0.15% Triton X-100 and 3 mM dithiothreitol, pH 9.2) on a glass slide. The slide was then incubated overnight at room temperature and counterstained and imaged by confocal microscopy. Tip-to-tip length of chromosome arms was measured using the LAS AF Lite software (Leica). To prevent underestimation of chromosome numbers, lower magnification pictures were taken before confocal imaging (10× objective with 63× lens and additional 5× digital zoom). For karyotyping experiments, two to three biological replicates were carried out.
Immunofluorescence (IF) imaging
Embryos were prepared for IF as previously described (Chang et al., 2005; Lin et al., 2010). The following antibodies were used: H3.3 (Abnova H00003021-M01; Abcam ab62642), H3 (Abcam ab1971), H4K16ac (Millipore 07-329), H3K36me2 (Abcam ab9049), H1 (Abcam ab1938; ab62884), H3K4me3 (Millipore 04-745), H3K9me2 (Abcam ab1220), H3K9me3 (Abcam ab8898), H3K27me3 (Millipore 07-449), HP1β (Santa Cruz sc-10212), H4K5/8/12ac (Upstate 04-557), Pan-H4ac (Millipore 06-866), Mof (Santa Cruz Biotechnology sc-13677), Cdx2 (Biogenex Laboratories MU392) and Oct4 (Pou5F1; Santa Cruz Biotechnology sc-9081). Embryos were imaged on a laser-scanning confocal microscope (CTR 6500, Leica). Individual confocal sections were electronically merged so that all nuclei were visible in a single image. Individual nuclear areas were outlined according to DAPI signal and within that area we measured mean intensity in each IF channel using ImageJ (NIH). All measured intensities were normalized to the respective channel in control embryos [Ctrl MO or non-targeting (NT) siRNA]. For IF experiments, two to three biological replicates were carried out.
RNA isolation and quantitative RT-PCR (qRT-PCR)
Total RNA was isolated from embryos using PicoPure RNA Extraction Kit (Arcturus). cDNA were generated using iScript kit (Bio-Rad) and qRT-PCR reactions were performed in duplicate using the SYBR Green quantitative RT-PCR Master Mix (Applied BioSystems) and ran on an Applied BioSystems 7900HT Sequence Detection System. Relative abundance of mRNAs was calculated by normalization to Hprt or H2A.1 (also known as Hist1h2ae) mRNA level. Primer sequences for this study are listed in supplementary material Table S1.
Statistical analyses
For analyses of percentage development to the blastocyst stage, the χ2 test was used as previously described (Wakayama et al., 1998; Paull et al., 2013). For analyses of IF intensity and chromosome arm lengths, two-tailed t-test with unequal variance was used. All error bars indicate s.d.
RESULTS
H3.3 is essential for mouse preimplantation development
Although the open reading frames of H3f3a and H3f3b are highly conserved, their 5′ untranslated regions (5′ UTR) are very divergent. We took advantage of this fact to design antisense morpholino oligonucleotides (MOs) aimed at inhibiting the translation of each mRNA (supplementary material Fig. S1A). Inhibition of translation by MOs is a widely used tool in developmental biology and we routinely apply it to study mouse preimplantation development (Chen et al., 2011; Oh et al., 2011). We note that, unlike in Xenopus (Szenker et al., 2012; Lim et al., 2013), 5′ UTR sequence divergence in mouse allows us to design MOs expected to target H3.3 but not H3.1 or H3.2. We microinjected MOs into early zygotes (Fig. 1A) to deplete both maternal and zygotic functions of H3.3 mRNAs. To assess the knockdown (KD) efficiency of H3.3 MOs, we measured the protein level via quantitative immunofluorescence (IF) by comparing the signal intensity after double staining for H3.3 and pan-H3 at the two-cell stage, after overnight culture of injected zygotes. Embryos injected with H3.3 MOs showed an ∼66% reduction in H3.3 intensity compared with embryos injected with Ctrl MO (P=2.3E-10), whereas no significant changes were observed for overall H3 signal (Fig. 1B). These results indicate that the H3.3 MOs disrupted the translation of H3.3 without altering total levels of H3, and that H3.3 must be a relatively small fraction of the total H3 in early mouse embryos, similar to observations in Drosophila Kc cells (McKittrick et al., 2004).
Fig. 1.

H3.3 is essential for preimplantation mouse development. (A) Schematic of experimental design. Early mouse zygotes were microinjected and subsequently analyzed at the molecular, cytological and developmental levels. (B) H3.3 morpholinos (MOs) specifically lead to the knockdown of H3.3 without affecting global H3 level. Immunofluorescence of single sections (left panel) and maximal projections (middle panel), and quantification (right panel) of H3.3 and H3 in control (Ctrl) MO- and H3.3 MO-treated embryos. Scale bar: 20 μm. (C) Development to the blastocyst stage is impaired in H3.3 MO-injected embryos. Upper panel: development of uninjected (Uninj.), Ctrl MO and H3.3 MOs groups at E3.5 and E4.5. Lower panel: Quantification of blastocyst formation percentage. (D) Exogenous expression of H3.3-GFP, but not H3.1-GFP, rescues development of H3.3 MO-treated embryos. Left panel: Brightfield (BF) and GFP fluorescence images of E1.5, E2.5 and E4.5 embryos injected at the zygote stage with H3.3 MOs, H3.3 MOs plus H3.1-GFP mRNA or H3.3 MOs plus H3.3-GFP mRNA. Scale bar: 100 μm. Right panel: Quantification of blastocyst formation percentage. In this and all subsequent figures, χ2 P-values are indicated for developmental rates, otherwise t-test P-values are shown (see Materials and methods). All error bars indicate s.d.
