

Background: No information is available about the effect of class-selective histone deacetylase inhibitors (DIs) following hindlimb ischemia.
Results: Class I and class IIa DIs prevent/delay ischemic muscle reconstruction at different stages.
Conclusion: Evidence is provided about a detrimental effect of DIs during normal muscle regeneration.
Significance: The therapeutic relevance of class-selective DIs in hindlimb ischemia may be limited by adverse effects.
Keywords: Epigenetics, Histone Deacetylase Inhibitors, Ischemia, Muscle Regeneration, Nitric Oxide, Protein Phosphatase 2A
Abstract
Histone deacetylase inhibitors (DIs) are promising drugs for the treatment of several pathologies including ischemic and failing heart where they demonstrated efficacy. However, adverse side effects and cardiotoxicity have also been reported. Remarkably, no information is available about the effect of DIs during tissue regeneration following acute peripheral ischemia. In this study, mice made ischemic by femoral artery excision were injected with the DIs MS275 and MC1568, selective for class I and IIa histone deacetylases (HDACs), respectively. In untreated mice, soon after damage, class IIa HDAC phosphorylation and nuclear export occurred, paralleled by dystrophin and neuronal nitric-oxide synthase (nNOS) down-regulation and decreased protein phosphatase 2A activity. Between 14 and 21 days after ischemia, dystrophin and nNOS levels recovered, and class IIa HDACs relocalized to the nucleus. In this condition, the MC1568 compound increased the number of newly formed muscle fibers but delayed their terminal differentiation, whereas MS275 abolished the early onset of the regeneration process determining atrophy and fibrosis. The selective DIs had differential effects on the vascular compartment: MC1568 increased arteriogenesis whereas MS275 inhibited it. Capillarogenesis did not change. Chromatin immunoprecipitations revealed that class IIa HDAC complexes bind promoters of proliferation-associated genes and of class I HDAC1 and 2, highlighting a hierarchical control between class II and I HDACs during tissue regeneration. Our findings indicate that class-selective DIs interfere with normal mouse ischemic hindlimb regeneration and suggest that their use could be limited by alteration of the regeneration process in peripheral ischemic tissues.
Introduction
Acute peripheral ischemia is a major health problem associated with complications of age, diabetes, and atherosclerosis which frequently result in serious morbidity and mortality. Following the ischemic event, in fact, an initial massive reduction in blood flow is a prelude to muscular tissue necrosis. In healthy individuals, blood flow is restored in a relatively short time and the tissue is repaired, whereas in the presence of chronic cardiovascular alterations both processes are inefficient (1). Several therapeutic approaches have been investigated to promote blood flow recovery, including promotion of arteriogenesis, angiogenesis, and muscle reconstitution; however, an effective treatment is still lacking.
Histone deacetylase inhibitors (DIs)5 have been proven effective in cancer, neuromuscular disorders (2), and in several experimental cardiac disease models including heart ischemia and failure (3). However, their potential therapeutic use in the treatment of peripheral vascular diseases has not been evaluated. Histone deacetylases (HDACs) are a family of 18 molecules divided in four subclasses (I-II-III-IV) according to their structural similarities and are involved in several biological processes including development, differentiation, and proliferation. Through modulation of the acetylation level of histone and nonhistone proteins they act as global regulators of gene expression (4) and protein function (5). Among HDACs, classes I and II have been described as important regulators of tissue regeneration. Class I HDACs (HDAC1, 2, 3, and 8) show a prevalent intranuclear distribution and target predominantly chromatin. Class II HDACs, further divided into IIa and IIb, shuttle between the cytoplasm and nucleus and show different tissue specificity. Specifically, the class IIa members HDAC4, 5, and 9 are expressed prevalently in heart, cardiac, and skeletal muscle whereas HDAC7 is found in CD4/CD8 double-positive thymocytes (6) as well as in endothelial and smooth muscle cells (7). Class IIa members shuttle between the nucleus and cytoplasm (8–10) depending on their phosphorylation status, which is catalyzed by Ca2+/calmodulin-dependent protein kinases, and determine nuclear export and inactivation (11). On the contrary, their dephosphorylation, induced by protein phosphatase 2A activation, mediates nuclear import (12). Interestingly, the latter process may occur as a consequence of nitric oxide (NO) production and is, in fact, impaired when NO is reduced as in the degenerative context of dystrophic mdx mice, an animal model of Duchenne muscular dystrophy (12, 13).
Because class IIa members do not bind chromatin directly, their function requires the formation of multiprotein complexes containing transcription factors and other HDACs such as HDAC3. In skeletal muscle, class IIa members associate with MEF2 and repress myogenesis (14, 15). However, other evidence suggests that their selective inhibition may arrest myogenesis in isolated satellite cells (13, 16).
Whereas pharmacological treatment with class I HDAC inhibitors promoted muscle fibers maturation in mdx mice (17), no information is available about the efficacy of these or other epigenetic drugs in the postischemic regeneration of normal muscular tissue. In light of these considerations this study was aimed at evaluating the therapeutic potential of class-selective DIs in a mouse model of post-ischemic hindlimb regeneration.
Evidence is provided about a detrimental effect of selective DIs on the ischemic hindlimb regeneration process. The DIs interfered with processes important for triggering muscle fiber maturation, including class I and class IIa activation and satellite cell differentiation, raising important questions about the opportunity of their use in post-ischemia hindlimb regeneration.
EXPERIMENTAL PROCEDURES
Animal Models and Surgical Procedures
All experimental procedures complied with the Guidelines of the Italian National Institute of Health and were approved by the Institutional Animal Care of the Istituto Dermopatico dell'Immacolata (IRCCS) (Rome, Italy). Male 2-month-old C57BL/10 mice were used. Before surgical procedures, mice were anesthetized with an intraperitoneal injection of 1 mg/kg medetomidine (Domitor; Vetem, Milan, Italy) and 75 mg/kg ketamine (Ketavet 100; Intervet Farmaceutici, Aprilia, Italy). The femoral artery dissection procedure (n = 6/time point) was described previously (18). Mice received daily intraperitoneal injections of the class IIa HDAC-specific inhibitor MC1568 (40 mg/kg) or the class I HDAC-specific inhibitor MS275 (5 mg/kg) starting 3 days before the ischemia. Doses were chosen accordingly to our previous in vivo studies (17, 19).
Control mice were treated with vehicle (saline plus dimethyl sulfoxide; n = 6/treatment). Animals were analyzed at 3, 7, 14, and 21 days after ischemia. The nonspecific NO synthase inhibitor N-(ω)-nitro-l-arginine methyl ester (l-NAME) was given to mice daily in the drinking water at a dose of 12.5 mg/100 ml (20) for 28 days before the ischemic procedure and then maintained.
Infections
Adductor muscles were infected with either adenoviruses CMV-GFP encoding the constitutively active isoform of endothelial nitric-oxide synthase (eNOS S1177D) or CMV-GFPnull (18). Muscles were injected with 1 × 108 plaque-forming units/animal immediately after ischemia. Five mice were used for each experimental condition. Adenovirus transfection efficiency was evaluated by measuring the percentage of GFP-positive fibers.
Confocal Microscopy
Confocal analysis was performed as reported previously (19). The following antibodies were used: anti-HDAC4, anti-HDAC5, and anti-HDAC9 (1:100, polyclonal; Abcam); anti-HDAC2 (1:100, polyclonal; Santa Cruz Biotechnology); anti-SMA (1:100, monoclonal; Santa Cruz Biotechnology); anti-dystrophin (1:100, polyclonal; Abcam); anti-nNOS (1:100, polyclonal; Santa Cruz Biotechnology), anti-iNOS (1:50, polyclonal; Abcam); anti-desmin (1:100, polyclonal; Abcam); and anti-GFP-FITC (1:500, polyclonal; Abcam). Samples were analyzed using a Zeiss LSM510 Meta confocal microscope.
