

Abstract
Vitamin B-6 deficiency is associated with impaired tryptophan metabolism because of the coenzyme role of pyridoxal 5′-phosphate (PLP) for kynureninase and kynurenine aminotransferase. To investigate the underlying mechanism, we developed a mathematical model of tryptophan metabolism via the kynurenine pathway. The model includes mammalian data on enzyme kinetics and tryptophan transport from the intestinal lumen to liver, muscle, and brain. Regulatory mechanisms and inhibition of relevant enzymes were included. We simulated the effects of graded reduction in cellular PLP concentration, tryptophan loads and induction of tryptophan 2,3-dioxygenase (TDO) on metabolite profiles and urinary excretion. The model predictions matched experimental data and provided clarification of the response of metabolites in various extents of vitamin B-6 deficiency. We found that moderate deficiency yielded increased 3-hydroxykynurenine and a decrease in kynurenic acid and anthranilic acid. More severe deficiency also yielded an increase in kynurenine and xanthurenic acid and more pronounced effects on the other metabolites. Tryptophan load simulations with and without vitamin B-6 deficiency showed altered metabolite concentrations consistent with published data. Induction of TDO caused an increase in all metabolites, and TDO induction together with a simulated vitamin B-6 deficiency, as has been reported in oral contraceptive users, yielded increases in kynurenine, 3-hydroxykynurenine, and xanthurenic acid and decreases in kynurenic acid and anthranilic acid. These results show that the model successfully simulated tryptophan metabolism via the kynurenine pathway and can be used to complement experimental investigations.
Introduction
Tryptophan is mainly degraded via the kynurenine (Kyn)7 pathway (1). Alterations of the Kyn pathway have been extensively studied and discussed in several contexts including inflammation (2), cancer (3), pregnancy (4, 5), oral contraceptive (OC) usage (5–9), and vitamin B-6 deficiency (10–12) in animals and humans. In the liver, tryptophan is first oxidized to N-formylkynurenine by tryptophan 2,3-dioxygenase (TDO) (1, 13), whereas in nonhepatic tissues, indoleamine 2,3-dioxygenase (IDO) catalyzes this reaction (14, 15). TDO and IDO play an important role in tryptophan homeostasis (16). Whereas TDO has been reported to be induced by tryptophan (16–18), glucocorticoids (18–20), glucagon (21), and estrogens (22), IDO is induced primarily by interferon (IFN)-γ (3, 12), and therefore IDO has been extensively studied in the immune response (2, 14).
Vitamin B-6 plays an important role in the Kyn pathway because pyridoxal 5′-phosphate (PLP) functions as a coenzyme for the bifunctional enzymes kynureninase (KYN) and kynurenine aminotransferase (KAT; Fig. 1) (23, 24). KYN catalyzes the hydrolysis of Kyn to anthranilic acid (AA) and 3-hydroxykynurenine (HK) to 3-hydroxyanthranilic acid (HaA). KAT catalyzes the conversion of kynurenine to kynurenic acid (KA) and HK to xanthurenic acid (XA) (25–27) (Fig. 1). Studies of the effects of vitamin B-6 deficiency on the activity of these 2 enzymes suggest that KYN is more susceptible than KAT (24).
FIGURE 1.

Diagram of the model. The rectangles represent substrates, and the ellipses contain the acronyms of enzymes. The 2 instances of KYN and KAT are labeled differently because they have different substrates and (possibly) different velocities. Transport of tryptophan from the gut to the serum and from the serum to the muscle or liver compartments is by the l-amino acid transporter. Steady state concentrations (μmol/L) are shown. AA, anthranilic acid; AFD, aryl formamidase; gtrp, tryptophan in the gut; HaA, 3-hydroxyanthranilic acid; HAO, 3-hydroxynalic acid oxygenase; HK, 3-hydroxykynurenine; KA, kynurenic acid; KAT, kynurenine aminotransferase; KMO, kynurenine 3-monooxygenase; Kyn, kynurenine; KYN, kynureninase; Lpool, tryptophan incorporated into proteins in liver; Mpool, tryptophan incorporated into proteins in muscle; Mtrp, tryptophan in muscle; NFK, N-formyl-kynurenine; Qa, quinolinic acid; pertrp, tryptophan in the peripheral circulation; PLP, pyridoxal 5′-phosphate; portrp, tryptophan in portal circulation; TDO, tryptophan 2,3- dioxygenase; trp, tryptophan; XA, xanthurenic acid.
The tryptophan load test has been used for many years as a functional assessment of vitamin B-6 status (28). Elevation of urinary concentrations of XA, Kyn, and HK after tryptophan loads in vitamin B-6–deficient conditions has been consistently reported in the literature (12, 25, 29, 30). Relatively little is known about the relationship between low cellular concentrations of PLP and the patterns of the other metabolites, including quinolinic acid (QA), HaA, AA, and KA under basal and post–tryptophan load conditions. Moreover, variation in the concentrations of the tryptophan catabolites is found in the published data. For example, Shibata et al. (29) reported an increase in urinary XA and a decrease in KA in vitamin B-6–deficient rats. Bender et al. (30) did not find a change in urinary KA in mice fed a vitamin B-6–deficient diet but reported an increase in XA, Kyn, and HK with HK > XA > Kyn. Yeh and Brown (12) found that, in rats, the percentage of tryptophan load excreted as XA, Kyn, HK, and QA was greater in the vitamin B-6–deficient group than in controls. However, the percentage excreted as KA and HaA did not change in the deficient group. In the same study, humans examined before and after 28 d of a vitamin B-6–deficient diet showed that the percentage of the tryptophan load excreted as XA, Kyn, HK, HaA, KA, and QA all increased in the order HK > Kyn > XA > KA > QA. Similar abnormalities in tryptophan metabolite excretion have been reported in OC users. Miller et al. (31) reported a greater increase in urinary excretion of XA, followed by HK, Kyn, HaA, and KA after a tryptophan load in OC users compared with controls. Donald and Bosse (32) found a higher increase in Kyn followed by XA, HK, and KA in OC users compared with nonusers after a tryptophan load.
The literature is incomplete regarding the basal range of tryptophan metabolites in fasting and fed states and the effect of vitamin B-6 status on their concentrations in plasma and urine. In addition, much of the literature regarding vitamin B-6 and tryptophan metabolism deals with responses to a tryptophan load, rather than with more physiologic dietary intake amounts. Mathematical models constructed by using published experimental kinetic data are excellent tools for the study of nutritional influences on a metabolic system (33). Importantly, such models often can extend, clarify, and provide new insights in metabolism (33, 34). Here, we present the development and initial applications of a mathematical model that simulates tryptophan metabolism via the Kyn pathway in the liver. Our model includes tryptophan input in the intestinal lumen, transport of tryptophan from intestinal lumen to circulation by the l-transporter, delivery of tryptophan to the liver, and exchange of circulating tryptophan with muscle and brain. The model was based on Michaelis-Menten constants, Vmax and Km, for all the enzymes and included provisions for PLP as a coenzyme for KYN and KAT and regulation and inhibition of enzymes.
We report here a mathematical model of whole-body tryptophan metabolism and the results of validation studies that show good agreement of the model with published data. In addition, we report the results of in silico experiments with the model to investigate the effects of various levels of vitamin B-6 deficiency, the effects of TDO induction, and the interaction of vitamin B-6 deficiency.
Materials and Methods
The model.
The model was built on the basis of information obtained from published mammalian data on concentrations of tryptophan and its metabolites in the liver, kinetics of the enzymes involved in the Kyn pathway, kinetics of the transporter involved in the transport of tryptophan to tissues, and enzyme regulatory mechanisms (e.g., substrate activation and inhibition of TDO by tryptophan). The kinetic constants used were obtained from different species, including rats, mice, and humans (Table 1). To better approximate whole-body tryptophan metabolism, and to be able to do experiments to examine the effects of different tryptophan inputs (e.g., daily average, oral tryptophan loads), we included 6 compartments in the model: liver, intestinal lumen (gut), portal circulation, peripheral circulation, muscle, and brain. The volume assumptions for the gut and liver compartments were 3 L each and for portal circulation, peripheral circulation, and muscle were 0.33 L, 2.66 L, and 30 L, respectively.
TABLE 1.
