

Chondrosarcoma (CHS) is a heterogeneous collection of malignant bone tumours and is the second most common primary malignancy of bone after osteosarcoma. Recent work has identified frequent, recurrent mutations in IDH1/2 in nearly half of central CHS. However, there has been little systematic genomic analysis of this tumour type and thus the contribution of other genes is unclear. Here we report comprehensive genomic analyses of 49 cases of CHS. We identified hypermutability of the major cartilage collagen COL2A1 with insertions, deletions and rearrangements identified in 37% of cases. The patterns of mutation were consistent with selection for variants likely to impair normal collagen biosynthesis. In addition we identified mutations in IDH1/2 (59%), TP53 (20%), the RB1 pathway (33%) and hedgehog signaling (18%).
Forty-nine cases of untreated CHS including 30 central, 4 peripheral, 14 dedifferentiated and 1 arising in a patient with synovial chondromatosis, along with matching normal tissue were subjected to whole exome sequencing as previously described1. Dedifferentiated CHS is characterised by biphasic histology represented by conventional CHS abutting a high-grade non-cartilaginous component. For the dedifferentiated CHS, sequence was obtained from the non-cartilaginous high-grade component of the tumour in each case. Case distribution by grade is indicated in Supplementary Table 1. The exomic sequence coverage at a minimum depth of 30× was 65-70% and data was processed as previously described1. Sequence variants were confirmed as somatic utilising custom targeted capture and sequencing combined with manual data inspection. Candidate genes were evaluated in an extension series of 26 cases (Supplementary Table 1). Allele specific copy number was assessed with SNP6.0 arrays and direct analyses of exome reads as previously described1,2.
In total, 1428 somatic mutations were identified, with somatic mutation burden ranging from 1 to 115 (Supplementary Table 2). Mutations comprised 944 missense, 61 nonsense, 37 essential splice, 80 indels, 301 synonymous changes and 5 substitutions in miRNAs. Somatic mutation burden showed a significant association with increasing grade. High grade (grade II, grade III and dedifferentiated) had on average more than double the somatic mutations per sample as grade I CHS (Wilcoxon rank sum test, p=0.00017).
The most striking finding of this study was the identification of mutations in COL2A1 that encodes the alpha chain of type II collagen fibers – the major collagen constituent of articular cartilage. Somatic mutations were initially identified in 8 cases from the exome screen, all small indels. This remarkable concentration of mutations prompted a more thorough investigation of COL2A1 to look for rearrangements in addition to point mutations. The entire gene footprint was tiled with baits for custom capture and all exome cases plus the 26 follow-up tumours were subjected to capture and sequencing. In total 44% (33/75) of cases had at least one mutation which would be predicted to affect coding sequence (Figure 1, Supplementary Table 3). The mutations consisted of splice site (n=2), indel (n=23), missense (n=2) and large-scale rearrangements (n=36). No synonymous mutations were identified (Supplementary Table 3). The majority of rearrangements had both breakpoints located within the footprint of COL2A1 (Supplementary Figure 1) and none were predicted to form in frame fusion events. Whole genome shotgun sequencing at low depth in 6 of the CHS cases demonstrated that breakpoints were clustered in COL2A1 and that chromosome 12 was not highly rearranged elsewhere (Supplementary Figure 1). In 13 cases a single COL2A1 mutation was identified, however 20 tumours had more than 1 mutation. A one-sided Fisher exact test indicated that amongst all cases, high grade tumours (grade II, III and dedifferentiated) were significantly more likely to contain a COL2A1 mutation than low grade (grade I) tumours (p=0.029). This association however was not significant when restricted to only to central CHS cases (p=0.093).
Figure 1.
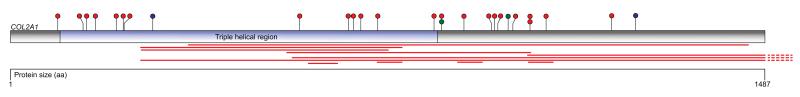
Representation of the protein coding sequence and major domain in the COL2A1 gene. Somatic mutations identified in the primary and extension investigations are indicated as circles; truncating (red), essential splice site (blue) and missense (green). Large deletions are depicted by red bars beneath the gene.
