

1. Introduction
Individuals with immunodeficiency have a vastly increased cancer risk. This risk appears to be largely due to a loss of immune surveillance to tumor antigens, although other contributing factors exist. Among tumor antigens are proteins expressed by oncogenic viruses. This review focuses on two gammaherpesviruses, namely Epstein Barr Virus (EBV) and Kaposi’s sarcoma herpesvirus (KSHV, also called human herpesvirus 8 [HHV-8]). Both of these viruses can infect B-lymphocytes, and largely coexist with their human host without causing disease. Like all other members of the herpesvirus family, they can establish a latent infection, in which no viral particles are produced, and only a few viral transcripts and proteins are made. Both EBV and KSHV have the capacity of expressing proteins and non-coding RNAs during latent infection that promote cellular proliferation, which is necessary for the virus to take hold in the appropriate cellular reservoir as part of their life cycle. Normally, the immune system limits this proliferation because it will recognize some of the viral proteins. If this process fails because of abnormal immune responses, the likelihood that the viral promotion of cellular proliferation will go unchecked, leading to a malignancy, increases greatly. Infection of B cells by EBV and KSHV can thereby result in the development of a variety of lymphoproliferative disorders (LPDs), which are illustrated in Figure 1 and reviewed in the following sections.
Figure 1. Infection with EBV or KSHV in malignancies occurring in patients with immunodeficiency.

The major subtypes of lymphoproliferative are shown in boxes, with arrows indicating the cause of immunodeficiency and the most common associations. Percentage of EBV infection is an approximation based on a compilation of the literature and the author’s personal experience, and reflects only the proportion of cases in the context of HIV infection; these numbers are much lower for MCD, DLBCL and HL in individuals without overt immunodeficiencies. Abbreviations: MCD: Multicentric Castleman’s disease, PH: plasmacytic hyperplasia, PBCH: polymphrphic B cell hyperplasia, PEL: primary effusion lymphoma, BL: Burkitt lymphoma, PBL: plasmablastic lymphoma, DLBCL: Diffuse large B cell lymphoma.
2. Lymphoproliferation and HIV infection
2.1 Multicentric Castleman’s disease (MCD)
Castleman’s disease is a poorly understood atypical lymphoproliferative disorder. Two distinct histopathologic subtypes had been reported before the identification of KSHV, the more common hyaline vascular type and the plasma cell type. Clinically, Castleman’s disease can be localized, or the patient may have multiple enlarged lymph nodes, therefore called “multicentric” Castleman’s disease (MCD) [1]. Approximately 90% of patients with MCD have the plasma cell type morphology. Systemic consititutional symptoms in MCD include fevers, malaise, wasting, hypoalbuminemia, cytopenias, and hyponatremia [2]. These patients frequently develop malignancies, most commonly KS and NHL, and soon after the discovery of KSHV, and association of this virus with MCD was described [3]. While in approximately half of MCD cases from immunocompetent individuals were associated with KSHV infection, this number is much higher in individuals with HIV, approaching 100%. In fact, the presence of a single lymph node containing KS and Castleman’s disease is not uncommon in HIV-positive patients. Notably, MCD, also called multicentric angiofollicular hyperplasia, is characterized by a vascular proliferation, which is reminiscent of KS. While KSHV has been reported in MCD with both hyaline vascular and plasma cell morphology [4], the majority of cases more closely resemble the plasma cell type of MCD. One report indicated that the KSHV-positive cases showed the highest intensity of angiosclerosis and germinal center and perifollicular vascular proliferation, while plasmacytosis is less pronounced than in the KSHV-negative cases of the plasma cell type [5]. It now appears that the KSHV-positive cases represent a distinct morphologic variant, resembling more the plasma cell type, but in addition showing the presence of larger cells in the mantle zones, which are have been called plasmablasts, and contain KSHV [6], that can be detected with monoclonal antibodies to the latent KSHV protein LANA1 [7]. These cells can be numerous, coalesce and form microlymphomas or frank lymphomas (described below). KSHV-infected plasmablasts are B cells that for some unknown reason are monotypic but polyclonal, almost invariably expressing IgMl [8]. One study showed that KSHV-positive endothelial cells can also be found in MCD lymph nodes, in both HIV-positive and –negative patients [9].
Systemic symptoms in MCD are thought to be due to production of excess cytokines, and in particular interleukin-6 (IL-6). Notably, KSHV produces a protein that is a homologue of IL-6, called viral IL-6 (vIL-6), which could possibly contribute to the symptoms, as it is expressed in MCD in scattered plamablasts surrounding the lymphoid follicles [10; 11; 12], and expression of this viral cytokine may confer a worse prognosis [13]. Lytic antigens are also expressed more frequently in KSHV-infected cells in MCD that in other disorders associated with this virus, suggesting that lytic viral replication may be a feature of MCD [14]. It is possible that MCD falls in the spectrum of KSHV-associated lymphadenopathies, as we have identified patients with HIV-associated lymphadenopathy that have KSHV-infected cells appreciated in lymph node biopsies, without all the defining pathologic characteristics of MCD. Furthermore, an IL-6-related systemic inflammatory syndrome has been reported in patients with HIV and KS, but without a pathologic diagnosis of MCD [15].
2.2 AIDS-Related Lymphoma (ARL)
Non-Hodgkin lymphomas (NHL) are considered an AIDS-defining condition and represent the second most common malignancy in individuals with HIV infection. The incidence has been decreasing with combination antiretroviral therapy (ART) but remains a problem. Recently a standardized incidence ratio (SIR) of around 15 was reported for the United States (US) [16], where it remains the most common cancer-related cause of death in HIV-infected individuals (36% of deaths during 1996–2006)[17]. Although not an AIDS-defining condition, the incidence of Hodgkin lymphoma (HL) is also increased in HIV-infected individuals [16]. The pathogenesis of NHL in the context AIDS is complex and thought to be related to disrupted immune surveillance to tumor antigens, as well as to genetic alterations, chronic antigenic stimulation, and cytokine dysregulation [18; 19; 20]. Also playing an important role in the pathogenesis of AIDS-related lymphoma is infection with EBV and/or KSHV.
Although HIV-related lymphomas are almost always of B cell origin, they are morphologically diverse. Several subtypes are similar to lymphomas occurring in immunocompetent patients, while others preferentially develop in the context of AIDS. HIV-related lymphomas can be classified by morphology, and/or by primary site of presentation (i.e. systemic, primary central nervous system, body cavity) [20; 21]. They include lymphomas that also occur in immunocompetent patients, those that are more common in HIV-positive patients and those occurring in other immunodeficient states.
The most common types of AIDS-related lymphoma are:
HIV-related Burkitt lymphomas (BL)
These include cases exhibiting the features of classical BL, those showing plasmacytoid differentiation and those exhibiting greater nuclear polymorphism, which in the past were referred to as atypical Burkitt or Burkitt-like lymphoma. Translocation of MYC into one of the immunoglobulin (Ig) loci is a hallmark of this disease. The most common translocation is a t(8;14), involving the MYC and immunoglobulin heavy chain (IGH) genes, but in 10% of the cases it can involve c-myc and one of the Ig light chain genes. The method of choice to demonstrate this translocation is fluorescent in situ hybridization (FISH) using a break-apart probe, which will show a split signal regardless of the translocation partner. It is thought that this translocation leads to deregulated expression of the c-myc gene. While in a small minority of cases this translocation cannot be demonstrated by FISH, it is likely that MYC is nevertheless deregulated in true BL. In fact, mutations in the c-myc locus also occur in BL [22], and may also lead to abnormal expression or prolonged protein stability. In the absence of a demonstrable MYC translocation, the histology and phenotype must be otherwise completely typical for a diagnosis of BL. Critical immunophenotypic criteria for a diagnosis of BL are positivity for B cell antigens, CD10, BCL6, and negativity or only weak positivity for BCL2. Importantly, BL is one of the fastest growing tumors in humans, which can be shown with Ki67 immunohistochemistry that will stain practically all the tumor cells.
Diffuse large B cell lymphomas (DLBCL)
These can be divided into centroblastic (CB) and immunoblastic (IB) categories. The IB type is more frequently associated with EBV infection, with reported rates approaching 90%. Patients with the IB variant have advanced AIDS and are significantly immunosuppressed, so the frequency of this subtype has dramatically decreased with the widespread use of ART in the US and Europe. Primary central nervous system (CNS) lymphoma is very similar to the IB variant in that it has an immunoblastic morphology, frequent EBV infection, and has been decreasing in frequency. CB-DLBCL in HIV+ patients is similar to DLBCL in immunocompetent individuals, with the exception of more frequent association with EBV in the former (30% vs. <5%). They can be similarly divided into germinal center and non-germinal center subtypes, although the clinical significance of this subclassification remains controversial and may be treatment-dependent [23; 24; 25].