Parameter values used in the model1
| Parameter | Units | Model value | Literature values | References |
| l-transporter (Vtrpgs, Vtrpsm, Vtrpsl) | ||||
| Km | μmol/L | 64 | 64 | (41) |
| Vmax | μmol/(L · h) | 400, 840, 2000 | —2 | |
| VTDO | ||||
| Km | μmol/L | 43 | 24.6–250 | (50, 82, 83) |
| Vmax | μmol/(L · h) | 100.5 | — | |
| VAFD | ||||
| Km | μmol/L | 50 | 50–180 | (53, 54) |
| Vmax | μmol/(L · h) | 2000 | — | |
| VKMO | ||||
| Km | μmol/L | 25 | 25 | (84) |
| Vmax | μmol/(L · h) | 78.4 | — | |
| VKYN1 | ||||
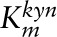
|
μmol/L | 240 | 240–1000 | (56, 85) |
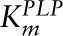
|
μmol/L | 0.88 | 0.88 | (24) |
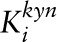
|
μmol/L | 20 | 20 | (57) |
| Vmax | μmol/(L · h) | 128.46 | — | |
| VKYN2 | ||||
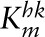
|
μmol/L | 3 | 3–45 | (57) (56, 85, 86) |
|
|
μmol/L | 0.88 | 0.88 | (24) |
|
|
μmol/L | 20 | 20 | (57) |
| Vmax | μmol/(L · h) | 512 | — | |
| VKAT1 | ||||
|
|
μmol/L | 4700 | 2000–5000 | (87–90) |
|
|
μmol/L | 0.85 | 0.85–7 | (24, 88) |
| Vmax | μmol/(L · h) | 1998 | — | |
| VKAT2 | ||||
|
|
μmol/L | 5000 | 2000–5000 | (87–90) |
|
|
μmol/L | 0.85 | 0.85–7 | (24, 88) |
| Vmax | μmol/(L · h) | 47,952 | — | |
| VHAO | ||||
|
|
μmol/L | 10 | 2–21 | (91, 92) |
| Vmax | μmol/(L · h) | 2000 | — | |
| Removal coefficients3 | ||||
| d3 | h−1 | 0.2 | ||
| d4 | h−1 | 3 | ||
| d5 | h−1 | 240 | ||
| d6 | h−1 | 8 | ||
| d7 | h−1 | 6 | ||
| d8 | h−1 | 40 | ||
| d9 | h−1 | 0.2 | ||
| Other coefficients3 | ||||
| k2 | h−1 | 0.0002 | ||
| k13 | h−1 | 1 | ||
| k14 | h−1 | 1.2 | ||
| m | h−1 | 7 |
Vmax values used in constructing the model and conducting simulations are shown (see text for explanations). AFD, aryl formamidase; HAO, 3-hydroxynalic acid oxygenase; hk, 3-hydroxykynurenine; KAT, kynurenine aminotransferase; KMO, kynurenine 3-monooxygenase; kyn, kynurenine; KYN, kynureninase; PLP, pyridoxal 5′-phosphate; TDO, tryptophan 2,3- dioxygenase; trpgs, tryptophan from gut to portal circulation; trpsl, tryptophan from portal circulation to liver; trpsm, tryptophan from peripheral circulation to muscle.
Dashes indicate that values for Vmax were not taken from the literature.
See text for Literature values.
The differential equations (Supplemental Table 1) were derived by using the following approach: If we suppose that tryptophan is being transported from compartment A to compartment B, if V is the rate of decrease in tryptophan concentration in A per unit of time, then the rate of increase of its concentration in B is:
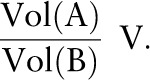 |
All of the explicit numbers in the differential equations are due to such volume factors. The parameter choices and references for reactions that have Michaelis-Menten kinetics are shown in Table 1 and are in either of the following standard forms:
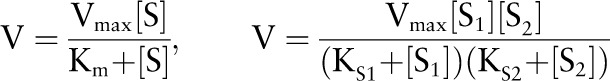 |
In the model, the gut compartment directly receives tryptophan from dietary intake or tryptophan loads. For the purpose of this model, the steady state concentration of tryptophan in the liver is maintained by not only dietary tryptophan but the tryptophan that comes from proteolysis in the tissues (muscle and liver). We did not consider other sources of amino acids or the effects of plasma albumin on tryptophan concentration. We considered both steady state and variable tryptophan inputs. The daily human intake of tryptophan, which can be calculated by using either catabolism measurements or intake from food, is ∼0.5 g/d (35). This is the equivalent of ∼2.5 mmol/L per day, or ∼100 μmol/h. The gut compartment in the model has a volume of 3 L, so our steady state input to the gut is 33.3 μmol/L per hour. Tryptophan (trp) is removed from the gut and enters the portal circulation by the l-transporter (Vtrpgs) with a Km = 64 μmol/L (36). We estimated the Vmax of the transporter to correspond to 33.3 μmol leaving the gut and entering the portal circulation per hour. Because the portal circulation is assumed to have a volume of 0.33 L, and the gut is assumed to have a volume of 3 L, the molar concentration of tryptophan in portal circulation increases by 9 times the amount that its molar concentration in the gut decreases, which explains the number 9 preceding Vtrpgs in equation 10.
We assumed that the plasma has a volume of 3 L and is divided into 2 compartments: the portal circulation (0.33 L) and the peripheral circulation, which has a volume of 2.66 L. We divided the plasma volume into these 2 compartments because it is known that the portal concentrations are substantially higher after meals (37, 38). During tryptophan loading experiments it is important that much of the tryptophan that is imported from the gut goes first to the portal circulation, then to the liver, and then to the peripheral circulation by mixing of the portal and peripheral circulations (equations 10 and 14). The model also provides for tryptophan to leave the liver and return to the portal circulation (the term k1 · trp in equation 1). Tryptophan is exported from the peripheral circulation into muscle and brain. The transport into the brain is small (39, 40), only ∼3–5% of the tryptophan whole-body tryptophan utilization, so we represent this simply as a sink for tryptophan indicated by the term k2 · pertrp in equation 14.
The terms for the mixing of the portal and peripheral circulations are derived as follows. The portal and peripheral circulations are connected as indicated in Figure 1. For any designated time, pertrp and portrp are the current concentrations of tryptophan in the 2 compartments, respectively. The portal flow of absorbed tryptophan is calculated as follows: In time Δt, the portal plasma advances a distance Δx into the peripheral compartment. Because the volume of the peripheral compartment is 2.66 L, then the new peripheral tryptophan concentration (newpertrp) is given by the following equation:
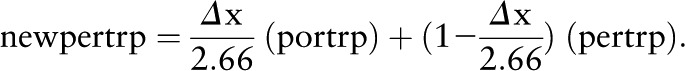 |
If m is the velocity of the fluid, then mΔt = Δx. So rearranging, we get
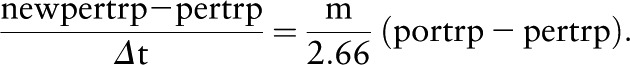 |
Taking the limit as Δt→0, we see that
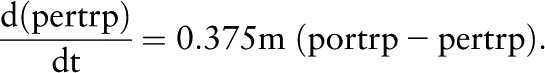 |
Similarly,
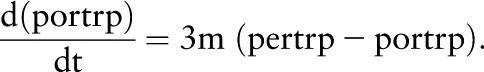 |
We choose the mixing constant m so that the portal and peripheral circulations equilibrate after ∼8 h (37).
The muscle compartment receives tryptophan from the peripheral circulation and exchanges free tryptophan with the tryptophan-incorporated into protein (Mpool). We include the muscle compartment in our model because the large amount of protein in muscle is a store of tryptophan and between meals some tryptophan leaves the muscle compartment into the plasma to maintain the plasma concentration. Thus, free tryptophan is used for protein synthesis and, conversely, free tryptophan becomes available from proteolysis. We model these processes as simple reactions whose rates are proportional to concentrations of substrates (tryptophan and protein, respectively). We do not attempt to model the details of protein synthesis, which involves necessarily all the other amino acids. Tryptophan is imported into the muscle compartment by the l-transporter, Vtrpsm, with Km = 64 μmol/L (41). To calculate the molarity of protein in the body, we used the estimate that 20% of the body is protein (42). For a 60-kg person, then 200 g/kg body mass is protein. Assuming a typical protein molecular weight of 50,000 (43), then 200 g is equivalent to 4 mmol, and therefore 200 g/L is ∼4 mmol/L or 4000 μmol/L. Our model calculated the steady state concentrations in muscle and liver to be 7232 and 6695 μmol/L, respectively, which is reasonable because muscle and liver should have higher protein concentrations than many other tissues.