Histology
Sections from adductor muscles were deparaffinized and processed for hematoxylin and eosin (H&E) staining. Degenerating muscle fibers were identified by morphology, differential eosin staining, and the presence of infiltrating cells near the degenerating fibers. Assessment of muscle fiber regeneration was performed by measuring centrally nucleated fibers indicating the presence of immature muscle fibers. The data are expressed as total muscle fibers containing central nuclei present in complete cross-sections of entire adductor muscles. Degenerative areas were calculated by measuring the inflammatory/degenerative regions on H&E-stained sections. A Zeiss Axioplan 2 fluorescence microscope with an image analyzer and KS300 software (Carl Zeiss, Oberkochen, Germany) was used to acquire images. Immature regenerating fibers were also evaluated by desmin staining. The extent of fibrosis was evaluated on Masson trichrome-stained sections. Quantification was expressed as the percentage of fibrotic tissue present on the total section. Assessment of arteriogenesis and capillarogenesis was performed on smooth muscle actin or H&E-stained sections as described previously (18).
Blood Flow Measure
Blood flow was measured in ischemic and contralateral hindlimbs by laser Doppler perfusion imaging (Lisca) at base line, immediately after induction of ischemia (day 0, to confirm efficient induction of ischemia), and 7, 14, and 21 days after ischemia. Before imaging, mice were placed on a heating mat at 37 °C. Low or no perfusion was displayed in dark blue, whereas high perfusion was displayed in red. To avoid the influence of ambient light and temperature, results were expressed as the ratio between perfusion in the left (ischemic) versus right (nonischemic) limb.
Chromatin Immunoprecipitation
Adductor muscles from ischemic (7 and 14 days) mice treated or not with MC1568 were used for the experiment. Chromatin cross-linking and ChIP assays (n = 3) were performed as described previously (13, 21) using specific antibodies to HDAC1 (Sigma), HDAC3 (Abcam, Santa Cruz Biotechnology), HDAC4 (Abcam), H3K9m3 (Abcam), and H3ac (Abcam). Negative controls were generated omitting antibody (NoAb) or using normal IgG (Santa Cruz). DNA fragments were recovered and analyzed by quantitative RT-PCR as described previously (13). Briefly, standard curves were generated by serially diluting the input (5-log dilutions in triplicate). Quantitative RT-PCR was performed in the ABI Prism 7500 and 7900HT fast PCR instruments (Applied Biosystems) using SYBR Master mix (Applied Biosystems) with evaluation of dissociation curves. The quantitative RT-PCR analyses were performed in duplicate or triplicate, and the data obtained were normalized to the corresponding DNA input control. Data are represented as relative enrichment. Primer sequences used for ChIP are as indicated in Table 1 and in Ref. 21 (m-telomerase reverse transcriptase (TERT)).
TABLE 1.
Sequences of primers used for ChIP and mRNA analysis
| Primer sequences for ChIP |
||
|---|---|---|
| Forward | Reverse | |
| mCCNB1 | 5′-AGCTTGGACAGCACACAAGTGA-3′ | 5′-CGCGAGGTCAGGCTCTATG-3′ |
| m-fos | 5′-TCCAGTTCCGCCCAGTGA-3′ | 5′-TCAGCTGGCGCCTTTATAGAA-3′ |
| mTNFα | 5′-TGTCCCCAACTTTCCAAACC-3′ | 5′-GAAAACTTCCTTGGTGGAGAAAAC-3′ |
| mHDAC2 | 5′-CCACACCTATAAGGAGATGGACATG-3′ | 5′-CAGCATCCATACCTGCTCGTAAT-3′ |
| mHDAC1 | 5′-CTCGGCGTATTCTCGGTAAAA-3′ | 5′-CTCTGCAGCCCGCTTCAC-3′ |
| mMir206 | 5′-CCCGGATGCCCCTAAAAA-3′ | 5′-AGCACTGACCAGTTCTGAGTGTTG-3′ |
| mDys | 5′-TCTCCATGTTCTACAGGTTGTGAAG-3′ | 5′-GCTACAGGTTGCTCTCCACAAA-3′ |
| nNOS | 5′-CCTGGGCAGCAAGATGCTAA-3′ | 5′-TCACGCTGTGGCATTTCTCT-3′ |
| eNOS | 5′-CCAATCCTCTACACCCAAATACCT-3′ | 5′-TGCGTGTTGGAGTTTATAGTTCTGT-3′ |
| MYH2 | 5′-CCCAGCGACGCCAAAA-3′ | 5′-TGAGTATGTGGGTAAATCTCCTATGG-3′ |
| MYOG | 5′-TCACATGTAATCCACTGGAAACG-3′ | 5′-AATATAGCCAACGCCACAGAAAC-3′ |
| Primer sequences for mRNA |
||
|---|---|---|
| Forward | Reverse | |
| c-fos | 5′-CGGAGGAGGGAGCTGACA-3′ | 5′-CAAACGCAGACTTCTCATCTTCAA-3′ |
| Aldolase | 5′-CGCTGCCAGTATGTTACTGAGAA-3′ | 5′-TGGTCGCTCAGAGCCTTGT-3′ |
RNA Extraction and Real-time PCR Analysis
Total RNA was extracted using the TRIzol Reagent (Invitrogen) following the manufacturer's instructions. cDNA preparation and real-time PCR conditions were as described previously (22). Primer sequences used for mRNA analysis were as indicated in Ref. 23 (armadillo repeat containing X-linked 2 (Armcx2)) and in Table 1.
Western Blotting and Immunoprecipitation
Western blotting was performed according to standard procedures. Adductor muscles were lysed in Laemmli buffer. Results were analyzed by ImageJ v1.28 software. Optical density values of specific proteins were normalized to that of tubulin and corrected for those obtained from controls that were considered equal to 1. Data represent the mean of at least three independent experiments ± S.E. The following antibodies were used: anti-HDAC5 (1:1000, polyclonal; Abcam), anti-HDAC2 (1:500, polyclonal; Santa Cruz Biotechnology), anti-phospho-HDAC5 (1:500, polyclonal; Abcam), anti-nNOS (1:500, polyclonal; Abcam), anti-eNOS (1:1000, polyclonal; Abcam), anti-iNOS (1:200, polyclonal; Abcam), anti-H4 trimethylated lysine 20 (1:1000, polyclonal; Abcam), anti-H3 trimethylated lysine 4 (1:1000, polyclonal; Abcam), anti-H3 phosphorylated serine 10 (1:1000, monoclonal; Abcam), anti-H3 acetylated lysine 9 (1:1000, polyclonal; Abcam), anti-GAPDH (1:2000, monoclonal; Abcam), and anti-tubulin (1:4000, monoclonal; Sigma). Co-immunoprecipitations were performed using 500-μg extracts after lysis of samples in 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1% Triton X-100, 2 mm MgCl2, and 1% sodium deoxycholate supplemented with 1 mm PMSF and protease inhibitor mix using 4 μg of anti-S-nitrosocysteine (polyclonal; Alpha Diagnostic) or anti-acetylated lysine (polyclonal; Abcam). The Ademtech Bioadembeads paramagnetic beads system was used to immunoprecipitate the specific proteins according to the manufacturer's instructions.
Phosphatase Assay
Phosphatase assays were performed using the Ser/Thr Phosphatase Assay System (Promega) according to the manufacturer's instructions as described previously (13).
miRNA Analysis
Total RNA was obtained homogenizing adductor muscles in TRIzol. miRNA levels were analyzed using the TaqMan real-time PCR (quantitative PCR) method (3.4 ng/assay) and quantified with a 7900HT Fast Real-time PCR system (Applied Biosystems). Primers for miRNAs and the reagents for reverse transcriptase and real-time PCR reactions were all obtained from Applied Biosystems. Relative expression was calculated using the comparative cycle threshold (Ct) method (2DCt). Different samples were normalized to miR-16 or to U6 snRNA expression.
Statistics
Data are presented as mean ± S.E. Differences among groups were assessed by analysis of variance. The Student's unpaired t test was used to compare 2 groups. A 95% confidence interval (p < 0.05) was considered significant.
