

Background: The TGR5 bile acid receptor controls energy balance, inflammation, and digestion, but TGR5 signaling is poorly understood.
Results: TGR5 does not interact with β-arrestins, internalize, or desensitize, but signals from plasma membrane rafts.
Conclusion: TGR5 transmits sustained signals close to the cell surface.
Significance: Understanding TGR5 signaling will facilitate design of TGR5 agonists for metabolic, inflammatory, and digestive disorders.
Keywords: Arrestin, Bile Acid, Endocytosis, G Protein-coupled Receptors (GPCR), Lipid Raft
Abstract
TGR5 is a G protein-coupled receptor that mediates bile acid (BA) effects on energy balance, inflammation, digestion, and sensation. The mechanisms and spatiotemporal control of TGR5 signaling are poorly understood. We investigated TGR5 signaling and trafficking in transfected HEK293 cells and colonocytes (NCM460) that endogenously express TGR5. BAs (deoxycholic acid (DCA), taurolithocholic acid) and the selective agonists oleanolic acid and 3-(2-chlorophenyl)-N-(4-chlorophenyl)-N, 5-dimethylisoxazole-4-carboxamide stimulated cAMP formation but did not induce TGR5 endocytosis or recruitment of β-arrestins, as assessed by confocal microscopy. DCA, taurolithocholic acid, and oleanolic acid did not stimulate TGR5 association with β-arrestin 1/2 or G protein-coupled receptor kinase (GRK) 2/5/6, as determined by bioluminescence resonance energy transfer. 3-(2-chlorophenyl)-N-(4-chlorophenyl)-N, 5-dimethylisoxazole-4-carboxamide stimulated a low level of TGR5 interaction with β-arrestin 2 and GRK2. DCA induced cAMP formation at the plasma membrane and cytosol, as determined using exchange factor directly regulated by cAMP (Epac2)-based reporters, but cAMP signals did not desensitize. AG1478, an inhibitor of epidermal growth factor receptor tyrosine kinase, the metalloprotease inhibitor batimastat, and methyl-β-cyclodextrin and filipin, which block lipid raft formation, prevented DCA stimulation of ERK1/2. Bioluminescence resonance energy transfer analysis revealed TGR5 and EGFR interactions that were blocked by disruption of lipid rafts. DCA stimulated TGR5 redistribution to plasma membrane microdomains, as localized by immunogold electron microscopy. Thus, TGR5 does not interact with β-arrestins, desensitize, or traffic to endosomes. TGR5 signals from plasma membrane rafts that facilitate EGFR interaction and transactivation. An understanding of the spatiotemporal control of TGR5 signaling provides insights into the actions of BAs and therapeutic TGR5 agonists/antagonists.
Introduction
During feeding, the presence of lipids in the small intestine stimulates the release of cholecystokinin, a hormone that induces contraction of the gall bladder and secretion of bile into the intestine. Bile acids (BAs)3 are a major component of bile. The two primary bile acids in humans, chenodeoxycholic acid and cholic acid, facilitate the emulsification and absorption of dietary fats and the lipid-soluble vitamins A, D, E, and K (1, 2). The bulk of the primary BAs are absorbed in the ileum by the sodium bile acid transporter. Primary BAs that escape ileal absorption pass into the colon and are converted by bacterial enzymes to secondary BAs, including deoxycholic acid (DCA), lithocholic acid, taurolithocholic acid (TLCA), and ursodeoxycholic acid, which are absorbed passively. Absorbed BAs enter the portal vein and recycle to the liver via the enterohepatic circulation. Because bile is secreted into the intestine episodically, the circulating levels of BAs wax and wane during feeding and fasting, similar to the levels of gut hormones, and BAs have hormone-like effects on many cell types (1, 3, 4). Moreover, the synthesis, secretion, absorption, and metabolism of BAs are under tight physiological control, and defects in these processes lead to abnormal circulating and intestinal concentrations of BAs, which can cause disease (1, 3, 4).
BAs exert their hormone-like effects by activating receptors in the nucleus or at the plasma membrane (1, 3, 4). Nuclear BA receptors include the farnesoid X receptor, which regulates BA homeostasis (5, 6), and the pregnane X receptor and vitamin D receptor, which protect against the hepatotoxic actions of certain BAs (7, 8). TGR5 (GpBAR1 or M-BAR1) is a G protein-coupled receptor (GPCR) of the plasma membrane that interacts with multiple BAs that activate TGR5 with graded potencies (4, 9, 10). TGR5 is expressed in brown adipose tissue; skeletal muscle; immune, epithelial, and endothelial cells; and by enteric and primary spinal afferent neurons, where activation has biologically important consequences. In brown adipose tissue and skeletal muscle, TGR5 agonism results in the activation of thyroxine, leading to increased energy expenditure and weight loss (11). TGR5 activation on intestinal L cells stimulates release of glucagon-like peptide 1, an incretin that controls insulin secretion and glucose homeostasis and suppresses food intake and gastrointestinal transit (12, 13). In the gall bladder, TGR5 controls the composition and secretion of bile (14–17). Activation of TGR5 on macrophages and Kupffer cells inhibits cytokine release, phagocytosis, and lipid loading of macrophages (4, 9, 10) and diminishes hepatic and intestinal inflammation (4, 9, 18). In the intestine, luminal BAs can activate TGR5 on enterochromaffin cells and enteric neurons to release 5-hydroxytryptamine and calcitonin gene-related peptide, which induce peristalsis and mediate the well described prokinetic effects of bile (19, 20). BAs can also activate TGR5 expressed by primary spinal afferent and spinal neurons, and TGR5 mediates BA-evoked itch and analgesia, which may contribute to pruritus and painless jaundice that are observed in some patients with cholestatic disease (21). The identification of the biological actions of TGR5 has provided an impetus for the development of TGR5 agonists and antagonists as treatments for metabolic, inflammatory, digestive, and sensory disorders (4). However, the signal transduction mechanisms that give rise to the biological actions of TGR5 are not fully understood.
GPCR signals arise from multiprotein signaling complexes at the plasma membrane, in plasma membrane-associated compartments such as lipid rafts and caveolae, and in endosomes (22–24). The same receptor can transmit distinct signals from these different subcellular domains, with divergent physiological outcomes. Thus, an understanding of the mechanisms that regulate the subcellular location of GPCRs and the assembly and disassembly of these signaling complexes is key to understanding GPCR functions. For many GPCRs, G protein-coupled receptor kinases (GRKs) and β-arrestins play a central role in the spatiotemporal control of signaling (23). GRKs phosphorylate agonist-occupied receptors at the plasma membrane, which increases their affinity for β-arrestins, cytosolic proteins that translocate to the plasma membrane and interact with phosphorylated receptors. β-arrestins sterically uncouple GPCRs from heterotrimeric G proteins, which desensitizes plasma membrane signaling, and also couple receptors to clathrin and AP2, which mediate receptor endocytosis. By recruiting components of the MAPK cascade to internalized receptors, β-arrestins can also mediate sustained G protein-independent signaling from receptors in endosomes (23). TGR5 couples to Gαs, resulting in the activation of adenylate cyclase and formation of cAMP (9). TGR5 agonists also induce activation of MAPKs, including ERK1/2 and c-Jun-N-terminal kinase (25, 26), and TGR5 transactivates the epidermal growth factor receptor (EGFR), which, in turn, activates ERK1/2 (25, 27). However, it is unclear whether these signals derive from TGR5 that is localized to the plasma membrane, plasma membrane microdomains, or endosomes, and the mechanisms that control the duration of these signals are unknown.
In this study, we investigated TGR5 signaling in response to diverse endogenous and synthetic agonists. These included BAs (DCA, TLCA), a naturally occurring TGR5-selective agonist (oleanolic acid, OA) (28), and a highly potent synthetic agonist, 3-(2-chlorophenyl)-N-(4-chlorophenyl)-N, 5-dimethylisoxazole-4-carboxamide (CCDC) (29). We examined signaling and regulation of TGR5 expressed in a model cell line (HEK293 cells) and in non-transformed human colonocytes that naturally express this receptor (NCM460 cells). Our results show that TGR5 is a highly unusual GPCR that does not interact with β-arrestin1/2 or GRK2/5/6 when activated by its endogenous agonists. Consistent with these findings, BA-activated TGR5 does not traffic to endosomes. Activation results in the formation of cAMP at the plasma membrane and in the cytosol, but these signals do not undergo tachyphylaxis or desensitize to repeated stimulation. Instead, BA-activated TGR5 redistributes to plasma membrane microdomains, where association of TGR5 with the EGFR mediates efficient ERK1/2 signaling. This information about the spatiotemporal control of TGR5 signaling provides insight into the biological effects of BAs and is relevant to the mechanism of action of TGR5 agonists and antagonists that are under development for the treatment of diverse and debilitating diseases.