Although finding 20 cases with more than one COL2A1 mutation is formally compatible with a loss of function mechanism, the presence of multiple cases with a single heterozygous mutation suggests that functional consequences at the protein level may be more complex. We therefore performed immunohistochemistry for COL2A1 protein on 43 available tumours (21 mutation positive and 22 wild-type, Supplementary Table 4). Abnormal staining (absent or focal compared to diffuse staining seen in normal cartilage, see Supplementary Figure 2) was identified in 31/43 tumours, with absence of staining in 8 tumours and focal staining in 23. Of the eight cases with no staining, 6 were mutant with only one of those having two demonstrable mutations. For the 23 cases showing focal staining, 12 were mutant (2 with more than 1 mutation) and 11 had no detectable mutation. It is possible that COL2A1 mutations in a proportion of the “wildtype” cases have not been detected, in particular given the complex nature of the rearrangements that predominate, or have otherwise aberrant type II collagen deposition for reasons that remain obscure. These data argue against complete loss of protein being the predominant outcome of COL2A1 mutation.
The entire COL2A1 gene was also sequenced in 56 osteosarcomas, 24 chordomas, 10 other cartilaginous tumours and 73 meningiomas to compare patterns of mutation. The results revealed an accumulation of mutations across the gene footprint of COL2A1 that was restricted to CHS (Supplementary Table 5). This pattern suggested that COL2A1 is hypermutable in CHS. Given that COL2A1 is transcribed at a substantial rate in chondrocytes the data may support a transcription-associated mutation/recombination (TAM/TAR) mechanism3. An elevated COL2A1 mutation rate does not preclude biological significance of the mutations. Analyses of the patterns of mutation provided evidence of selection. No silent coding mutations were found. There was a preponderance of frame-shifting indels in the exons compared to substitution mutations (Supplementary Table 6a). In the introns these mutation classes were in equal proportion. Considering just indels there are very few mutations in coding sequence that were multiples of three (“in-frame”) as opposed to the non-coding portions of the gene (Supplementary Table 6b). These data suggest positive selection for those mutations that would substantially disrupt COL2A1 coding potential. To further explore the specificity of COL2A1 mutation in CHS, mutations across all collagen genes were assessed in multiple bone and cartilage tumours including chondroblastoma (6), chondromyxoid fibroma (2), chordoma (24) and osteosarcoma (56). The results clearly indicate an enrichment for non-synonymous mutations that was restricted to COL2A1 in chondrosarcoma (Supplementary Figure 3).
Mature collagen fibrils are formed via the assembly of pro-collagen alpha chains into triple helix formations involving post-translational modifications that stabilise the triple helical conformation. Constitutional mutations of COL2A1 lead to a variety of skeletal and ocular disorders that range from lethal perinatal dwarfism due to de novo missense mutations in achondrogenesis type II (OMIM 200610) to Stickler syndrome type I (OMIM 108300) caused primarily by familial truncating mutations. The variety of COL2A1 somatic mutations reported here in CHS would likely lead to disruption of the collagen maturation process via production of aberrant pro-collagen alpha chains – with possible dominant negative effects given the heterozygous nature of many of the mutations. In a mouse model carrying a dominant negative Col2a1 mutation found in the human disease spondyloepiphyseal dysplasia congenita (OMIM 183900), depletion of collagen fibrils was found to significantly impair chondrocyte differentiation4. However, there is no reported increased predisposition to CHS in these human developmental disorders nor in their mouse models – similar to the lack of cancer predisposition found for germline alleles of FGFRs that give rise to other skeletal disorders. Of note, targeted Pten deficiency in chondrocytes leads to dyschondroplasia resembling human enchondroma5. A dedifferentiated CHS (PD6363a) found to have PTEN homozygous deletion did not have a COL2A1 or IDH mutation. Lastly, amongst various matrix components, it was type 2 collagen that was found to restore cartilaginous features of otherwise dedifferentiated primary chondrocytes in monolayer culture6. Thus, there is a rational basis to hypothesise that COL2A1 mutations in CHS may not be merely passenger events but that they bring about fundamental perturbation of matrix deposition and signaling which may contribute to oncogenesis through abrogation of normal differentiation programs. These data indicate that development of in vivo approaches for mechanistic investigation of COL2A1 in CHS will be important.
As we previously reported, IDH mutations are prevalent in central chondrosarcoma7 and absent from peripheral CHS. IDH1/2 were recurrently point mutated in 29/49 (59%) (Figure 2). A Fisher exact test for a difference in the proportion of tumours of different grades bearing an IDH mutation gave no significant result (p=0.22). Given the active development of inhibitors for mutant IDH8, CHS would be a compelling indication as there are currently no effective treatments other than radical surgical resection. In addition, the evaluation of 2-hydroxyglutarate, the ‘oncometabolite’ produced by mutant IDH, as a biomarker9 in CHS management is warranted.