RESULTS
Class IIa HDAC Nuclear Localization Is Lost during Early Muscle Regeneration following Hindlimb Ischemia
Class IIa-selective inhibitors have never been tested in skeletal muscle regeneration. To understand their potential application in this context, we analyzed the function of class IIa during post-ischemic regeneration. Mice were made ischemic by femoral artery excision (18, 24), and class IIa HDAC distribution was analyzed by confocal microscopy. In normo-perfused or sham-operated animals, HDAC5, HDAC4, and HDAC9 (Fig. 1A) were localized predominantly in muscle fiber nuclei. Soon after ischemia (3–7 days) they were exported to the cytoplasm and reacquired nuclear localization at later time points (days 14 and 21) (b–e and insets).
FIGURE 1.

Alteration in the dystrophin-nitric oxide pathway following ischemia determines class IIa nuclear export. A, confocal microscopy showing HDAC5, HDAC4, and HDAC9 distribution (green) in control (a) and post-ischemic adductor muscle at 3 (b), 7 (c), 14 (d), and 21 days (e). Nuclei were counterstained with TOPRO3 (blue). Magnification, ×40. Insets show HDAC5 distribution from control and ischemia 3-day panels; arrows indicate nuclei with low HDAC5 content. B, measurement of cGMP levels during ischemia. C, Western blotting showing the level of HDAC5 and its phosphorylation in control and in ischemic muscle. The graph shows the relative increase in HDAC5 phosphorylation normalized to total HDAC5 protein and to loading control (Tubulin). D, phosphatase activity in control and ischemic muscles. *, p < 0.05 versus control. Error bars, S.E. AU, arbitrary units.
NO is a potent epigenetic regulator active on class I and IIa HDACs (12, 13). Thus, we reasoned that after hindlimb ischemia the nuclear export of class IIa HDACs could be a consequence of the transient down-regulation of dystrophin and nNOS reducing local NO production and the activity of protein phosphatase 2A (12). In agreement with this hypothesis, we observed that NO-dependent production of cGMP decreased at 3 and 7 days after ischemia and recovered at 14 days (Fig. 1B). Coincidently, HDAC5 nuclear export and phosphorylation occurred (Fig. 1C), paralleled by reduction in protein phosphatase 2A activity (Fig. 1D). As expected, as early as 3 days after ischemia, dystrophin expression was lost, regaining normal levels around the 14-day time point as shown in Fig. 2A. This process coincided with a transient down-regulation of nNOS between 3 and 7 days after damage (Fig. 2, B and C). Further experiments revealed that, during the early phases after acute ischemia, iNOS expression was significantly increased, consistent with its role in the stress response to injury and inflammation (Fig. 2D), whereas eNOS and nNOS were down-regulated (Fig. 2E).
FIGURE 2.

A, representative images of dystrophin expression (red) in control and post-ischemic muscle at 3 and 14 days. Nuclei were stained with TOPRO3 (red). Magnification, ×40. B and C, analysis of nNOS expression in control and ischemic muscles by confocal microscopy (B; magnification, ×63) and Western blotting (C). Nuclei were stained with TOPRO3 (blue). The graph shows the relative level of nNOS normalized to loading control (GAPDH). *, p < 0.05 versus control. D, evaluation of iNOS expression in control and ischemic muscles at 3, 7, 14, and 21 days by Western blotting and confocal microscopy. E and F, evaluation of eNOS and nNOS (E) and dystrophin (F) in ischemic muscles from control (vehicle) and MC1568- or MS275-treated mice at 3 and 14 days after ischemia. Error bars, S.E. AU, arbitrary units.
Class IIa Inhibition Delays Muscle Fiber Maturation
The data reported above indicate the nuclear export/import of class IIa HDACs as timely regulated during post-ischemia regeneration. To explore the effect of their inhibition on tissue recovery we treated ischemic mice with the class IIa-selective inhibitor MC1568 (25). Histological analyses revealed that the prolonged inhibition of class IIa had no effect on the extension of ischemic areas (see Fig. 3B). However, although MC1568 increased the number of newly formed fibers, it clearly delayed their maturation-determining accumulation of central nucleated fibers. Their persistence was, in fact, still evident at 14 (Fig. 3, C and D, and graph in A) and 21 days after ischemia, when the adductor muscle fibers in untreated ischemic controls were almost completely regenerated (Fig. 3D). Accordingly, in ischemic muscles treated with MC1568, myosin heavy chain (MHC) expression was reduced compared with vehicle-treated mice (Fig. 3E), suggesting a persistent delay of the regeneration process. To assess whether MC1568 treatment could have impact on the dystrophin-NOS-NO pathway, we investigated the expression of these proteins. In the presence of MC1568 nNOS and dystrophin were down-regulated at 3 days and recovered to normal levels at 14 days as in ischemic control. eNOS had a similar expression pattern in vehicle- and MC1568-treated mice at 3 days whereas its recovery at 14 days was slightly delayed (Fig. 2, E and F).
FIGURE 3.

Effect of class IIa inhibition on muscle differentiation and epigenetic modifications. A–D, histological analysis of control (A) and ischemic muscle (3, 14, and 21 days, B–D, respectively) treated with vehicle or the class IIa HDAC-specific inhibitor MC1568 (H&E-stained sections). The graph (A, right panel) shows that the increase in immature centronucleated fibers is higher in MC1568-treated ischemic muscles than in vehicle-treated ischemic muscles. *, p < 0.05 versus control. E, Western blot showing MHC levels during post-ischemic regeneration in adductor muscles from untreated and MC1568-treated mice. The graph shows the relative level of MHC normalized to loading control (GAPDH). *, p < 0.05 versus control. F, Western blot of histone modifications showing an increase, soon after ischemia, in the level of H3K9ac, H3S10p, and H3K4m3 whereas H3K20m3 level was decreased. G, Western blot of histone modifications (H3K9ac, H3S10p) showing the effect of MC1568 treatment in ischemic samples. Band density analyses are shown in the bottom panels. *, p < 0.05 versus control; #, versus ischemia 3 days; °, versus ischemia 14 days. Error bars, S.E.
Class IIa Inhibition Amplifies Ischemia-dependent Epigenetic Changes
HDACs typically repress gene expression by reducing the acetylation level of core histones and nonhistone proteins, including transcription factors. We noted that in untreated ischemic tissue, the nuclear export of class IIa HDACs was paralleled by an increase in histone H3 lysine 9 acetylation (H3K9ac) well detectable at 3 and 7 days after damage. Other H3 epigenetic marks associated with gene activation, including serine 10 phosphorylation (H3S10p) and lysine 4 trimethylation (H3K4m3), were also increased. In contrast to this, the global level of lysine 20 trimethylation on histone H4 (H4K20m3), typically associated with chromatin compaction and gene repression, was markedly decreased (Fig. 3F). In untreated animals, these ischemia-associate changes were transient and fully reversed between 14 and 21 days post-damage. Conversely, the presence of increased H3S10p or H3K9ac levels was significantly prolonged in ischemic mice treated with MC1568, suggesting persistence of cellular activation in response to the drug (Fig. 3G).
Prior studies from our group demonstrated overexpression of genes involved in proliferation and differentiation, including c-fos, cyclin A, cyclin B, and TNFα, in chronically regenerating muscle of dystrophic mdx mice (13). This alteration was associated with the presence of abundant epigenetic marks of active transcription (13). To mechanistically explore the role of HDACs in this condition, chromatin immunoprecipitations (ChIP) were performed in ischemic muscle. The results revealed that the association of class IIa HDAC4 with the c-fos promoter (vehicle-treated control muscle, CTR), was undetectable 7 days post-ischemia in both untreated and MC1568-treated animals (Fig. 4A). However, at 14 days post-ischemia there was a significant increase of HDAC4 occupancy in control mice that was prevented by MC1568. During the progress of these experiments, we noted that, following HDAC4 occupancy, the trimethylation of histone H3 lysine 9 (H3K9m3) in the c-fos promoter region increased significantly, fostering a potential transcription repression role of this molecule. To address whether the epigenetic control mediated by class IIa HDACs correlates with c-fos expression we evaluated its mRNA level at 14 days when the MC1568 effect was very evident. In agreement with the ChIP results, we found that c-fos expression was repressed, an effect abolished by the DI (Fig. 4B). ChIP performed on other genes involved in proliferation and inflammation including mTERT, TNFα, and Cyclin B1, revealed similar epigenetic regulation by class IIa (Fig. 4, C–E). Because on chromatin the catalytic activity of class IIa HDACs relies on their association with co-factors and other members of the class I HDAC family, we analyzed the presence of HDAC3 on c-fos, mTERT, TNFα, and Cyclin B1 promoters. HDAC3 recruitment paralleled that of HDAC4. Conversely, analysis of HDAC3 and 4 occupancy on promoters of genes implicated in muscle differentiation, such as MHC, myogenin, dystrophin, eNOS, and nNOS, revealed that the chromatin binding of these molecules was not induced by ischemia and remained unchanged in MC1568-treated samples (Fig. 5). On the contrary, the parallel analysis of HDAC1 occupancy on the same genomic regions showed recruitment after ischemia. Nevertheless, this occurred only in the presence of the class IIa inhibitor MC1568, suggesting hierarchical relationships among HDACs of different classes (Fig. 5). Taken together, these results indicate that a HDAC3/4 complex is present on chromatin and is possibly involved in the regulation of gene expression during post-ischemia muscle regeneration.