The developmental potential of H3.3 KD embryos was examined by culture up to the blastocyst stage. Microinjection of either control (Ctrl), H3f3a or H3f3b MOs did not cause significant developmental defects compared with uninjected (Uninj.) embryos (Fig. 1C; data not shown). However, a severe developmental arrest was observed when we microinjected both H3f3a MO and H3f3b MO (H3.3 MOs) simultaneously (24.1% of blastocyst rate, P=3.7E-11 compared with Ctrl MO group, with total MO concentration maintained constant; Fig. 1C). Interestingly, the majority of the H3.3 KD embryos (∼79%) reached the morula stage but then failed to form blastocysts (Fig. 1C). We counted the number of cells in embryos from Ctrl MO and H3.3 MOs groups after 3 days of culture. Both groups reached compact morula morphology and had at least eight cells. Cell counts indicate that at this stage 52% of the Ctrl MO embryos had completed the fourth mitosis (have 16 or more cells), whereas only 38% of H3.3 MOs embryos had done so (n=31 for Ctrl MO, n=53 for H3.3 MOs, P=2.0E-06). These data indicate that although H3.3 KD embryos can reach the compacted morula stage, they are delayed at this stage and do not transition to blastocysts.
To assess the specificity of the H3.3 MOs, rescue experiments were performed by co-microinjection of MOs with in vitro transcribed, capped and polyadenylated H3.1-GFP or H3.3-GFP mRNAs. These mRNAs lack the H3.3 5′ UTRs and therefore are not targeted by the H3.3 MOs. GFP fluorescence allowed us to control for varying levels of expression of the rescue transcripts, by selecting two-cell embryos with similar GFP intensity to continue to culture overnight. As shown in Fig. 1D, embryos injected with H3.1- or H3.3-GFP expressed equally stable and visible GFP until embryonic day (E)4.5, indicating that the fusion proteins were expressed at similar levels. We obtained a nearly complete rescue of the H3.3 MOs phenotype and an average of ∼77% of blastocyst formation by introducing H3.3-GFP, but no rescue using H3.1-GFP (Fig. 1D). These data demonstrate that H3.3 is essential for preimplantation mouse development.
Given the association of H3.3 with transcriptionally active chromatin, we monitored the potential effects of H3.3 KD on major zygotic genome activation (ZGA), which occurs in mouse at the two-cell stage, by qRT-PCR. Erv4, Mt1 and Eif1a are diagnostic markers of mouse ZGA (Zeng and Schultz, 2005; Macfarlan et al., 2012). All of these genes are strongly induced in two-cell embryos relative to zygotes in both Ctrl MO and H3.3 MO groups (supplementary material Fig. S1B). This result is consistent with the fact that H3.3 KD embryos can develop to the morula stage, whereas embryos with defective ZGA tend to arrest at the two- to four-cell stage (Bultman et al., 2006). Furthermore, no major differences in expression were detected in H3.3 MO KD embryos at the morula stage in genes related to lineage commitment [including Oct4 (Pou5f1 - Mouse Genome Informatics), Sox2, Nanog, Klf2 (ICM/epiblast markers)], Gata6 (primitive endoderm marker) and Cdx2 (trophectoderm marker) (supplementary material Fig. S1C). In agreement with the qRT-PCR results, we detected normal levels and distribution of Cdx2 and Oct4 proteins by IF in H3.3 KD morulae (supplementary material Fig. S1D). As expected, Oct4 was evenly distributed in all nuclei and Cdx2 was heterogeneously expressed, with stronger signal on the outer cells of embryos. These results indicate that inhibition of translation of H3.3 in early mouse embryos impedes development to the blastocyst stage but does not appear to affect ZGA at the two-cell stage nor the expression of lineage commitment genes at the morula stage. Although we could not detect staining for cleaved Parp1 or cleaved Caspase 3 in H3.3 KD morulae (data not shown), we do not exclude the possibility that apoptosis may underlie their embryonic arrest.
Loss of H3.3 leads to over-condensation and mis-segregation of chromosomes
Our findings reveal a specific requirement for H3.3 in early mouse development, in contrast with Drosophila, in which double-null mutants for both H3.3 genes are viable and survive to adulthood, although both females and males are sterile (Sakai et al., 2009). The lack of H3.3 in the Drosophila germline was shown to result in defects in chromosome segregation at meiosis (Sakai et al., 2009). To address whether deficiency of H3.3 affects mitotic chromosome integrity during mouse embryogenesis, we analyzed various stages of the KD embryos. Indeed, mitotic aberrations such as micronuclei and lagging chromosomes, were visible in ∼30% of the H3.3 KD embryos from E1.5 to E3.5 (Fig. 2A,B), whereas none were detected in control embryos. The GFP fluorescence in the rescue experiments revealed that the micronuclei phenotype in H3.3 KD embryos was rescued by introduction of H3.3-GFP RNA, but not H3.1-GFP mRNA (Fig. 2C). In addition, a significantly higher rate of aneuploidy was detected in H3.3 KD two-cell embryos compared with controls (73% in H3.3 MOs and 31% in Ctrl MO, P=0.0047; Fig. 2D).
Fig. 2.