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL
These are aggressive lymphomas that do not fit cleanly into the DLBCL or BL categories [26]. Previously, some of these cases were classified as Burkitt-like lymphoma. The presence of MYC translocations in an otherwise typical DLBCL or the lack of MYC in a typical BL, is not sufficient to place into this category. Rather, an unusual morphology or phenotype, or a combination, should be used. This appears to be a heterogeneous category, useful only for classification purposes. Some of these may belong to a separate molecular category, which is that lymphomas with MYC translocations plus a complex karyotype. In addition, some of these cases may correspond to those cases classified as BL, but upon gene expression profiling do not have a Burkitt signature [27; 28]. The true frequency of EBV in this category is unclear, but both EBV positive and negative cases can be found.
Hodgkin’s lymphoma (HL)
This disease is increased in incidence in AIDS[29], and the proportion of HL appears to be further increasing with patients surviving longer and with higher CD4 counts [29]. Approximately 30% of HLs are positive for EBV in immunocompetent individuals, but this proportion is increased in underdeveloped countries and in immunodeficient patients. In the context of HIV, nearly all cases of HL are associated with EBV infection. While the nodular sclerosis subtype is more common in the general population, in HIV+ patients, most correspond to either the mixed cellularity or lymphocyte depleted forms of HL [21; 30].
Primary effusion lymphoma (PEL)
This is a lymphoma subtype occurring primarily, albeit not exclusively, in HIV-positive patients. PELs are characterized by the presence of KSHV within the tumor cells, and the vast majority also contain EBV [31; 32]. Another important feature is the lack of expression of B cell-associated antigens, in spite of a B cell genotype. These are rare tumors, accounting for only approximately 4% of all HIV-related NHLs. They usually present as an effusion involving one or more of the pleural, peritoneal and pericardial spaces. While the majority lacks an associated tissue mass at presentation, in about one third of the cases dissemination to extracavitary sites occurs. In addition to the typical primary effusion lymphomas, KSHV is present in the tumor cells of a subset of AIDS-related lymphomas with no evidence of body cavity involvement. In our experience, these represent approximately 5% of all AIDS-NHLs and they typically have the morphology of immunoblastic lymphoma but like PELs, they frequently lack of expression of B cell antigens and are commonly co-infected with EBV [33].
Plasmablastic lymphoma (PBL)
This is an aggressive malignancy first reported in the oral cavity of HIV-infected individuals, but can also occur in other sites, as well as in conjunction with other immunodeficient states. The vast majority of cases in the oral cavity are EBV-infected, but in other sites EBV can be found in around 75% of cases. The immunophenotype is that of plasma cells, with expression of monotypic cytoplasmic immunoglobulin in the majority, but not all cases. The stringency of the criteria used for classification of PBL has varied over time, leading some studies to use a very strict definition (including presentation in the oral cavity and presence of EBV) which has resulted in this tumor being considered extremely rare [34], to a more general definition provided by the WHO that accepts EBV-negative cases and extra-oral presentation, as long as the morphology and immunophenotype is that of B immunoblasts or plasma cells [35].
Lymphoma arising in KSHV-associated multicentric Castleman’s disease (MCD)
These lymphomas been described mainly as occurring in HIV positive patients [6], and have also been called plasmablastic lymphomas. However, they differ from the PBL category in that they are KSHV, rather than EBV, positive. These lymphomas are different from PEL in a number of ways. Lymphomas arising in KSHV-associated MCD are EBV negative, do not contain mutations in the immunoglobulin genes, and are thought to arise from naïve IgM lambda-expressing B cells rather than terminally differentiated B cells. A different entity has also been reported, called germinotropic lymphoproliferative disorder, in which germinal center B cells are co-infected with EBV and KSHV [36].
Polymorphic B-cell lymphomas
These are extremely rare lesions but morphologically resemble polymorphic post-transplantation lymphoproliferative disorders (PTLDs).
2.2 Post-transplant lymphoproliferative disorders (PTLD)
Post-transplantation lymphoproliferative disorders (PTLDs) develop in the setting of immunosuppression following solid organ transplantation (SOT) or allogeneic bone marrow transplantation (BMT). The incidence of this disease rages between 0.3 to 12.5%, based on the type of organ transplanted as well as on the type and amount of immunosuppression employed. Children have a higher incidence than adults. Patients with SOT PTLDs, have a variable clinical outcome, and some of the lesions can regress following a reduction of immunosuppression and others requiring aggressive treatment. In contrast, patients who develop PTLDs following allogeneic BMT usually have an aggressive, frequently fatal, clinical course. As with other immunodeficiency-related LPDs, the development of PTLDs in both SOT and BMT recipients is highly associated with EBV infection. Furthermore, the relative incidence of these lesions is higher in patients who are EBV negative at the time of transplantation and don’t have underlying immunity to the virus [37].
Studies of PTLDs occurring in SOT recipients (primarily heart, kidney and lung recipients) have shown that most of these lesions can be separated into three categories based on morphologic and molecular genetic criteria. These categories correlate with the biologic behavior of the lesions. Because of the special clinical setting, i.e. that of iatrogenic immunosuppression that can be modulated, a unique classification scheme was originally developed by Nalesnik, et al. [38], to describe the clinical course and outcome of SOT PTLD patients, and subsequently modified by Knowles, et al. [39] and updated by the WHO [37]. This classification divides cases into three major categories:
Plasmacytic hyperplasia (PH) and infectious mononucleosis (IM)-like PTLD
These lesions show retention of the overall architecture of the tissue. They are more common in children than adults, and the tonsils or adenoids are the most common site of presentation. Genotypically these lesions in general are polyclonal based on Ig rearrangement studies, or have small monoclonal/oligoclonal populations. The majority of cases are EBV positive. These lesions do not contain structural alterations of known oncogenes or tumor suppressor genes. Most of these regress following a reduction of immunosuppression os spontanelusly in SOT patients, but some IM-like lesions can be fatal [40].
Polymorphic PTLD
These lesions histologically show destruction of the underlying architecture and are composed of a heterogeneous (polymorphic) cell population. At the genetic level these lesions are monoclonal based both on immunoglobulin studies and the presence of clonal EBV. Polymorphic PTLDs lack structural alterations in oncogenes and tumor suppressor genes except for the presence of BCL6 gene mutations, which have been identified in approximately half of the cases studied [41]. A variable proportion of these cases in SOT patients regress with reduction of immunosuppressive therapy, but others require chemotherapy [40].
Monomorphic PTLD lesions are composed of cytologically malignant cells and should be classified according to standard lymphoma classification criteria. They are most frequently of B cell origin, and the majority fit the classification for DLBCL, but less frequently can be BL or plasma cell neoplasm, like multiple myeloma or plasmacytoma. They are monoclonal based both on Ig and EBV clonality studies. Additionally, cytogenetic abnormalities are common and they contain structural alterations in oncogenes and tumor suppressor genes frequently involved in lymphomagenesis such as P53, N-RAS and MYC [39; 42]. Approximately half of these cases have hypermethylation of O-methylguanine-DNA methyl- transferase (MGMT) [43]. MGMT is a DNA repair gene, so its lack of expression due to hypermethylation could result in tumorigenesis by generating genetic instability and acquisition of p53 and RAS point mutations. While the majority of monomorphic PTLDs are associated with EBV infection, the proportion of negative cases increases with time after organ transplantation, and most EBV-negative cases belong to this category [44].
2.3 Lymphoproliferation in other immunodeficiencies
LPDs have also been reported in the context of other iatrogenic immunodeficiencies (for example in patients with autoimmune diseases). These are most commonly of B cell origin, and can range from resemble polymorphic PTLDs, or lymphomas, most commonly DLBCLs.
Patients with primary immune disorders also have an increased risk of developing EBV-associated LPDs. Since there are many different primary immune disorders, the presentation and appearance of the lesions can vary greatly, and range from IM-like lesions to those resembling those seen in AIDS patients, including DLBCL, HL and BL [45]. A particular entity is lymphomatoid granulomatosis, an EBV-driven B cell tumor with extensive T cell infiltration, which is increased in patients with Wiskott-Aldrich syndrome [46].
2.4 EBV-associated T and NK cell malignancies
In addition to the presence of EBV in LPDs of B cell origin, there is a strong association between EBV infection and several non-B cell malignancies. More than 90% of cases of aggressive NK-cell leukemia harbour EBV. This is a rare disease with higher prevalence in Asians [47]. Extranodal NK-T cell lymphoma, nasal type, is another EBV-associated tumor. This malignancy has also been called angiocentric T-cell lymphoma and lethal midline granuloma. It is characterized by vascular damage, necrosis and a cytotoxic T cell phenotype. It most commonly presents in the upper respiratory tract but can also occur in other extranodal and nodal sites, with a higher incidence in Asians and Native Latin Americans [48]. In children, two different T cell LPDs have been found to be associated with EBV infection: 1) Systemic EBV-positive lymphoproliferative disease of childhood, characterized by proliferation of EBV-infected T cells with an activated cytotoxic phenotype; and 2) Hydroa vacciniforme-like lymphoma, an EBV-positive cutaneous T-cell lymphoma associated with sun sensitivity [49].