To determine the rate of protein synthesis we proceed as follows. In adult rats, ∼5–12% of total protein weight is synthesized each day in the muscle (44–47). Thus, ∼0.05 × 7000 μmol/L should be synthesized in our muscle compartment per day or ∼15 μmol/L per hour. Although the rate of protein turnover in rats is higher than in humans (48), the steady state concentration is not affected by the rate of protein synthesis, which generally will be in balance with the rate of proteolysis. Therefore, a higher or lower protein synthesis rate did not affect the predictions of the model. In the model, protein synthesis (PRO) was described (depending on available tryptophan) by the following formula:
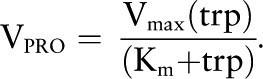 |
where we choose Km = 25 μmol/L, a typical cellular tryptophan concentration, and Vmax = 30 μmol/L per hour. Because free tryptophan in muscle is ∼25 μmol/L, the protein synthesis rate in the model is ∼15 μmol/L per hour.
In the liver, tryptophan is imported from the portal circulation by the l-transporter (Vtrpsl) and leaves the liver into the peripheral circulation via the term k1 · trp in equation 1. Here, tryptophan is not just catabolized but is used in a variety of reactions including protein synthesis. Thus, we have reactions representing the use of free tryptophan for protein synthesis and the release of free tryptophan from proteolysis (Lpool). This is important because these processes buffer the free tryptophan in the liver. Without these processes, free liver tryptophan would be excessively sensitive to input from the gut. We take these reactions to be the same as in the muscle compartment discussed above. The velocities of kynurenine 3-monooxygenase (VKMO), 3-hydroxyanthranilic acid (VHAO), kynurenine aminotransferase catalyzing Kyn→ KA (VKAT1), and kynurenine aminotransferase catalyzing HK→ XA (VKAT2) have standard Michaelis-Menten kinetics; constants are given in Table 1.
The enzyme TDO shows both substrate activation and substrate inhibition:
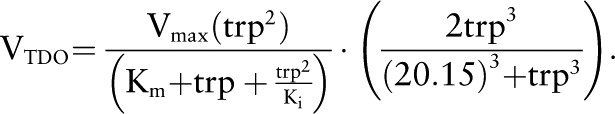 |
The first term expresses Michaelis-Menten kinetics modified for uncompetitive substrate inhibition (49). We take Km = 43 μmol/L from ref 50 and the inhibition constant Ki = 170 μmol/L from ref 51. The second term requires more discussion. Sainio and Sainio (52) showed that the enzyme TDO exhibits substantial substrate activation. Increasing tryptophan concentration causes the TDO to become more saturated with the iron-porphyrin–containing coenzyme, making the more active holoenzyme. This effect approximately doubles the activity of the enzyme because the tryptophan concentration is increased above normal by a factor of 4. In our model, the normal liver concentration of tryptophan is 20.2 μmol/L, so we chose the last factor in the formula for VTDO so that it is equal to 1 when tryptophan is normal and to ∼2 when tryptophan increases by a factor of 4.
VAFD.
The enzyme aryl formamidase (AFD) has a Km = 50 μmol/L (53, 54) for N-formyl-kynurenine (nfk) and shows product inhibition by Kyn with Ki = 250 μmol/L (55).
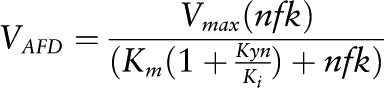 |
VKYN1.
The enzyme kynureninase (KYN) shows substrate inhibition for Kyn. We take the Km of the reaction for Kyn to be 240 μmol/L (56) and the inhibition constant Ki = 20 μmol/L from ref 57. PLP is an activator in these reactions. Thus, if A is an activator of an enzymatic reaction, the most natural way to model its effect is to multiply the Vmax of the reaction by a term of the form
This activation function is monotone increasing and saturates (at level c), both reasonable assumptions. Then, c/K is just the slope of the activation curve for low [A] and K is the level of the activator that produces half-maximal activation. The form of this term is exactly the same as if A were a substrate, and thus we believe that it is reasonable to call K a Km as has been done in the literature. Therefore, our PLP is a cofactor with a 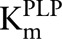 of 0.88 μmol/L (24).
of 0.88 μmol/L (24).
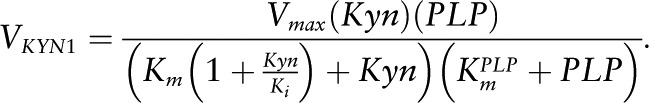 |
VKYN2.
Similarly, KYN shows a Km for HK of Km = 3 μmol/L (58).
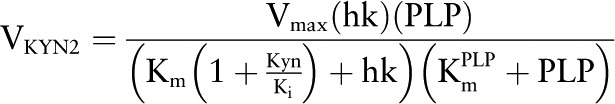 |
Finally, the major downstream metabolites of tryptophan metabolism in the liver, namely Kyn, KA, AA, HK, XA, HaA, and QA, are released from the liver into the circulation and are excreted into the urine in the kidney. We simplify this process by having a linear removal term in each of the differential equations 3–9 of the form diyi, where yi is the variable. Thus, predicted plasma concentrations will parallel liver concentrations of these metabolites. The coefficients di, whose values appear in Table 1, were chosen so these metabolites have their normal concentrations at steady state.
Results and Discussion
Predicted steady state concentrations and velocities.
The steady state concentrations of tryptophan and its metabolites are shown in Figure 1. The full names of the variables in the model are indicated in Table 2, and the 15 differential equations formulated for the reactions shown in Figure 1 are provided in Supplemental Table 1. The model prediction for steady state tryptophan concentration in the peripheral circulation was slightly lower than in the portal circulation, as found in refs 37 and 38. Predicted steady state concentrations of tryptophan and tryptophan metabolites in the liver were close to values reported in the literature, which included hepatic tryptophan of 12.3 μmol/L reported in ref 59 and 8.2 μmol/L for Kyn (59). We estimated from other published data the following concentrations for hepatic tryptophan metabolites: 0.06–3 μmol/L for HK (60–62), 0.16–2.5 μmol/L for KA (60, 62, 63), 1 μmol/L for HaA (60), 0.16 μmol/L for AA (62), and 0.13 μmol/L for XA (62). The velocity split at Kyn corresponds to the velocities of the catabolic reactions catalyzed by kynurenine 3-monooxygenase, kynurenine aminotransferase catalyzing Kyn→ KA, and KYN1. Model predictions for the fluxes of these reactions were ∼80, 10, and 10%, respectively, consistent with the reported dominance of the hydroxylation process forming HK (64, 65).
TABLE 2.
Names and abbreviations used for variables
| In equations | In text | Full name |
| trp | tryptophan | Free tryptophan in liver |
| nfk | NFK | N-formyl-kynurenine |
| kyn | Kyn | Kynurenine |
| Ka | KA | Kynurenic acid |
| aa | AA | Anthranilic acid |
| hk | HK | 3-Hydroxykynurenine |
| xa | XA | Xanthurenic acid |
| haa | HaA | 3-Hydroxyanthranilic acid |
| qa | QA | Quinolinic acid |
| portrp | portrp | Tryptophan in the portal circulation |
| lpool | Lpool | Tryptophan incorporated into proteins in liver |
| mtrp | Mtrp | Free tryptophan in muscle |
| mpool | Mpool | Tryptophan incorporated into proteins in muscle |
| pertrp | pertrp | Tryptophan in the peripheral circulation |
| gtrp | gtrp | Tryptophan in the gut |
Simulation: predicted dependence of steady state metabolite concentrations and metabolic rates on vitamin B-6 status.
