

Abstract
Measures of structural similarity between known protein structures provide an objective basis for classifying protein folds and for revealing a global view of the protein structure universe. Here, we describe a rapid method to measure structural similarity based on the profiles of representative local features of Cα distance matrices of compared protein structures. We first extract a finite number of representative local feature (LF) patterns from the distance matrices of all protein fold families by medoid analysis. Then, each Cα distance matrix of a protein structure is encoded by labeling all its submatrices by the index of the nearest representative LF patterns. Finally, the structure is represented by the frequency distribution of these indices, which we call the LF frequency (LFF) profile of the protein. The LFF profile allows one to calculate structural similarity scores among a large number of protein structures quickly, and also to construct and update the “map” of the protein structure universe easily. The LFF profile method efficiently maps complex protein structures into a common Euclidean space without prior assignment of secondary structure information or structural alignment.
Keywords: protein structural similarity, protein distance matrix, local protein structural features profile, protein fold, protein fold space
Recent advances of experimental techniques and automation in molecular and structural biology have led to the rapid increase in the determination of many protein structures. The number of structures deposited in the Protein Data Bank (PDB) (1) is now >20,000 and the contents are growing rapidly. Furthermore, the ongoing structural genomics projects, which aim to determine representative structures in protein fold space, have begun to produce, in a high throughput way, a large number of structures (2), including many structures of the proteins encoded by genes of unknown functions, the “hypothetical” proteins. Over half of all of the proteins of sequenced genomes has no inferable molecular (biochemical and biophysical) functions. As sequence similarity infers functional similarity, structural similarity also infers similarity in molecular function: if a hypothetical protein has a structure similar to one or more protein structures of known function, the structural similarity infers a powerful clue to the molecular function of the hypothetical protein (3).
Measures of structural similarity, assessed computationally or visually, between pairs of proteins are also the foundation for classifying protein structures. Many systems have been proposed for structural classification, such as structural classification of proteins (SCOP) (4), class architecture topology homology (CATH) (5), families of structurally similar proteins (FSSP) (6), and others. Measuring structural fold similarity is usually done by structural alignment algorithms such as dali (7), ce (8), vast (9), ssap (10), and others. Most of these methods are computationally intensive and time-consuming, especially when searching large databases, due to intrinsic complexity of structural alignment. To shorten computational time, several methods have been developed that do not depend on the structural alignment, such as the methods based on graph theory (11), secondary structure matching (www.ebi.ac.uk/msd-srv/ssm/ssmstart.html), and Cα-Cα distances (12).
Methods
In developing our method for quickly assessing structural similarity, we start with the distance matrix representation of protein structure. The distance matrix of a protein structure is a square matrix consisting of the distances between all pairs of Cα atoms in the protein. It not only represents the overall 3D folding of polypeptide chains in two dimensions, but also provides a simple description of information about secondary structure and tertiary interactions between parts spatially distant in the structure. Furthermore, the matrix contains sufficient information to reproduce the original 3D backbone structure by using the distance geometry method (13, 14). Because of its fluent information content, the distance matrix has been exploited in diverse studies such as domain recognition (15), structure alignment (dali) (7), protein folding studies (contact energy function) (16, 17), and protein database searching (18).
We subdivide the distance matrix of each protein structure into many overlapping submatrices, each describing a local feature (secondary and/or tertiary feature). We use a collection of these submatrices from a large number of distance matrices to extract a set of K representative local features (medoid submatrices) from K clusters of submatrices by medoid analysis (19). Then, any given protein structure can be represented by a profile, a vector of a common length K, containing the frequencies of occurrence of these representative local features (medoid submatrices) in the structure. Thus, we can now treat protein structures as points in K-dimensional Euclidean space (RK). After converting each protein structure into a local feature frequency (LFF) profile, the fold similarity between a pair of proteins can be computed very easily as Euclidean distance or cosine distance between two corresponding LFF profile vectors. This enables quick computation of an all-against-all structural similarity matrix of a very large set of proteins, which, in turn, can be used for objectively clustering protein structures of similar fold, for constructing a map of the “protein structure universe,” and for exploring protein fold space.