FIGURE 4.

Epigenetic control of class IIa HDACs at gene-specific level. Analysis of HDAC4 and 3 occupancy onto the promoter of c-fos (A), TERT (C), TNFα (D), and CCNB1 (E) by chromatin immunoprecipitations. The enrichment of H3K9m3 on these promoters is also shown. *, p < 0.05 versus control; #, versus ischemia 7 days; °, versus ischemia 14 days. B, mRNA expression level of c-fos. *, p < 0.05 versus ischemia 14 days. Error bars, S.E.
FIGURE 5.
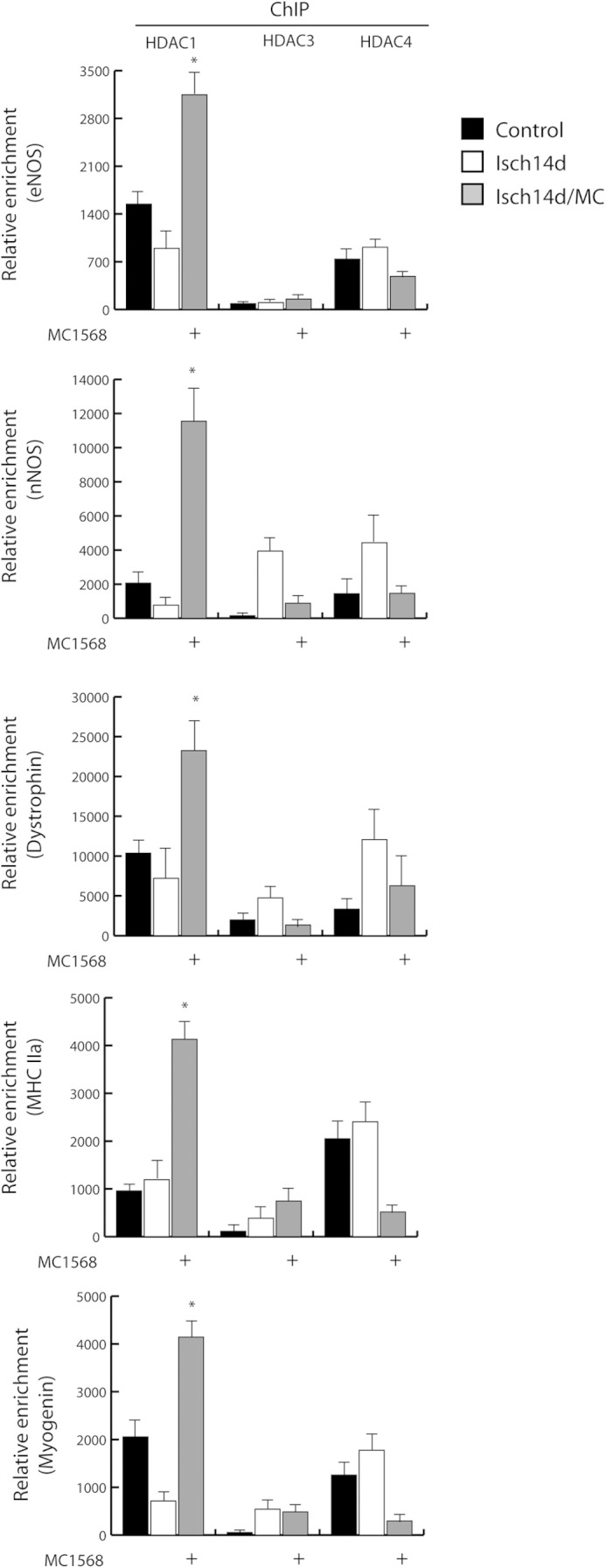
Evaluation of HDAC1, 3, and 4 recruitment on eNOS, dystrophin, nNOS, MHC, and myogenin promoters by ChIP. *, p < 0.05 versus ischemia 14 days. Error bars, S.E.
HDAC1 and 2 Expression Is Class IIa-dependent
Although transiently, ischemic muscle shows features similar to those of the chronically regenerating tissue of mdx mice where NO deficiency contributes to class IIa inactivation and class I overexpression and activation (13, 26). We show here, in fact, that during post-ischemic regeneration HDAC2 levels increase between 3 and 14 days. Surprisingly, MC1568 induced HDAC2 overexpression as assessed by confocal microscopy and Western blotting (Fig. 6A). Further, we measured the level of HDAC2 cysteine nitrosylation, which is NO-dependent and known to negatively regulate enzyme activity (26, 27) (Fig. 6B) and found that this post-translational modification was increased in ischemic samples at 14 days compared with untreated controls. In the presence of MC1568 the level of HDAC2 S-nitrosylation was significantly reduced compared with untreated ischemic controls. The levels of acetylated HDAC2 remained unchanged in all experimental conditions (Fig. 6B). HDAC1 expression was also evaluated and found increased in the presence of ischemia and further up-regulated by MC1568 treatment (Fig. 6C). On the contrary, there were no detectable changes in HDAC3 levels (data not shown). Of note, whereas the inhibition of class IIa HDACs induced overexpression of class I members HDAC1 and 2 during ischemia, inhibition of class I HDACs by MS275 did not alter the expression of class IIa members HDAC4 and 5 (data not shown), supporting the possibility of a hierarchical relationship between the two classes.
FIGURE 6.

The expression level of HDAC1 and 2 is under the control of class IIa HDACs during ischemia. A, analysis of HDAC2 protein levels by confocal microscopy or Western blotting in control and ischemic muscle treated with vehicle or MC1568. The graph shows the relative level of HDAC2 normalized to loading control (GAPDH). Error bars, S.E. B, analysis of HDAC2 post-translational modifications. Western blots show the level of cysteine nitrosylation and acetylation of HDAC2 in control and ischemic muscle, treated or not with MC1568, at 14 days. The graph on the right indicates the levels of these PTM relative to total unmodified protein. *, p < 0.05 versus control; #, versus ischemia 14 days. C, evaluation of HDAC1 protein level. The graph shows the relative level of HDAC1 normalized to loading control (GAPDH). *, p < 0.05 versus control. D and E, recruitment of HDAC4, HDAC3, and H3K9m3 onto HDAC1 (D) and 2 (E) promoters by ChIP. *, p < 0.05 versus control; #, versus ischemia 7days; °, versus ischemia 14 days. F and G, relative expression of miR-1 (F) and miR-206 (G) in normal and ischemic muscle treated or not with MC1568. *, p < 0.05 versus ischemia 7 days; #, versus ischemia 14 days; °, versus ischemia 21 days. H, measurement of HDAC1 and H3ac enrichment on miR-206 promoter by chromatin immunoprecipitation. º, p < 0.05 versus ischemia 14 days.