H3.3 loss leads to chromosome over-condensation and segregation defects. (A) H3.3-deficient embryos display abnormal nuclear morphology. Immunofluorescence images of two-cell and morula stage of Ctrl MO and H3.3 MO embryos. H3.3 (red); DNA (DAPI, blue). Arrowhead indicates a micronucleus and arrow indicates a lagging chromosome. Scale bars: 20 μm. (B) Quantification of chromosome mis-segregation percentage in Ctrl MO and H3.3 MO embryos. Incidence of micronuclei or lagging chromosomes is displayed. No defects were detected in Ctrl MO-treated embryos. (C) The micronuclei phenotype in H3.3-deficient embryos can be rescued by exogenous H3.3-GFP but not H3.1-GFP mRNA. GFP fluorescence images of E1.5 embryos injected at the zygote stage with H3.3 MOs plus H3.1-GFP or H3.3 MOs plus H3.3-GFP. Arrows indicate micronuclei. (D) Aneuploidy in H3.3-deficient embryos. Upper panel: Representative metaphase spreads of Ctrl MO and H3.3 MO embryos. Scale bar: 10 μm. Lower panel: Stacked columns for percentage of euploidy (2N=40) and hypoploidy (2N<40) of Ctrl MO and H3.3 MO embryos. No hyperploidy was detected. (E) H3.3-deficient embryos have significantly shorter chromosomes at metaphase. Upper panel: Representative metaphase spreads of Ctrl MO and H3.3 MO embryos. Scale bar: 10 μm. Lower panel: Scatter plot of percentage of mean chromosome arm length, relative to Ctrl MO embryos. Each dot represents a tip-to-tip chromosome arm length normalized to the mean arm length of Ctrl MO chromosomes. All error bars indicate s.d.
We also observed an intriguing phenotype at metaphase: chromosome spreads of two-cell embryos synchronized by nocodazole treatment revealed that depletion of H3.3 results in chromosome over-condensation. We measured every countable tip-to-tip chromosome arm length in embryos injected with either Ctrl MO or H3.3 MOs, and found that H3.3 KD embryos have significantly shorter chromosomes than controls (Fig. 2E). The mis-segregation and chromosome over-condensation seen in H3.3 KD embryos suggest that H3.3 is required for maintaining normal chromosome architecture. These results further suggest that chromosomes are not maximally condensed during mitosis in mouse embryos, a suggestion for which there is precedent in scid (Prkdcscid) cells and adult mice (Wong et al., 2004). It appears, however, that chromosome over-condensation comes at the expense of segregation fidelity.
H3.3 is required for high levels of the open chromatin marks H3K36me2 and H4K16ac
To investigate further the molecular mechanism responsible for the cytological and developmental defects in H3.3 KD embryos, we first conducted a screen for H3 post-translational modification residues by IF. Mouse embryos double mutant for H3K9 histone methyltransferases Suv39h1 and Suv39h2 also display chromosome segregation defects (Peters et al., 2001). However, the over-condensation phenotype in the H3.3 KD embryos (Fig. 2E) appears to be the opposite of that in Suv39h double mutant embryos, in which the chromosomes have enlarged and more relaxed heterochromatin (Peters et al., 2001). We did not observe a significant difference in IF intensity after analysis of heterochromatin markers H3K9me2 or H3K9me3 (supplementary material Fig. S2). The H3.3K27 residue has been implicated in heterochromatin formation and HP1β localization in early mouse embryogenesis (Santenard et al., 2010). In agreement with that study, we observed that the level of HP1β was significantly decreased in H3.3 KD embryos. In contrast with that study, however, we found no significant difference in H3K27me3 IF intensity in KD embryos. These divergent data may be due to the different experimental approaches used in the two studies: Santenard et al. (Santenard et al., 2010) used the introduction of an exogenous H3.3K27R mutant mRNA, whereas here we used a MO-based approach that blocks translation of the entire endogenous H3.3 protein.
Overall, H3.3 depletion did not appear to affect the silencing/heterochromatin marks H3K9me2, H3K9me3 or H3K27me3. Therefore, we considered the possibility that the residues involved in the defects observed in H3.3 KD embryos could carry active/euchromatin marks. H3K4me3 remained at similar levels between Ctrl MO and H3.3MOs embryos (supplementary material Fig. S2). We next measured H4K16ac because this mark is able to inhibit higher order chromatin formation (Shogren-Knaak et al., 2006) and embryos with reduction of H4K16ac fail to develop beyond the blastocyst stage (Thomas et al., 2008). It was also recently shown that loss of H4K16ac causes massive chromatin compaction in embryonic stem cells (Li et al., 2012). Interestingly, H3.3 KD embryos show a significant reduction (∼56%) of signal for H4K16ac (P=2.0E-08; Fig. 3A). The reduction of H4K16ac levels is not due to dysregulation of enzymes involved in H4K16 acetylation/deacetylation after H3.3 KD: the levels of Mof (Kat8 - Mouse Genome Informatics), Ep300 and Ncoa1 (acetyltransferases), or Hdac1 and Hdac2 (deacetylases) remain essentially unchanged in H3.3 KD embryos as assessed by qRT-PCR (supplementary material Fig. S3A).
Fig. 3.