Changes in urinary metabolites (Fig. 2) are presented as a function of decreasing relative cellular PLP concentrations. Relative values for vitamin B-6 status were 1 to 0.7 (adequate status), 0.7 to 0.3 (moderate deficiency), and 0.3 to 0.01 (deficiency). The results showed little or no change in the metabolites until cellular PLP concentrations reached ≤50% of the normal value. It is known that in low vitamin B-6 status an increase in urinary Kyn, XA, and HK occurs. In the model predictions, urinary HK increased in moderate deficiency and markedly increased in more severe deficiency, as reported previously (66). The model also predicted that HK increased to the greatest extent, with the increase of Kyn about half that of HK, whereas XA increased almost as much as Kyn, as reported earlier (10). Data on urinary excretion of KA and AA vary in the literature. The predictions of our model for these 2 metabolites suggested that a slight decrease occurred in moderate vitamin B-6 deficiency, which became more pronounced in severe deficiency. The model also predicted that HaA excretion increased slightly in moderate deficiency and then decreased slightly in more pronounced deficiency, as reported (9). These results can be explained by considering that KYN2 is largely saturated with PLP but KAT2 is not (presumably) at typical cellular PLP concentrations, so the first effect in vitamin B-6 deficiency is a reduction in KAT2 but not in KYN2 activity. Thus, urinary HK and HaA both increased. However, more severe vitamin B-6 deficiency and consequentially further cellular PLP depletion also caused the rate of KYN2 to be reduced, which yielded a reduction in HaA production and excretion. Concentrations of urinary tryptophan metabolites excreted in a 24-h period in adequate vitamin B-6 status (steady state) and vitamin B-6 deficiency (PLP concentration decreased to 5% of normal value in the model) were compared and are shown in Supplemental Table 2. HK, Kyn, XA, HaA, and QA excretion were 70, 10, 10, 4, and 3% higher and KA and AA were 27 and 30% lower, respectively, in deficiency compared with adequate status. Predictions of urinary excretion rates of total tryptophan catabolites with and without simulated tryptophan load were unchanged by the deficiency, even though a change in the proportion of the metabolites occurred. These predictions showed that overall tryptophan degradation flux via TDO was not affected by vitamin B-6 deficiency (Supplemental Table 2).
FIGURE 2.

Dependence of urinary metabolites on vitamin B-6 status. Vitamin B-6 deficiency was simulated by decreasing PLP concentration in the model. The relative vitamin B-6 status was designated as follows: adequate, 0.7 to 1; moderate deficiency, 0.3 to 0.7; and deficiency, 0.01 to 0.3. AA, anthranilic acid; HaA, 3-hydroxyanthranilic acid; HK, 3-hydroxykynurenine; KA, kynurenic acid; Kyn, kynurenine; PLP, pyridoxal 5′-phosphate; XA, xanthurenic acid.
Simulation: dynamic changes of tryptophan and tryptophan metabolites under tryptophan load with or without low vitamin B-6 status.
Measurements of Kyn metabolites after tryptophan loads have been used to assess vitamin B-6 deficiency (28). Here, we simulated a tryptophan load in the presence of adequate and low vitamin B-6 status (Figs. 3 and 4, respectively). The tryptophan load experiments were simulated by delivering an extra 15/24 of the normal total daily input of tryptophan for 1 h. Vitamin B-6 deficiency was simulated by decreasing PLP concentration to 5% of the normal value in the model. Peripheral plasma tryptophan increased from 30 to ∼125 μmol/L at ∼2 h after the load (Fig. 3), which is consistent with the findings by Vignau et al. (67) in which tryptophan concentration increased by ∼300% and peaked at ∼2 h after the oral tryptophan load. The plasma Kyn concentration increased by ∼600%, peaking at ∼3 h after the load in their study. In our simulation, Kyn excretion (Kynout) peaked at 2.5 to 3 h after the load but increased by only 280%. Møller (68) also measured plasma concentrations of tryptophan and Kyn and found that they peaked at 1.5 and 4 h after the load, respectively. In his study, plasma Kyn peaked at ∼10% of the tryptophan maximum, whereas our Kyn peak was ∼25% of the peak plasma tryptophan concentration. Finally, the author’s HK in the plasma also peaked at 4 h after the load and at approximately one-sixth of the Kyn concentration, whereas our simulation predicted an HK peak that was 70% of the plasma Kyn peak.
FIGURE 3.

Dynamic changes of tryptophan and tryptophan metabolites after simulation of a tryptophan load test. Oral tryptophan load was simulated by delivering an extra amount of the normal total daily input for 1 h. The word “out” denotes urinary metabolites. AA, anthranilic acid; bptrp, tryptophan in peripheral circulation; gut, tryptophan in gut; HaA, 3-hydroxyanthranilic acid; HK, 3-hydroxykynurenine; hptrp, tryptophan in portal circulation; KA, kynurenic acid; Kyn, kynurenine; Ltrp, tryptophan in liver; Mtrp, tryptophan in muscle; QA, quinolinic acid; XA, xanthurenic acid.
FIGURE 4.

Dynamic changes of tryptophan and tryptophan metabolites after simulation of a tryptophan load test in the presence of vitamin B-6 deficiency. The tryptophan load was simulated as described in Figure 3. Vitamin B-6 deficiency was simulated by decreasing PLP concentration to 5% of the normal value in the model. The word “out” denotes urinary metabolites. AA, anthranilic acid; bptrp, tryptophan in peripheral circulation; gut, tryptophan in gut; HaA, 3-hydroxyanthranilic acid; HK, 3-hydroxykynurenine; hptrp, tryptophan in portal circulation; KA, kynurenic acid; Kyn, kynurenine; Ltrp, tryptophan in liver; Mtrp, tryptophan in muscle; QA, quinolinic acid; PLP, pyridoxal 5′-phosphate; XA, xanthurenic acid.
Wolf et al. (69) measured many metabolites in the urine over a 24-h period throughout a tryptophan load of 9800 μmol. Their results showed that excretion of KA increased by 300%, whereas in our model it increased by 300%. Kyn increased by 166%, and in our model it increased by 250%; XA increased by almost 272%, whereas in our model it increased slightly more than a factor of 3 (200%); HK increased by almost 166%, whereas the model predicted an increased HK excretion of 250%; and HaA increased by 93%, which was similar to the model prediction.
In a simulated tryptophan load concurrent with vitamin B-6 deficiency (5% of normal PLP concentration), the main changes were the decrease in KA and the increases in Kyn, XA, and HK (Fig. 4). Urinary KA decreased by 25% with the deficiency, whereas Kyn, XA, and HK were higher by 15, 10, and 70%, respectively, compared with their response after the tryptophan load in adequate vitamin B-6 status. This was also demonstrated by predictions of 24-h urinary excretion calculated as an AUC (Supplemental Table 2).
Simulation: dynamic changes of tryptophan and tryptophan metabolites due to meals.
Amino acid concentrations after meals and the effects of the composition of the meals on those concentrations have been a target of study for many years. It is known that the protein content of a meal and the secretion of insulin after a meal influence plasma amino acid concentrations (70). For example, insulin has been shown to increase the uptake of branched-chain amino acids by the muscle decreasing their concentration in plasma (70). A meal with protein increases amino acid concentrations in plasma, whereas a protein-free diet causes a decrease in the concentrations of all amino acids (71, 72).
Simulations of tryptophan inputs due to meals and the dynamic changes of tryptophan and its metabolites were conducted over a period of 24 h (Fig. 5). To simulate meals, we introduced into the gut one-fourth of the daily dose of tryptophan between 0700 and 0800 h, one-fourth of the daily dose of tryptophan between 1200 and 1300 h, and one-half of the daily dose of tryptophan from 1800 to 1900 h. The model showed increased tryptophan in plasma after each meal and an increase in its metabolite concentrations due to the increase in its oxidation. The model also showed a decline in plasma tryptophan overnight from ∼30 to 20 μmol/L, consistent with the decline reported after a meal with no protein (73).
FIGURE 5.

Dynamic changes of tryptophan and tryptophan metabolites after simulation of tryptophan input due to meals in a period of 24 h. The tryptophan inputs were simulated by delivering one-fourth of the daily dose of tryptophan between 0700 and 0800 h, one-fourth of the daily dose of tryptophan between 1200 and 1300 h, and one-half of the daily dose of tryptophan from 1800 to 1900 h. The word “out” denotes urinary metabolites. AA, anthranilic acid; bptrp, tryptophan in peripheral circulation; gut, tryptophan in gut; HaA, 3-hydroxyanthranilic acid; HK, 3-hydroxykynurenine; hptrp, tryptophan in portal circulation; KA, kynurenic acid; Kyn, kynurenine; Ltrp, tryptophan in liver; Mtrp, tryptophan in muscle; QA, quinolinic acid; XA, xanthurenic acid.