EXPERIMENTAL PROCEDURES
Materials
Primary antibodies were obtained from the following sources. Anti-HA11 (rabbit or rat) and anti-FLAG (mouse) were from Sigma-Aldrich (St. Louis, MO), anti-GFP (rabbit) was from Clontech (Mountainview, CA), anti-β-arrestin1/2 (rabbit) was from Abcam, Cambridge, MA), anti-EGFR (mouse) was from BD Biosciences), and anti-phospho-ERK1/2 (pERK, mouse) and anti-ERK2 (rabbit) were from Santa Cruz Biotechnology (Santa Cruz, CA). Goat anti-rabbit or anti-mouse IgG conjugated to FITC or Rhodamine Red-X were from Jackson ImmunoResearch (West Grove, PA); goat anti-rabbit, anti-rat, and anti-mouse conjugated to Alexa Fluor 488, 594, or 647 were from Invitrogen; and goat anti-rabbit IgG conjugated to 10 nm of gold particles was from Ted Pella Inc. (Redding, CA). The protease-activated receptor 2 (PAR2) agonists SLIGRL-NH2 and 2-furoyl-LIGRLO-NH2 were from American Peptide (Sunnyvale, CA), and the PAR2 agonist AC 264613 was from Tocris (Ellisville, MO). The TGR5 agonist CCDC (29) was from BioVision (Milpitas, CA). Other reagents were from Sigma-Aldrich unless stated otherwise.
cDNAs
Human TGR5 with an N-terminal extracellular HA11 epitope (HA-TGR5) or C-terminal intracellular GFP (TGR5-GFP) and human PAR2 with an N-terminal FLAG epitope (FLAG-PAR2) have been described (20, 30). To generate luciferase-tagged receptors, cDNAs encoding human TGR5 or PAR2 flanked by 5′ BamH1 and 3′ Not1 restriction sites were generated by PCR using Phusion high-fidelity PCR (New England Biosciences, Ipswich, MA) with the following primers: TGR5 sense, cgc gga tcc atg acg ccc aac agc act g and antisense, ata gtt tag cgg ccg cag ggc gta gtc ggg cac gtc; PAR2 sense, cgc gga tcc atg ccg aga tcg tgc tgc ag and antisense, tag ttt agc ggc cgc agg gca taa tcg gga aca tc. PCR products were cut with BamH1 and Not1 (New England Biosciences) and ligated into pcDNA3-RLuc8 using the BamH1 and Not1 sites. Human β-arrestin1 and β-arrestin2 with N-terminal GFP or YFP have been described (31). GRK2-Venus was amplified by PCR with the following primers to generate attB sites at either end of the DNA sequence: sense, ggg gac aag ttt gta caa aaa agc agg ctt cac cat ggc gga cct gga ggc ggt g and antisense, ggg gac cac ttt gta caa gaa agc tgg gtc tta ctt gta cag ctc gtc cat. The PCR product was gel-purified and transferred into pDONR201 using BP Clonase II (Invitrogen). The construct was subcloned into pcDNA5-frt-TO using LR Clonase II (Invitrogen). GRK5-Venus and GRK6-Venus were amplified by PCR with the following primers to generate GRK5 flanked by the 5′ BamH1 and 3′ XbaI sites and GRK6 flanked by the 5′ BamH1 and 3′ Not1 sites: GRK5 sense, cgg gat ccc gat gga gct gga aaa cat cgt and antisense, gga cga gct gta caa gta atc tag agc; GRK6 sense, cgg gat ccc gat gga gct cga gaa cat cgt and antisense, gga cga gct gta caa gta agc ggc cgc taa act at. PCR products were excised and ligated into pcDNA3.1 (32). KRas-Venus, Rab5a-Venus, and Rab7a-Venus have been described (33). EGFR-Venus was provided by Dr. Kevin Pfleger (University of Western Australia). Gγ2-Venus, Gβ1, and Gα were generated as described (34). The cAMP BRET sensor CAMYEL (MBA-277) was from the ATCC. cAMP sensors on the basis of an exchange factor directly regulated by cAMP (Epac2) for measurement of global (glEpac2 and plasma membrane (pmEpac2)) cAMP levels were provided by Dr. Martin J. Lohse (University of Würzburg) and have been characterized previously (35). All constructs were sequenced to confirm identity.
Generation and Maintenance of Stably Transfected Cell Lines
Human embryonic kidney (HEK293) cells stably expressing HA-TGR5, TGR5-GFP, or FLAG-PAR2 were generated using the FLP-Intm system (Invitrogen) and have been described (20, 36). HEK cell lines were cultured in DMEM supplemented with 10% heat-inactivated FBS and 100 μg/ml zeocin or hygromycin B. Human non-transformed colonic epithelial NCM460 colonocytes (Incell, San Antonio, TX) were cultured in M3D medium (Incell) containing 10% FBS. HA-TGR5 was stably expressed in NCM460 cells using a similar approach. Cell lines were cultured at 37 °C in 95% air, 5% CO2.
TGR5, PAR2, and β-Arrestin Trafficking
HEK-HA-TGR5 cells, HEK-TGR5-GFP cells, and NCM460-HA-TGR5 cells (3 × 105/35-mm dish) were plated onto glass coverslips coated with poly-D-lysine (100 μg/ml) and were studied 24–48 h later. To assess trafficking of TGR5 and PAR2 in the same cells, HEK-HA-TGR5 cells were transiently transfected with cDNA encoding FLAG-PAR2 using LipofectamineTM 2000 (Invitrogen) as described. To examine trafficking of receptors and β-arrestins, HEK-HA-TGR5 cells or HEK-FLAG-PAR2 cells were transiently transfected with cDNAs encoding β-arrestin1 or β-arrestin2 with GFP or YFP tags. Cells were studied 24–48 h after transfection and plating. For trafficking assays, cells were incubated in DMEM containing 0.1% BSA at 37 °C and then challenged with TGR5 or PAR2 agonists for 0–60 min. Receptors and β-arrestins were detected by immunofluorescence and confocal microscopy. To examine TGR5 trafficking in real time, HEK-TGR5-GFP cells grown on coverslips were incubated in Hanks' balanced salt solution (HBSS) containing 20 mm HEPES at 37 °C. Cells were exposed to TGR5 agonists, and confocal images were collected at 5-min intervals for 60 min.
Immunofluorescence Labeling and Confocal Microscopy
Cells were washed in 100 mm PBS (pH 7.4) and fixed in 4% paraformaldehyde in PBS for 20 min at 4 °C. Cells were washed in PBS and incubated in blocking buffer (PBS containing 0.1% saponin and 1% normal goat serum) (3 × 10 min). Cells were incubated in blocking buffer with the following primary antibodies (all overnight at 4 °C): anti-HA11 (rabbit or rat, 1:200), anti-FLAG (mouse, 1:500), and anti-EGFR (mouse, 1:100). Cells were washed and incubated with fluorescent secondary antibodies (1:200, 2 h, room temperature). Cells were washed and mounted in Vectashield (Vector Laboratories, Burlingame, CA) or Prolong Gold (Invitrogen). Cells were observed with a Zeiss LSM Meta 510 laser-scanning confocal microscope (Carl Zeiss, Thornwood, NY) using a Fluor Plan Apochromat ×63 oil immersion objective (numerical aperture 1.4) or a Plan Apochromat ×100 oil immersion objective (numerical aperture 1.4) or were observed with a Leica TCS SP8 laser-scanning confocal microscope (North Ryde, New South Wales, Australia) using a Leica HCX PL APO ×63 oil immersion objective (numerical aperture 1.4).