Nonredundant Protein Structure Set. The test of the method was implemented on a representative SCOP fold set from the SCOP database release 1.61 (November 2002). The PDB-style files for the SCOP nonredundant set (a sub-SCOP set filtered at 40% sequence identity) were downloaded from the ASTRAL compendium database (20), and LFF profiles were computed for all 3,792 structural domains in this set, which includes all α, all β, α/β, and α+β classes of proteins.
Representative Local Feature Patterns in Distance Matrix. In this test 100 proteins randomly selected from 3,792 in the nonredundant SCOP fold set were indexed by p = 1,..., P (P = 100). When there are np residues in protein p, its distance matrix is the matrix Dp = {dp(i, j):i, j = 1,..., np}, where dp(i, j) is the Cα-Cα distance (in Å) between residues i and j. The overlapping submatrices presenting local features involving m-residues by m-residues in the protein is the collection expressed by
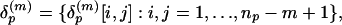 |
of m × m submatrices described by
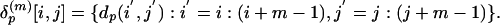 |
To emphasize the importance of the close contacts in the protein structures, all Cα-Cα distances ≥20 Å are set to 20 Å. The collection of these submatrices over P proteins is 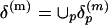 . They are grouped into K clusters, and each cluster is represented by a medoid in the space δ(m) metrized by the Euclidean distance by using the partitioning around medoids (PAM) analysis of Kaufman and Rousseeuw (19). Algorithmically, the PAM procedure searches K representative objects or medoids among the observations and then constructs K clusters by assigning each observation to the nearest medoid. PAM can be applied to general data types and tends to be more robust than k-means algorithm (19). In this study, we use K = 100 and m = 10 (see Fig. 3). Thus, we use the 100 m × m medoid submatrices as the reference to which all m × m submatrices from all protein distance matrices will be compared.
. They are grouped into K clusters, and each cluster is represented by a medoid in the space δ(m) metrized by the Euclidean distance by using the partitioning around medoids (PAM) analysis of Kaufman and Rousseeuw (19). Algorithmically, the PAM procedure searches K representative objects or medoids among the observations and then constructs K clusters by assigning each observation to the nearest medoid. PAM can be applied to general data types and tends to be more robust than k-means algorithm (19). In this study, we use K = 100 and m = 10 (see Fig. 3). Thus, we use the 100 m × m medoid submatrices as the reference to which all m × m submatrices from all protein distance matrices will be compared.
Fig. 3.

One hundred medoid submatrices obtained from partitioning around medoids (PAM) analysis of distance matrices of 100 sampled proteins. They reflect 100 representative local structural features. Various combination of these features can reconstruct the original distance matrices of all 100 proteins. The medoid submatrices are indexed arbitrarily (from 1 to 100).
Generation of the LFF Profile and Calculation of Similarity/Dissimilarity Scores. To express the distance matrix of a protein p in terms of the representative local feature patterns (medoid submatrices), each of its submatrices 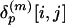 is labeled by the index of the nearest medoid submatrix. Again, the space of submatrices is metrized by the Euclidean distance. Then, the frequency of the medoid submatrices assigned to label k,
is labeled by the index of the nearest medoid submatrix. Again, the space of submatrices is metrized by the Euclidean distance. Then, the frequency of the medoid submatrices assigned to label k, 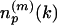 , is counted. The count vector
, is counted. The count vector 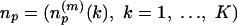 , summarizes the frequency distribution of local feature patterns of the protein. We call this decoding process profiling of the protein structure by LFF and the final feature vector np, or its transformation Ap, the structural profile or simply the profile of protein p. Here, we normalize frequency of local interaction pattern k in protein p by
, summarizes the frequency distribution of local feature patterns of the protein. We call this decoding process profiling of the protein structure by LFF and the final feature vector np, or its transformation Ap, the structural profile or simply the profile of protein p. Here, we normalize frequency of local interaction pattern k in protein p by
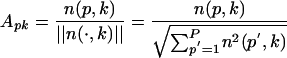 |
and use Ap = [Ap1... ApK] ∈ RK as the profile of protein p. The collection of profiles, or the protein-by-pattern matrix
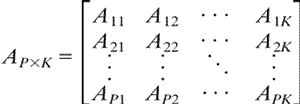 |
is our raw data matrix for computing similarity. As a measure of structural similarity between two proteins p and q with profiles Ap and Aq in RK, we use their cosine
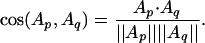 |
It is also called the normalized inner product, because the cosine is simply the dot product if vectors are normalized. The cosine distance is defined as 1 - cos(Ap, Aq) and used to represent structural dissimilarity or structural distance. Note that the cosine distance ranges from 0 (closest) to 1 (farthest).