Simulation: effects of induction of tryptophan oxidation and OC use on tryptophan metabolites.
The increase of tryptophan oxidation due to an increase in the activity of TDO, the first enzyme in the hepatic Kyn pathway, has been shown by many investigators. Knox and Mehler (17) showed the induction of TDO activity by its substrate tryptophan, and Schütz et al. (74) showed that glucocorticoids increase the activity of the enzyme by increasing the synthesis of new mRNA and TDO protein. Additionally, the effects of estrogen on the activity of TDO were shown by Braidman and Rose (22) when they found that administration of 10 μg estradiol benzoate to female rats yielded an increase of TDO activity by 90%. Thus, it is believed that the use of OC agents exerts a similar induction of TDO.
We first simulated the induction of TDO activity by increasing the TDO Vmax to 200% of the basal level (i.e., a doubling). The effects of increased TDO in the urinary excretion of Kyn metabolites in urine are shown in Table 3. Here, the model predictions were consistent with published findings from a study of TDO induction by hydrocortisone, which yielded an increase of urinary Kyn (16). Second, we simulated the increase of TDO activity in the presence of low vitamin B-6 status as has been reported for OC users (Fig. 6) (75, 76). In these simulations, the Vmax of TDO was increased by 80% and PLP concentration was lowered to 50% of the normal value in the model (moderate deficiency). The model predicted that TDO induction concurrent with vitamin B-6 deficiency augments the elevation of urinary Kyn, HK, and XA and a decrease in the other metabolites that are otherwise associated with vitamin B-6 deficiency. These effects were more pronounced when a simulation of tryptophan load was given in this simulation of OC users, as has been reported in the literature (32).
TABLE 3.
Model prediction of the effect of extent of induction of TDO activity on urinary excretion of tryptophan metabolites1
| TDO percentage of basal activity |
|||||
| Trp metabolites in urine | 100 (baseline) | 120 | 150 | 180 | 200 |
| μmol/L | |||||
| Kyn | 2.00 | 2.12 | 2.25 | 2.35 | 2.41 |
| HK | 1.43 | 1.52 | 1.62 | 1.70 | 1.74 |
| XA | 1.40 | 1.48 | 1.58 | 1.65 | 1.69 |
| KA | 3.44 | 3.64 | 3.87 | 4.05 | 4.15 |
| HaA | 3.21 | 3.33 | 3.47 | 3.57 | 3.62 |
| AA | 3.37 | 3.49 | 3.63 | 3.73 | 3.79 |
| QA | 15.9 | 16.4 | 17.1 | 17.5 | 17.7 |
AA, anthranilic acid; HaA, 3-hydroxyanthranilic acid; HK, 3-hydroxykynurenine; KA, kynurenic acid; Kyn, kynurenine; QA, quinolinic acid; TDO, tryptophan 2,3-dioxygenase; Trp, tryptophan; XA, xanthurenic acid.
FIGURE 6.

Relative concentrations of tryptophan metabolites after simulation of low vitamin B-6 concentrations, OC use, and tryptophan load test. Vitamin B-6 deficiency was simulated by lowering PLP to 50% of the normal value in the model. OC usage was simulated by inducing TDO activity by 80% and decreasing PLP by 50% of normal value in the model. The trytophan load test was simulated as described in Figure 3. AA, anthranilic acid; HaA, 3-hydroxyanthranilic acid; HK, 3-hydroxykynurenine; KA, kynurenic acid; Kyn, kynurenine; OC, oral contraceptive; PLP, pyridoxal 5′-phosphate; TDO, tryptophan 2,3-dioxygenase; Trp, tryptophan; XA, xanthurenic acid.
Further considerations regarding the model.
We presented here a mathematical model that simulates tryptophan metabolism via the Kyn pathway. The model is compartmentalized in tissues that are involved in tryptophan metabolism, including intestinal lumen, liver, muscle, and brain. The model uses kinetic constant values and concentrations that are based on values reported in the literature for rodents and humans. We used values reported in rodents when information on metabolite concentrations or enzyme kinetics in humans was not available. This approach has been previously used in the development of other metabolism models in which predictions have been highly satisfactory [e.g., (33,34)]. Although this model uses simplified assumptions (e.g., the metabolite concentrations in plasma are considered to parallel those in liver and the excretion of the metabolites in urine is assumed to be linear, thus skipping the pass of the metabolites through the blood and the uptake by the kidney), we consider that this does not affect the predictions of the model because it successfully reproduces experimental data.
We focused most of our experiments in simulating the effects of induced low vitamin B-6 on the Kyn pathway in the liver, which allowed comparisons of model predictions with the results of studies in which dietary vitamin B-6 deficiency has been associated with an impaired Kyn pathway. Early studies showed the effects of dietary vitamin B-6 deficiency on trytophan metabolism by measuring tryptophan metabolite concentrations in urine after tryptophan loads. However, Schaeffer et al. (77) showed that a tryptophan load test of 24.5 μmol (5 mg) of l-tryptophan/100 g body weight was not effective in detecting vitamin B-6 marginal deficiency in adult female Long-Evans rats. Our model was useful in assessing changes in HK, KA, and AA in the marginal (moderate) deficiency range, which shows the usefulness of the model to study the effects of a mild vitamin B-6 deficiency. Indeed, the model predictions agreed well with human data obtained in our laboratory from marginal vitamin B-6–deficient participants in whom plasma HK was significantly increased and KA was significantly decreased without a prior tryptophan load (V. da Silva, L. Rios-Avila, Y. Lamers, M. Ralat, Ø. Midttun, E.P. Quinlivan, T.J. Garrett, B. Coats, M.N. Shankar, S.S. Percival, Y.-Y. Chi, K.E. Muller, P.M. Ueland, P.W. Stacpoole, J.F. Gregory, unpublished results). However, to observe the effects on the other metabolites, the model requires a simulation of a more pronounced deficiency.
On the basis of the structure of the Kyn pathway (Fig. 1) we can predict that the urinary excretion of KA and AA would decline during vitamin B-6 deficiency and that Kyn would increase because the exit processes are being restricted. The cases of XA and HaA in vitamin B-6 deficiency are more interesting because there are 2 conflicting effects. First, HK will clearly increase because the flux in will be higher and KAT2 and KYN2 will be somewhat dysfunctional due to reduced PLP availability. The effects of vitamin B-6 deficiency on XA and HaA are due to the reduced activity of both KAT2 and KYN2. These concentrations are not independent because as KYN2 becomes more dysfunctional, the resulting increase in HK will tend to increase XA. In general, the lack of effect on vitamin B-6 status over a large range is because the Km values of KYN2 and KAT2 for PLP are very low (0.88 and 0.85 μmol/L, respectively) so that their rates are only weakly affected by a reduction in cellular PLP concentration that is generally well above the Km values.
We used the model to simulate tryptophan load tests in the presence or absence of vitamin B-6 deficiency. In adequate vitamin B-6 status, the tryptophan load induced an increase in all the tryptophan metabolites in blood and urine as expected. In the presence of vitamin B-6 deficiency, the increase in HK, Kyn, and XA was consistent with the effects of deficiency on the PLP-dependent enzymes. However, our XA response to vitamin B-6 deficiency (under tryptophan load or not) does not seem to be as high as in the published literature. The effect of vitamin B-6 deficiency on XA may be underestimated in the model. Low PLP has been reported to yield increased expression of KAT (78), which would tend to compensate for the loss of KAT activity due to the reduced coenzyme availability.
Our model is also useful to predict effects of induction or inhibition of enzyme activities. For example, our model was used to simulate the induction of the activity of TDO as reported for glucocorticoids and sex hormones (18, 22) and the induction of TDO activity concurrent with low plasma concentrations of PLP as has been reported in OC users (22, 76, 79). Inhibitory effects of enzymes also can be simulated, thus providing useful information to interpret abnormalities in the metabolites under different conditions including dietary and genetic disorders.