Immunogold Labeling and Electron Microscopy
HEK293 cells were transiently transfected with TGR5-GFP or empty vector (vector control). Cells were stimulated with DCA (100 μm) or vehicle (control) for 30 min. In some experiments, cells were also incubated with methyl-β-cyclodextrin (20 μm) or vehicle (control). Cells were washed twice with PBS, fixed in 2% paraformaldehyde/0.1% glutaraldehyde/0.1 M cacodylate fixative (pH 7.4) (3 h, room temperature), dehydrated through ethanol, and embedded in L. R. White resin (Electron Microscopy Sciences, Hatfield, PA) according to the recommendations of the manufacturer. Ultrathin sections were prepared using a Leica Ultracut UCT microtome (Leica Microsystems Inc., IL). Sections were incubated with rabbit anti-GFP (1:200) or normal serum (overnight, room temperature). Sections were washed and incubated with goat anti-rabbit IgG conjugated to 10-nm particles (1:100, 45 min, room temperature). Sections were stained with 5% aqueous uranyl acetate as described and examined with a Philips Tecnai 10 electron microscope (FEI, Hillsboro, OR).
cAMP BRET CAMYEL assay
The BRET CAMYEL (cAMP sensor using YFP-Epac-RLuc) cAMP sensor permits quantification of intracellular cAMP levels with high sensitivity and a broad dynamic range (37). HEK293 and HEK-HA-TGR5 cells (4 × 106/10-cm plate) were transfected with 4 μg of cDNA encoding CAMYEL. Similarly, HEK293 cells were transfected with 1 μg of cDNA encoding TGR5-RLuc8 and 4 μg of CAMYEL. Cells were transfected using polyethylenimine with a 6:1 polyethylenimine:cDNA ratio in 500 μl of 0.15 m NaCl. Cells were incubated overnight in DMEM 10% FBS, washed in PBS, and incubated in 1 ml of versene for 10 min. Cells were plated onto poly-D-lysine-treated 96-well plates and maintained overnight. Cells were washed in HBSS and then incubated in HBSS for 30 min at 37 °C. Coelenterazine H (Promega, Madison, WI; 5 μm final) was added, and cells were incubated in the dark for 5 min at 37 °C. Luminescence for RLuc8 (480 nm) and YFP fluorescence (530 nm) was measured using a microplate reader (LUMIstar Omega, BMG Labtech, Mornington, Australia) for a 2-min basal period and for 10 min after addition of TGR5 agonists, forskolin (10 μm), or vehicle. Base-line and vehicle control values were subtracted, and the BRET signal was normalized as a percentage of the forskolin response.
FRET cAMP Assay
The FRET Epac-based cAMP sensors glEpac2 and pmEpac2 permit sensitive quantification of total cytoplasmic (global, glEpac2) and membrane-associated (plasma membrane, pmEpac2) cAMP levels and are suitable for studies of dynamic alterations in cAMP levels at the single cell level (38). HEK-HA-TGR5 cells were transfected with 4 μg cDNA encoding pmEpac2 or glEpac2 using polyethylenimine and were plated onto 96-well plates. Cells were incubated in HBSS and observed using an IN Cell Analyzer 2000 (GE Healthcare Bio-Sciences, Uppsala, Sweden) to measure the emission ratio of cyan fluorescent protein (470 nm) and yellow fluorescent protein (YFP; 535 nm). To assess the duration of cAMP signals, cells were incubated with the TGR5 agonist DCA (1 or 10 μm) or with isoprenaline (100 nm), an agonist of the endogenous β2 adrenergic receptors in HEK cells. To assess desensitization to a repeated challenge, cells were incubated with DCA (10 μm) or vehicle (control) for 10 min, washed six times in HBSS, and challenged with DCA (10 μm) 10 min after washing. At the end of the experiment, cells were challenged with 10 μm forskolin in the presence of 100 μm isobutylmethylxanthine. Images of individual cells were analyzed using ImageJ software (National Institutes of Health) as described (38, 39). The base line was subtracted from stimulated ratios, and results were normalized to the maximal response to the forskolin challenge. The criterion for exclusion of single cells was a maximal response smaller than 5% of the base-line ratio.
ERK1/2 Assays
For Western blot analysis of pERK, HEK-HA-TGR5 cells (5 × 105 cells/6 wells) and NCM460 cells (2 × 105/well in a 12-well plate) were plated 48 h before treatments, and serum was replaced with 0.1% BSA for 16 h before assays. Cells were incubated in serum-free medium containing 0.1% BSA at 37 °C and were challenged with TGR5 agonists for 0–30 min. At specified times, cells were lysed in 50 mm Tris/HCl (pH 7.4), 150 mm NaCl, 5 mm MgCl2, 1 mm EGTA, 10 mm NaF, 10 mm Na4P2O7, 10 mm Na3VO4, and 1% Nonidet P-40. Lysates were separated by SDS-PAGE (12%). Proteins were transferred to polyvinylidene difluoride membranes (Immobilon-FL, Millipore, Billerica, MA) and blocked for 1 h at room temperature (Odyssey blocking buffer, LiCOR, Lincoln, NE). Membranes were incubated with antibodies to pERK1/2 (1:1000) and ERK2 (1:5000). Membranes were washed (1× PBS, 0.1% Tween 20, 30 min) and incubated with secondary antibodies coupled to Alexa Fluor 680 or IRDyetm 800 (1:20,000, 1 h, room temperature). Blots were analyzed with the Odyssey infrared imaging system (LiCOR).
The AlphaScreen SureFire phospho-ERK assay (PerkinElmer Life Sciences, Waltham MA, USA) was also used to quantify pERK. HEK-HA-TGR5 cells (5 × 104 cells/well) or NCM460 cell (6 × 104 cells/well) were plated in 96-well plates 48 h before treatments. Serum was replaced with 0.1% BSA for 16 h before assays. Cells were washed and incubated in serum-free medium containing 0.1% BSA at 37 °C. Cells were challenged with DCA (100 μm), CCDC (10 μm), or EGF (10 ng/ml) for 0–60 min. At specified times, cells were lysed, and pERK was quantified according to the protocols of the manufacturer.
To determine the mechanism of ERK1/2 activation, cells were preincubated with vehicle (control), the EGFR tyrosine kinase inhibitor AG1478 (1 μm, Calbiochem), the metalloprotease inhibitor batimastat (10 μm), or with methyl-β-cyclodextrin (500 μm) or filipin (1 μm), which deplete membrane cholesterol and block formation of lipid rafts. Inhibitors were preincubated with cells 30–60 min prior to stimulation with BAs and were present throughout the experiments.
BRET Assays to Assess Interactions of TGR5 and PAR2 with β-Arrestins, GRKs, Plasma Membrane and Endosomal Markers, G Proteins, and EGFR
To study the interactions of TGR5 and PAR2 with β-arrestins, GRKs, and plasma membrane and endosomal markers, HEK293 cells (4 × 106 cells/10-cm plate) were transiently transfected with 1 μg of cDNA encoding TGR5-RLuc8 or PAR2-RLuc8 plus 4 μg of β-arrestin1-YFP, β-arrestin2-YFP, GRK2-Venus, GRK5-Venus, GRK6-Venus, KRas-Venus, Rab5a-Venus, or Rab7a-Venus using polyethylenimine (33, 40–42). Control cells were transfected with receptor-RLuc8 vector and pcDNA3.1 vector without insert. Cells were harvested 24 h post-transfection in HEPES-buffered phenol red-free complete medium containing 10% FBS (PRF/FBS), plated onto poly-L-lysine-coated 96-well plates, and maintained at 37 °C, 5% CO2 for 24 h. Cells were incubated in fresh PRF/FBS at 37 °C with coelenterazine h (5 μm final) and challenged with agonists of TGR5 or PAR2. Luminescence for RLuc8 (480 nm) and YFP or Venus fluorescence (530 nm) were measured using a microplate reader (LUMIstar Omega) at various times after stimulation. The net BRET ratio was determined by subtracting the BRET ratio of the RLuc8/pcDNA3.1 control from the experimental groups. The net BRET ratio was normalized to the base line and vehicle control.
To study interactions of TGR5 with G proteins, HEK293 cells (6 × 105 cells/6-well plate) were transiently transfected with 0.18 μg of cDNA encoding TGR5-RLuc8 plus 0.4 μg of Gγ2-Venus, 0.27 μg of Gβ1, and 0.27 μg of Gα using GeneJuice (Novagen, Victoria, Australia). BRET was determined as described for other binding partners.
To study the interactions between TGR5 and EGFR by saturation analysis, HEK293 cells were transiently transfected with 0.25 μg of cDNA encoding TGR5-RLuc8 plus graded amounts of EGFR-Venus (0.05, 0.075, 0.1, 0.125, 0.175, 0.5, 1, 1.25, 1.5, 1.625, and 1.75 μg) or empty vector using GeneJuice. Cells were plated, and BRET was measured as described. The expression levels of TGR5-RLuc8 and EGFR-Venus were quantified by measurement of luminescence (LUMIstar Omega) and fluorescence (Envision, PerkinElmer Life Sciences), respectively. The BRET ratios were then compared with the protein expression ratios. In some experiments, cells were incubated with methyl-β-cyclodextrin (500 μm) or filipin (1.5 μm) for 60 min before the BRET assays.
TGR5 RT-PCR
Total RNA was isolated from HEK-TGR5-GFP and NCM460 cells and human colonic smooth muscle cells (ScienCell Research Laboratories, Carlsbad CA) using TRIzoltm (Invitrogen) and treated with DNase I (Ambion, Austin, TX). RNA (2 μg) was reverse-transcribed with Oligo d(T)15 and TaqMan reverse transcription reagents. PCR reactions used primers specific to human TGR5 (forward, 5′-tcgtctacttggctcccaac-3′, +260 to +279; reverse, 5′-ctcagtcctggcctatgagc-3′, +757 to +776). To exclude contamination by genomic DNA, control reactions omitted reverse transcriptase. PCR products were separated by electrophoresis on a 2% gel, detected with ethidium bromide, and sequenced to confirm identity.