3. Role of EBV in lymphomagenesis
3.1 Patterns of EBV latency in B cells
EBV is generally found in its latent form in lymphomas and lymphoproliferative disorders. Although the pattern of EBV gene expression can vary significantly during latency, and probably represents a spectrum, a simplified classification establishing three patterns of latency has been used (Figure 2) [50]. In latency I, EBNA1 (necessary for maintaining the episomal EBV) is the major viral protein expressed. This latency pattern is established in infected B cells in healthy individuals. Latency III involves the unrestricted expression of all 9 latent genes including six EBV-encoded nuclear antigens (EBNA1-6), and three latent membrane proteins (LMP1, LMP2A, and LMP2B). Latency II is an intermediate pattern with expression of EBNA1 and varying and the other latent proteins, but lacking expression of one of the two critical EBV transforming proteins (EBNA 2 in type IIa and LMP1 in type IIb latencies) [51]. More recently, a different latency pattern was reported in BL in vivo; approximately 15% of BL have a mutant EBV genomes that are deleted for EBNA2 and use the Wp promoter, not usually used in BL, leading to expression of EBNA 3A, 3B, 3C proteins, in addition to EBNA1, in the absence of EBNA2 and the LMPs. This type of latency has been called Wp-restricted latency [52]. BL cells that have these deleted version of EBV have a higher resistance to apoptosis, and this was found to be due to expression of a viral BCL2 homologue, called BHRF1, which is normally a lytic gene [53]. Infection of B cell in vitro leads to their transformation to lymphoblastoid cells (LCLs), which are immortal in culture. These cells have a Latency III gene expression pattern, and have been used extensively to study the function of many EBV genes.
Figure 2. Major patterns of EBV and KSHV latent gene expression in lymphoproliferative disorders.
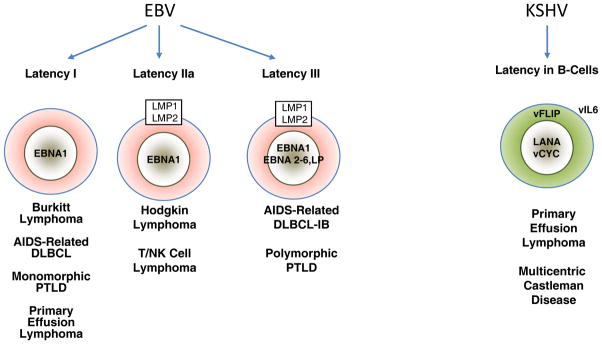
The main EBV latency patterns and the most common lymphoproliferative disorders in which these patterns are seen are illustrated. The KSHV proteins that are uniformly expressed in PEL and MDC are shown in the right.
In addition to these proteins, EBV expresses many non-coding RNAs. Among these classes of RNA are the EBER 1 and EBER2, which are small non-coding RNAs expressed ubiquitously and abundantly in EBV-infected cells, and because of these features they have become a useful diagnostic technique for EBV infection using in situ hybridization. The function of the EBERs has remained enigmatic. They appear to protect cells from apoptosis, although they are not essential for transformation or maintenance of a transformed phenotype in vitro [54; 55; 56]. In B cells, EBERs can upregulate the amount of interleukin 10 mRNA [57], so they may play a role in transcriptional or posttranscriptional regulation of this and other genes, and thereby indirectly provide tumorigenic functions effects.
MicroRNAS (miRs) are also expressed during EBV latency, and at least 22 EBV miRs have been identified [56]. Specifically which miRs are expressed varies according to cell type and experimental system. Different patterns of miR expression have been reported in BL, LCLs and nasopharyngeal carcinomas (NPC). A cluster of miRs, encoded in the BamHI H region of the genome, therefore called BHRF1 miRs, is not detected in NPC cells, but is expressed in latency III B cells, but only at low levels in cells with type I latency I (Cai et al., 2006; Xing and Kieff, 2007). While the functions of these multiple miRs are just beginning to be elucidated, some interesting functions have started to appear. For example, it has been recently demonstrated that the viral miRs of the BHRF1 locus inhibit apoptosis and favor cell cycle progression and proliferation during the early phase of infected human primary B cells [58].
3.2 EBV latency in lymphomas and immune recognition
With the exception of EBNA1, the EBNA proteins are immunogenic. Therefore, an important feature of Latency III is the recognition and elimination of the EBV infected cells by the immune system. Therefore, lymphomas with unrestricted EBV latency are mainly encountered in immunodeficient individuals. In contrast, most lymphomas in immunocompetent hosts will have Latency I or IIa, as down-regulation of the immunogenic EBNA proteins is thought to be an important mechanism of immune evasion by EBV [59]. BL is characterized by type I latency, and HL by type IIa latency. Both of these diseases can also be found in immunocompetent individuals. In fact, EBNA I, expressed in types I and II latencies, have been shown to actively suppress antigen presentation [60; 61].
3.3 Molecular mechanisms of EBV mediated transformation
Lymphoproliferative disorders with type I latency include BL, some ARL and some monomorphic PTLDs. In these, only EBNA1 and EBERs are expressed, and other driving oncogenic alterations are present. EBNA1 is clearly essential for viral genome maintenance [62], and thereby has a critical function. While its role in transformation has been long debated, EBNA1 transgenic mice have an increased incidence of lymphomas [63], and infection of B cells with EBNA1-deleted EBV leads to the development of LCLs with markedly reduced efficiency as compared to wild type EBV [64]. Two studies have shown that EBNA1 can provide growth and survival effects to BL cells in culture [65; 66]. Lymphomas with Latency I bear cellular oncogenic alterations, such as translocations involving the c-myc oncogene characteristic of BLs, as well as frequent mutation of P53 [67; 68; 69]. It is likely that EBNA1 protects from apoptosis in the context of deregulated cMYC expression. This protection from apoptosis would be further enhanced by expression of the BCL2 homolog BHRF1 in cases with Wp-restricted latency [53].
Other types of lymphoproliferative disorders with type I EBV latency are PEL and DLBCL in patients with AIDS and transplant recipients [70; 71; 72]. In these lymphomas, additional transforming events exist. In PEL, the KSHV genome produces several potentially oncogenic proteins, and in DLBCL, BCL-6 is frequently translocated and other genetic alterations are present, including cMYC and BCL2 translocations, as well as P53, N-RAS and A20 mutations.
In tumors with type II and III latency, EBNA2 and/or LMP1 proteins are expressed, both of which are essential for transformation by EBV in vitro. EBNA2 is thought to represent a constitutively active member of the Notch signaling pathway [73; 74]. The LMP1 protein is transforming and tumorigenic in vitro [75], and in vivo. Transgenic mice expressing LMP1 under the control of immunoglobulin gene regulatory elements develop B-cell lymphomas [76]. LMP1 functions as a constitutively active TNF (tumor necrosis factor)-receptor. LMP1 aggregates in the membrane as its cytoplasmic tail interacts with TNF receptor-associated factors (TRAFs) and TNFR-1-associated death domain protein (TRADD), leading to activation of nuclear factor (NF-κB) and the c-Jun amino-terminal kinase (JNK) [77; 78; 79]. In ARL, LMP2A also contributes to NF-κB signaling by controlling TRAF2 expression, which in turn is important for LMP1 signaling [80]. Both LMP1 and LMP2A are important for the survival of type III lymphoma cells: knock down of either of these proteins with RNA interference results in NF-kB downregulation and apoptosis [80].
4. Mechanisms of transformation by KSHV
The consistent presence of KSHV in PELs suggests that it is necessary for their development. However, PELs are rare tumors, even in populations with high KSHV seroprevalence, so it is evident that KSHV infection represents only one of several events involved in the development of PEL. Another co-factor appears to be EBV, because most PELs contain both viral genomes. The majority of PELs in vivo, as well as in culture, are latently infected with both viruses. As mentioned above, analysis of the pattern of EBV gene expression in PELs revealed that only EBNA1 was expressed, corresponding to Latency I [70; 71]. In addition, PELs lack structural alterations in most cellular-transforming genes frequently involved in lymphomagenesis, with the possible exception of mutation in the regulatory region of Bcl-6 [32; 81]. Our current understanding supports the concept that KSHV plays a transforming role in PELs, and that expression of a handful of genes is important for proliferation and survival of infected tumor cells. Three viral gene products are clearly expressed in all latently infected cells from a single promoter in a tricistronic transcript, which are LANA, vCYC and vFLIP. However, other viral gene products have bee shown to be expressed in selected cell types of subsets of cells in some lesions. The viral product that appear to be important in PEL and MCD pathogenesis are the following:
LANA-1
This protein is essential for episome maintenance, but also has the ability to affect several pathways that are involved in tumorigenesis: it can bind and inactivate the retinoblastoma (Rb) protein [82], and similarly bind and inactivate p53 [83]. LANA-1 can also bind and inactivate GSK3b, leading to activation of the b-catenin pathway involed in solid tumors [84], and through this pathway stabilize the cMYC protein [85]. LANA-1 has also been show to have transcriptional effects on a variety of viral and cellular genes, including IL-6, hTERT, Pim1, and the TGFbeta type II receptor [86; 87; 88; 89; 90]. Monoclonal antibodies to LANA are commercially available, and these have been very useful for immunohistochemical diagnosis of KS, PEL and MCD [91].