A computer simulation of tryptophan metabolism via the Kyn pathway in the liver was previously published (80). However, this model did not account for the distribution of metabolites in other tissues nor for enzyme regulation. This previous model predicted the effects of vitamin B-6 adequacy and deficiency (incorrectly designated by that author as “pyridoxal” deficiency) on metabolite concentrations in conditions of presence or absence of the PLP coenzyme. Changes in enzyme activities were simulated by eliminating the coenzyme-dependent rate constants. The results showed a decrease in HaA, KA, and XA after simulations in which both KYN and KAT activities were lost. However, because the enzymes are not equally affected by vitamin B-6 deficiency, simulation of the reduction in the activity of each enzyme separately showed different results for each metabolite analyzed. For example, HaA increased when KYN sensitivity for the coenzyme was lost and the activity of KYN aminotransferase was kept normal, whereas it decreased when KAT activity was reduced and KYN was kept normal. In that model, Kyn activity was not affected by the reduction in the enzyme activities. Our model predicted an increase in Kyn, HK, XA, and HaA and a decrease in KA and AA as a result of the simulation of a general vitamin B-6 deficiency in which the activities of the enzymes were not completely reduced but rather responded to the varying PLP concentrations.
In summary, the results from the model simulations reliably reproduce experimental data and confirm the validation of the model to be used as a tool to study tryptophan metabolism. Moreover, the model predictions confirmed the increases in Kyn, HK, and XA and the decreases in AA and KA in vitamin B-6 deficiency. A further application of the model will be its use as a platform to develop a model to study extrahepatic tryptophan metabolism, the specific role of IDO, and effects of potential redistribution of PLP in inflammatory tissues (81).
Supplementary Material
Acknowledgments
J.F.G., H.F.N., and M.C.R. designed the study; H.F.N. and M.C.R. constructed the model; H.F.N., M.C.R., and L.R.-A. conducted simulation studies; H.F.N., M.C.R., L.R.-A., and J.F.G. wrote the manuscript; J.F.G had the primary responsibility for the final content; and all authors contributed data on which the model is based. All authors read and approved the final manuscript.
Footnotes
Abbreviations used: AA, anthranilic acid; HaA, 3-hydroxyanthranilic acid; HK, 3-hydroxykynurenine; IDO, indoleamine 2,3-dioxygenase; IFN, interferon; KA, kynurenic acid; KAT, kynurenine aminotransferase; KAT2, kynurenine aminotransferase catalyzing HK→ XA; Kyn, kynurenine; KYN, kynureninase; KYN1, kynureninase catalyzing Kyn→ AA; KYN2, kynureninase catalyzing HK→ HaA; OC, oral contraceptive; PLP, pyridoxal 5′-phosphate; QA, quinolinic acid; TDO, tryptophan 2,3-dioxygenase; XA, xanthurenic acid.
Literature Cited
- 1.Wolf H. Studies on tryptophan metabolism in man: the effects of hormones and vitamin B6 on urinary excretion of metabolites of the kynurenine pathway. Scand J Clin Lab Invest Suppl. 1974;33:11–87. [PubMed] [Google Scholar]
- 2.Oxenkrug GF. Interferon-gamma-inducible kynurenines/pteridines inflammation cascade: implications for aging and aging-associated psychiatric and medical disorders. J Neural Transm. 2011;118:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suzuki Y, Suda T, Furuhashi K, Suzuki M, Fujie M, Hahimoto D, Nakamura Y, Inui N, Nakamura H, Chida K. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung Cancer. 2010;67:361–5. [DOI] [PubMed] [Google Scholar]
- 4.Sprince H, Lowy RS, Folsome CE, Behrman JS. Studies on the urinary excretion of “xanthurenic acid” during normal and abnormal pregnancy: a survey of the excretion of “xanthurenic acid” in normal nonpregnant, normal pregnant, pre-eclamptic, and eclamptic women. Am J Obstet Gynecol. 1951;62:84–92. [DOI] [PubMed] [Google Scholar]
- 5.Rose DP, Braidman IP. Excretion of tryptophan metabolites as affected by pregnancy, contraceptive steroids, and steroid hormones. Am J Clin Nutr. 1971;24:673–83. [DOI] [PubMed] [Google Scholar]
- 6.Rose DP. Excretion of xanthurenic acid in the urine of women taking progestogen-oestrogen preparations. Nature. 1966;210:196–7. [DOI] [PubMed] [Google Scholar]
- 7.Rose DP, Adams PW. Oral contraceptives and tryptophan metabolism: effects of oestrogen in low dose combined with a progestagen and of a low-dose progestagen (megestrol acetate) given alone. J Clin Pathol. 1972;25:252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bender DA, Tagoe CE, Vale JA. Effects of oestrogen administration on vitamin B6 and tryptophan metabolism in the rat. Br J Nutr. 1982;47:609–14. [DOI] [PubMed] [Google Scholar]
- 9.Price SA, Rose DP, Toseland PA. Effects of dietary vitamin B 6 deficiency and oral contraceptives on the spontaneous urinary excretion of 3-hydroxyanthranilic acid. Am J Clin Nutr. 1972;25:494–8. [DOI] [PubMed] [Google Scholar]
- 10.Yess N, Price JM, Brown RR, Swan PB, Linkswiler H. Vitamin B6 depletion in man: urinary excretion of tryptophan metabolites. J Nutr. 1964;84:229–36. [DOI] [PubMed] [Google Scholar]
- 11.Brown RR, Yess N, Price JM, Linkswiler H, Swan P, Hankes LV. Vitamin B6 depletion in man: urinary excretion of quinolinic acid and niacin metabolites. J Nutr. 1965;87:419–23. [DOI] [PubMed] [Google Scholar]
- 12.Yeh JK, Brown RR. Effects of vitamin B-6 deficiency and tryptophan loading on urinary excretion of tryptophan metabolites in mammals. J Nutr. 1977;107:261–71. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka T, Knox WE. The nature and mechanism of the tryptophan pyrrolase (peroxidase-oxidase) reaction of pseudomonas and of rat liver. J Biol Chem. 1959;234:1162–70. [PubMed] [Google Scholar]
- 14.Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991;5:2516–22. [PubMed] [Google Scholar]
- 15.Shimizu T, Nomiyama S, Hirata F, Hayaishi O. Indoleamine 2,3-dioxygenase: purification and some properties. J Biol Chem. 1978;253:4700–6. [PubMed] [Google Scholar]
- 16.Altman K, Greengard O. Correlation of kynurenine excretion with liver tryptophan pyrrolase levels in disease and after hydrocortisone induction. J Clin Invest. 1966;45:1527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knox WE, Mehler AH. The adaptive increase of the tryptophan peroxidase-oxidase system of liver. Science. 1951;113:237–8. [DOI] [PubMed] [Google Scholar]
- 18.Schutz G, Killewich L, Chen G, Feigelson P. Control of the mRNA for hepatic tryptophan oxygenase during hormonal and substrate induction. Proc Natl Acad Sci USA. 1975;72:1017–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knox WE, Auerbach VH. The hormonal control of tryptophan peroxidase in the rat. J Biol Chem. 1955;214:307–13. [PubMed] [Google Scholar]
- 20.Wolf H, Brown RR. Studies on tryptophan metabolism in male subjects treated with hydrocortisone. J Clin Endocrinol Metab. 1971;33:838–43. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura T, Shinno H, Ichihara A. Insulin and glucagon as a new regulator system for tryptophan oxygenase activity demonstrated in primary cultured rat hepatocytes. J Biol Chem. 1980;255:7533–5. [PubMed] [Google Scholar]
- 22.Braidman IP, Rose DP. Effects of sex hormones on three glucocorticoid-inducible enzymes concerned with amino acid metabolism in rat liver. Endocrinology. 1971;89:1250–5. [DOI] [PubMed] [Google Scholar]