β-Arrestin Western Blotting
NCM460 cells were lysed, and lysates were separated by SDS-PAGE and blotted as described for ERK1/2 assays. Membranes were incubated with rabbit anti-arrestin 1/2 (1:1000, overnight, 4 °C), washed, incubated with secondary antibodies, and then blots were analyzed with an Odyssey infrared imaging system (LiCOR Biosciences).
Data and Statistical Analyses
Quantitative analyses are from n ≥ 3 independent experiments. Results were analyzed using PRISM 5 (GraphPad Software Inc., La Jolla, CA). Data are presented as mean ± S.E. Results were compared using two-way analysis of variance with Bonferroni post-test. A value of p < 0.05 was considered significant. Micrographs are representative of n ≥ 3 independent experiments.
RESULTS
TGR5 Expressed in HEK Cells Couples to Generation of cAMP
To verify that TGR5 expressed in HEK cells is fully functional, we measured agonist-stimulated formation of cAMP by using the Epac-based BRET sensor CAMYEL, which permits the quantification of intracellular cAMP levels with high sensitivity and a broad dynamic range. In HEK cells stably expressing HA-TGR5 and transiently expressing CAMYEL, the endogenous TGR5 agonists DCA and TLCA (1 nm-100 μm, 5 min) stimulated a concentration-dependent increase in cAMP formation (Fig. 1A) (EC50: DCA, 70 nm; TLCA, 2.2 nm). OA, a naturally occurring component of leaves from the olive tree Olea europaea that potently activates TGR5 but not the nuclear farnesoid X BA receptor (43), also stimulated cAMP formation (EC50, 586 nm). The synthetic TGR5 agonist CCDC was the most potent stimulant of cAMP formation (EC50, 2.3 pm). DCA, TLCA, OA, and CCDC did not stimulate cAMP formation in HEK cells that did not express TGR5 (Fig. 1B). However, all agonists stimulated cAMP formation in HEK cells expressing TGR5 with C-terminal tags that were used for BRET assays (RLuc8, Fig. 1C) and trafficking studies (GFP, Ref. 20). These results indicate that the agonists stimulate cAMP generation by a TGR5-dependent process and that tagged versions of TGR5 are fully functional.
FIGURE 1.

TGR5-mediated cAMP formation. cAMP levels were measured using the BRET-based CAMYEL cAMP sensor in HEK-HA-TGR5 cells (A), untransfected HEK293 cells (B), and HEK cells transiently transfected with TGR5-RLuc8 (C). Cells were incubated with the indicated concentrations of TGR5 agonists for 5 min, and BRET signals were measured. Results are normalized to the maximal forskolin response. Shown are triplicate observations from n = 3 experiments.
TGR5 Is Localized to the Plasma Membrane and Does Not Undergo Agonist-induced Endocytosis
Upon activation, many GPCRs traffic from the plasma membrane to endosomes by a clathrin- and dynamin-dependent mechanism (22, 23). To determine whether BAs that activate TGR5 also stimulate receptor endocytosis, we used sequential confocal imaging to study the trafficking in real time of TGR5-GFP expressed in HEK cells. In unstimulated cells, TGR5-GFP was present at the plasma membrane with minimal intracellular pools (Fig. 2A). Exposure to DCA (100 μm, 37 °C) did not alter the subcellular distribution of TGR5, which remained at the plasma membrane for up to 60 min of stimulation. We also examined the subcellular localization of HA-TGR5 expressed in HEK cells. Cells were incubated with vehicle (0.1% ethanol) or DCA, TLCA, OA, or CCDC (100 μm, 0–60 min); fixed at defined times; and then HA-TGR5 was localized by immunofluorescence using an HA11 antibody and confocal microscopy. In vehicle-treated cells, TGR5 was present at the plasma membrane. DCA, TLCA, OA, or CCDC did not stimulate a detectable alteration in the subcellular localization of TGR5, which remained at the plasma membrane at all time points (Fig. 2B, 10 min). To ensure that the HEK cell line was capable of supporting endocytosis of other GPCRs, we coexpressed TGR5-GFP with FLAG-PAR2, which undergoes agonist-dependent endocytosis (44). In unstimulated cells, TGR5-GFP and FLAG-PAR2 were colocalized at the plasma membrane. DCA (100 μm, 30 min) did not alter the subcellular distribution of TGR5-GFP or FLAG-PAR2 (Fig. 2C). In contrast, the PAR2-AP (SLIGRL-NH2, 50 μm, 30 min) stimulated endocytosis of FLAG-PAR2 but not TGR5-GFP (Fig. 2D). These results indicate that agonists of TGR5 that potently activate the receptor and stimulate cAMP formation do not cause detectable redistribution of the receptor to endosomes. In contrast, agonists stimulate robust endocytosis of PAR2 in the same cell line, which indicates that the cells are capable of supporting agonist-stimulated internalization of other GPCRs in the expected manner.
FIGURE 2.

Agonist-stimulated trafficking of TGR5 and PAR2. A and B, trafficking of TGR5. A, real-time trafficking of TGR5-GFP in HEK cells. The images show the localization of TGR5-GFP at the plasma membrane (arrowheads) of the same two cells before and after stimulation with DCA (100 μm) and indicate that DCA did not affect the subcellular localization of TGR5. B, localization of HA-TGR5 after incubation with vehicle, DCA, TLCA, OA, or CCDC (all 100 μm) for 10 min, showing the retention of TGR5 at the plasma membrane (arrowheads). C and D, trafficking of TGR5 and PAR2. TGR5-GFP and FLAG-PAR2 were coexpressed in HEK cells. Cells were incubated with DCA (100 μm, C) or PAR2-AP (SLIGRL-NH2, 50 μm, D) for 0 or 30 min, and both receptors were localized simultaneously. DCA did not affect the subcellular localization of TGR5 or PAR2, which remained at the plasma membrane (arrowheads). In contrast, PAR2-AP stimulated the trafficking of PAR2 to endosomes (arrows). Scale bars = 10 μm. Shown are representative images of n ≥ 3 experiments.
Activated TGR5 Does Not Traffic through the Endosomal Network
To further investigate the subcellular location of TGR5 upon agonist stimulation, we used a recently described BRET assay that allows the detection of energy transfer between receptors and BRET partners targeted to defined subcellular compartments (33). This approach allows quantitative assessment of receptor trafficking from the plasma membrane to defined populations of endosomes. To do so, TGR5-RLuc8 or PAR2-RLuc8 were coexpressed in HEK293 cells with Venus-tagged BRET acceptors that are localized to different compartments: KRas-Venus at the plasma membrane, Rab5a-Venus in early endosomes, and Rab7a-Venus in late endosomes/lysosomes. Cells were incubated with agonists of TGR5 or PAR2 for 30 min at 37 °C. Coelenterazine h was added in the last 10 min of the agonist incubation, and then BRET was measured. The potent PAR2 agonists 2-Fluoryl-LIGRLO-NH2 and AC 264613 (both 10 μm) resulted in a significant decrease of the BRET signal between PAR2-RLuc8 and Venus-KRas, indicative of receptor endocytosis (Fig. 3A). The decline in BRET signals between PAR2 and the plasma membrane acceptor coincided with a significant increase in the BRET signals between PAR2-RLuc8 and Rab5a-Venus and Rab7a-Venus (Fig. 3, B and C). These results are consistent with the expected accumulation of PAR2 in Rab5a-positive early endosomes and Rab7a-positive late endosomes/lysosomes. In contrast, when cells were incubated with the TGR5 agonists DCA, TLCA, CCDC (all 100 μm), or OA (10 μm), there was no significant change in BRET between TGR5-RLuc8 and KRas-Venus, suggesting lack of internalization (Fig. 3A). Incubation with TLCA and CCDC caused a small increase in BRET between TGR5-RLuc8 and Rab5a-Venus or Rab7a-Venus, although this was significant only for Rab7a-Venus and CCDC (Fig. 3, B and C). Considered together, these results are consistent with the microscopy and indicate that TGR5 does not undergo robust agonist-stimulated endocytosis.
FIGURE 3.