H3.3 maintains histone marks associated with open chromatin. (A) H4K16ac is significantly reduced in H3.3-deficient embryos. Immunofluorescence (upper panel) and quantification (lower panel) of H3.3 (green) and H4K16ac (red) in Ctrl MO- and H3.3 MO-treated embryos. Scale bar: 20 μm. (B) H3K36me2 is significantly reduced in H3.3-deficient embryos. Immunofluorescence (upper panel) and quantification (lower panel) of H3.3 (green) and H3K36me2 (red) in Ctrl MO- and H3.3 MO-treated embryos. Scale bar: 20 μm. All error bars indicate s.d.
There are four lysines that can undergo acetylation in the histone H4 tail, namely K5, K8, K12 and K16 (supplementary material Fig. S3B). To determine whether the decrease of acetylation is K16-specific or shared with other residues, we tested two additional antibodies, one that can recognize acetylation at K5, K8 and K12 (H4K5/8/12ac), and another that can recognize acetylation at all four lysine residues (pan-H4ac). No significant difference in staining was detected using the H4K5/8/12ac-antibody (supplementary material Fig. S3C). By contrast, staining using the pan-H4ac antibody was significantly reduced in H3.3 KD embryos (supplementary material Fig. S3D). Taken together, these results indicate that H3.3 KD causes a specific decrease of H4K16ac, not a general loss of acetylation on H4.
The lysine 36 residue is crucial for H3.3 function and embryonic development
We next assessed the status of another euchromatin mark, H3K36me2, which is enriched at H3.3 (Hake et al., 2006; Loyola et al., 2006; Bell et al., 2007; Szenker et al., 2011) and associated with H4K16ac (Bell et al., 2007). In agreement with these reports, H3K36me2 is also significantly reduced in H3.3 KD embryos (Fig. 3B), and this was not caused by changes in expression of H3K36 demethylase (Setd3) or methyltransferases (Kdm2a or Kdm8) (supplementary material Fig. S3A).
To assess whether the loss of methylation at the K36 residue was involved in the phenotypes described above, we compared the rescue ability of wild-type H3.3-GFP with a mutant H3.3K36R-GFP (which cannot be methylated at position 36). Interestingly, only wild-type H3.3-GFP can restore blastocyst formation in H3.3 KD embryos, not H3.3K36R-GFP (Fig. 4A). Injection of either the wild-type H3.3-GFP or the H3.3K36R-GFP mRNAs alone (without MOs) at the concentrations used had no detectable deleterious effects on development to the blastocyst stage compared with Uninj. embryos (∼80% among the three groups; Fig. 4B). Therefore, the H3.3K36R-GFP mRNA is neither toxic nor does it act as a dominant negative, but it cannot rescue the phenotype of the H3.3 MOs. We conclude that the K36 residue, and probably its methylation, is essential for H3.3 function in early mouse embryos. We propose that H3.3K36me2 promotes H4K16ac as part of a euchromatic histone code, as it does in Drosophila (Bell et al., 2007), and that this code antagonizes chromatin condensation.
Excessive H1 incorporation mediates the phenotype of H3.3-deficient embryos
Linker H1 has the opposite effect of H4K16ac and promotes chromatin condensation (Matsuoka et al., 1994) and higher order chromatin formation (Robinson et al., 2008). Triple knockout of H1c, H1d and H1e isotypes causes global chromatin decondensation in mouse embryonic stem cells (Fan et al., 2005). Several lines of evidence point to a potential antagonistic relationship between H3.3 and H1. In Drosophila Kc167 cells, the genome-wide binding of H1 is negatively correlated with H3.3, and depletion of H3.3 by RNAi leads to increased H1 binding at promoters and regulatory elements (Braunschweig et al., 2009). During differentiation of C2C12 cells, H3.3 is deposited at regulatory regions of the MyoD gene (Myod1) and promotes gene activation with concomitant loss of H1 (Yang et al., 2011). Moreover, gastrulation defects in H3-deficient Xenopus embryos can be partially rescued by knockdown of H1 (Lim et al., 2013). To determine whether this antagonistic cross-talk between H3.3 and H1 operates in mouse embryos, we measured the H1 levels in H3.3 KD embryos. Interestingly, we found by IF that the nuclear level of H1 increased about twofold in H3.3 KD embryos at both the two-cell (P=0.000778) and four-cell (P=1.6E-09) stages relative to controls (Fig. 5A). These data indicate that, similarly to Drosophila cultured cells, depletion of H3.3 leads to excessive incorporation in early mouse embryos.
Fig. 5.

Antagonism of linker H1 by H3.3 is essential for proper chromosome condensation and early mouse development. (A) Knockdown of H3.3 leads to increased levels of H1. Immunofluorescence (upper panel) and quantification (lower panel) of H3.3 (red) and H1 (green) in Ctrl MO and H3.3 MO embryos at two-cell (left) and four-cell (right) stages. Scale bars: 20 μm. (B) Knockdown of H1 partially rescues the development of H3.3-deficient embryos. Representative brightfield images of E4.5 embryo development (left panel) and quantification of blastocyst formation percentage (right panel) of embryos injected at the zygote stage with H3.3MOs plus non-targeting (NT) siRNA or H1 siRNA. Scale bar: 100 μm. (C) Knockdown of H1 rescues the chromosome over-condensation defect of H3.3-deficient embryos. Left panel: Representative chromosomal spreads of Ctrl MO, H3.3 MOs plus NT siRNA, and H3.3 MOs plus H1 siRNA embryos. Scale bar: 10 μm. Right panel: Scatter plot of percentage of mean chromosome length of Ctrl MO embryos. Each dot represents a tip-to-tip chromosome arm length normalize to mean of Ctrl MO chromosomes. All error bars indicate s.d.