Singular Value Decomposition (SVD) and Biplots of Protein-by-Pattern Matrix. SVD is used for deriving a set of uncorrelated indexing variables or factors, whereby each pattern and protein is represented as a vector in RK using elements of the left and right singular vectors. For a P × K matrix A, with P ≥ K and rank(A) = r, the SVD of A is defined as A= U∑VT, where UTU = VTV = IK (K × K identity matrix) and ∑ = diag(σ1,..., σK), σ1 ≥ · · · ≥ σr > 0 = σr+1 = · · · = σP. The columns ui and vi of U and V, respectively, are referred to as left and right singular vectors. Matrices U, V, ∑ reflect a breakdown of the original relationships into linearly independent vectors or factor values. The use of the κ factors with the largest singular values is equivalent to approximating the original protein by pattern matrix by
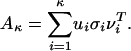 |
We compute the truncated SVD with κ = 3 to obtain rank-three approximation A3 of the protein by pattern matrix, because the first three σ values are significantly greater than the rest. We can represent proteins and patterns in the same R3 space by their first three principal coordinates
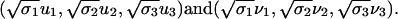 |
In this paper, (1st, 2nd) and (2nd, 3rd) principal coordinates pairs are plotted as biplots (21) in R2.
Results
One Hundred Representative Local Features of Protein Structures. In the distance matrix of a protein structure, many local structural features can be recognized as various contact patterns in submatrices. Secondary structure elements such as α helices and β sheets are visually identifiable as specific local features in the matrix as thick line patterns and thin line patterns on and off diagonal areas, respectively, and the tertiary interactions between them appear as patches of contacts in off-diagonal areas of the matrix. Among β strands, parallel β-strands appear as thin line patterns parallel to the main diagonal, and antiparallel β-strands appear as thin line patterns perpendicular to the main diagonal. Other tertiary features, like α-β interactions and coils, also emerge as specific patterns in the distance matrix (Fig. 1).
Fig. 1.

Representation of protein structures by their distance matrices and representative local structure feature patterns (medoid submatrices). The procedure is illustrated by using 3D protein structures, distance matrices, and 50 representative patterns (medoids) of four proteins sampled one each from all-α, all-β, α/β, and α + β classes. Among the patterns, “null feature patterns” (with no Cα-Cα distance <20 Å, light pink background only) are the most abundant in all proteins.
There are millions of different local feature patterns (submatrices) in all protein structures. However, we expect that most of these are common in many protein structures, and the majority of the local feature patterns are null patterns without any contact within a threshold of 20 Å (i.e., all submatrix elements have the Cα-Cα distance >20 Å). Thus, we expect that a finite number, K, of representative local features (K medoid submatrices) will adequately represent all observed local features in all proteins. Then, all local feature patterns can be labeled according to the index (from 1 to K) of the closest medoids, where “closeness” can be defined in terms of Euclidean distance or other distance metrics.
To determine the optimum submatrix size (m) and number of medoids K, 100 protein structures were randomly chosen from the SCOP representative folds. Lengths of the proteins in the set range from 29 to 595, with the average of 165. We then varied K = 10-300 and m = 8-16 while doing the medoid analysis. After replacing all observed submatrices by the representative medoid submatrices, the reconstructed distance map was calculated by averaging the overlapping medoid submatrices. The distance matrix error (DME), which is a root-mean square difference between original distance map and reconstructed one, is used to plot Fig. 2. Based on this test, the size of K and m was set to 100 and 10, respectively.
Fig. 2.

Optimization of K, the number of representative local feature patterns (medoid submatrices) and m, the size of the submatrix. The distance matrices of 100 chosen protein structures were reconstructed by using K closest representative medoid submatrices of size m. There is no significant error reduction over K = 100 medoids, and the best reconstruction condition is at m = 10 and K = 100. The dissimilarity between original distance matrix and reconstructed distance matrix was measured by distance matrix error, 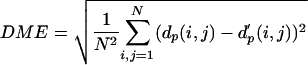 where dp(i, j) and d′p(i, j) are the Cα-Cα distances (in Å) between residue i and j in the original and reconstructed distance matrix, respectively. N is the number of residues in the protein.
where dp(i, j) and d′p(i, j) are the Cα-Cα distances (in Å) between residue i and j in the original and reconstructed distance matrix, respectively. N is the number of residues in the protein.