Figure 2.

Known and likely driver variants identified in the 49 primary tumours. Mutated genes are shown in each row and the 49 tumours presented in each column. Clinical grade for each tumour is indicated above the columns. The tumours are classified as ‘Central’, ‘Peripheral’ or ‘S’, indicating a malignant chondrosarcoma that arose in a patient with synovial chondoromatosis. Statistical anaylsis to look for non-random accumulation of non-synonymous substitutions confirmed IDH1 as significant (see Supplementary Methods). Variants are depicted with shaded squares; missense (green), truncating and rearrangements (red), homozygous deletions (blue with triangle), amplifications (purple with triangle), in frame deletion (orange) and germline truncations (black). Boxes with crosses indicate the samples with more than 1 COL2A1 mutation.
We identified frequent involvement of the RB pathway (Figure 2, Figure 3a). Copy number analyses identified 13 CHS with homozygous deletions of CDKN2A consistent with previous reports10 (Supplementary Table 7, Supplementary Figure 4). A p.D108G missense mutation with loss of the wild-type allele was also identified as well as two further truncating mutations in the extension series. We found focal amplifications of CDK4 (1 case) and CDK6 (2 cases) one of which also had a CDKN2A mutation. CDK4 was co-amplified with MDM2 as has been previously reported11. CDK6, to the best of our knowledge, has not been previously identified as amplified in CHS. Taken together 27% (13/49) exome series cases have mutation of RB regulatory constituents. TP53 mutation plays an important role in CHS, with 20% (10/49) of cases having coding mutations. Mutations of other known cancer genes included SETD2, KDM6A, NF2, SF3B1, TET2, DNMT3A and TSC1. Germline EXT1/2 mutations were identified in three grade I peripheral CHS. PD7299a, the case with the largest substitution burden, had a somatic MUTYH missense mutation previously reported in the context of colorectal cancer susceptibility in FAP212-15 (OMIM 608456).
Figure 3.

(a) RB1 pathway indicating genes with mutations in this series. Genes in green are activated by mutations, while genes in red are inactivated. (b) Hedgehog pathway with known cancer genes mutated in this series indicated. Genes in green are activated by mutations, while genes in red are inactivated.
We identified evidence for Indian hedgehog (IHH) signaling pathway involvement in CHS (Figure 2, Figure 3b). IHH signaling is crucial for normal chondrocyte differentiation. Mutations resulting in constitutive hedgehog signaling are causal in benign cartilage tumours16-18. We identified 4 mutations of PTCH1 in the exome screen (2 missense and 2 truncating) and a missense in the extension series (Figure 2, Supplementary Table 2). An inactivating SUFU mutation and a GLI1 amplification was identified in two additional cases. Further, we identified focal amplifications of RUNX2 and HHIP- both tightly linked to IHH signaling16,17. Taken together there is mutation evidence for IHH pathway activation in 18% (9/49) of exome cases. Hedgehog pathway inhibition via the small molecule Vismodegib targeting SMO has recently been shown to have significant activity in advanced basal cell carcinoma, including patients with basal-cell nevus syndrome due to hedgehog pathway mutation18. Assessment of hedgehog pathway inhibitors in that subset of CHS patients with demonstrable pathway mutations may be an important line of therapeutic investigation.
Combined with our previous efforts identifying IDH mutation in central CHS, the comprehensive analysis presented here begins to paint a more complete picture of CHS genomics for the most common and most lethal subtypes. Involvement of the RB, IHH pathways and the novel finding of fundamental perturbation of matrix biology via COL2A1 mutation have been identified as likely contributory to CHS. In addition to opportunities for exploiting IDH mutations in therapeutic and biomarker contexts, delineating contributions of RB and IHH pathway mutations present opportunities for focused therapeutic exploration. Further, if the production of mature collagen fibrils is indeed compromised by mutant COL2A1, then there may be an opportunity to explore therapeutic strategies exploiting cellular and endoplasmic reticulum stress responses to improperly folded proteins. This study illustrates the necessity to characterise comprehensively all cancer types/subtypes to generate maximum opportunity for comparative oncogenomics and, crucially, expedited development of therapeutic and diagnostic strategies.