vCYC (v-cyclin)
This protein is a functional cyclin that can associate with CDK6 and induce phosphorylation of retinoblastoma (Rb) protein and overcome Rb-mediated cell-cycle arrest [92; 93]. vCYC differs from the cellular cyclin D in its ability to induce degradation of the CDK inhibitor p27Kip when complexed with CDK6 [94; 95]. Therefore, vCYC is likely to modulate cell cycle in PEL cells, avoiding normal regulatory checkpoints. Transgenic mice with vCYC develop lymphomas, but only in the absence of p53, and a model has been proposed whereby vCYC induces genome instability and that loss of p53 subsequently allows expansion of tumorigenic clones [96; 97]. Genomic instability may be an inevitable consequence of latent KSHV infection, as a result of vCyc–CDK6-mediated phosphorylation of nucleophosmin (NPM1) [98; 99].
vFLIP (K13)
This protein is homologous to cellular FLICE/caspase 8 inhibitory protein (cFLIP) [100], which can inhibit apoptosis induced by death receptors. vFLIP can activate NF-κB though both the classical and alternative pathways by binding to IKKγ and TRAF2 [101; 102; 103; 104]. Through this mechanism, vFLIP induces expression of anti-apoptotic genes and protect cells from Fas-induced cell death in vitro and in vivo [105; 106]. vFLIP protects PEL cells from spontaneous apoptosis is essential for tumor cell survival in vitro [106; 107]. It also protects B cells from autophagy by binding to Atg3 [108]. vFLIP transgenic mice develop B cell malignancies when expressed in B cells, and accelerates the development of cMYC induced lymphomas [109; 110; 111]. In addition to its anti-apoptotic function, NF-κB prevents lytic reactivation [112], and therefore vFLIP may play a role in maintaining viral latency [113].
LANA-2 (vIRF-3)
This protein is expressed in latency in PEL, although not in Kaposi’s sarcoma. It was shown to potently inhibit p53 in reporter assays [114], thereby inactivating this tumor-suppressor pathway. While LANA-2 is not a DNA-binding protein, it is recruited to the interferon promoters via its interaction with cellular IRF-3 and IRF-7, and stimulates their transcriptional activity [115].
vIL-6
This viral protein is a homolog of cellular IL-6, but differs from the latter in that it is selectively glycosylated and can bind the gp130 receptor in the absence of the high affinity IL-6 receptor to activate IL-6-responsive genes and promote B-cell survival [116; 117; 118]. vIL-6 is considered to be a lytic gene because its expression is increased upon lytic reactivation. However, it is expressed by a variable but significant proportion of latently-infected PEL cells as well as in MCD. Considering the fact that this protein can be secreted, it may affect other tumor and reactive cells that don’t express it, and play a role in their proliferation and clinical manifestations.
K1
This protein has an ITAM motif that can activate cytoplasmic tyrosine kinases and mimic signaling by the B-cell antigen receptor [119]. K15 contains a variable number of transmenbrane regions and cytoplasmic SH2 and SH3 domains. K1 can activate the MAP kinase and NF-κB signaling pathways and have potential antiapoptotic functions [120; 121]. K15 has also been shown to activate the Akt and Src kinases [122; 123]. Therefore, it appears to have very broad signaling capabilities. While this protein is clearly upregulated during lytic replication, it was also reported to be expressed at low levels during latency [124]. K1 has been shown to be tumorigenic in a model where it was found to induce lymphomas in common marmosets when replaced for the saimiri transforming protein (STP) [125], as well as tumors in transgenic mice [126].
Kaposins
The kaposin region is complex; it contains a small coding region (K12) preceded by two families of two direct repeats (DR1 and DR2), and is transcribed potentially encoding three proteins, called Kaposin A, B, and C [127]. Kaposin B, encompassing DR1 and DR2, appears to be expressed in KS and some PEL cells [127], but not others [128]. This protein has potentially transforming functions, including activation of the p38 pathway and stabilization of cytokine mRNAs: kaposin B-mediated activation of MK2 blocks the decay of mRNAs with AU-rich elements (AREs) in their 3′ untranslated regions, and because several cytokine mRNAs have ARE elements, kaposin B expression results in an increase in the production of pro-inflammatory cytokines [129]. Kaposin A also has been reported to be transforming [130], and can signal by recruiting cytohesin 1 to the plasma membrane [131]. Further complexity in this locus arises from RNA editing that can render the transcript encoding Kaposins A and B non-transforming [132].
K15
This open reading frame encodes a protein with complex splicing and multiple transmembrane motifs, structurally resembling EBV LMP-2A. It has SH2 and SH3 motifs, and also a TRAF binding motif, and can also activate signaling cascades that include NF-κB and MAP kinase [120]. Also like EBV LMP-2A, K15 can inhibit B cell receptor signaling [133]. K15 protein has been found to be present in most latently infected cells by one study [121], but the RNA expression data is controversial, most studies arguing for lytic expression. K15 has been shown to have anti-apoptotic functions [121].
KSHV microRNAs
At least 12 miRNAs are encoded by KSHV, which cluster in the K12/Kaposin genomic locus and are expressed in latently infected cells [134; 135; 136; 137]. The function of these miRs is rapidly being elucidated, and recent studies have revealed interesting functions that are very relevant to KSHV pathogenesis. For example, miR-K1, targets IκBα, an inhibitor of NF-κB. Therefore, it may strengthen the NF-κB activity induced by vFLIP, thereby promoting cellular survival [138]. miR-K1 also targets cellular mRNAs encoding the cellular cyclin-dependent kinase inhibitor p21, thereby preventing cell cycle arrest [139]. The KSHV-encoded miR-K12-11 is an ortholog of cellular miR-155, which plays an important role in B cell transformation and differentiation [140; 141].
5. Conclusions and future perspectives
EBV and KSHV are both lymphotropic herpesviruses, and as such, they are involved in they are capable of infecting lymphocytes, and causing lymphoproliferative disorders. These are more frequently of B cell origin reflecting the natural viral tropism. Which evolution has led to a balance where many of us are infected with these viruses without even knowing it, problems arise when this balance is broken. The mechanisms that EBV and KSHV have acquired to ensure the survival of the cell they call home, can also lead to the uncontrolled proliferation of this cells, leading to the development of lymphoproliferations that can range from controllable disease to a highly aggressive malignancy. Understanding the mechanisms involved in this fine balance, and developing the tool to manipulate it, have already resulted in improved treatment of patients with EBV-associated malignancies. For example, adoptive immunotherapy is already in use for the treatment of patients with PTLD, where EBV-specific cytotoxic T cells are infused [142]. Understanding the mechanisms whereby viral gene products affect tumor cell survival and proliferation are shedding light about important cellular proteins and pathways that could be targeted, such as NF-kB, mTOR, notch and NF-kB, potentially leading to improved treatment. In addition, this understanding provides a unique opportunity to develop new therapies that directly target the virus. High throughput screening methodologies have begun to lead to the discovery of inhibitors of potential viral oncoproteins, such as for EBNA1 [143]. The approach of specifically targeting EBV or KSHV, hopefully eliminating all cellular toxicities, is likely to be the therapy of choice in the future.
Acknowledgments
EC is funded by NIH grant R01-CA068939, 3UO1CA12194, RC2CA148317, UL1 RRO24996, and the Starr Cancer Consortium.
Footnotes
Conflict of interest
EC has no conflicts to declare.