For the submatrix size m = 10, ≈1.6 × 106, different local patterns (submatrices) were retrieved from the training set of 100 protein structures. One hundred representative local feature patterns were identified as 100 “medoids.” They can be considered as the centers of 100 clusters from 1.6 × 106 input patterns. Then, all input patterns can be labeled from 1 to 100 according to the index of the closest medoids, where closeness is defined in terms of Euclidean distance in this study. Fig. 3 shows the 100 representative patterns found by this medoid analysis.
LFF Profile as a Representation of Protein Structure. The method we use here is analogous to that used in text information retrieval (22), in which each document is represented as a vector of word counts. In our approach, each protein structure is considered as a document, consisting of many words (different medoid submatrices representing different local features). A protein structure as represented by its distance matrix is treated as a collection of overlapping submatrices (local feature patterns), and each of them is labeled by the index of the closest medoid submatrix. Thus, a protein structure can be represented by the profile of the frequency distribution of the medoid pattern indices. We call this the LFF profile, or simply the profile of the protein.
Structural Similarity Calculation Using LFF Profile. After the profiling described above, protein structures can be mapped into a common space where the similarity or dissimilarity between any two protein structures can be computed easily as a cosine or cosine distance (or Euclidean distance), respectively, between two profile vectors. However, because the abundance of local patterns varies considerably from one pattern to another, some normalization of the profile is necessary, as shown in Methods. For example, the “null” pattern (15th submatrix in Fig. 3) is most abundant of all, and, without normalization, such an abundant pattern will dominate when computing structural similarity or dissimilarity distances. This is not desirable because the frequency of the void pattern contains little structural information. As can be seen in Fig. 4, similarity between structural profiles reflects, in general, the similarity between 3D structures according to SCOP classification.
Fig. 4.

LFF profiles of protein structures from the globin family (a.1.1.2), the Ig V set domain family (b.1.1.1), the α-amylases N-terminal domain family (c.1.8.1), and the microbial ribonuclease family (d.1.1.1) in the SCOP database. (upper four plots) The raw counts of LFF are plotted as a function of 100 different representative medoids (shown in Fig. 3) in red, blue, yellow, and green, respectively, of the four protein families. The highest peak in each family corresponds to the medoid index 15 of Fig. 3, which is the “null” medoid submatrix, with all of the matrix elements having a distance >20 Å. LFF profiles for five proteins sampled from each family are shown. The quality of clustering of local features is difficult to discern because of the domination of the null medoid and low signal to noise ratio of the rest of the modoids (lower four plots). However, after normalization by the spread of the counts in each representative medoid, the similarity among LFF profiles within each family is evident.
A Global Presentation of the Protein Fold Universe. Analogous to the physical universe map, mapping of the protein fold universe provides a global view of distribution of different protein structures in fold space, of unbiased classification of protein structures, and of evolution of protein structures (23). First, the structural profile of all 3,792 nonredundant SCOP domains was computed. The profiles were assembled into a protein-by-local pattern matrix of size 3,792 (proteins) by 100 (patterns). The matrix is processed by SVD as described in Methods. We compute the truncated SVD with K = 3 to obtain rank-three approximation A3 of the protein-by-pattern matrix. This approximation is justified by the fact that the first three eigenvalues are significantly greater than the rest. Fig. 5 shows biplots of 100 representative patterns (medoid submatrices) and 3,792 representative SCOP proteins, using the 1st-2nd and the 2nd-3rd principal axes pairs. From the plots, a correlation between representative patterns and structure classes are clearly visible. We also observe that the first three principal coordinates are approximately related to the length of protein, type of secondary structural elements (SSEs), and parallelism of β strands, respectively. One embedding of protein fold universe in 3D space using the SVD analysis of the profile matrx A is shown in Fig. 5.
Fig. 5.

Biplots of 3,792 protein structure profiles and 100 representative medoid patterns after SVD of the protein-by-pattern matrix. The 1st-2nd and the 2nd-3rd principal axes pairs are drawn. The 1st, 2nd, and 3rd principal coordinates can be interpreted as approximately related to the length of protein, types of secondary structure elements (SSEs), and parallelism of β strands, respectively. Proteins belonging to all-α, all-β, α/β, and α+β classes according to SCOP are colored red, blue, yellow, and green, respectively. The overall 3D plot is also shown.