Supplementary Material
Acknowledgements
This work was supported by funding the Wellcome Trust (grant reference 077012/Z/05/Z), and the Skeletal Cancer Action Trust (SCAT), UK. The material was obtained from the RNOH Musculoskeletal Research Programme and Biobank. Support was provided to AMF (UCL) by the National Institute for Health Research, UCLH Biomedical Research Centre, and the UCL Experimental Cancer Centre. We are grateful to the patients for participating in the research and to the clinicians and support staff of the London Sarcoma Service involved in their care. P.J.C. is personally funded through a Wellcome Trust Senior Clinical Research Fellowship (grant reference WT088340MA). P.V.L. is a postdoctoral researcher of the Research Foundation - Flanders (FWO). SB is funded through the Wellcome Trust PhD Programme for Clinicians.
Footnotes
Informed consent was obtained from all subjects and ethical approval obtained from Cambridgeshire 2 Research Ethics Service (ref 09/H0308/165). Genome data have been deposited at the European Genome-Phenome Archive (EGA, http://www.ebi.ac.uk/ega/), which is hosted by the European Bioinformatics Institute (EBI), under accession number EGAD00001000358 (exome; SNP6 data submitted – accession number pending).
The authors declare no competing financial interests.
References
- 1.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–4. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Loo P, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A. 2010;107:16910–5. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim N, Jinks-Robertson S. Transcription as a source of genome instability. Nat Rev Genet. 2012;13:204–14. doi: 10.1038/nrg3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbieri O, et al. Depletion of cartilage collagen fibrils in mice carrying a dominant negative Col2a1 transgene affects chondrocyte differentiation. Am J Physiol Cell Physiol. 2003;285:C1504–12. doi: 10.1152/ajpcell.00579.2002. [DOI] [PubMed] [Google Scholar]
- 5.Yang G, et al. PTEN deficiency causes dyschondroplasia in mice by enhanced hypoxia-inducible factor 1alpha signaling and endoplasmic reticulum stress. Development. 2008;135:3587–97. doi: 10.1242/dev.028118. [DOI] [PubMed] [Google Scholar]
- 6.Chiu LH, et al. Differential effect of ECM molecules on re-expression of cartilaginous markers in near quiescent human chondrocytes. J Cell Physiol. 2011;226:1981–8. doi: 10.1002/jcp.22530. [DOI] [PubMed] [Google Scholar]
- 7.Amary MF, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334–43. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 8.Popovici-Muller J, et al. Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo. Acs Medicinal Chemistry Letters. 2012;3:850–855. doi: 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fathi AT, et al. Prospective serial evaluation of 2-hydroxyglutarate, during treatment of newly diagnosed acute myeloid leukemia, to assess disease activity and therapeutic response. Blood. 2012;120:4649–4652. doi: 10.1182/blood-2012-06-438267. [DOI] [PubMed] [Google Scholar]
- 10.Hallor KH, et al. Genomic Profiling of Chondrosarcoma: Chromosomal Patterns in Central and Peripheral Tumors. Clinical Cancer Research. 2009;15:2685–2694. doi: 10.1158/1078-0432.CCR-08-2330. [DOI] [PubMed] [Google Scholar]
- 11.Schrage YM, et al. Central chondrosarcoma progression is associated with pRb pathway alterations: CDK4 down-regulation and p16 overexpression inhibit cell growth in vitro. Journal of Cellular and Molecular Medicine. 2009;13:2843–2852. doi: 10.1111/j.1582-4934.2008.00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aceto G, et al. Mutations of APC and MYH in unrelated Italian patients with adenomatous polyposis coli. Hum Mutat. 2005;26:394. doi: 10.1002/humu.9370. [DOI] [PubMed] [Google Scholar]
- 13.Ahn J, et al. Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1) Nat Genet. 1995;11:137–43. doi: 10.1038/ng1095-137. [DOI] [PubMed] [Google Scholar]
- 14.Hopyan S, et al. A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat Genet. 2002;30:306–10. doi: 10.1038/ng844. [DOI] [PubMed] [Google Scholar]
- 15.Stickens D, et al. The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nat Genet. 1996;14:25–32. doi: 10.1038/ng0996-25. [DOI] [PubMed] [Google Scholar]
- 16.Bruce SJ, et al. Inactivation of Patched1 in the mouse limb has novel inhibitory effects on the chondrogenic program. J Biol Chem. 2010;285:27967–81. doi: 10.1074/jbc.M109.091785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wuelling M, Vortkamp A. Transcriptional networks controlling chondrocyte proliferation and differentiation during endochondral ossification. Pediatr Nephrol. 2010;25:625–31. doi: 10.1007/s00467-009-1368-6. [DOI] [PubMed] [Google Scholar]
- 18.Tang JY, et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med. 2012;366:2180–8. doi: 10.1056/NEJMoa1113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.