References
- 1.Ballon G, Cesarman E. Castleman’s Disease. In: Volberding PA, Palefsky J, editors. Viral and Immunolgogical Malignancies. BC Decker Inc; Hamilton, Ontario, Canada: 2006. [Google Scholar]
- 2.Waterston A, Bower M. Fifty years of multicentric Castleman’s disease. Acta Oncol. 2004;43:698–704. doi: 10.1080/02841860410002752. [DOI] [PubMed] [Google Scholar]
- 3.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d’Agay M-F, Clauvel J-P, Raphael M, Degos L, Sigaux F. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86:1275–1280. [PubMed] [Google Scholar]
- 4.Larroche C, Cacoub P, Soulier J, Oksenhendler E, Clauvel JP, Piette JC, Raphael M. Castleman’s disease and lymphoma: report of eight cases in HIV-negative patients and literature review. Am J Hematol. 2002;69:119–26. doi: 10.1002/ajh.10022. [DOI] [PubMed] [Google Scholar]
- 5.Suda T, Katano H, Delsol G, Kakiuchi C, Nakamura T, Shiota M, Sata T, Higashihara M, Mori S. HHV-8 infection status of AIDS-unrelated and AIDS-associated multicentric Castleman’s disease. Pathol Int. 2001;51:671–9. doi: 10.1046/j.1440-1827.2001.01266.x. [DOI] [PubMed] [Google Scholar]
- 6.Dupin N, Diss TL, Kellam P, Tulliez M, Du MQ, Sicard D, Weiss RA, Isaacson PG, Boshoff C. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood. 2000;95:1406–12. [PubMed] [Google Scholar]
- 7.Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc Natl Acad Sci U S A. 1999;96:4546–51. doi: 10.1073/pnas.96.8.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du MQ, Liu H, Diss TC, Ye H, Hamoudi RA, Dupin N, Meignin V, Oksenhendler E, Boshoff C, Isaacson PG. Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in Castleman disease and associated lymphoproliferative disorders. Blood. 2001;97:2130–6. doi: 10.1182/blood.v97.7.2130. [DOI] [PubMed] [Google Scholar]
- 9.Brousset P, Cesarman E, Meggetto F, Lamant L, Delsol G. Colocalization of the viral interleukin-6 with latent nuclear antigen-1 of human herpesvirus-8 in endothelial spindle cells of Kaposi’s sarcoma and lymphoid cells of multicentric Castleman’s disease. Hum Pathol. 2001;32:95–100. doi: 10.1053/hupa.2001.21131. [DOI] [PubMed] [Google Scholar]
- 10.Parravicini C, Corbellino M, Paulli M, Magrini U, Lazzarino M, Moore PS, Chang Y. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman’s disease. Am J Pathol. 1997;151:1517–1522. [PMC free article] [PubMed] [Google Scholar]
- 11.Cannon JS, Nicholas J, Orenstein JM, Mann RB, Murray PG, Browning PJ, DiGiuseppe JA, Cesarman E, Hayward GS, Ambinder RF. Heterogeneity of viral IL-6 expression in HHV-8-associated diseases. J Inf Dis. 1999;180:824–828. doi: 10.1086/314956. [DOI] [PubMed] [Google Scholar]
- 12.Staskus KA, Sun R, Miller G, Racz P, Jaslowski A, Metroka C, Brett-Smith H, Haase AT. Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. J Virol. 1999;73:4181–7. doi: 10.1128/jvi.73.5.4181-4187.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menke DM, Chadbum A, Cesarman E, Green E, Berenson J, Said J, Tiemann M, Parwaresch R, Thome SD. Analysis of the human herpesvirus 8 (HHV-8) genome and HHV-8 vIL-6 expression in archival cases of castleman disease at low risk for HIV infection. Am J Clin Pathol. 2002;117:268–75. doi: 10.1309/7243-AV50-KJ28-V6J9. [DOI] [PubMed] [Google Scholar]
- 14.Katano H, Sato Y, Kurata T, Mori S, Sata T. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease. Virology. 2000;269:335–44. doi: 10.1006/viro.2000.0196. [DOI] [PubMed] [Google Scholar]
- 15.Uldrick TS, Wang V, O’Mahony D, Aleman K, Wyvill KM, Marshall V, Steinberg SM, Pittaluga S, Maric I, Whitby D, Tosato G, Little RF, Yarchoan R. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin Infect Dis. 2010;51:350–8. doi: 10.1086/654798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simard EP, Pfeiffer RM, Engels EA. Spectrum of cancer risk late after AIDS onset in the United States. Arch Intern Med. 2010;170:1337–45. doi: 10.1001/archinternmed.2010.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simard EP, Engels EA. Cancer as a cause of death among people with AIDS in the United States. Clin Infect Dis. 2010;51:957–62. doi: 10.1086/656416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carbone A. Emerging pathways in the development of AIDS-related lymphomas. Lancet Oncol. 2003;4:22–9. doi: 10.1016/s1470-2045(03)00957-4. [DOI] [PubMed] [Google Scholar]
- 19.Gaidano G, Carbone A, Dalla-Favera R. Genetic basis of acquired immunodeficiency syndrome-related lymphomagenesis. J Natl Cancer Inst Monogr. 1998:95–100. doi: 10.1093/oxfordjournals.jncimonographs.a024181. [DOI] [PubMed] [Google Scholar]
- 20.Knowles DM. Neoplastic Hematopathology. Williams and Wilkins; Baltimore: 2001. [Google Scholar]
- 21.Raphael M, Said J, Borisch B, Cesarman E, Harris NL. Lymphomas associated with HIV infection. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press; Lyon, France: 2008. pp. 340–342. [Google Scholar]
- 22.Cesarman E, Dalla-Favera R, Bentley D, Groudine M. Mutations in the first exon are associated with altered transcription of c-myc in Burkitt lymphoma. Science. 1987;238:1272–1275. doi: 10.1126/science.3685977. [DOI] [PubMed] [Google Scholar]
- 23.Chadburn A, Chiu A, Lee JY, Chen X, Hyjek E, Banham AH, Noy A, Kaplan LD, Sparano JA, Bhatia K, Cesarman E. Immunophenotypic analysis of AIDS-related diffuse large B-cell lymphoma and clinical implications in patients from AIDS Malignancies Consortium clinical trials 010 and 034. J Clin Oncol. 2009;27:5039–48. doi: 10.1200/JCO.2008.20.5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunleavy K, Wilson WH. Role of molecular subtype in predicting outcome of AIDS-related diffuse large B-cell lymphoma. J Clin Oncol. 2010;28:e260. doi: 10.1200/JCO.2009.27.7087. [DOI] [PubMed] [Google Scholar]
- 25.Chadburn A, Noy A, Lee JY, Hyjek E, Banham AH, Sparano JA, Bhatia K, Cesarman E. Reply to K. Dunleavy et al. J Clin Oncol. 2010;28:e261–e262. [Google Scholar]
- 26.Kluin PM, Harris NL, Stein H, Leoncini L, Raphael M, Campo E, Jaffe ES. B-cell lymphoma, unclassifiable, with features intermediate betwwwn diffuse large B-cell lymphoma and Burkitt lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press; Lyon, France: 2008. pp. 265–266. [Google Scholar]
- 27.Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ, Greiner TC, Weisenburger DD, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Delabie J, Rimsza LM, Braziel RM, Grogan TM, Campo E, Jaffe ES, Dave BJ, Sanger W, Bast M, Vose JM, Armitage JO, Connors JM, Smeland EB, Kvaloy S, Holte H, Fisher RI, Miller TP, Montserrat E, Wilson WH, Bahl M, Zhao H, Yang L, Powell J, Simon R, Chan WC, Staudt LM. Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med. 2006;354:2431–42. doi: 10.1056/NEJMoa055759. [DOI] [PubMed] [Google Scholar]
- 28.Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TF, Bernd HW, Cogliatti SB, Dierlamm J, Feller AC, Hansmann ML, Haralambieva E, Harder L, Hasenclever D, Kuhn M, Lenze D, Lichter P, Martin-Subero JI, Moller P, Muller-Hermelink HK, Ott G, Parwaresch RM, Pott C, Rosenwald A, Rosolowski M, Schwaenen C, Sturzenhofecker B, Szczepanowski M, Trautmann H, Wacker HH, Spang R, Loeffler M, Trumper L, Stein H, Siebert R. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354:2419–30. doi: 10.1056/NEJMoa055351. [DOI] [PubMed] [Google Scholar]
- 29.Glaser SL, Clarke CA, Gulley ML, Craig FE, DiGiuseppe JA, Dorfman RF, Mann RB, Ambinder RF. Population-based patterns of human immunodeficiency virus-related Hodgkin lymphoma in the Greater San Francisco Bay Area, 1988–1998. Cancer. 2003;98:300–9. doi: 10.1002/cncr.11459. [DOI] [PubMed] [Google Scholar]
- 30.Carbone A, Gloghini A, Serraino D, Spina M. HIV-associated Hodgkin lymphoma. Curr Opin HIV AIDS. 2009;4:3–10. doi: 10.1097/COH.0b013e32831a722b. [DOI] [PubMed] [Google Scholar]
- 31.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s Sarcoma-associated Herpesvirus-like DNA sequences in AIDS-related body cavity-based lymphomas. N Eng J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 32.Nador RG, Cesarman E, Chadburn A, Dawson DB, Ansari MQ, Said J, Knowles DM. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpesvirus. Blood. 1996;88:645–656. [PubMed] [Google Scholar]
- 33.Chadburn A, Hyjek E, Mathew S, Cesarman E, Said J, Knowles DM. KSHV-positive solid lymphomas represent an extra-cavitary variant of primary effusion lymphoma. Am J Surg Pathol. 2004;28:1401–16. doi: 10.1097/01.pas.0000138177.10829.5c. [DOI] [PubMed] [Google Scholar]
- 34.Delecluse HJ, Anagnostopoulos I, Dallenbach F, Hummel M, Marafioti T, Schneider U, Huhn D, Schmidt-Westhausen A, Reichart PA, Gross U, Stein H. Plasmablastic lymphomas of the oral cavity: a new entity associated with the human immunodeficiency virus infection. Blood. 1997;89:1413–20. [PubMed] [Google Scholar]