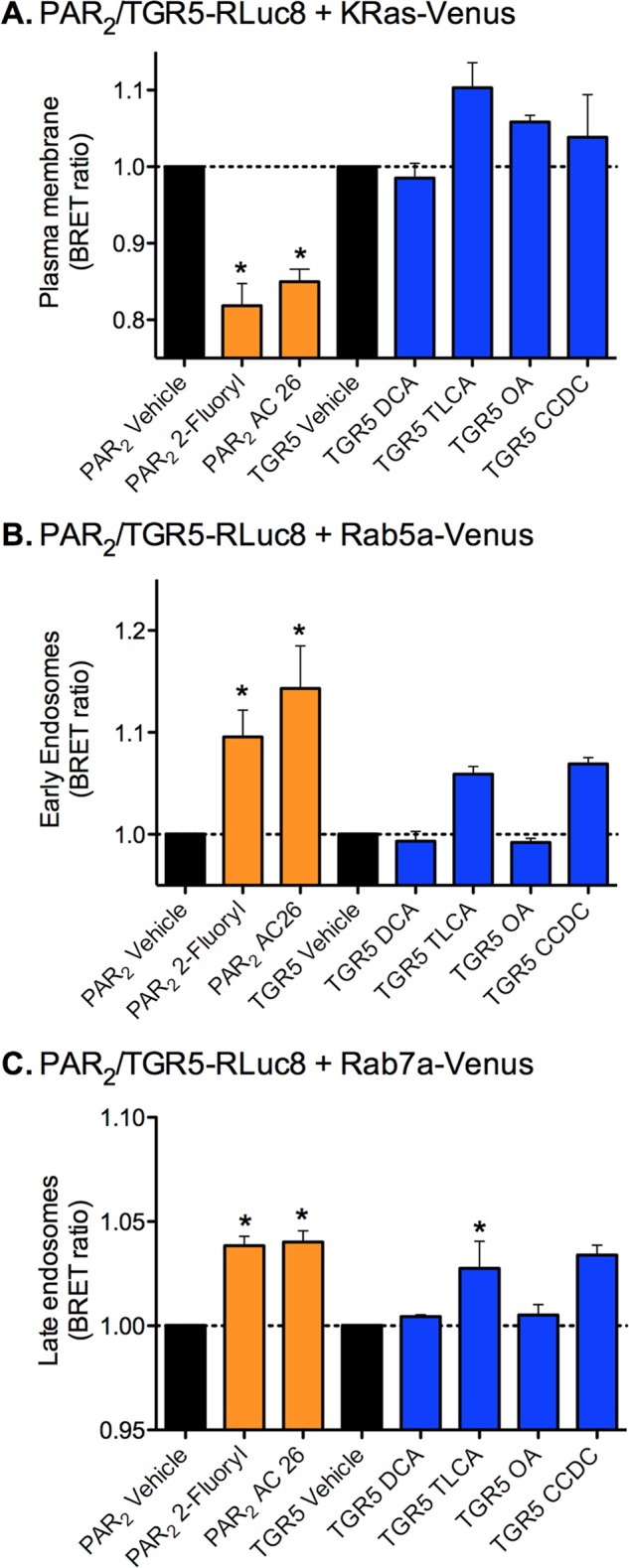
BRET analysis of interactions between PAR2 or TGR5 with plasma membrane and endosomal markers. PAR2-RLuc8 or TGR5R-Luc8 was coexpressed with the plasma membrane BRET acceptor KRas-Venus (A), the early endosomal BRET acceptor Rab5a-Venus (B), or the late endosomal BRET acceptor Rab7a-Venus (C) in HEK293 cells. Cells were incubated with vehicle or with agonists of PAR2 (orange) or TGR5 (blue) for 30 min, and BRET signals were measured. The PAR2 agonists 2-furoyl-LIGRLO (2-furoyl) and AC264613 (AC26) (both 10 μm) induced a decline in PAR2-RLuc8 and KRas-Venus BRET and an increase in PAR2-RLuc8 and Rab5a-Venus and Rab7a-Venus BRET, indicative of trafficking of PAR2 from the plasma membrane to early and late endosomes. The TGR5 agonists DCA, TLCA, CCDC (100 μm), or OA (10 μm) did not affect TGR5-RLuc8 and KRas-Venus BRET and had no significant effect on the TGR5-RLuc8 and Rab5a-Venus and Rab7a-Venus BRET, with the exception of CCDC, which increased TGR5-RLuc8 and Rab7a-Venus BRET. Thus, TGR5 agonists do not cause a robust trafficking of TGR5 from the plasma membrane to early and late endosomes. *, p < 0.05 to vehicle. Shown are triplicate observations from n = 3 experiments.
Activated TGR5 Does Not Recruit β-Arrestin1 or β-Arrestin2
Agonists of many GPCRs promote the recruitment of β-arrestins to GRK-phosphorylated receptors at the plasma membrane, where β-arrestins uncouple receptors from heterotrimeric G proteins and desensitize plasma membrane signaling (22, 23, 44). β-arrestins also couple receptors to clathrin and AP2, which mediate endocytosis of the receptor and β-arrestin complex. To determine whether TGR5 activation similarly induces the recruitment of β-arrestins to the plasma membrane or endosomes, we transiently expressed β-arrestin1 or β-arrestin2 with GFP or YFP tags in HEK-HA-TGR5 cells. Cells were incubated with TGR5 agonists, and HA-TGR5 and β-arrestin1 or β-arrestin2 GFP/YFP were localized by immunofluorescence and confocal microscopy. In unstimulated cells, TGR5 was at the plasma membrane, and β-arrestin1-GFP and β-arrestin2-GFP were distributed uniformly in the cytosol (Fig. 4, A and B). Incubation with DCA (100 μm) for 5 or 30 min did not alter this subcellular distribution of TGR5 or cause trafficking of β-arrestin1-GFP or β-arrestin2-GFP from the cytosol to the plasma membrane or endosomes. Similarly, exposure of cells to TLCA, OA, or CCDC (100 μm) for 5 or 30 min did not affect the subcellular distribution of TGR5, β-arrestin1-YFP (not shown), or β-arrestin2-YFP (Fig. 4C, 30 min). In marked contrast, PAR2-AP (SLIGRL-NH2, 50 μm, 30 min) stimulated the trafficking of FLAG-PAR2 and β-arrestin1-GFP (Fig. 4D) and β-arrestin2-GFP (not shown) to the same endosomes. These results suggest that agonists do not promote the interaction of TGR5 with β-arrestin1 or β-arrestin2 in HEK cells.
FIGURE 4.

Agonist-stimulated trafficking of TGR5, PAR2, and β-arrestins (βARR). A–C, HEK-HA-TGR5 cells were transiently transfected with β-arrestin1-GFP (A), β-arrestin2-GFP (B), or β-arrestin2-YFP (C). Cells were incubated with DCA, TLCA, OA, or CCDC (all 100 μm) for 0–30 min, and TGR5 and β-arrestins were localized. The merged images in C show HA-TGR5 (red) and β-arrestin2-YFP (green). DCA, TLCA, OA, and CCDC did not alter the subcellular distribution of TGR5, which remained at the plasma membrane (arrowheads), or β-arrestins, which remained in the cytosol (arrows) at every time point. D, HEK-FLAG-PAR2 cells were transiently transfected with β-arrestin1-GFP. Cells were incubated with PAR2-AP (SLIGRL-NH2, 50 μm) for 30 min, which induced trafficking of PAR2 and β-arrestin-1-GFP to the same endosomes (arrows). Scale bars = 10 μm. Shown are representative images of n ≥ 3 experiments.
Activated TGR5 Does Not Internalize or Interact with β-Arrestins in Human Colonocytes That Naturally Express This Receptor
To ascertain whether agonists promote TGR5 and β-arrestin trafficking in cells that naturally express these proteins, we studied NCM460 cells, a non-transformed human colonic epithelial cell line that has been used to assess the trafficking and signaling of GPCRs (45). By using RT-PCR, transcripts encoding TGR5 mRNA of the anticipated size (498 bp) were amplified from NCM460 cells (Fig. 5A). TGR5 was similarly amplified from HEK-TGR5-GFP cells and human colonic smooth muscle cells, which served as positive controls. Analysis of lysates from NCM460 cells by Western blotting revealed that these cells expressed both β-arrestin1 and β-arrestin2, although β-arrestin1 was more abundant (Fig. 5B). Thus, NCM460 cells naturally express TGR5 and β-arrestins. To facilitate detection, HA-TGR5 was stably expressed in NCM460 cells, which were then transiently transfected with β-arrestin1-GFP or β-arrestin2-GFP. Incubation of cells with DCA (100 μm, 0–30 min) did not affect the subcellular localization of TGR5, which remained at the plasma membrane, or of β-arrestin1-GFP, which remained distributed uniformly in the cytosol (Fig. 5C). Similarly, DCA did not affect the subcellular localization of β-arrestin2-GFP in NCM460 cells (not shown). Thus, activated TGR5 neither internalizes nor recruits β-arrestins in a human colonocyte cell line that endogenously expresses these proteins and that supports agonist-dependent trafficking of other GPCRs and β-arrestins (45).
FIGURE 5.