Class I and II Interplay Modulates myomiR Expression
A potential hierarchical regulation of transcription among HDACs of different classes during post-ischemia regeneration was investigated by ChIP. HDAC4 and HDAC3 were found present on HDAC1 and HDAC2 promoters in nonischemic control samples (Fig. 6, D and E). Ischemic damage significantly reduced this association at 7 days after ischemia regardless of DI treatment. However, at 14 days after ischemia, HDAC1 and 2 promoter occupancy by the HDAC3/4 complex significantly increased in untreated ischemic animals, whereas it was virtually abrogated in the presence of MC1568. Further, ChIP analysis for the transcriptional repression chromatin marker H3K9m3 showed similar kinetics on HDAC1 and 2 promoters (Fig. 6, D and E), indicating the lack of repression in the presence of MS1568 at 14 days. This evidence suggests a regulatory role exerted by class IIa HDACs on HDAC1 and 2 and possibly on their targets.
HDAC1 and 2 regulate muscle differentiation through the modulation of different targets, including microRNAs (miRs) such as miR-1 and miR-206. These miRs are important for muscle development and regeneration (28, 29), and their alteration contributes to the progression of different types of muscular dystrophy (30, 31). Prior studies demonstrated that in mdx mice reconstitution of NO signaling or treatment with the specific class I HDAC inhibitor MS275 normalized myomiR expression including miR-1 and miR-206 (32). Based on this evidence, we evaluated whether the increase in HDAC1 and 2 expression, occurring in ischemic muscle and sustained by the class IIa inhibitor, could alter miR-1 and miR-206 levels. In this context, miR-1, which showed a progressive increase during normal post-ischemic muscle regeneration (32) (Fig. 6F), was significantly down-regulated by MC1568 at 14 days. Expression of miR-206, increasing in normal post-ischemic muscle, was also down-regulated by MC1568 between 14 and 21 days of post-ischemia regeneration (Fig. 6G). To determine whether this down-regulation was mediated directly by HDAC1 we performed ChIP assays. We observed detachment of HDAC1 from the miR-206 promoter as early as 7 days after ischemia in both vehicle- and MC1568-treated animals (Fig. 6F). On the contrary, at 14 days after damage, a significant HDAC1 recruitment was observed only in the presence of MC1568 (Fig. 6H). Evaluation of acetylated H3 enrichment onto the miR-206 promoter indicated that an open, transcriptionally competent chromatin was present in this region at 14 days after ischemia but absent in the presence of MC1568.
Class I Restricted Inhibitor MS275 Prevents Proliferation and Promotes Fibrosis after Hindlimb Ischemia
Inhibition of class IIa HDACs determined a change in the post-ischemic response of muscle precursors that were kept proliferating without undergoing terminal differentiation (Fig. 3). Similar to dystrophic mdx mice, this condition was characterized by increased levels of HDAC2. In light of the fact that HDAC2 inhibition, by means of selective HDAC inhibitors, NO donors, or siRNAs, induced maturation and functional amelioration of the dystrophic muscle (17, 26), we reasoned that class I HDAC inhibition could have a beneficial effect in the post-ischemic regeneration. Surprisingly, inhibition of class I HDAC function by MS275 was detrimental and amplified the ischemic damage to hindlimb muscles, preventing the regeneration process (Fig. 7A). In agreement with this evidence, we found a significant delay in dystrophin, eNOS, and nNOS expression in animals treated with MS275 compared with controls (Fig. 2, E and F). This phenomenon was paralleled by a dramatic reduction of centrally nucleated fibers and a significant accumulation of fibrosis (Fig. 7, A and B). Accordingly, the expression of desmin, which increases in regenerating fibers, was higher in MC1568-treated mice and reduced in MS275-treated mice (Fig. 7C).
FIGURE 7.

Evaluation of class I- and class IIa-selective DIs on post-ischemic regeneration. A, comparative analysis of tissue recovery after ischemia (14 days) in adductor muscles treated with the class IIa HDAC-specific inhibitor MC1568 or the class I-specific inhibitor MS275 (H&E-stained sections). B, comparative analysis of the same samples for the presence of fibrosis (Masson trichrome staining). *, p < 0.05 versus control. C, representative images showing desmin expression in ischemic muscles (14 days) treated with vehicle, MC1568, or MS275. D–F, evaluation of arteriogenesis (D and E) and capillarogenesis (F) in ischemic muscles at 3, 14, and 21 days from mice treated with vehicle, MC1568, or MS275. *, p < 0.05 versus control. Representative images of arterioles are shown for ischemic muscles treated with vehicle, MC, or MS at 14 days. G, measurements of limb perfusion by Lisca immediately after ischemia (T0) and at 2, 7, 14, and 21 days after ischemia. Error bars, S.E.
Because recovery of dystrophin/NO signaling appears delayed in MS275-treated mice we evaluated whether this could affect HDAC4, 5, and 9 nuclear relocalization. We found that in the presence of MC1568, class IIa nuclear shuttling occurred normally at 14 days after ischemia whereas it was significantly delayed in the presence of MS275 (Fig. 8, A–C). Further, we found that class IIa inactivation by chemical treatment (MC1568) or by cytoplasmic sequestration, secondary to MS275 treatment, was associated with epigenetic global alteration. In fact, at 14 days after ischemia H3K4m3 and H4K20m3, which were up-regulated and down-regulated, respectively, soon after ischemia, return to normal in ischemic control mice (Fig. 3F). However, in the presence of MC1568 or MS275 (Fig. 8D) the level of H3K4m3 remained high at 14 days whereas the level of H4K20m was still low.
FIGURE 8.
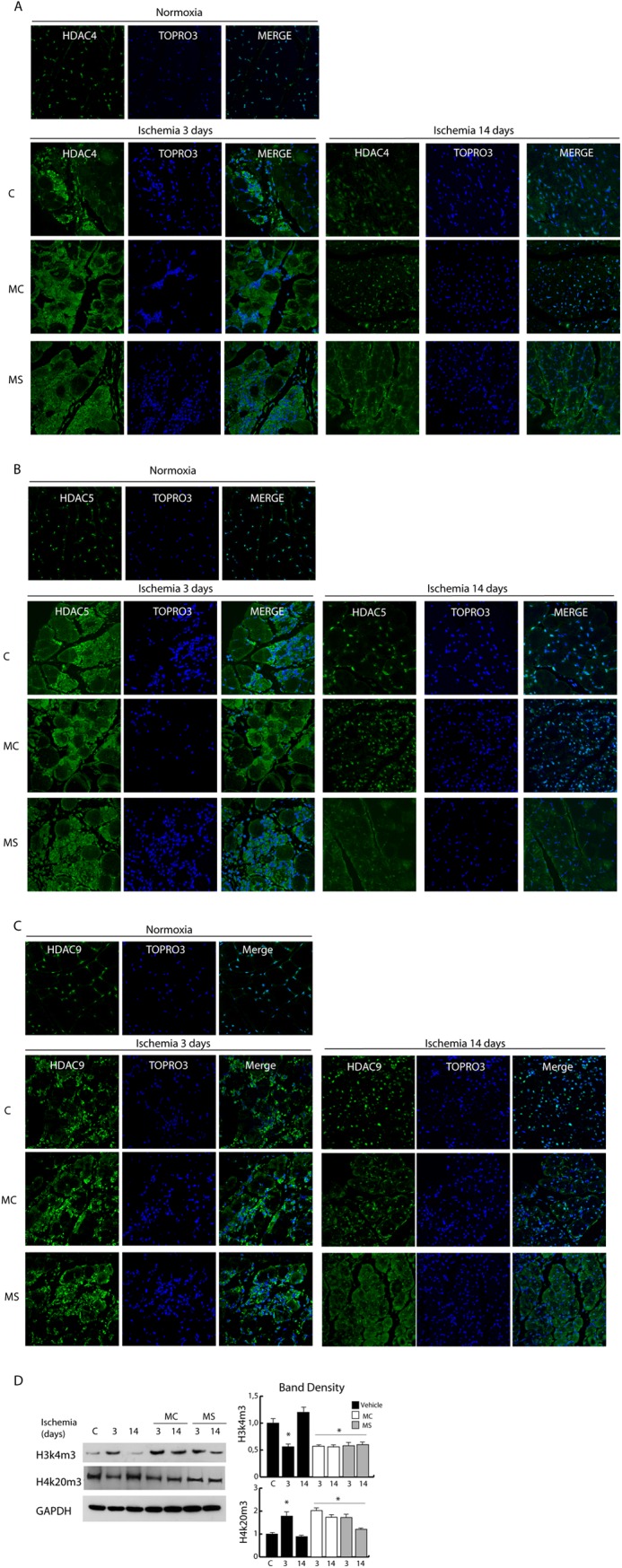
Evaluation of class I- and class IIa-selective DIs on class IIa HDAC nuclear export. A–C, confocal analysis of HDAC4 (A), HDAC5 (B), and HDAC9 (C) distribution (green) in control and post-ischemic adductor muscles at 3 and 14 days in untreated or MC- or MS-treated mice. Nuclei were counterstained with TOPRO3. D, Western blots of histone modifications (H3K4m3, H3K20m3) in control and ischemic muscles at 3 and 14 days of untreated or MC- or MS-treated mice. Band densities are shown in the right panels. *, p < 0.05 versus control. Error bars, S.E.