- 35.Stein H, Harris NL, Campo E. Plasmablastic lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press; Lyon, France: 2008. pp. 256–257. [Google Scholar]
- 36.Du MQ, Diss TC, Liu H, Ye H, Hamoudi RA, Cabecadas J, Dong HY, Harris NL, Chan JK, Rees JW, Dogan A, Isaacson PG. KSHV- and EBV-associated germinotropic lymphoproliferative disorder. Blood. 2002;100:3415–8. doi: 10.1182/blood-2002-02-0487. [DOI] [PubMed] [Google Scholar]
- 37.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Webber SA, Chadburn A, Ferry JA. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press; Lyon, France: 2008. Post-transplant lymphoproliferative disorders. [Google Scholar]
- 38.Nalesnik MA, Jaffe R, Starzl TE, Demetris AJ, Porter K, Burnham JA, Makowka L, Ho M, Locker J. The pathology of posttransplant lymphoproliferative disorders occurring in the setting of cyclosporine A-prednisone immunosuppression. Am J Pathol. 1988;133:173–192. [PMC free article] [PubMed] [Google Scholar]
- 39.Knowles DM, Cesarman E, Chadburn A, Frizzera G, Chen J, Rose EA, Michler RE. Correlative morphologic and molecular genetic analysis demonstrates three distinct categories of posttransplantation lymphoproliferative disorders. Blood. 1995;85:552–565. [PubMed] [Google Scholar]
- 40.Chadburn A, Cesarman E, Knowles DM. Classification of post-transplantation lymphoproliferative disorders (PT-LPDs) based on morphologic and molecular genetic features has clinical relevance. Lab Invest. 1997;76:121A. [Google Scholar]
- 41.Cesarman E, Chadburn A, Liu YF, Migliazza A, Dalla-Favera R, Knowles DM. BCL-6 gene mutations in posttransplantation lymphoproliferative disorders predict response to therapy and clinical outcome. Blood. 1998;92:2294–302. [PubMed] [Google Scholar]
- 42.Vakiani E, Nandula SV, Subramaniyam S, Keller CE, Alobeid B, Murty VV, Bhagat G. Cytogenetic analysis of B-cell posttransplant lymphoproliferations validates the World Health Organization classification and suggests inclusion of florid follicular hyperplasia as a precursor lesion. Hum Pathol. 2007;38:315–25. doi: 10.1016/j.humpath.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 43.Rossi D, Gaidano G, Gloghini A, Deambrogi C, Franceschetti S, Berra E, Cerri M, Vendramin C, Conconi A, Viglio A, Muti G, Oreste P, Morra E, Paulli M, Capello D, Carbone A. Frequent aberrant promoter hypermethylation of O6-methylguanine-DNA methyltransferase and death-associated protein kinase genes in immunodeficiency-related lymphomas. Br J Haematol. 2003;123:475–8. doi: 10.1046/j.1365-2141.2003.04644.x. [DOI] [PubMed] [Google Scholar]
- 44.Leblond V, Davi F, Charlotte F, Dorent R, Bitker MO, Sutton L, Gandjbakhch I, Binet JL, Raphael M. Posttransplant lymphoproliferative disorders not associated with Epstein-Barr virus: a distinct entity? J Clin Oncol. 1998;16:2052–9. doi: 10.1200/JCO.1998.16.6.2052. [DOI] [PubMed] [Google Scholar]
- 45.Van Krieken JH, Onciu M, Elenitoba-Johnson KSJ, Jaffe ES. Lymphoproliferative diseases associete with primary immune disorders. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press; Lyon, France: pp. 336–339. [Google Scholar]
- 46.Ilowite NT, Fligner CL, Ochs HD, Brichacek B, Harada S, Haas JE, Purtilo DT, Wedgwood RJ. Pulmonary angiitis with atypical lymphoreticular infiltrates in Wiskott-Aldrich syndrome: possible relationship of lymphomatoid granulomatosis and EBV infection. Clin Immunol Immunopathol. 1986;41:479–84. doi: 10.1016/0090-1229(86)90018-8. [DOI] [PubMed] [Google Scholar]
- 47.Akashi K, Mizuno S. Epstein-Barr virus-infected natural killer cell leukemia. Leuk Lymphoma. 2000;40:57–66. doi: 10.3109/10428190009054881. [DOI] [PubMed] [Google Scholar]
- 48.Aozasa K, Takakuwa T, Hongyo T, Yang WI. Nasal NK/T-cell lymphoma: epidemiology and pathogenesis. Int J Hematol. 2008;87:110–7. doi: 10.1007/s12185-008-0021-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quintanilla-Martinez L, Kimura H, Jaffe ES. EBV-positive T-cell lymphoproliferative disorders of childhood. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press; Lyon, France: 2008. pp. 278–280. [Google Scholar]
- 50.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004;4:757–68. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 51.Klein E, Kis LL, Klein G. Epstein-Barr virus infection in humans: from harmless to life endangering virus-lymphocyte interactions. Oncogene. 2007;26:1297–305. doi: 10.1038/sj.onc.1210240. [DOI] [PubMed] [Google Scholar]
- 52.Kelly G, Bell A, Rickinson A. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat Med. 2002;8:1098–104. doi: 10.1038/nm758. [DOI] [PubMed] [Google Scholar]
- 53.Kelly GL, Long HM, Stylianou J, Thomas WA, Leese A, Bell AI, Bornkamm GW, Mautner J, Rickinson AB, Rowe M. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog. 2009;5:e1000341. doi: 10.1371/journal.ppat.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Komano J, Maruo S, Kurozumi K, Oda T, Takada K. Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt’s lymphoma cell line Akata. J Virol. 1999;73:9827–31. doi: 10.1128/jvi.73.12.9827-9831.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ruf IK, Rhyne PW, Yang C, Cleveland JL, Sample JT. Epstein-Barr virus small RNAs potentiate tumorigenicity of Burkitt lymphoma cells independently of an effect on apoptosis. J Virol. 2000;74:10223–8. doi: 10.1128/jvi.74.21.10223-10228.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swaminathan S. Noncoding RNAs produced by oncogenic human herpesviruses. J Cell Physiol. 2008;216:321–6. doi: 10.1002/jcp.21480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kitagawa N, Goto M, Kurozumi K, Maruo S, Fukayama M, Naoe T, Yasukawa M, Hino K, Suzuki T, Todo S, Takada K. Epstein-Barr virus-encoded poly(A)(-) RNA supports Burkitt’s lymphoma growth through interleukin-10 induction. Embo J. 2000;19:6742–50. doi: 10.1093/emboj/19.24.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seto E, Moosmann A, Gromminger S, Walz N, Grundhoff A, Hammerschmidt W. Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. 2004;350:1328–37. doi: 10.1056/NEJMra032015. [DOI] [PubMed] [Google Scholar]
- 60.Yin XM, Oltvai ZN, Veis-Novack DJ, Linette GP, Korsmeyer SJ. Bcl-2 gene family and the regulation of programmed cell death. Cold Spring Harbor Symp Quant Biol. 1994;59:387–394. doi: 10.1101/sqb.1994.059.01.043. [DOI] [PubMed] [Google Scholar]
- 61.Levitskaya J, Sharipo A, Leonchiks A, Ciechanover A, Masucci MG. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc Natl Acad Sci U S A. 1997;94:12616–21. doi: 10.1073/pnas.94.23.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kieff E, Rickinson A. Epstein-Barr Virus and its replication. In: Fields B, Knipe D, HPM, editors. Fields Virology. Wolters Kluwer Health/Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2603–2655. [Google Scholar]
- 63.Wilson JB, Bell JL, Levine AJ. Expression of Epstein-Barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice. Embo J. 1996;15:3117–26. [PMC free article] [PubMed] [Google Scholar]
- 64.Humme S, Reisbach G, Feederle R, Delecluse HJ, Bousset K, Hammerschmidt W, Schepers A. The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc Natl Acad Sci U S A. 2003;100:10989–94. doi: 10.1073/pnas.1832776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kennedy G, Komano J, Sugden B. Epstein-Barr virus provides a survival factor to Burkitt’s lymphomas. Proc Natl Acad Sci U S A. 2003;100:14269–74. doi: 10.1073/pnas.2336099100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yin Q, Flemington EK. siRNAs against the Epstein Barr virus latency replication factor, EBNA1, inhibit its function and growth of EBV-dependent tumor cells. Virology. 2006;346:385–93. doi: 10.1016/j.virol.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 67.Dalla-Favera R, Bregni M, Erickson M, Patterson D, Gallo RC, Croce CM. Human c-myc oncogene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 1982;79:7824–7827. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gaidano G, Ballerini P, Gong JZ, Inghirami G, Neri A, Newcomb EW, Magrath IT, Knowles DM, Dalla-Favera R. p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 1991;88:5423–5417. doi: 10.1073/pnas.88.12.5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nakamura H, Said JW, Miller CW, Koeffler HP. Mutation and protein expression of p53 in acquired immunodeficiency syndrome-related lymphomas. Blood. 1993;82:920–926. [PubMed] [Google Scholar]
- 70.Horenstein MG, Nador RG, Chadburn A, Hyjek EM, Inghirami G, Knowles DM, Cesarman E. Epstein-Barr virus latent gene expression in primary effusion lymphomas containing Kaposi’s sarcoma-associated herpesvirus human herpesvirus-8. Blood. 1997;90:1186–1191. [PubMed] [Google Scholar]