Expression and trafficking of TGR5 in NCM460 colonocytes. A, detection of mRNA encoding TGR5 by RT-PCR in HEK-TGR5-GFP cells, NCM460 cells, and human colonic smooth muscle cells. Transcripts of the anticipated size (498 bp) were amplified from all cell lines and identified by sequencing. B, detection of endogenous β-arrestin1 (βARR1) and β-arrestin2 (βARR2) in NCM460 cells by Western blotting. Note that β-arrestin1 was more highly expressed. C, agonist-stimulated trafficking of TGR5 and β-arrestin1-GFP in NCM460 cells. β-arrestin1-GFP was transiently expressed in NCM460-HA-TGR5 cells. Cells were incubated with DCA (100 μm, 0–30 min), and TGR5 and β-arrestin1-GFP were localized. Note that DCA did not alter the subcellular distribution of TGR5, which remained at the plasma membrane (arrowheads) or β-arrestin1-GFP, which remained cytosolic (arrows). Scale bar = 10 μm. Shown are representative images of n ≥ 3 experiments.
BRET Analysis Indicates That TGR5 Agonists Do Not Promote Interactions between TGR5 and β-Arrestin1/2 or GRK2/5/6
BRET is a sensitive and quantifiable technique that is widely used to assess the interactions between GPCRs and accessory proteins, such as β-arrestins (31). To quantitatively assess the interactions between TGR5 and β-arrestin1 or β-arrestin2, HEK cells were cotransfected with RLuc8-tagged TGR5 and YFP-tagged β-arrestins. As a positive control, RLuc8-tagged PAR2 and YFP-tagged β-arrestins were coexpressed similarly. Cells were incubated with coelenterazine h for 5 min, and BRET signals were measured before and after agonist stimulation. The PAR2 agonists 2-furoyl-LIGRLO-NH2 and AC 264613 (both 10 μm) stimulated an immediate and robust BRET signal between PAR2-RLuc8 and β-arrestin1-YFP or β-arrestin2-YFP, consistent with interactions between PAR2 and β-arrestins (Fig. 6, A and B). Stimulation with graded concentrations of these agonists (1 nm-1 mm, 5 min) revealed a concentration-dependent BRET signal (Fig. 6, C and D). In marked contrast, stimulation of cells with the TGR5 agonists DCA, OA, or TLCA (1 nm-1 mm) did not result in an increase in the BRET signal between TGR5-RLuc8 and β-arrestin1-YFP or β-arrestin2-YFP over the base line (Fig. 6, A and B).
FIGURE 6.

BRET analysis of interactions between PAR2 or TGR5 with β-arrestins or GRKs. A–D, PAR2-RLuc8 or TGR5RLuc8 was coexpressed with β-arrestin1-YFP or β-arrestin2-YFP in HEK cells. When compared with prestimulated values, the PAR2 agonists AC264613 (AC26) and 2-furoyl-LIGRLO (2-Furoyl) (both 10 μm, arrows) induced a rapid and sustained increase in the BRET signal from PAR2-RLuc8 to β-arrestin1-YFP (A) and β-arrestin2-YFP (B) (p < 0.0001 for all time points after 2 min). The interaction of PAR2-RLuc8 and β-arrestin1-YFP (C) and β-arrestin2-YFP (D) was concentration-dependent (measured at 5 min). In contrast, DCA, TLCA, and OA failed to induce an increase in BRET between TGR5-RLuc8 and β-arrestin1-YFP (A and C, p > 0.05) or β-arrestin2-YFP (B and D, p > 0.05). E–G, PAR2-RLuc8 or TGR5RLuc8 was coexpressed with GRK2-Venus, GRK5-Venus, or GRK6-Venus in HEK cells. When compared with prestimulated values, the PAR2 agonists AC264613 and 2-furoyl-LIGRLO (both 1 μm) induced a rapid and sustained increase in the BRET signal from PAR2-RLuc8 to GRK2-Venus (E), GRK5-Venus (F), and GRK6-Venus (G) (p < 0.0001 for all time points after 2 min). In contrast, DCA, TLCA, and OA did not evoke an increase in the BRET signal between TGR5-RLuc8 and GRK2-Venus, GRK5-Venus, or GRK6-Venus (p > 0.05 for all time points after 2 min). Shown are triplicate observations from n = 3 experiments.
GRK2, GRK5 and GRK6 can interact with and phosphorylate many agonist-occupied GPCRs, which promotes the recruitment of β-arrestins. To assess whether agonists of PAR2 and TGR5 induce the interaction between receptors and GRKs, we cotransfected HEK cells with RLuc8-tagged receptors and GRK2-Venus, GRK5-Venus or GRK6-Venus. The PAR2 agonists 2-furoyl-LIGRLO-NH2 and AC 264613 (both 10 μm) stimulated an immediate and robust BRET signal between PAR2-RLuc8 and GRK2-Venus (Fig. 6E), and 2-furoyl-LIGRLO-NH2 also induced BRET between PAR2-RLuc8 and GRK5-Venus (F) and GRK6-Venus (G). In contrast, DCA, OA, or TLCA (all 100 μm) did not increase the BRET signal between TGR5-RLuc8 and GRK2-Venus (Fig. 6E), and DCA did not cause an increased BRET signal between TGR5-RLuc8 and GRK5-Venus (F) and GRK6-Venus (G). The DCA-evoked decline in BRET between TGR5-RLuc8 and GRK5-Venus may reflect a conformational change in TGR5 at the plasma membrane.
Thus, although PAR2 agonists promote the interaction between PAR2 and GRK2/5/6 and β-arrestin1/2, TGR5 agonists do not stimulate interactions between TGR5 and GRK2/5/6 and β-arrestin1/2. These results are consistent with the inability of TGR5 agonists to stimulate detectable endocytosis of the receptor or redistribution of the subcellular distribution of β-arrestins.
Activation of TGR5 with a Synthetic Agonist Results in β-Arrestin2 and GRK2 Recruitment
CCDC is a synthetic agonist of TGR5 (29) that is a more potent stimulant of cAMP formation than endogenous BA agonists (Fig. 1). We used BRET to determine whether CCDC could promote the interaction of TGR5 with β-arrestin1/2 or GRK2/5/6. Strikingly, CCDC (100 μm) stimulated the preferential interaction between TGR5-RLuc8 and β-arrestin2-YFP but not β-arrestin1-YFP (Fig. 7, A and B). CCDC also stimulated the interaction between TGR5-RLuc8 and GRK2-Venus (Fig. 7C) but not between TGR5-RLuc8 and GRK5-Venus or GRK6-Venus (D). However, although CCDC induced a significant increase in the BRET signal between TGR5-RLuc8 and β-arrestin2-YFP and GRK2-Venus, the BRET signals were ∼2-fold lower than those for PAR2-RLuc8 and β-arrestin2-YFP and 3-fold lower than those for PAR2-RLuc8 and GRK2-Venus. Moreover, CCDC did not induce a detectable redistribution of β-arrestin2-GFP to the plasma membrane or endosomes and did not stimulate endocytosis of TGR5 (Figs. 3A and 4C). Thus, the highly potent agonist CCDC can induce the preferential interaction between TGR5 and β-arrestin2 and GRK2 but not β-arrestin1 or GRK5/6. These interactions are insufficient to support endocytosis of TGR5.
FIGURE 7.

CCDC-induced interaction of TGR5 with β-arrestins and GRKs. TGR5-RLuc8 was coexpressed with β-arrestin1-YFP, β-arrestin2-YFP, GRK2-Venus, GRK5-Venus, or GRK6-Venus in HEK cells. A, when compared with prestimulated values, the synthetic TGR5 agonist CCDC (100 μm, arrow) did not affect the BRET signal between TGR5-RLuc8 and β-arrestin1-YFP (p > 0.05 for all time points after 2 min) but induced a rapid and sustained increase in the BRET signal between TGR5-RLuc8 and β-arrestin2-YFP (p < 0.0001 for all time points after 2 min). B, CCDC stimulated a BRET signal between TGR5-RLuc8 and β-arrestin2-YFP only at the highest concentrations (measured after 5 min) C, CCDC (100 μm, arrow) also stimulated a BRET signal between TGR5-RLuc8 and GRK2-Venus (p < 0.0001 for all time points after 2 min). D, CCDC (100 μm, arrow) did not stimulate a BRET signal between TGR5-RLuc8 and GRK5-Venus or GRK6-venus (p > 0.05 for all time points after 2 min). Shown are triplicate observations from n = 3 experiments.