To investigate whether the developmental arrest in H3.3 KD embryos is directly mediated by excessive H1 incorporation, we sought to test whether depletion of H1 can rescue the H3.3 KD phenotype. There are at least eight isotypes of H1 encoded by different genes, and therefore it would not be feasible to knock down each one individually. To circumvent this, we aligned five major H1 isotypes (H1a to H1e) and designed a pan-H1 small interfering RNA (siRNA) aimed at targeting an identical sequence conserved in all five isotypes (supplementary material Fig. S4A). We first validated by qRT-PCR that this H1 siRNA leads to knockdown of all five isotypes (∼67-80%; supplementary material Fig. S4B), relative to a non-targeting (NT) control siRNA. We further verified that the pan-H1 siRNA leads to a ∼45-80% reduction in H1 protein IF signal after microinjection of two different dosages of H1 siRNAs (supplementary material Fig. S4C). Interestingly, we found that the knockdown of H1 does not alter H3.3 levels (supplementary material Fig. S4C), suggesting that H3.3 lies upstream of H1. We then simultaneously microinjected zygotes with H3.3 MOs and either H1 siRNA or non-targeting (NT) siRNA. Remarkably, H1 siRNA injection partially rescues blastocyst formation in H3.3 KD embryos to ∼51%, a significantly greater proportion compared with H3.3MOs plus NT siRNA control (Fig. 5B). We next investigated the effect of H1 RNAi on the chromosome over-condensation phenotype of H3.3 KD embryos. As shown in Fig. 5C, simultaneous knockdown of H3.3 and H1 restores the chromosome arm length of H3.3 KD embryos to control levels. Moreover, the micronuclei phenotype in H3.3 KD embryos is reduced from ∼30% to 10% after H1 RNAi, which is a level similar to that observed in the rescue experiments using H3.3-GFP mRNA (supplementary material Fig. S5).
Finally, we investigated a possible direct role for H4K16ac in antagonizing H1. We knocked down Mof (an acetyltransferase specfic for H4K16; also known as Myst1 or Kat8) using siRNAs. Of note, zygotic deletion of Mof has been reported to lead to early embryonic arrest (Gupta et al., 2008; Thomas et al., 2008), although the underlying mechanism remains unclear. We validated that both Mof and H4K16ac levels were decreased after Mof siRNA-injected zygotes were cultured to the four-cell stage (supplementary material Fig. S6). We then compared the level of H4K16ac and H1 by double staining of NT siRNA- or Mof siRNA-injected four-cell embryos. We found a significantly reduced level of H4K16ac (P=6.5E-05) and a significantly increased level of H1 (P=1.3E-11) in Mof siRNA-injected embryos, relative to controls (Fig. 6).
Fig. 6.
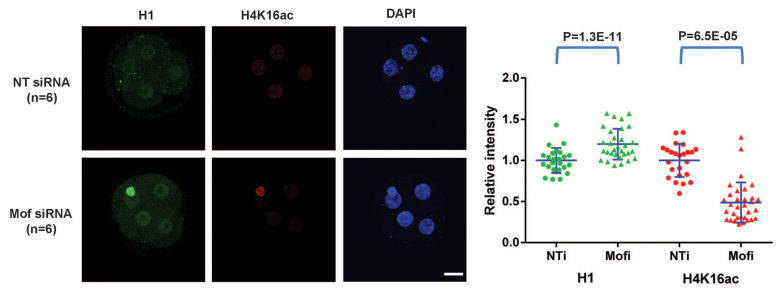
Knockdown of Mof and H4K16ac leads to increased levels of H1. Immunofluorescence (left panel) and scatter plots of quantification (right panel) of H1 (red) and H4K16ac (green) in NT siRNA and Mof siRNA embryos at the four-cell stage. Scale bar: 20 μm. All error bars indicate s.d.
Taken together, our data provide compelling evidence for the functional relevance of the loss of open chromatin marks (H3K36me2 and H4K16ac) and the excessive H1 incorporation for the chromosome over-condensation/segregation defects and the developmental arrest of H3.3 KD embryos.
DISCUSSION
We report here that the histone variant H3.3 is required for early mouse development, and specifically for the transition between the morula and the blastocyst stage. We found that H3.3 maintains a decondensed chromatin state and is essential for fidelity of chromosome segregation during mitosis. Our data support a mechanistic model (Fig. 7) whereby K36me2 is a key epigenetic mark at H3.3 that may drive H4K16Ac. Our data on the role of H4K16ac in antagonizing H1 incorporation (Fig. 6) lends further support to this model. We propose that this epigenetic code imparted by H3.3 antagonizes linker H1 incorporation to maintain a decondensed chromatin state (Fig. 7). Studies with Drosophila Kc cells support this model: it has been shown that K36me2 is enriched at H3.3 and promotes cross-histone acetylation of H4 at H4K16 (Bell et al., 2007), and that H3.3 inhibits incorporation of H1 (Braunschweig et al., 2009). In further agreement with this model, recruitment of Sirt1 to a luciferase reporter in human cell lines leads to deacetylation of H4K16, binding of H1, formation of heterochromatin and gene silencing (Vaquero et al., 2004). This model is also supported by findings in Xenopus on the importance of a balance between H3 (including H3.3) and H1 for gastrulation (Lim et al., 2013). We note that the epigenetic effects (loss of K36me2 and H4K16Ac, gain of H1) and cytogenetic effects (chromosome over-condensation and mis-segregation) of H3.3 knockdown in zygotes are observed very rapidly and are already clear at the two-cell stage (1 day after MO injection). These findings indicate that that a dynamic balance of the opposing influences of H3.3 and H1 maintains an epigenetic landscape required for the developmental competence of early mouse embryos. We note that it is not feasible to assess development past the blastocyst stage in this system, not just because this would require transfer to foster dams, but also owing to dilution of the MOs or siRNAs with embryonic growth. While the amount of material obtained from preimplantation mouse embryos is very limiting, a challenge for future studies will be to attempt to perform ChIP-Seq and determine the genomic locations that abnormally gain H1 upon loss of H3.3.