- 71.Szekely L, Chen F, Teramoto N, Ehlin-Henriksson B, Pokrovskaja K, Szeles A, Manneborg-Sandlund A, Lowbeer M, Lennette ET, Klein G. Restricted expression of Epstein-Barr virus (EBV)-encoded, growth transformation-associated antigens in an EBV- and human herpesvirus type 8-carrying body cavity lymphoma line. Gen Virol. 1998;79:1445–1452. doi: 10.1099/0022-1317-79-6-1445. [DOI] [PubMed] [Google Scholar]
- 72.Rea D, Delecluse HJ, Hamilton-Dutoit SJ, Marelle L, Joab I, Edelman L, Finet JF, Raphael M. Epstein-Barr virus latent and replicative gene expression in post-transplant lymphoproliferative disorders and AIDS-related non-Hodgkin’s lymphomas. French Study Group of Pathology for HIV-associated Tumors. Ann Oncol. 1994;5(Suppl 1):113–6. doi: 10.1093/annonc/5.suppl_1.s113. [DOI] [PubMed] [Google Scholar]
- 73.Grossman SR, Johannsen E, Tong X, Yalamanchili R, Kieff E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc Natl Acad Sci U S A. 1994;91:7568–72. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hsieh JJ, Hayward SD. Masking of the CBF1/RBPJ kappa transcriptional repression domain by Epstein-Barr virus EBNA2. Science. 1995;268:560–3. doi: 10.1126/science.7725102. [DOI] [PubMed] [Google Scholar]
- 75.Wang D, Liebowitz D, Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43:831–840. doi: 10.1016/0092-8674(85)90256-9. [DOI] [PubMed] [Google Scholar]
- 76.Kulwichit W, Edwards RH, Davenport EM, Baskar JF, Godfrey V, Raab-Traub N. Expression of the Epstein-Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice. Proc Natl Acad Sci U S A. 1998;95:11963–8. doi: 10.1073/pnas.95.20.11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kieser A, Kilger E, Gires O, Ueffing M, Kolch W, Hammerschmidt W. Epstein-barr-virus latent membrane protein-1 triggers ap-1 activity via the c-jun n-terminal kinase cascade. Embo Journal. 1997;16:6478–6485. doi: 10.1093/emboj/16.21.6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kilger E, Kieser A, Baumann M, Hammerschmidt W. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J. 1998;17:1700–1709. doi: 10.1093/emboj/17.6.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eliopoulos AG, Blake SM, Floettmann JE, Rowe M, Young LS. Epstein-Barr virus-encoded latent membrane protein 1 activates the JNK pathway through its extreme C terminus via a mechanism involving TRADD and TRAF2. J Virol. 1999;73:1023–35. doi: 10.1128/jvi.73.2.1023-1035.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guasparri I, Bubman D, Cesarman E. EBV LMP2A affects LMP1-mediated NF-kappaB signaling and survival of lymphoma cells by regulating TRAF2 expression. Blood. 2008;111:3813–20. doi: 10.1182/blood-2007-03-080309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carbone A, Gaidano G, Gloghini A, Larocca LM, Capello D, Canzonieri V, Antinori A, Tirelli U, Falini B, Dalla-Favera R. Differential expression of BCL-6, CD138/syndecan-1, and Epstein-Barr virus-encoded latent membrane protein-1 identifies distinct histogenetic subsets of acquired immunodeficiency syndrome-related non-Hodgkin’s lymphomas. Blood. 1998;91:747–755. [PubMed] [Google Scholar]
- 82.Radkov SA, Kellam P, Boshoff C. The latent nuclear antigen of kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene hras transforms primary rat cells. Nat Med. 2000;6:1121–7. doi: 10.1038/80459. [DOI] [PubMed] [Google Scholar]
- 83.Friborg J, Jr, Kong W, Hottiger MO, Nabel GJ. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature. 1999;402:889–94. doi: 10.1038/47266. [DOI] [PubMed] [Google Scholar]
- 84.Fujimuro M, Hayward SD. Manipulation of glycogen-synthase kinase-3 activity in KSHV-associated cancers. J Mol Med. 2004 doi: 10.1007/s00109-003-0519-7. [DOI] [PubMed] [Google Scholar]
- 85.Bubman D, Guasparri I, Cesarman E. Deregulation of c-Myc in primary effusion lymphoma by Kaposi’s sarcoma herpesvirus latency-associated nuclear antigen. Oncogene. 2007;26:4979–86. doi: 10.1038/sj.onc.1210299. [DOI] [PubMed] [Google Scholar]
- 86.Renne R, Barry C, Dittmer D, Compitello N, Brown PO, Ganem D. Modulation of cellular and viral gene expression by the latency- associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2001;75:458–68. doi: 10.1128/JVI.75.1.458-468.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bajaj BG, Verma SC, Lan K, Cotter MA, Woodman ZL, Robertson ES. KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology. 2006;351:18–28. doi: 10.1016/j.virol.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 88.Verma SC, Borah S, Robertson ES. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J Virol. 2004;78:10348–59. doi: 10.1128/JVI.78.19.10348-10359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.An J, Lichtenstein AK, Brent G, Rettig MB. The Kaposi sarcoma-associated herpesvirus (KSHV) induces cellular interleukin 6 expression: role of the KSHV latency-associated nuclear antigen and the AP1 response element. Blood. 2002;99:649–54. doi: 10.1182/blood.v99.2.649. [DOI] [PubMed] [Google Scholar]
- 90.Di Bartolo DL, Cannon M, Liu YF, Renne R, Chadburn A, Boshoff C, Cesarman E. KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood. 2008;111:4731–40. doi: 10.1182/blood-2007-09-110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rainbow L, Platt GM, Simpson GR, Sarid R, Gao SJ, Stoiber H, Herrington CS, Moore PS, Schulz TF. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J Virol. 1997;71:5915–5921. doi: 10.1128/jvi.71.8.5915-5921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Godden-Kent D, Talbot SJ, Boshoff C, Chang Y, Moore P, Weiss RA, Mittnacht S. The cyclin encoded by Kaposi’s sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J Virol. 1997;71:4193–4198. doi: 10.1128/jvi.71.6.4193-4198.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li M, Lee H, Yoon DW, Albrecht JC, Fleckenstein B, Jung J. Kaposi’s sarcoma-associated herpesvirus encodes a functional cyclin. J Virol. 1997;71:1984–1991. doi: 10.1128/jvi.71.3.1984-1991.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ellis M, Chew YP, Fallis L, Freddersdorf S, Boshoff C, Weiss RA, Lu X, Mittnacht S. Degradation of p27(Kip) cdk inhibitor triggered by Kaposi’s sarcoma virus cyclin-cdk6 complex [In Process Citation] Embo J. 1999;18:644–53. doi: 10.1093/emboj/18.3.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mann DJ, Child ES, Swanton C, Laman H, Jones N. Modulation of p27Kip1 levels by the cyclin encoded by Kaposi’s sarcoma-associated herpesvirus. EMBO J. 1999;18:654–663. doi: 10.1093/emboj/18.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Verschuren EW, Klefstrom J, Evan GI, Jones N. The oncogenic potential of Kaposi’s sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell. 2002;2:229–41. doi: 10.1016/s1535-6108(02)00123-x. [DOI] [PubMed] [Google Scholar]
- 97.Verschuren EW, Hodgson JG, Gray JW, Kogan S, Jones N, Evan GI. The role of p53 in suppression of KSHV cyclin-induced lymphomagenesis. Cancer Res. 2004;64:581–9. doi: 10.1158/0008-5472.can-03-1863. [DOI] [PubMed] [Google Scholar]
- 98.Cuomo ME, Knebel A, Morrice N, Paterson H, Cohen P, Mittnacht S. p53-Driven apoptosis limits centrosome amplification and genomic instability downstream of NPM1 phosphorylation. Nat Cell Biol. 2008;10:723–30. doi: 10.1038/ncb1735. [DOI] [PubMed] [Google Scholar]
- 99.Sarek G, Jarviluoma A, Moore HM, Tojkander S, Vartia S, Biberfeld P, Laiho M, Ojala PM. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathog. 2010;6:e1000818. doi: 10.1371/journal.ppat.1000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 101.Chaudhary PM, Jasmin A, Eby MT, Hood L. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene. 1999;14:5738–5746. doi: 10.1038/sj.onc.1202976. [DOI] [PubMed] [Google Scholar]
- 102.Matta H, Chaudhary PM. Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP) Proc Natl Acad Sci U S A. 2004;101:9399–404. doi: 10.1073/pnas.0308016101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guasparri I, Wu H, Cesarman E. The KSHV oncoprotein vFLIP contains a TRAF-interacting motif and requires TRAF2 and TRAF3 for signalling. EMBO Rep. 2006;7:114–9. doi: 10.1038/sj.embor.7400580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Field N, Low W, Daniels M, Howell S, Daviet L, Boshoff C, Collins M. KSHV vFLIP binds to IKK-gamma to activate IKK. J Cell Sci. 2003;116:3721–3728. doi: 10.1242/jcs.00691. [DOI] [PubMed] [Google Scholar]