TGR5 Couples to Gαs and Induces Sustained cAMP Signals That Do Not Desensitize Robustly
To investigate the interaction between TGR5 and G proteins, we examined BRET between TGR5-RLuc8 and Gγ2-Venus (34). In HEK293 cells expressing TGR5-RLuc8, Gγ2-Venus, Gβ1, and Gαs, DCA (100 μm), OA (10 μm), and CCDC (10 μm) stimulated BRET between TGR5-RLuc8 and Gγ2-Venus (Fig. 8A). These agonists did not stimulate BRET in cells that were not transfected with Gαs (Fig. 8B). The results are consistent with an agonist-induced reorientation of the TGR5 Gγ2 complex, leading to the activation of Gαs.
FIGURE 8.

TGR5 coupling to Gαs and sustained TGR5-mediated cAMP formation. A and B, TGR5 and Gαs interactions. A, TGR5-RLuc8, Gγ2-Venus, Gβ1, and Gαs were coexpressed in HEK293 cells. When compared with prestimulated values, DCA (100 μm), OA (10 μm), and CCDC (10 μm) (arrows) induced a rapid and sustained increase in the BRET signal between TGR5-RLuc8 and Gγ2-Venus (p < 0.0001 for all time points after 2 min). B, ligand-induced BRET was not observed in the absence of Gαs, consistent with a ligand-evoked interaction between TGR5 and Gαs. Shown are triplicate observations from n = 3 experiments. C–F, duration of DCA-stimulated cAMP formation. cAMP levels were measured in HEK-HA-TGR5 cells expressing the plasma membrane (pmEpac2, C and E) and global (glEpac2, D and F) cAMP FRET sensors. C and D, continuous incubation with DCA (1 or 10 μm) stimulated a rapid increase in cAMP formation at the plasma membrane (C) and in the cytoplasm (D) compared with vehicle. The increase in cAMP was sustained during the time of exposure to DCA (100 min), indicating a lack of tachyphylaxis. In contrast, the response to isoprenaline (Iso, 100 nm) was of a similar magnitude but underwent tachyphylaxis as the cAMP levels quickly returned toward the base line. E and F, addition of DCA (10 μm at 10 min) resulted in a rapid increase in cAMP formation at the plasma membrane (E) and in the cytosol (F), which rapidly returned to base line after washing (W) at 20 min. The response to a second exposure to DCA (10 μm at 30 min) was the same as to the first exposure or to that of vehicle-treated (Veh) cells, indicating a lack of desensitization. At the end of the experiments, all cells were challenged with forskolin (FSK, 10 μm). Base line-subtracted FRET ratios are expressed as a percentage of the maximal response to forskolin. Results are from n ≥ 15 cells/experiment with n = 3 (isoprenaline), n = 6 (vehicle), and n = 5–9 (DCA) separate experiments.
The signals that emanate from many activated GPCRs fade over time (tachyphylaxis) and are attenuated in response to repeated stimulation with the same agonist, illustrative of homologous desensitization of receptors (46). Homologous desensitization largely depends on GRK-mediated receptor phosphorylation and interaction of the phosphorylated receptors with β-arrestins, which sterically impede the interaction of GPCRs with heterotrimeric G proteins and, thereby, uncouple receptors from the signaling machinery. To assess whether TGR5 desensitizes similarly, we measured the formation of intracellular cAMP in HEK-HA-TGR5 cells using the FRET cAMP sensors glEpac2 and pmEpac2. These sensors permit the sensitive quantification of total cytoplasmic (global, glEpac2) and membrane-associated (plasma membrane, pmEpac2) cAMP levels and are suitable for studies of dynamic alterations in cAMP levels at the single cell level. DCA (1 or 10 μm) stimulated a rapid (within minutes), robust, and concentration-dependent increase in cAMP formation both within the cytoplasm and at the plasma membrane (Fig. 8, C and D). Notably, cAMP levels were sustained for at least 100 min in the presence of both concentrations of DCA with no sign of tachyphylaxis. Thus, after 100 min, the cytoplasmic and plasma membrane cAMP levels were, respectively, 110 and 115% of the maximal DCA-evoked (10 μm) levels measured after 4 min of stimulation. Isoprenaline (100 nm), which activates endogenous β2 adrenergic receptors in HEK cells, induced a similar maximal increase in cytoplasmic and plasma membrane cAMP levels (Fig. 8, C and D). However, in sharp contrast to the sustained response to DCA, isoprenaline-stimulated cAMP levels exhibited marked tachyphylaxis over the same time period. After 100 min, the cytoplasmic and plasma membrane cAMP levels were, respectively, 28 and 46% of the maximal isoprenaline levels measured after 4 min of stimulation. Although the DCA-evoked increase in cAMP levels declined within 5 min of washing and removal of the agonist (10 μm), repeated application of DCA (10 μm) stimulated an identical increase in cytoplasmic and plasma membrane cAMP levels, with no indication of desensitization (Fig. 8, E and F).
These results indicate that DCA-evoked cAMP formation in the cytoplasm and at the plasma membrane does not undergo tachyphylaxis and is not attenuated in response to a repeated challenge. Thus, in contrast to the β2 adrenergic receptor, which undergoes well documented desensitization, TGR5 is remarkably resistant to desensitization and is capable of continuing to signal from the plasma membrane for prolonged periods. This lack of desensitization is consistent with the inability of TGR5 to interact with GRK2/5/6 and β-arrestin1/2, which desensitize plasma membrane signaling of many other GPCRs, including PAR2 and the β2 adrenergic receptor.
TGR5 Transactivates the EGFR within Plasma Membrane Microdomains to Stimulate ERK1/2
GPCRs can activate ERK1/2 by G protein-dependent and independent mechanisms. A prominent G protein-dependent mechanism that operates from the plasma membrane involves G protein-mediated activation of membrane metalloproteases that cleave and liberate membrane-tethered agonists of the EGFR, leading to EGFR-mediated ERK1/2 activation (i.e. EGFR transactivation) (47). By recruiting components of the MAP kinase cascade such as src and raf-1 to internalized GPCRs, β-arrestins can mediate G protein-independent ERK1/2 activation (23, 48). To determine whether TGR5 activates ERK1/2 by these mechanisms, we challenged HEK-HA-TGR5 cells with DCA (100 μm) and assessed ERK1/2 activation by Western blotting for pERK1/2 and ERK2. DCA stimulated a robust increase in levels of pERK1/2 that was maximal after 2 min and sustained for at least 30 min (Fig. 9, A and C). The EGFR tyrosine kinase inhibitor AG1478 strongly inhibited DCA-stimulated ERK1/2 activation (Fig. 9, B and C).
FIGURE 9.

Mechanisms of TGR5-induced ERK1/2 activation in HEK-TGR5 cells. A–C, HEK-HA-TGR5 cells were incubated with DCA (100 μm) or vehicle (0.1% ethanol) (0–30 min, 37 °C). Levels of pERK1/2 and total ERK2 were determined by Western blotting (A and B) and were quantified by densitometry (C). DCA increased levels of pERK1/2 compared with vehicle. The EGFR inhibitor AG1478 significantly inhibited DCA-stimulated levels of pERK1/2 at 2 and 5 min. *, p < 0.05; ***, p < 0.0001; DCA + AG1478 compared with DCA. Shown are triplicate observations from n = 3 experiments. D–G, HEK-HA-TGR5 cells were incubated with DCA (100 μm) plus vehicle, methyl-β-cyclodextrin (MβCD), or filipin (0–30 min, 37 °C). Levels of pERK1/2 and total ERK2 were determined by Western blotting (D, E, and F) and were quantified by densitometry (G). DCA increased levels of pERK1/2. Depletion of membrane cholesterol with methyl-β-cyclodextrin or filipin inhibited DCA-stimulated levels of pERK1/2. * p < 0.01; ** p > 0.001; *** p > 0.0001; DCA + methyl-β-cyclodextrin or filipin compared with DCA + vehicle. Shown are triplicate observations from n = 4 (DCA), n = 5 (methyl-β-cyclodextrin), or n = 3 (filipin) experiments.
The compartmentalization of GPCRs within microdomains at the plasma membrane, including lipid rafts and caveolae, can enhance the efficiency of signal transduction (24). Cholesterol is enriched in lipid rafts and caveolae, and agents that deplete the plasma membrane of cholesterol, including methyl-β-cyclodextrin and filipin, disrupt assembly of lipid rafts and caveolae. To determine whether compartmentalization is necessary for TGR5-mediated transactivation of the EGFR and activation of ERK1/2, we treated cells with methyl-β-cyclodextrin or filipin and examined DCA-stimulated ERK1/2 activation by Western blotting. Both treatments strongly inhibited DCA-stimulated activation of ERK1/2 (Fig. 9, D–G).