Vascular Regeneration Is Differentially Regulated by Class I and IIa HDAC Inhibitors
To explore further whether HDAC inhibitors could have an impact on vascular regeneration, arteriogenesis and capillarogenesis were measured after ischemia. The results show that in the presence of MC1568 arteriogenesis increased as indicated by the number of arterioles detected at 14 and 21 days, whereas the MS275 compound significantly reduced this effect (Fig. 7, D and E). Capillary density was unaffected by either drug and undistinguishable from control (Fig. 7F). Consistently, hindlimb perfusion showed that MC1568 determined a more efficient flow recovery at 14 and 21 days (Fig. 7G).
NO production is very important during muscle and vascular regeneration (33). In line with this consideration, our data on muscle regeneration showed that negative effects of class-selective DIs were associated with impaired eNOS/nNOS reexpression. To investigate whether the effect of DIs could be modulated by NO, ectopic expression of a constitutively active form of eNOS was achieved by adenoviral infection. As described previously, the adenoviral transfer of eNOS reduced ischemic damage (Fig. 9, A–C) and improved blood flow recovery (Fig. 9E). Of note, eNOS overexpression counteracted the effect of MC1568 and MS275, reducing the number of central nucleated fibers and the extent of degenerative areas. On the contrary, the inhibition of NOS by l-NAME, which interferes with the class I HDAC inactivation mediated by NO (34), determined a significant increase of immature central nucleated fibers. Accordingly, limb perfusion analysis revealed that the eNOS gene transfer strongly improved blood flow recovery at 14 days with no difference between control and MC1568- or MS275-treated mice (Fig. 9E).
FIGURE 9.

Evaluation of the effect of eNOS (adenovirus eNOS-S1177D) overexpression in ischemic mice treated or not with MC1568 and MS275 at 14 days. Adenovirus GFP-CMVnull-injected mice were used as controls. A, representative images of H&E-stained sections. B, analysis of centronucleated fibers. C, evaluation of degenerative areas. *, p < 0.05 versus control; §, p < 0.05 versus MC; °, p < 0,05 versus MS. D, evaluation of centronucleated fibers at 14 days of ischemia in vehicle and l-NAME-treated mice. E, measurement of limb perfusion by LISCA immediately after ischemia and at 14 days after ischemia in AdGFP-CMV-eNOS- or AdGFP-CMVnull-infected adductors treated or not with MC1568 or MS275. *, p < 0.05 versus control; §, p < 0.05 versus MC. Representative images are also provided of animals soon after ischemia and at 14 days. F, representative images of GFP-positive fibers in ischemic adductor muscles infected with AdGFP-CMV-eNOS or AdGFP-CMVnull. The graph shows the percentage of GFP-positive fibers in ischemic untreated or MC1568- or MS275-treated mice. Error bars, S.E.
DISCUSSION
The present study addresses the efficacy of class-restricted DIs in post-ischemic tissue regeneration. Our data show that the class IIa-selective inhibitor MC1568 significantly increases immature muscle fibers and new arteriole formation preventing, however, complete tissue maturation, possibly as a consequence of HDAC1 and 2 overexpression in the nuclei of surviving/regenerating muscle precursors and newly formed fibers. Conversely, the inhibition of class I HDACs by the selective compound MS275 caused a dramatic detrimental effect characterized by early growth arrest of muscle precursors, prevention of post-damage tissue regeneration, and significant accumulation of fibrosis. This evidence clearly puts into a negative light the potential use of DIs in tissue regeneration after hindlimb ischemia. The molecular basis of this detrimental effect may rely on different functions of the two classes of HDACs selectively inhibited by the drugs (Fig. 10). Our experimental evidence, in fact, indicates that the nuclear export of class IIa, which occurs early after post-ischemic damage, is strictly timely regulated and is required for class I HDAC overexpression, chromatin modification, and gene regulation in the early phases of regeneration. Of interest, the effect of MS275 was remarkably different from that obtained in nonischemic chronically regenerating mdx mice where class I inhibitors ameliorated the process of muscle regeneration (17). We reasoned that this divergence could reflect intrinsic differences in the molecular mechanisms involved in acute versus chronic tissue regeneration. Class I HDAC inhibitors were beneficial, in fact, when it was necessary to inhibit the chronically ongoing muscle regeneration process typical of Duchenne muscular dystrophy characterized by a constant rate of precursor activation and proliferation.
FIGURE 10.
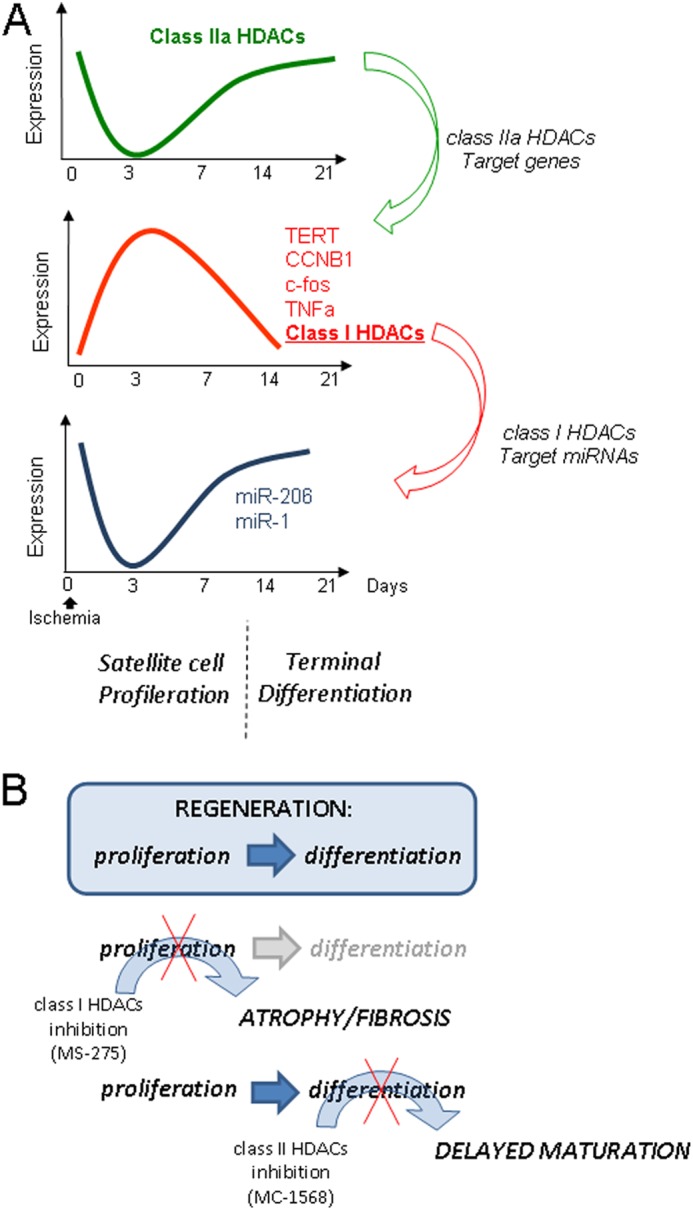
Scheme illustrating the temporal activation of class I and IIa HDACs during post-ischemic regeneration, their interplay, and the effect of class-selective DIs. A, during the early phase of this process, characterized by satellite cell activation and proliferation, class IIa HDAC is inactivated, and this allows the derepression of class IIa-dependent genes (c-fos, TERT, CycB, TNFα, HDAC1, and HDAC2). Class I HDAC activation in this phase, in turn, regulates the expression of miR-1 and 206. In the second phase of post-ischemic repair (from 14 to 21 days) characterized by terminal differentiation, class IIa is reactivated, and class I is repressed. B, effects of class I (MS275) and IIa (MC1568) DIs on muscle regeneration process during ischemia. Block of class I determines muscle atrophy and fibrosis, whereas block of class IIa induces delayed muscle maturation.