Fig. 7.

Model for an H3.3-mediated decondensed chromatin histone code essential for early mouse development. Incorporation of H3.3 in wild-type early mouse embryos maintains H3.3K36me2, which promotes cross-histone acetylation of H4K16 and antagonizes excessive H1 incorporation. This balance keeps early embryos in a decondensed chromatin state required for faithful chromosome segregation and developmental competence. Knockdown of H3.3 causes loss of H3.3K36me2 and associated acetyl-H4K16, and leads to abnormally high incorporation of H1. Excessive levels of H1 cause over-condensation of chromosomes and result in developmental arrest. See text for details.
Our data do not exclude a role for maternal H3.3 protein in the regulation of ZGA, as the MOs only block de novo translation from the zygote onwards. Genetic ablation of the genes coding for H3.3 or its chaperones specifically in oocytes will be required to achieve removal of H3.3 protein prior to fertilization. Nevertheless, the cytogenetic defects observed as early as the two-cell stage, together with the lack of detectable defects in ZGA or expression of lineage commitment genes at the morula stage, indicate that regulation of chromosome structure is a primary role of H3.3 during early mouse development.
This study has several implications for future work. From a basic biology point of view, it will be interesting to investigate whether the antagonistic epistatic relationship between H3.3 and H1, mediated by H3K36me2 and H4K16Ac, and the roles in chromatin condensation and genomic stability revealed here also operate in other settings of reported H3.3 roles, such as paternal genome reprogramming (Loppin et al., 2005), gastrulation (Szenker et al., 2012; Lim et al., 2013), neural crest lineage commitment (Cox et al., 2012) or meiosis (Sakai et al., 2009). Although in some of these studies H3.3 has been proposed to play a role in the regulation of gene expression (Cox et al., 2012; Szenker et al., 2012; Lim et al., 2013), an alternative role in genomic stability and cell survival deserves further exploration. In addition, it will be of interest to assess whether chromatin reprogramming events reported to occur in primordial germ cells (Hajkova et al., 2008) involve H3.3. Furthermore, a dissection of the roles of different H3.3 chaperones, such as Hira, Atrx, Daxx and Dek (Goldberg et al., 2010; Sawatsubashi et al., 2010), may reveal differential contributions to aspects of H3.3 function during mouse preimplantation development. Of note in this regard, RNAi knockdown of Atrx in mouse oocytes disrupts meiosis and leads to chromosome mis-segregation and reduced fertility, although effects on H3.3 incorporation were not analyzed (Baumann et al., 2010). The novel tools reported here, namely MOs for double knockdown of H3f3a and H3f3b, an H3.3K36R mutant and an siRNA for simultaneous knockdown of five isotypes of H1, should be useful for studies of H3.3 and H1 in other contexts.
From a biomedical point of view, cytogenetic abnormalities are thought to be the main cause of failure of human embryonic development past the morula stage during in vitro fertilization (Mantikou et al., 2012). It has been hypothesized (Mantikou et al., 2012) that a ‘cytogenetic integrity’ checkpoint at the morula stage selects the embryos capable of supporting later stages of development. Although there are differences between mouse and human preimplantation development, notably on the timing of ZGA, it will be interesting to determine whether epigenetic abnormalities, such as deficits in H3.3 incorporation, underlie some of the chromosomal instability observed in human morulae. In addition, driver mutations in H3.3, including at G34, have recently been reported in pediatric glioblastoma (Schwartzentruber et al., 2012; Sturm et al., 2012; Wu et al., 2012), and it may prove fruitful to explore whether these mutations affect K36 methylation or the ability to antagonize H1.
Supplementary Material
Acknowledgments
We are grateful to Maria-Elena Torres Padilla for providing the H3.1-GFP and H3.3-GFP plasmids; to Anderj Susor for help with chromosome spreads; and to Yuhong Fan for providing qPCR primer sequences for H1 isotypes. We thank Robert Blelloch and Diana Laird for input during the course of the work; and members of the Santos and Conti labs for critical reading of the manuscript.