- 23.Dalgliesh CE, Knox WE, Neuberger A. Intermediary metabolism of tryptophan. Nature. 1951;168:20–2. [DOI] [PubMed] [Google Scholar]
- 24.Ogasawara N, Hagino Y, Kotake Y. Kynurenine-transaminase, kynureninase and the increase of xanthurenic acid excretion. J Biochem. 1962;52:162–6. [DOI] [PubMed] [Google Scholar]
- 25.Takeuchi F, Shibata Y. Kynurenine metabolism in vitamin-B-6-deficient rat liver after tryptophan injection. Biochem J. 1984;220:693–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiss O, Fuchs H. [Splitting of kynurenine, oxykynurenine and related substances by the rat liver enzyme.] Experientia. 1950;6:472–3. [DOI] [PubMed] [Google Scholar]
- 27.Nakatani M, Morimoto M, Noguchi T, Kido R. Subcellular distribution and properties of kynurenine transaminase in rat liver. Biochem J. 1974;143:303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown A. The tryptophan load test as an index of vitamin B-6 nutrition. In: Leklem JE, Reynolds RD, editors. Methods in vitamin B-6 nutrition: analysis and status assessment. New York: Plenum Press; 1981. p. 321–340. [Google Scholar]
- 29.Shibata K, Mushiage M, Kondo T, Hayakawa T, Tsuge H. Effects of vitamin B6 deficiency on the conversion ratio of tryptophan to niacin. Biosci Biotechnol Biochem. 1995;59:2060–3. [DOI] [PubMed] [Google Scholar]
- 30.Bender DA, Njagi EN, Danielian PS. Tryptophan metabolism in vitamin B6-deficient mice. Br J Nutr. 1990;63:27–36. [DOI] [PubMed] [Google Scholar]
- 31.Miller LT, Benson EM, Edwards MA, Young J. Vitamin B6 metabolism in women using oral contraceptives. Am J Clin Nutr. 1974;27:797–805. [DOI] [PubMed] [Google Scholar]
- 32.Donald EA, Bosse TR. The vitamin B6 requirement in oral contraceptive users. II. Assessment by tryptophan metabolites, vitamin B6, and pyridoxic acid levels in urine. Am J Clin Nutr. 1979;32:1024–32. [DOI] [PubMed] [Google Scholar]
- 33.Reed MC, Nijhout HF, Neuhouser ML, Gregory JF, 3rd, Shane B, James SJ, Boynton A, Ulrich CM. A mathematical model gives insights into nutritional and genetic aspects of folate-mediated one-carbon metabolism. J Nutr. 2006;136:2653–61. [DOI] [PubMed] [Google Scholar]
- 34.Nijhout HF, Gregory JF, Fitzpatrick C, Cho E, Lamers KY, Ulrich CM, Reed MC. A mathematical model gives insights into the effects of vitamin B-6 deficiency on 1-carbon and glutathione metabolism. J Nutr. 2009;139:784–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pollack R, Kravitz E. Effects of tryptophan ingestion. J Nutr. 1987;117:1315–6. [DOI] [PubMed] [Google Scholar]
- 36.Kilberg M, Haussinger D. Mammalian amino acid transport, mechanisms and control. New York: Plenum Press; 1992.
- 37.Reverter M, Lundh T, Gonda HL, Lindberg JE. Portal net appearance of amino acids in growing pigs fed a barley-based diet with inclusion of three different forage meals. Br J Nutr. 2000;84:483–94. [PubMed] [Google Scholar]
- 38.Karlsson A, Eliason EJ, Mydland LT, Farrell AP, Kiessling A. Postprandial changes in plasma free amino acid levels obtained simultaneously from the hepatic portal vein and the dorsal aorta in rainbow trout (Oncorhynchus mykiss). J Exp Biol. 2006;209:4885–94. [DOI] [PubMed] [Google Scholar]
- 39.Jans LA, Lieben CK, Smits LT, Blokland A. Pharmacokinetics of acute tryptophan depletion using a gelatin-based protein in male and female Wistar rats. Amino Acids. 2009;37:349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richard DM, Dawes MA, Mathias CW, Acheson A, Hill-Kapturczak N, Dougherty DM. L-tryptophan: basic metabolic functions, behavioral research and therapeutic indications. Int J Tryptophan Res. 2009;2:45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saito K, Nagamura Y, Ohta Y, Sasaki E, Ishiguro I. Characterization of the L-tryptophan transport system in the liver of growing rats. Life Sci. 1991;49:527–34. [DOI] [PubMed] [Google Scholar]
- 42.Forbes RM, Cooper AR, Mitchell HH. The composition of the adult human body as determined by chemical analysis. J Biol Chem. 1953;203:359–66. [PubMed] [Google Scholar]
- 43.Lodish HBA, Matsudaira P, Kaiser C, Krieger M, Scott M, Zipursky SL, Darnell J. Molecular cell biology. 5th ed. New York: W.H. Freeman and Company; 2003.
- 44.Preedy VR, Garlick PJ. Protein synthesis in skeletal muscle of the perfused rat hemicorpus compared with rates in the intact animal. Biochem J. 1983;214:433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dardevet D, Sornet C, Bayle G, Prugnaud J, Pouyet C, Grizard J. Postprandial stimulation of muscle protein synthesis in old rats can be restored by a leucine-supplemented meal. J Nutr. 2002;132:95–100. [DOI] [PubMed] [Google Scholar]
- 46.Glick Z, McNurlan MA, Garlick PJ. Protein synthesis rate in liver and muscle of rats following four days of overfeeding. J Nutr. 1982;112:391–7. [DOI] [PubMed] [Google Scholar]
- 47.Garlick PJ, Millward DJ, James WP. The diurnal response of muscle and liver protein synthesis in vivo in meal-fed rats. Biochem J. 1973;136:935–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phillips SM, Glover EI, Rennie MJ. Alterations of protein turnover underlying disuse atrophy in human skeletal muscle. J Appl Physiol. 2009;107:645–54. [DOI] [PubMed] [Google Scholar]
- 49.Reed MC, Lieb A, Nijhout HF. The biological significance of substrate inhibition: a mechanism with diverse functions. Bioessays. 2010;32:422–9. [DOI] [PubMed] [Google Scholar]
- 50.Ferry G, Ubeaud C, Lambert PH, Bertin S, Coge F, Chomarat P, Delagrange P, Serkiz B, Bouchet JP, Truscott RJ, et al. Molecular evidence that melatonin is enzymatically oxidized in a different manner than tryptophan: investigations with both indoleamine 2,3-dioxygenase and myeloperoxidase. Biochem J. 2005;388:205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu C, Lin Y, Yeh SR. Inhibitory substrate binding site of human indoleamine 2,3-dioxygenase. J Am Chem Soc. 2009;131:12866–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sainio EL, Sainio P. Comparison of effects of nicotinic acid or tryptophan on tryptophan 2,3-dioxygenase in acute and chronic studies. Toxicol Appl Pharmacol. 1990;102:251–8. [DOI] [PubMed] [Google Scholar]
- 53.Shinohara R, Ishiguro I. The purification and properties of formamidase from rat liver. Biochim Biophys Acta. 1970;198:324–31. [DOI] [PubMed] [Google Scholar]
- 54.Pabarcus MK, Casida JE. Cloning, expression, and catalytic triad of recombinant arylformamidase. Protein Expr Purif. 2005;44:39–44. [DOI] [PubMed] [Google Scholar]
- 55.Menge U. [Formamidase—microheterogeneity, catalytic properties and inhibitors (author's translation).] Hoppe Seylers Z Physiol Chem. 1979;360:185–96. [PubMed] [Google Scholar]
- 56.Takeuchi F, Otsuka H, Shibata Y. Purification and properties of kynureninase from rat liver. J Biochem. 1980;88:987–94. [DOI] [PubMed] [Google Scholar]
- 57.Walsh HA, O'Shea KC, Botting NP. Comparative inhibition by substrate analogues 3-methoxy- and 3-hydroxydesaminokynurenine and an improved 3 step purification of recombinant human kynureninase. BMC Biochem. 2003;4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walsh HA, Botting NP. Purification and biochemical characterization of some of the properties of recombinant human kynureninase. Eur J Biochem. 2002;269:2069–74. [DOI] [PubMed] [Google Scholar]