Peripheral ischemia determines muscular and vascular damage. For this reason we evaluated the effects of MS275 and MC1568 on newly formed arterioles and capillaries. As in muscle fibers, we found a different effect of the two inhibitors, with MC1568 increasing arteriogenesis and MS275 counteracting it. On the contrary, neither treatment altered capillarogenesis. In agreement with this evidence, MC1568 induced a faster blood perfusion recovery whereas MS275 delayed it compared with controls. Arteriogenesis and angiogenesis are qualitative, quantitative, and temporally distinct processes. A large body of knowledge indicates that arteriogenesis is the enlargement of preexisting vessels based on smooth muscle cell proliferation and driven mainly by shear stress, NO, and inflammatory cytokines such as MCP-1 (35). Conversely, angiogenesis is essentially a de novo generation of capillaries driven by hypoxia and regulated by several factors, including VEGF and Notch signaling (36). Tissue-specific differences in HDAC expression, not all of them being expressed in endothelial, smooth muscle, or satellite cells (37), may explain the discrepancies. It cannot be excluded, however, that proangiogenic and proarteriogenic factors could be differentially regulated by MC1568 and MS275. In this regard, we observed a class IIa-dependent regulation of the inflammatory factor TNFα in MC1568-treated mice. In addition to these considerations, we reasoned that the post-ischemic revascularization process might be under the influence of NO synthesis because it occurs poorly in eNOS-deficient mice (33). NO is also very important in muscle regeneration process, and its production is compromised in mdx mice. In the progress of our work we accumulated evidence that the reduced/delayed eNOS and nNOS reexpression after ischemic damage could be one of the key elements at the basis of the detrimental DI effect on muscle regeneration. Indeed, the ectopic expression of an eNOS transgene, known to reduce oxidative stress and to improve perfusion and tissue recovery (38), overcame the regeneration limitations induced by MC1568 and MS275 by promoting a faster recovery of limb perfusion in controls and in DI-treated animals. Therefore, the alteration in gene expression leading to prevention or delayed reactivation of the dystrophin-NOS-NO axis determined by both DIs may provide the molecular basis to explain our findings.
Taken together, these results indicate that interfering with the timely regulated function of class I and IIa HDACs during ischemic muscle tissue regeneration by means of class-selective DIs was detrimental rather than beneficial for tissue regeneration. Although DIs are now in clinical use as valuable anticancer therapy, cardiac adverse side effects (39, 40) and cardiac hypertrophy (41) have been recently described associated with their use. Thus, the evaluation of their safety and toxicity is of particular relevance for potential therapeutic application in regenerative medicine. In conclusion, our data raise a concern about potential risks associated with the use of DIs in the treatment of post-ischemic damage.
Footnotes
- DI
- histone deacetylase inhibitor
- eNOS
- endothelial nitric-oxide synthase
- H3K9ac
- histone H3 lysine 9 acetylation
- H3K9m3
- histone 3 lysine 9 trimethylation
- H3S10p
- histone 3 serine 10 phosphorylation
- HDAC
- histone deacetylase
- iNOS
- inducible nitric-oxide synthase
- l-NAME
- N-(ω)-nitro-l-arginine methyl ester
- MHC
- myosin heavy chain
- nNOS
- neuronal nitric-oxide synthase.
REFERENCES
- 1. Sneider E. B., Nowicki P. T., Messina L. M. (2009) Regenerative medicine in the treatment of peripheral arterial disease. J. Cell. Biochem. 108, 753–761 [DOI] [PubMed] [Google Scholar]
- 2. Colussi C., Illi B., Rosati J., Spallotta F., Farsetti A., Grasselli A., Mai A., Capogrossi M. C., Gaetano C. (2010) Histone deacetylase inhibitors: keeping momentum for neuromuscular and cardiovascular diseases treatment. Pharmacol. Res. 62, 3–10 [DOI] [PubMed] [Google Scholar]
- 3. McKinsey T. A. (2012) Therapeutic potential for HDAC inhibitors in the heart. Annu. Rev. Pharmacol. Toxicol. 52, 303–319 [DOI] [PubMed] [Google Scholar]
- 4. Narlikar G. J., Fan H. Y., Kingston R. E. (2002) Cooperation between complexes that regulate chromatin structure and transcription. Cell 108, 475–487 [DOI] [PubMed] [Google Scholar]
- 5. Hildmann C., Riester D., Schwienhorst A. (2007) Histone deacetylases: an important class of cellular regulators with a variety of functions. Appl. Microbiol. Biotechnol. 75, 487–497 [DOI] [PubMed] [Google Scholar]
- 6. Ficner R. (2009) Novel structural insights into class I and II histone deacetylases. Curr. Top. Med. Chem. 9, 235–240 [DOI] [PubMed] [Google Scholar]
- 7. Margariti A., Zampetaki A., Xiao Q., Zhou B., Karamariti E., Martin D., Yin X., Mayr M., Li H., Zhang Z., De Falco E., Hu Y., Cockerill G., Xu Q., Zeng L. (2010) Histone deacetylase 7 controls endothelial cell growth through modulation of β-catenin. Circ. Res. 106, 1202–1211 [DOI] [PubMed] [Google Scholar]
- 8. Grozinger C. M., Schreiber S. L. (2000) Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. U.S.A. 97, 7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang A. H., Kruhlak M. J., Wu J., Bertos N. R., Vezmar M., Posner B. I., Bazett-Jones D. P., Yang X. J. (2000) Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol. Cell. Biol. 20, 6904–6912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McKinsey T. A., Zhang C. L., Olson E. N. (2000) Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. U.S.A. 97, 14400–14405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McKinsey T. A., Zhang C. L., Lu J., Olson E. N. (2000) Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Illi B., Dello Russo C., Colussi C., Rosati J., Pallaoro M., Spallotta F., Rotili D., Valente S., Ragone G., Martelli F., Biglioli P., Steinkuhler C., Gallinari P., Mai A., Capogrossi M. C., Gaetano C. (2008) Nitric oxide modulates chromatin folding in human endothelial cells via protein phosphatase 2A activation and class II histone deacetylases nuclear shuttling. Circ. Res. 102, 51–58 [DOI] [PubMed] [Google Scholar]
- 13. Colussi C., Gurtner A., Rosati J., Illi B., Ragone G., Piaggio G., Moggio M., Lamperti C., D'Angelo G., Clementi E., Minetti G., Mozzetta C., Antonini A., Capogrossi M. C., Puri P. L., Gaetano C. (2009) Nitric oxide deficiency determines global chromatin changes in Duchenne muscular dystrophy. FASEB J. 23, 2131–2141 [DOI] [PubMed] [Google Scholar]
- 14. McKinsey T. A., Zhang C. L., Olson E. N. (2001) Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev. 11, 497–504 [DOI] [PubMed] [Google Scholar]
- 15. Lu J., McKinsey T. A., Nicol R. L., Olson E. N. (2000) Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 97, 4070–4075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nebbioso A., Manzo F., Miceli M., Conte M., Manente L., Baldi A., De Luca A., Rotili D., Valente S., Mai A., Usiello A., Gronemeyer H., Altucci L. (2009) Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC-MEF2 complexes. EMBO Rep. 10, 776–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Minetti G. C., Colussi C., Adami R., Serra C., Mozzetta C., Parente V., Fortuni S., Straino S., Sampaolesi M., Di Padova M., Illi B., Gallinari P., Steinkühler C., Capogrossi M. C., Sartorelli V., Bottinelli R., Gaetano C., Puri P. L. (2006) Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat. Med. 12, 1147–1150 [DOI] [PubMed] [Google Scholar]