Footnotes
Funding
This project was supported in part by a Eunice Kennedy Shriver National Institute of Child Health and Human Development/National Institutes of Health (NIH) cooperative agreement [1U54HD055764], as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research. This work was funded by an NIH New Innovator Award [DP2OD004698 to M.R.-S.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Author contributions
M.R.-S. directed the project. C.-J.L. and M.R.-S., designed experiments, analyzed data and wrote the manuscript. C.-J.L. performed experiments. C.-J.L., M.C. and M.R.-S. interpreted the data.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.095513/-/DC1
References
- Ahmad K., Henikoff S. (2002). The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 9, 1191–1200 [DOI] [PubMed] [Google Scholar]
- Baumann C., Viveiros M. M., De La Fuente R. (2010). Loss of maternal ATRX results in centromere instability and aneuploidy in the mammalian oocyte and pre-implantation embryo. PLoS Genet. 6, e1001137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell O., Wirbelauer C., Hild M., Scharf A. N., Schwaiger M., MacAlpine D. M., Zilbermann F., van Leeuwen F., Bell S. P., Imhof A., et al. (2007). Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J. 26, 4974–4984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U., Hogan G. J., Pagie L., van Steensel B. (2009). Histone H1 binding is inhibited by histone variant H3.3. EMBO J. 28, 3635–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman S. J., Gebuhr T. C., Pan H., Svoboda P., Schultz R. M., Magnuson T. (2006). Maternal BRG1 regulates zygotic genome activation in the mouse. Genes Dev. 20, 1744–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. C., Ma Y., Jacobs S., Tian X. C., Yang X., Rasmussen T. P. (2005). A maternal store of macroH2A is removed from pronuclei prior to onset of somatic macroH2A expression in preimplantation embryos. Dev. Biol. 278, 367–380 [DOI] [PubMed] [Google Scholar]
- Chen J., Melton C., Suh N., Oh J. S., Horner K., Xie F., Sette C., Blelloch R., Conti M. (2011). Genome-wide analysis of translation reveals a critical role for deleted in azoospermia-like (Dazl) at the oocyte-to-zygote transition. Genes Dev. 25, 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox S. G., Kim H., Garnett A. T., Medeiros D. M., An W., Crump J. G. (2012). An essential role of variant histone H3.3 for ectomesenchyme potential of the cranial neural crest. PLoS Genet. 8, e1002938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt S., Su I. H., Schneider R., Barton S., Bannister A. J., Perez-Burgos L., Jenuwein T., Kouzarides T., Tarakhovsky A., Surani M. A. (2003). Consequences of the depletion of zygotic and embryonic enhancer of zeste 2 during preimplantation mouse development. Development 130, 4235–4248 [DOI] [PubMed] [Google Scholar]
- Eustermann S., Yang J. C., Law M. J., Amos R., Chapman L. M., Jelinska C., Garrick D., Clynes D., Gibbons R. J., Rhodes D., et al. (2011). Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat. Struct. Mol. Biol. 18, 777–782 [DOI] [PubMed] [Google Scholar]
- Fan Y., Nikitina T., Zhao J., Fleury T. J., Bhattacharyya R., Bouhassira E. E., Stein A., Woodcock C. L., Skoultchi A. I. (2005). Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123, 1199–1212 [DOI] [PubMed] [Google Scholar]
- Goldberg A. D., Banaszynski L. A., Noh K. M., Lewis P. W., Elsaesser S. J., Stadler S., Dewell S., Law M., Guo X., Li X., et al. (2010). Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu T. P., Guo F., Yang H., Wu H. P., Xu G. F., Liu W., Xie Z. G., Shi L., He X., Jin S. G., et al. (2011). The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610 [DOI] [PubMed] [Google Scholar]
- Gupta A., Guerin-Peyrou T. G., Sharma G. G., Park C., Agarwal M., Ganju R. K., Pandita S., Choi K., Sukumar S., Pandita R. K., et al. (2008). The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Mol. Cell. Biol. 28, 397–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P., Ancelin K., Waldmann T., Lacoste N., Lange U. C., Cesari F., Lee C., Almouzni G., Schneider R., Surani M. A. (2008). Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 452, 877–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hake S. B., Allis C. D. (2006). Histone H3 variants and their potential role in indexing mammalian genomes: the ‘H3 barcode hypothesis’. Proc. Natl. Acad. Sci. USA 103, 6428–6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hake S. B., Garcia B. A., Duncan E. M., Kauer M., Dellaire G., Shabanowitz J., Bazett-Jones D. P., Allis C. D., Hunt D. F. (2006). Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J. Biol. Chem. 281, 559–568 [DOI] [PubMed] [Google Scholar]
- Hodges C. A., Hunt P. A. (2002). Simultaneous analysis of chromosomes and chromosome-associated proteins in mammalian oocytes and embryos. Chromosoma 111, 165–169 [DOI] [PubMed] [Google Scholar]
- Li X., Li L., Pandey R., Byun J. S., Gardner K., Qin Z., Dou Y. (2012). The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network. Cell Stem Cell 11, 163–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C. Y., Reversade B., Knowles B. B., Solter D. (2013). Optimal histone H3 to linker histone H1 chromatin ratio is vital for mesodermal competence in Xenopus. Development 140, 853–860 [DOI] [PubMed] [Google Scholar]
- Lin C. J., Amano T., Zhang J., Chen Y. E., Tian X. C. (2010). Acceptance of embryonic stem cells by a wide developmental range of mouse tetraploid embryos. Biol. Reprod. 83, 177–184 [DOI] [PubMed] [Google Scholar]
- Loppin B., Bonnefoy E., Anselme C., Laurençon A., Karr T. L., Couble P. (2005). The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature 437, 1386–1390 [DOI] [PubMed] [Google Scholar]
- Loyola A., Bonaldi T., Roche D., Imhof A., Almouzni G. (2006). PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol. Cell 24, 309–316 [DOI] [PubMed] [Google Scholar]