Comparison with Other Methods. Compared with other classification schemes, how similar is our structural similarity? As a test, we asked whether the nearest neighbor of a given protein structure by our profile method belongs to the same fold family as the protein structure in the manually curated SCOP, which is often considered to be the gold standard. The LFF profile-based classification agrees with SCOP classification in 93% and 71% of the cases at class and fold levels, respectively (Table 1). The method agrees less well with CATH classification: 70% (class) and 61% (architecture). These features are also shown in Fig. 6 by the dendogram constructed based on the structural similarity scores by the LFF profile method, with color coding of the classifications by SCOP and CATH methods.
Table 1. Overall comparison of agreement in classification of the LFF profile method to SCOP and CATH methods at different levels of structural features.
| SCOP hierarchy | Agreement, % | CATH hierarchy | Agreement, % |
|---|---|---|---|
| Class | 93.2 | Class | 70.0 |
| Fold | 70.5 | Architecture | 60.6 |
| Superfamily | 68.6 | Topology | 57.5 |
| Family | 67.0 | Homology | 55.3 |
Fig. 6.

The dendogram of 3,792 SCOP protein domains (in four classes, 40% sequence identity filtered) was constructed by the hierachical clustering method based on the LFF profile distances. The red (all α), blue (all β), yellow (α/β), and green (α+β) colors in the top bar indicate SCOP class designations. The CATH classification on 2,679 intact protein chains that have counterparts in SCOP domains was used above. Three CATH classes are color-coded red (mainly α), blue (mainly β), and yellow (mixed α-β) in the bottom bar.
When we compared our method with the SCOP classification, we found examples of discrepancies. One extreme example is the case of quinohemoprotein amine dehydrogenase C chain (SCOP id: d1jmxg_), which is classified as “nonglobular all-α subunits of globular proteins” fold in SCOP. Among other proteins in the same SCOP fold, the closest one (SCOP id: d1l8cb_) ranks 3,350th among 3,792 structures by our profile method. Furthermore, our method finds a DNA/RNA-binding three-helical bundle fold (SCOP id: d1opc_), which belongs to a SCOP fold different from that of d1jmxg_, as the nearest neighbor fold to d1jmxg_. The distance map shows that the contact pattern of d1jmxg_ is quite different from other structures in the same SCOP fold, whereas d1jmxg_ and d1opc_ share considerable similar contact patterns (Fig. 7). This difference illustrates the different criteria used by the two methods in assessing the similarity between two proteins structures: assessment based on visual similarity of 3D fold in SCOP and that based on computational similarity of distance matrix features in the LFF profile method.
Fig. 7.

An example of discrepancy between the LFF profile method and the SCOP classification. The distance matrix of Quinohemoprotein amine dehydrogenase C chain (SCOP ID: d1jmxg_) is visually quite different from those of other proteins in the same SCOP fold (a.137). However, the OmpR DNA binding domain (SCOP ID: d1opc_), which belongs to another SCOP fold (a.4), is detected by the profile method to be closest to d1jmxg_.
Discussion
For testing the concept of the structure profile method, we used a simplified approach: (i) in constructing the reference set of medoid submatrices, we extracted them from 100 protein structures randomly selected from 3,792 nonredundant folds in the SCOP database; (ii) instead of extracting 100 representative LFs (medoid submatrices) from all submatrices of the 100 distance matrices (which will be about several million submatrices), we first found 50 medoids from each distance matrix, collected them together (50 × 100), and then extracted 100 medoids from the 5,000 medoids. These 100 medoid's medoids were used as the representative local features of all 3,792 proteins in LFF profiling.
In addition to expanding the structure database, from which we can extract a better set of medoid submatrices, we expect that the accuracy of the structural similarity score is likely to improve with calibration of various parameters in our method: varying the size of the local submatrix window to be large enough to capture nontrivial 3D interactions but at the same time be small enough to be observable in many different proteins and computable. Also, the number K of representative local feature patterns or medoids can be increased beyond our test of 100 to achieve optimum signal-to-noise ratios. Furthermore, a statistical score function can be developed to recognize folds that have no statistically significant structural similarity with known structures.