- 59.Mason M, Berg CP. The metabolism of d- and l-tryptophan and d- and l-kynurenine by liver and kidney preparations. J Biol Chem. 1952;195:515–24. [PubMed] [Google Scholar]
- 60.Badawy AA, Bano S, Steptoe A. Tryptophan in alcoholism treatment I: kynurenine metabolites inhibit the rat liver mitochondrial low Km aldehyde dehydrogenase activity, elevate blood acetaldehyde concentration and induce aversion to alcohol. Alcohol Alcohol. 2011;46:651–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu P, Di Prospero NA, Sapko MT, Cai T, Chen A, Melendez-Ferro M, Du F, Whetsell WO, Jr, Guidetti P, Schwarcz R, et al. Biochemical and phenotypic abnormalities in kynurenine aminotransferase II-deficient mice. Mol Cell Biol. 2004;24:6919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pawlak D, Tankiewicz A, Matys T, Buczko W. Peripheral distribution of kynurenine metabolites and activity of kynurenine pathway enzymes in renal failure. J Physiol Pharmacol. 2003;54:175–89. [PubMed] [Google Scholar]
- 63.Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Grant R. Changes in kynurenine pathway metabolism in the brain, liver and kidney of aged female Wistar rats. FEBS J. 2011;278:4425–34. [DOI] [PubMed] [Google Scholar]
- 64.Bender DA, McCreanor GM. The preferred route of kynurenine metabolism in the rat. Biochim Biophys Acta. 1982;717:56–60. [DOI] [PubMed] [Google Scholar]
- 65.Bender DA, McCreanor GM. Kynurenine hydroxylase: a potential rate-limiting enzyme in tryptophan metabolism. Biochem Soc Trans. 1985;13:441–3. [DOI] [PubMed] [Google Scholar]
- 66.Midttun O, Ulvik A, Ringdal Pedersen E, Ebbing M, Bleie O, Schartum-Hansen H, Nilsen RM, Nygard O, Ueland PM. Low plasma vitamin B-6 status affects metabolism through the kynurenine pathway in cardiovascular patients with systemic inflammation. J Nutr. 2011;141:611–7. [DOI] [PubMed] [Google Scholar]
- 67.Vignau J, Soichot M, Imbenotte M, Jacquemont MC, Danel T, Vandamme M, Lhermitte M, Allorge D. Impact of tryptophan metabolism on the vulnerability to alcohol-related blackouts and violent impulsive behaviours. Alcohol Alcohol. 2010;45:79–88. [DOI] [PubMed] [Google Scholar]
- 68.Møller SE. Pharmacokinetics of tryptophan, renal handling of kynurenine and the effect of nicotinamide on its appearance in plasma and urine following L-tryptophan loading of healthy subjects. Eur J Clin Pharmacol. 1981;21:137–42. [DOI] [PubMed] [Google Scholar]
- 69.Wolf H, Brown RR, Arend RA. The kynurenine load test, an adjunct to the tryptophan load test. Scand J Clin Lab Invest. 1980;40:9–14. [DOI] [PubMed] [Google Scholar]
- 70.Schweiger U, Warnhoff M, Pahl J, Pirke KM. Effects of carbohydrate and protein meals on plasma large neutral amino acids, glucose, and insulin plasma levels of anorectic patients. Metabolism. 1986;35:938–43. [DOI] [PubMed] [Google Scholar]
- 71.Vaughan DA, Womack M, McClain PE. Plasma free amino acid levels in human subjects after meals containing lactalbumin, heated lactalbumin, or no protein. Am J Clin Nutr. 1977;30:1709–12. [DOI] [PubMed] [Google Scholar]
- 72.Bergström J, Furst P, Vinnars E. Effect of a test meal, without and with protein, on muscle and plasma free amino acids. Clin Sci (Lond). 1990;79:331–7. [DOI] [PubMed] [Google Scholar]
- 73.Fernstrom JD, Wurtman RJ, Hammarstrom-Wiklund B, Rand WM, Munro HN, Davidson CS. Diurnal variations in plasma concentrations of tryptophan, tryosine, and other neutral amino acids: effect of dietary protein intake. Am J Clin Nutr. 1979;32:1912–22. [DOI] [PubMed] [Google Scholar]
- 74.Schütz G, Beato M, Feigelson P. Steroid induction of the mRNA for hepatic tryptophan oxygenase. Hamatol Bluttransfus. 1974;14:293–9. [PubMed] [Google Scholar]
- 75.Brown RR, Rose DP, Leklem JE, Linkswiler H, Anand R. Urinary 4-pyridoxic acid, plasma pyridoxal phosphate, and erythrocyte aminotransferase levels in oral contraceptive users receiving controlled intakes of vitamin B6. Am J Clin Nutr. 1975;28:10–9. [DOI] [PubMed] [Google Scholar]
- 76.Salkeld RM, Knorr K, Korner WF. The effect of oral contraceptives on vitamin B6 status. Clin Chim Acta. 1973;49:195–9. [DOI] [PubMed] [Google Scholar]
- 77.Schaeffer MC, Sampson DA, Skala JH, O'Connor DK, Gretz D. Insensitivity of the tryptophan-load test to marginal vitamin B-6 intake in rats. J Nutr. 1991;121:1627–34. [DOI] [PubMed] [Google Scholar]
- 78.van de Kamp JL, Smolen A. Response of kynurenine pathway enzymes to pregnancy and dietary level of vitamin B-6. Pharmacol Biochem Behav. 1995;51:753–8. [DOI] [PubMed] [Google Scholar]
- 79.Lumeng L, Cleary RE, Li TK. Effect of oral contraceptives on the plasma concentration of pyridoxal phosphate. Am J Clin Nutr. 1974;27:326–33. [DOI] [PubMed] [Google Scholar]
- 80.Heinmets F. Computer simulation and analysis of tryptophan metabolism via kynurenine pathway in liver. Comput Biol Med. 1974;1:323–36. [DOI] [PubMed] [Google Scholar]
- 81.Paul L, Ueland PM, Selhub J. Mechanistic perspective on the relationship between pyridoxal 5′-phosphate and inflammation. Nutr Rev. 2013;71:239–44. [DOI] [PubMed] [Google Scholar]
- 82.Kudo Y, Boyd CA. Human placental indoleamine 2,3-dioxygenase: cellular localization and characterization of an enzyme preventing fetal rejection. Biochim Biophys Acta. 2000;1500:119–24. [DOI] [PubMed] [Google Scholar]
- 83.Fukumura E, Sugimoto H, Misumi Y, Ogura T, Shiro Y. Cooperative binding of L-trp to human tryptophan 2,3-dioxygenase: resonance Raman spectroscopic analysis. J Biochem. 2009;145:505–15. [DOI] [PubMed] [Google Scholar]
- 84.Saito Y, Hayaishi O, Rothberg S. Studies on oxygenases; enzymatic formation of 3-hydroxy-L-kynurenine from L-kynurenine. J Biol Chem. 1957;229:921–34. [PubMed] [Google Scholar]
- 85.Lima S, Kumar S, Gawandi V, Momany C, Phillips RS. Crystal structure of the homo sapiens kynureninase-3-hydroxyhippuric acid inhibitor complex: insights into the molecular basis of kynureninase substrate specificity. J Med Chem. 2009;52:389–96. [DOI] [PubMed] [Google Scholar]
- 86.Okuno E, Kido R. Kynureninase and kynurenine 3-hydroxylase in mammalian tissues. Adv Exp Med Biol. 1991;294:167–76. [DOI] [PubMed] [Google Scholar]
- 87.De Antoni A, Costa C, Allegri G. Studies on the kynurenine adminotransferase activity in rat liver and kidney. Hoppe Seylers Z Physiol Chem. 1976;357:1707–12. [DOI] [PubMed] [Google Scholar]
- 88.Tobes MC. Kynurenine-oxoglutarate aminotransferase from rat kidney. Methods Enzymol. 1987;142:217–24. [DOI] [PubMed] [Google Scholar]
- 89.Han Q, Li J. pH dependence, substrate specificity and inhibition of human kynurenine aminotransferase I. Eur J Biochem. 2004;271:4804–14. [DOI] [PubMed] [Google Scholar]
- 90.Han Q, Cai T, Tagle DA, Robinson H, Li J. Substrate specificity and structure of human aminoadipate aminotransferase/kynurenine aminotransferase II. Biosci Rep. 2008;28:205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cesura AM, Alberati-Giani D, Buchli R, Broger C, Kohler C, Vilbois F, Lahm HW, Heitz MP, Malherbe P. Molecular characterisation of kynurenine pathway enzymes: 3-hydroxyanthranilic-acid dioxygenase and kynurenine aminotransferase. Adv Exp Med Biol. 1996;398:477–83. [DOI] [PubMed] [Google Scholar]
- 92.Decker RH, Kang HH, Leach FR, Henderson LM. Purification and properties of 3-hydroxyanthranilic acid oxidase. J Biol Chem. 1961;236:3076–82. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.