- 18. Zaccagnini G., Martelli F., Fasanaro P., Magenta A., Gaetano C., Di Carlo A., Biglioli P., Giorgio M., Martin-Padura I., Pelicci P. G., Capogrossi M. C. (2004) p66ShcA modulates tissue response to hindlimb ischemia. Circulation 109, 2917–2923 [DOI] [PubMed] [Google Scholar]
- 19. Colussi C., Rosati J., Straino S., Spallotta F., Berni R., Stilli D., Rossi S., Musso E., Macchi E., Mai A., Sbardella G., Castellano S., Chimenti C., Frustaci A., Nebbioso A., Altucci L., Capogrossi M. C., Gaetano C. (2011) Nϵ-lysine acetylation determines dissociation from GAP junctions and lateralization of connexin 43 in normal and dystrophic heart. Proc. Natl. Acad. Sci. U.S.A. 108, 2795–2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Henke N., Schmidt-Ullrich R., Dechend R., Park J. K., Qadri F., Wellner M., Obst M., Gross V., Dietz R., Luft F. C., Scheidereit C., Muller D. N. (2007) Vascular endothelial cell-specific NF-κB suppression attenuates hypertension-induced renal damage. Circ. Res. 101, 268–276 [DOI] [PubMed] [Google Scholar]
- 21. Nanni S., Benvenuti V., Grasselli A., Priolo C., Aiello A., Mattiussi S., Colussi C., Lirangi V., Illi B., D'Eletto M., Cianciulli A. M., Gallucci M., De Carli P., Sentinelli S., Mottolese M., Carlini P., Strigari L., Finn S., Mueller E., Arcangeli G., Gaetano C., Capogrossi M. C., Donnorso R. P., Bacchetti S., Sacchi A., Pontecorvi A., Loda M., Farsetti A. (2009) Endothelial NOS, estrogen receptor β, and HIFs cooperate in the activation of a prognostic transcriptional pattern in aggressive human prostate cancer. J. Clin. Invest. 119, 1093–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nanni S., Priolo C., Grasselli A., D'Eletto M., Merola R., Moretti F., Gallucci M., De Carli P., Sentinelli S., Cianciulli A. M., Mottolese M., Carlini P., Arcelli D., Helmer-Citterich M., Gaetano C., Loda M., Pontecorvi A., Bacchetti S., Sacchi A., Farsetti A. (2006) Epithelial-restricted gene profile of primary cultures from human prostate tumors: a molecular approach to predict clinical behavior of prostate cancer. Mol. Cancer Res. 4, 79–92 [DOI] [PubMed] [Google Scholar]
- 23. Gurtner A., Fuschi P., Magi F., Colussi C., Gaetano C., Dobbelstein M., Sacchi A., Piaggio G. (2008) NF-Y-dependent epigenetic modifications discriminate between proliferating and postmitotic tissue. PloS One 3, e2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Straino S., Germani A., Di Carlo A., Porcelli D., De Mori R., Mangoni A., Napolitano M., Martelli F., Biglioli P., Capogrossi M. C. (2004) Enhanced arteriogenesis and wound repair in dystrophin-deficient mdx mice. Circulation 110, 3341–3348 [DOI] [PubMed] [Google Scholar]
- 25. Mai A., Massa S., Pezzi R., Simeoni S., Rotili D., Nebbioso A., Scognamiglio A., Altucci L., Loidl P., Brosch G. (2005) Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (aryloxopropenyl)pyrrolyl hydroxyamides. J. Med. Chem. 48, 3344–3353 [DOI] [PubMed] [Google Scholar]
- 26. Colussi C., Mozzetta C., Gurtner A., Illi B., Rosati J., Straino S., Ragone G., Pescatori M., Zaccagnini G., Antonini A., Minetti G., Martelli F., Piaggio G., Gallinari P., Steinkuhler C., Clementi E., Dell'Aversana C., Altucci L., Mai A., Capogrossi M. C., Puri P. L., Gaetano C. (2008) HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. U.S.A. 105, 19183–19187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nott A., Watson P. M., Robinson J. D., Crepaldi L., Riccio A. (2008) S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 455, 411–415 [DOI] [PubMed] [Google Scholar]
- 28. Goljanek-Whysall K., Pais H., Rathjen T., Sweetman D., Dalmay T., Münsterberg A. (2012) Regulation of multiple target genes by miR-1/miR-206 is pivotal for C2C12 myoblast differentiation. J. Cell Sci. 125, 3590–3600 [DOI] [PubMed] [Google Scholar]
- 29. Chen J. F., Tao Y., Li J., Deng Z., Yan Z., Xiao X., Wang D. Z. (2010) microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J. Cell Biol. 190, 867–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cacchiarelli D., Martone J., Girardi E., Cesana M., Incitti T., Morlando M., Nicoletti C., Santini T., Sthandier O., Barberi L., Auricchio A., Musarò A., Bozzoni I. (2010) MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 12, 341–351 [DOI] [PubMed] [Google Scholar]
- 31. Liu N., Williams A. H., Maxeiner J. M., Bezprozvannaya S., Shelton J. M., Richardson J. A., Bassel-Duby R., Olson E. N. (2012) microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J. Clin. Invest. 122, 2054–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Greco S., De Simone M., Colussi C., Zaccagnini G., Fasanaro P., Pescatori M., Cardani R., Perbellini R., Isaia E., Sale P., Meola G., Capogrossi M. C., Gaetano C., Martelli F. (2009) Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 23, 3335–3346 [DOI] [PubMed] [Google Scholar]
- 33. Murohara T., Asahara T., Silver M., Bauters C., Masuda H., Kalka C., Kearney M., Chen D., Symes J. F., Fishman M. C., Huang P. L., Isner J. M. (1998) Nitric-oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Invest. 101, 2567–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spallotta F., Cencioni C., Straino S., Nanni S., Rosati J., Artuso S., Manni I., Colussi C., Piaggio G., Martelli F., Valente S., Mai A., Capogrossi M. C., Farsetti A., Gaetano C. (2013) A nitric oxide-dependent cross-talk between class I and III histone deacetylases accelerates skin repair. J. Biol. Chem. 288, 11004–11012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Troidl K., Schaper W. (2012) Arteriogenesis versus angiogenesis in peripheral artery disease. Diabetes Metab. Res. Rev. 28, 27–29 [DOI] [PubMed] [Google Scholar]
- 36. Al Haj Zen A., Madeddu P. (2009) Notch signalling in ischaemia-induced angiogenesis. Biochem. Soc. Trans. 37, 1221–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou B., Margariti A., Zeng L., Xu Q. (2011) Role of histone deacetylases in vascular cell homeostasis and arteriosclerosis. Cardiovasc. Res. 90, 413–420 [DOI] [PubMed] [Google Scholar]
- 38. Yan J., Tie G., Hoffman A., Yang Y., Nowicki P. T., Messina L. M. (2010) Oral tetrahydrobiopterin improves the beneficial effect of adenoviral-mediated eNOS gene transfer after induction of hindlimb ischemia. Mol. Ther. 18, 1482–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu Q., Lin X., Andrews L., Patel D., Lampe P. D., Veenstra R. D. (2013) Histone deacetylase inhibition reduces cardiac connexin43 expression and gap junction communication. Front. Pharmacol. 4, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shah M. H., Binkley P., Chan K., Xiao J., Arbogast D., Collamore M., Farra Y., Young D., Grever M. (2006) Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin. Cancer Res. 12, 3997–4003 [DOI] [PubMed] [Google Scholar]
- 41. Karagiannis T. C., Lin A. J., Ververis K., Chang L., Tang M. M., Okabe J., El-Osta A. (2010) Trichostatin A accentuates doxorubicin-induced hypertrophy in cardiac myocytes. Aging 2, 659–668 [DOI] [PMC free article] [PubMed] [Google Scholar]