- Macfarlan T. S., Gifford W. D., Driscoll S., Lettieri K., Rowe H. M., Bonanomi D., Firth A., Singer O., Trono D., Pfaff S. L. (2012). Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487, 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantikou E., Wong K. M., Repping S., Mastenbroek S. (2012). Molecular origin of mitotic aneuploidies in preimplantation embryos. Biochim. Biophys. Acta 1822, 1921–1930 [DOI] [PubMed] [Google Scholar]
- Matsuoka Y., Takechi S., Nakayama T., Yoneda Y. (1994). Exogenous histone H1 injection into mitotic cells disrupts synchronous progression of mitotic events by delaying chromosome decondensation. J. Cell Sci. 107, 693–701 [PubMed] [Google Scholar]
- McKittrick E., Gafken P. R., Ahmad K., Henikoff S. (2004). Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl. Acad. Sci. USA 101, 1525–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J. S., Susor A., Conti M. (2011). Protein tyrosine kinase Wee1B is essential for metaphase II exit in mouse oocytes. Science 332, 462–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y., Yamagata K., Hong K., Wakayama T., Zhang Y. (2010). A role for the elongator complex in zygotic paternal genome demethylation. Nature 463, 554–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull D., Emmanuele V., Weiss K. A., Treff N., Stewart L., Hua H., Zimmer M., Kahler D. J., Goland R. S., Noggle S. A., et al. (2013). Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature 493, 632–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A. H., O’Carroll D., Scherthan H., Mechtler K., Sauer S., Schöfer C., Weipoltshammer K., Pagani M., Lachner M., Kohlmaier A., et al. (2001). Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107, 323–337 [DOI] [PubMed] [Google Scholar]
- Puschendorf M., Terranova R., Boutsma E., Mao X., Isono K., Brykczynska U., Kolb C., Otte A. P., Koseki H., Orkin S. H., et al. (2008). PRC1 and Suv39h specify parental asymmetry at constitutive heterochromatin in early mouse embryos. Nat. Genet. 40, 411–420 [DOI] [PubMed] [Google Scholar]
- Robinson P. J., An W., Routh A., Martino F., Chapman L., Roeder R. G., Rhodes D. (2008). 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J. Mol. Biol. 381, 816–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai A., Schwartz B. E., Goldstein S., Ahmad K. (2009). Transcriptional and developmental functions of the H3.3 histone variant in Drosophila. Curr. Biol. 19, 1816–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santenard A., Ziegler-Birling C., Koch M., Tora L., Bannister A. J., Torres-Padilla M. E. (2010). Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat. Cell Biol. 12, 853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawatsubashi S., Murata T., Lim J., Fujiki R., Ito S., Suzuki E., Tanabe M., Zhao Y., Kimura S., Fujiyama S., et al. (2010). A histone chaperone, DEK, transcriptionally coactivates a nuclear receptor. Genes Dev. 24, 159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J., Korshunov A., Liu X. Y., Jones D. T., Pfaff E., Jacob K., Sturm D., Fontebasso A. M., Quang D. A., Tönjes M., et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak M., Ishii H., Sun J. M., Pazin M. J., Davie J. R., Peterson C. L. (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847 [DOI] [PubMed] [Google Scholar]
- Sturm D., Witt H., Hovestadt V., Khuong-Quang D. A., Jones D. T., Konermann C., Pfaff E., Tönjes M., Sill M., Bender S., et al. (2012). Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437 [DOI] [PubMed] [Google Scholar]
- Szenker E., Ray-Gallet D., Almouzni G. (2011). The double face of the histone variant H3.3. Cell Res. 21, 421–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szenker E., Lacoste N., Almouzni G. (2012). A developmental requirement for HIRA-dependent H3.3 deposition revealed at gastrulation in Xenopus. Cell Rep. 1, 730–740 [DOI] [PubMed] [Google Scholar]
- Thomas T., Dixon M. P., Kueh A. J., Voss A. K. (2008). Mof (MYST1 or KAT8) is essential for progression of embryonic development past the blastocyst stage and required for normal chromatin architecture. Mol. Cell. Biol. 28, 5093–5105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Padilla M. E., Bannister A. J., Hurd P. J., Kouzarides T., Zernicka-Goetz M. (2006). Dynamic distribution of the replacement histone variant H3.3 in the mouse oocyte and preimplantation embryos. Int. J. Dev. Biol. 50, 455–461 [DOI] [PubMed] [Google Scholar]
- Vaquero A., Scher M., Lee D., Erdjument-Bromage H., Tempst P., Reinberg D. (2004). Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 16, 93–105 [DOI] [PubMed] [Google Scholar]
- Wakayama T., Perry A. C., Zuccotti M., Johnson K. R., Yanagimachi R. (1998). Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 394, 369–374 [DOI] [PubMed] [Google Scholar]
- Wong H. P., Mozdarani H., Finnegan C., McIlrath J., Bryant P. E., Slijepcevic P. (2004). Lack of spontaneous and radiation-induced chromosome breakage at interstitial telomeric sites in murine scid cells. Cytogenet. Genome Res. 104, 131–136 [DOI] [PubMed] [Google Scholar]
- Wu G., Broniscer A., McEachron T. A., Lu C., Paugh B. S., Becksfort J., Qu C., Ding L., Huether R., Parker M., et al. ; St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome Project (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. H., Song Y., Seol J. H., Park J. Y., Yang Y. J., Han J. W., Youn H. D., Cho E. J. (2011). Myogenic transcriptional activation of MyoD mediated by replication-independent histone deposition. Proc. Natl. Acad. Sci. USA 108, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng F., Schultz R. M. (2005). RNA transcript profiling during zygotic gene activation in the preimplantation mouse embryo. Dev. Biol. 283, 40–57 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.