- 105.Djerbi M, Screpanti V, Catrina AI, Bogen B, Biberfeld P, Grandien A. The inhibitor of death receptor signaling, FLICE-inhibitory protein defines a new class of tumor progression factors [see comments] J Exp Med. 1999;190:1025–32. doi: 10.1084/jem.190.7.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guasparri I, Keller SA, Cesarman E. KSHV vFLIP Is Essential for the Survival of Infected Lymphoma Cells. J Exp Med. 2004;199:993–1003. doi: 10.1084/jem.20031467. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 107.Godfrey A, Anderson J, Papanastasiou A, Takeuchi Y, Boshoff C. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood. 2005;105:2510–8. doi: 10.1182/blood-2004-08-3052. [DOI] [PubMed] [Google Scholar]
- 108.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11:1355–62. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chugh P, Matta H, Schamus S, Zachariah S, Kumar A, Richardson JA, Smith AL, Chaudhary PM. Constitutive NF-{kappa}B activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc Natl Acad Sci U S A. 2005 doi: 10.1073/pnas.0408577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ballon G, Chen K, Perez R, Tam W, Cesarman E. Kaposi Sarcoma Herpesvirus (KSHV) vFLIP Induces B-cell Reprogramming and Tumorigenesis in vivo Mimicking Human Pathogenesis. (Submitted) [Google Scholar]
- 111.Ahmad A, Groshong JS, Matta H, Schamus S, Punj V, Robinson LJ, Gill PS, Chaudhary PM. Kaposi’s sarcoma associated herpesvirus-encoded viral FLICE inhibitory protein (vFLIP) K13 cooperates with Myc to promote lymphoma in mice. Cancer Biol Ther. 2010;10 doi: 10.4161/cbt.10.10.13291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. NF-kappaB inhibits gammaherpesvirus lytic replication. J Virol. 2003;77:8532–40. doi: 10.1128/JVI.77.15.8532-8540.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.de Oliveira DE, Ballon G, Cesarman E. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol. 2010;18:248–57. doi: 10.1016/j.tim.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 114.Rivas C, Thlick AE, Parravicini C, Moore PS, Chang Y. Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J Virol. 2001;75:429–38. doi: 10.1128/JVI.75.1.429-438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lubyova B, Kellum MJ, Frisancho AJ, Pitha PM. Kaposi’s sarcoma-associated herpesvirus-encoded vIRF-3 stimulates the transcriptional activity of cellular IRF-3 and IRF-7. J Biol Chem. 2004;279:7643–54. doi: 10.1074/jbc.M309485200. [DOI] [PubMed] [Google Scholar]
- 116.Moore PS, Boschoff C, Weiss RA, Chang Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science. 1996;274:1739–1744. doi: 10.1126/science.274.5293.1739. [DOI] [PubMed] [Google Scholar]
- 117.Molden J, Chang Y, You Y, Moore PS, Goldsmith MA. A Kaposi’s sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J Biol Chem. 1997;272:19625–19631. doi: 10.1074/jbc.272.31.19625. [DOI] [PubMed] [Google Scholar]
- 118.Nicholas J, Ruvolo VR, Burns WH, Sandford G, Wan XY, Ciufo D, Hendrickson SB, Guo HG, Hayward GS, Reitz MS. Kaposi’s sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nature Med. 1997;3:287–292. doi: 10.1038/nm0397-287. [DOI] [PubMed] [Google Scholar]
- 119.Lee H, Guo J, Li M, Choi JK, DeMaria M, Rosenzweig M, Jung JU. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi’s sarcoma-associated herpesvirus. Mol Cell Biol. 1998;18:5219–28. doi: 10.1128/mcb.18.9.5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brinkmann MM, Glenn M, Rainbow L, Kieser A, Henke-Gendo C, Schulz TF. Activation of mitogen-activated protein kinase and NF-kappaB pathways by a Kaposi’s sarcoma-associated herpesvirus K15 membrane protein. J Virol. 2003;77:9346–58. doi: 10.1128/JVI.77.17.9346-9358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, Ye H, Du MQ, Boshoff C. K15 protein of Kaposi’s sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J Virol. 2002;76:802–16. doi: 10.1128/JVI.76.2.802-816.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tomlinson CC, Damania B. The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J Virol. 2004;78:1918–27. doi: 10.1128/JVI.78.4.1918-1927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Prakash O, Swamy OR, Peng X, Tang ZY, Li L, Larson JE, Cohen JC, Gill J, Farr G, Wang S, Samaniego F. Activation of Src kinase Lyn by the Kaposi sarcoma-associated herpesvirus K1 protein: implications for lymphomagenesis. Blood. 2005;105:3987–94. doi: 10.1182/blood-2004-07-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chandriani S, Ganem D. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J Virol. 2010;84:5565–73. doi: 10.1128/JVI.02723-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee H, Veazey R, Williams K, Li M, Guo J, Neipel F, Fleckenstein B, Lackner A, Desrosiers RC, Jung JU. Deregulation of cell growth by the K1 gene of Kaposi’s sarcoma-associated herpesvirus. Nat Med. 1998;4:435–440. doi: 10.1038/nm0498-435. [DOI] [PubMed] [Google Scholar]
- 126.Prakash O, Tang ZY, Peng X, Coleman R, Gill J, Farr G, Samaniego F. Tumorigenesis and aberrant signaling in transgenic mice expressing the human herpesvirus-8 K1 gene. J Natl Cancer Inst. 2002;94:926–35. doi: 10.1093/jnci/94.12.926. [DOI] [PubMed] [Google Scholar]
- 127.Sadler R, Wu L, Forghani B, Renne R, Zhong W, Herndier B, Ganem D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated herpesvirus. J Virol. 1999;73:5722–30. doi: 10.1128/jvi.73.7.5722-5730.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li H, Komatsu T, Dezube BJ, Kaye KM. The Kaposi’s sarcoma-associated herpesvirus K12 transcript from a primary effusion lymphoma contains complex repeat elements, is spliced, and initiates from a novel promoter. J Virol. 2002;76:11880–8. doi: 10.1128/JVI.76.23.11880-11888.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McCormick C, Ganem D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science. 2005;307:739–41. doi: 10.1126/science.1105779. [DOI] [PubMed] [Google Scholar]
- 130.Muralidhar S, Pumfery AM, Hassani M, Sadaie MR, Azumi N, Kishishita M, Brady JN, Doniger J, Medveczky P, Rosenthal LJ. Identification of kaposin (open reading frame K12) as a human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) transforming gene. J Virol. 1998;72:4980–8. doi: 10.1128/jvi.72.6.4980-4988.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kliche S, Nagel W, Kremmer E, Atzler C, Ege A, Knorr T, Koszinowski U, Kolanus W, Haas J. Signaling by human herpesvirus 8 kaposin A through direct membrane recruitment of cytohesin-1. Mol Cell. 2001;7:833–43. doi: 10.1016/s1097-2765(01)00227-1. [DOI] [PubMed] [Google Scholar]
- 132.Gandy SZ, Linnstaedt SD, Muralidhar S, Cashman KA, Rosenthal LJ, Casey JL. RNA editing of the human herpesvirus 8 kaposin transcript eliminates its transforming activity and is induced during lytic replication. J Virol. 2007;81:13544–51. doi: 10.1128/JVI.01521-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Choi JK, Lee BS, Shim SN, Li M, Jung JU. Identification of the novel K15 gene at the rightmost end of the Kaposi’s sarcoma-associated herpesvirus genome. J Virol. 2000;74:436–46. [PMC free article] [PubMed] [Google Scholar]
- 134.Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci U S A. 2005;102:5570–5. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Samols MA, Hu J, Skalsky RL, Renne R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2005;79:9301–5. doi: 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. Identification of microRNAs of the herpesvirus family. Nat Methods. 2005;2:269–76. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 137.Grundhoff A, Sullivan CS, Ganem D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. Rna. 2006;12:733–50. doi: 10.1261/rna.2326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol. 2010;12:193–9. doi: 10.1038/ncb2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gottwein E, Cullen BR. A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J Virol. 2010;84:5229–37. doi: 10.1128/JVI.00202-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Skalsky RL, Samols MA, Plaisance KB, Boss IW, Riva A, Lopez MC, Baker HV, Renne R. Kaposi’s sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol. 2007;81:12836–45. doi: 10.1128/JVI.01804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, Braich R, Manoharan M, Soutschek J, Ohler U, Cullen BR. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450:1096–9. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.DiNardo CD, Tsai DE. Treatment advances in posttransplant lymphoproliferative disease. Curr Opin Hematol. 2010;17:368–74. doi: 10.1097/MOH.0b013e328339018c. [DOI] [PubMed] [Google Scholar]
- 143.Li N, Thompson S, Schultz DC, Zhu W, Jiang H, Luo C, Lieberman PM. Discovery of selective inhibitors against EBNA1 via high throughput in silico virtual screening. PLoS One. 2010;5:e10126. doi: 10.1371/journal.pone.0010126. [DOI] [PMC free article] [PubMed] [Google Scholar]