One immediate utility of the LFF profile method is a quick “mapping” of a recently determined structure in relation to all other structures in PDB or any subset in protein fold space (23). Another application may be to search for structural homologs of a query structure. For example, one could screen whole PDB quickly by using LFF profile method to find, say, the top 20 structural homologs of the query protein structure, then do the dali search among the 20 to find the best alignment.
Acknowledgments
We are grateful to Drs. Chao Zhang, Stephen Holbrook, and Paul Adams for their comments and suggestions. This work was supported by National Science Foundation Grant DBI-0114707.
Abbreviations: LFF, local feature frequency; PDB, Protein Data Bank; SCOP, structural classification of proteins; CATH, class architecture topology homology; SVD, singular value decomposition.
References
- 1.Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000) Nucleic. Acids Res. 28, 235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Service, R. F. (2002) Science 298, 948-950. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett, G. J., Todd, A. E. & Thornton, J. M. (2003) Methods Biochem. Anal. 44, 387-407. [PubMed] [Google Scholar]
- 4.Murzin, A. G., Brenner, S. E., Hubbard, T. & Chothia, C. (1995) J. Mol. Biol. 247, 536-540. [DOI] [PubMed] [Google Scholar]
- 5.Orengo, C. A., Michie, A. D., Jones, S., Jones, D. T., Swindells, M. B. & Thornton, J. M. (1997) Structure (London) 5, 1093-1108. [DOI] [PubMed] [Google Scholar]
- 6.Holm, L. & Sander, C. (1996) Nucleic Acids Res. 24, 206-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holm, L. & Sander, C. (1993) J. Mol. Biol. 233, 123-138. [DOI] [PubMed] [Google Scholar]
- 8.Shindyalov, I. & Bourne, P. (1998) Protein Eng. 11, 739-747. [DOI] [PubMed] [Google Scholar]
- 9.Gibrat, J. F., Madej, T. & Bryant, S. H. (1996) Curr. Opin. Struct. Biol. 6, 377-385. [DOI] [PubMed] [Google Scholar]
- 10.Orengo, C. A. & Taylor, W. R. (1996) Methods Enzymol. 266, 617-635. [DOI] [PubMed] [Google Scholar]
- 11.Harrison, A., Pearl, F., Mott, R., Thornton, J. & Orengo, C. (2002) J. Mol. Biol. 323, 909-926. [DOI] [PubMed] [Google Scholar]
- 12.Carugo, O. & Pongor, S. (2002) J. Mol. Biol. 315, 887-898. [DOI] [PubMed] [Google Scholar]
- 13.Havel, T. F., Kuntz, I. D. & Crippen, G. M. (1983) J. Theor. Biol. 104, 359-381. [DOI] [PubMed] [Google Scholar]
- 14.Vendruscolo, M., Kussell, E. & Domany, E. (1997) Fold Des. 2, 295-306. [DOI] [PubMed] [Google Scholar]
- 15.Holm, L. & Sander, C. (1994) Proteins 19, 256-268. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka, S. & Scheraga, H. A. (1976) Macromolecules 9, 945-950. [DOI] [PubMed] [Google Scholar]
- 17.Mirny, L. & Domany, E. (1996) Proteins 26, 391-410. [DOI] [PubMed] [Google Scholar]
- 18.Aung, Z., Fu, W. & Tan, K. L. (2003) in Proceedings of the 8th International Symposium on Database systems for Advanced Application, Kyoto, Japan (IEEE Computer Society, Los Alamitos, CA), pp. 311-318.
- 19.Kaufman, L. & Rousseeuw, P. (1990) in Finding Groups in Data: An Introduction to Cluster Analysis (Wiley, New York), pp. 68-163.
- 20.Chandonia, J.-M., Walker, N. S., Conte, L. L., Koehl, P., Levitt, M. & Brenner, S. E. (2002) Nucleic Acids. Res. 30, 260-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gabriel, K. R. (1971) Biometrika 58, 453-467. [Google Scholar]
- 22.van Rijsbergen, C. J. (1979) in Information Retrieval (Butterworths, London).
- 23.Hou, J., Sims, G. E., Zhang, C. & Kim, S.-H. (2003) Proc. Natl. Acad. Sci. USA 100, 2386-2390. [DOI] [PMC free article] [PubMed] [Google Scholar]