To examine the mechanisms by which endogenously expressed TGR5 activates ERK1/2, we measured ERK1/2 activation in NCM460 cells using the highly sensitive AlphaScreen phospho-ERK assay. We first verified that this assay gave similar results in HEK-TGR5 cells as the Western blot assay for ERK1/2 activation. As expected, DCA (100 μm) and CCDC (10 μm) both stimulated ERK activation (Fig. 10A). The EGFR tyrosine kinase inhibitor AG1478 and the metalloprotease inhibitor batimastat both abolished DCA- and CCDC-evoked ERK activation. Similarly, in non-transfected NCM460 colonocytes, DCA and CCDC stimulated ERK1/2 activation, which was abolished by AG1478 or batimastat (Fig. 10B). In NCM460 cells, as in the HEK-TGR5 cells, DCA-stimulated (100 μm, 5 min) ERK activation was inhibited by 78% by filipin (p > 0.0001) and 50% by methyl-β-cyclodextrin (p > 0.01) (not shown). In marked contrast, filipin or methyl-β-cyclodextrin did not inhibit EGF-stimulated (10 ng/ml) ERK activation in NCM460 cells, and these agents did not inhibit DCA-evoked cAMP formation in HEK-TGR5 cells (not shown).
FIGURE 10.
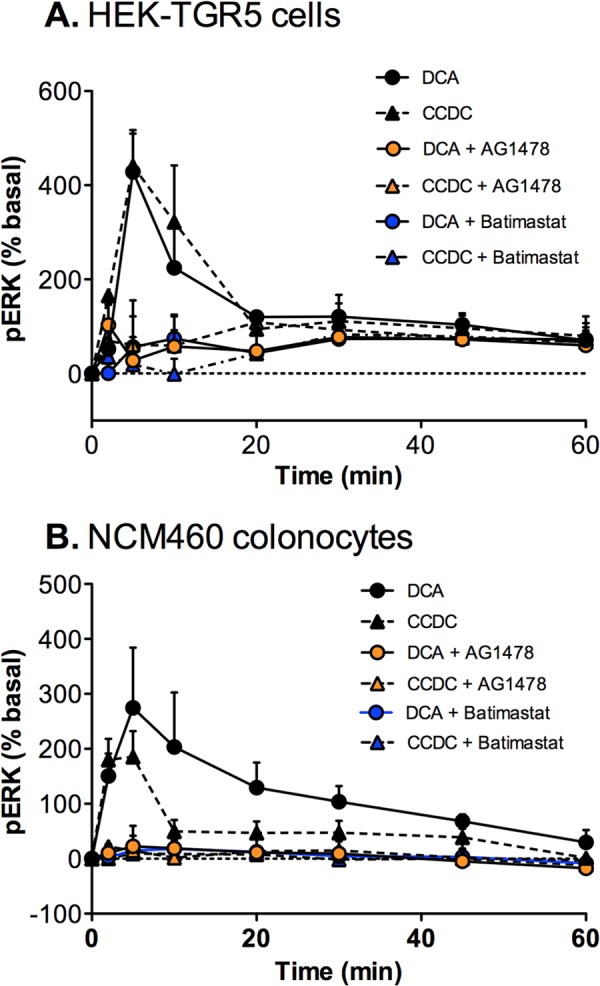
Mechanisms of TGR5-induced ERK1/2 activation in HEK-TGR5 and NCM460 cells. HEK-TGR5 cells (A) or NCM460 cells (B) were incubated with DCA (100 μm) or CCDC (10 μm) (0–60 min, 37 °C). pERK was measured using the AlphaScreen assay. DCA and CCDC stimulated ERK activation in both cell lines. When compared with agonist alone, AG1478 or batimastat abolished activation of ERK (all p < 0.0001 at 5 and 10 min). Shown are triplicate observations from n = 3 experiments.
These results indicate that TGR5 transactivates the EGFR to induce the activation of ERK1/2. This transactivation requires activity of a batimastat-sensitive proteases and occurs within plasma membrane microdomains, which probably serve to increase the efficiency of TGR5-stimulated EGFR transactivation and ERK1/2 signaling. However, TGR5-dependent cAMP formation does not require intact lipid rafts because it was not inhibited by filipin or methyl-β-cyclodextrin.
TGR5 and EGFR Form Heterodimers within Plasma Membrane Microdomains
The association between GPCRs and the EGFR within plasma membrane microdomains can enhance the efficiency of EGFR transactivation and ERK1/2 signaling (49–51). In HEK-HA-TGR5 cells, TGR5 and endogenous EGFR were colocalized at the plasma membrane (Fig. 11A). To determine whether TGR5 associates with EGFR, we examined interactions between TGR5-RLuc8 and EGFR-Venus using BRET. Saturation curves were generated by expressing a constant amount of TGR5-RLuc8 with increasing amounts of EGFR-Venus in the absence of TGR5 agonists. This analysis revealed that the BRET signal between TGR5-RLuc8 and EGFR-Venus saturated with increasing levels of EGFR expression, with a maximal BRET ratio of ∼0.03 (Fig. 11B). These data are consistent with the preassembly of TGR5 and EGFR heterodimers, even in the absence of the agonist. In cells treated with filipin, which depletes membrane cholesterol and blocks formation of lipid rafts, the TGR5 and EGFR interactions did not saturate (Fig. 11B). Methyl-β-cyclodextrin also blocked saturation, although to a lesser degree than filipin (not shown).
FIGURE 11.

TGR5 and EGFR interactions in HEK293 cells. A, localization of HA-TGR5 and EGFR in HEK-HA-TGR5 cells. TGR5 was at the plasma membrane (arrowheads), and EGFR was detected in vesicles located beneath the plasma membrane and also at the plasma membrane (arrowheads). Scale bar = 10 μm. B and C, BRET analysis between TGR5-RLuc8 and EGFR-Venus expressed in HEK293 cells. B, saturation analysis of ligand-independent interactions. A fixed amount of TGR5-RLuc8 and graded amounts of EGFR-Venus were coexpressed in HEK293 cells. The BRET ratios are plotted as a function of the protein expression ratio of [EGFR-Venus]/[TGR5-RLuc8]. The saturation curves were fitted using a non-linear regression equation assuming a single binding site (GraphPad Prism). Note that TGR5-RLuc8 and EGFR-Venus interactions saturate, and that filipin prevents saturation. C, real-time BRET ratios between TGR5-RLuc8 and EGFR-Venus before and after stimulation with DCA (100 μm), OA (10 μm), or CCDC (10 μm) (arrow). TGR5 agonists did not affect the TGR5-RLuc8 and EGFR-Venus BRET ratio. Shown are triplicate observations from n = 3 experiments.
Kinetic BRET assays were used to assess the effects of TGR5 agonists on TGR5 and EGFR interactions. DCA (100 μm), OA (10 μm), or CCDC (10 μm) had no effect on the BRET signal between TGR5-RLuc8 and EGFR-Venus (Fig. 11C). These results suggest that TGR5 and EGFR form a heterodimer that efficiently assembles in plasma membrane lipid rafts. The formation of this dimer is unaffected by TGR5 agonists.
Activated TGR5 Redistributes to Plasma Membrane Rafts
Confocal microscopy lacks the resolution to precisely localize receptors to plasma membrane microdomains. Therefore, we used immunogold labeling and electron microscopy to determine whether TGR5 agonists stimulated the redistribution of TGR5 to plasma membrane rafts. HEK-TGR5-GFP cells were incubated with DCA (100 μm) or vehicle (control) for 30 min, and TGR5 was detected using a GFP antibody and immunogold labeling. In vehicle-treated cells, TGR5 was present at the cell surface (Fig. 12A). DCA caused the redistribution of TGR5 to vacuoles close to the cell surface (Fig. 12B). Treatment with methyl-β-cyclodextrin prevented the DCA-stimulated redistribution of TGR5 (Fig. 12, C and D). These results are consistent with the agonist-evoked redistribution of TGR5 to microdomains close to the cell surface which could correspond to lipid rafts.
FIGURE 12.

Agonist-induced redistribution of TGR5 to plasma membrane rafts. HEK-TGR5-GFP cells were incubated with vehicle (A and C) or DCA (100 μm, 30 min, B and D). TGR5 was localized by immunogold labeling and electron microscopy using an antibody to GFP. In some experiments, cells were treated with vehicle or methyl-β-cyclodextrin (MβCD). A, in vehicle-treated cells, TGR5 was present predominantly at the cell surface (arrowheads). B, DCA evoked the redistribution of TGR5 to vacuoles located close to the cell surface. C, in vehicle- and methyl-β-cyclodextrin-treated cells, TGR5 was at the cell surface (arrowheads). D, methyl-β-cyclodextrin prevented DCA-induced redistribution of TGR5 to vacuoles and caused retention of TGR5 at the cell surface (arrowheads). Scale bar = 1 μm. Shown are representative images from n > 3 experiments.
DISCUSSION
We have investigated the spatiotemporal control of signaling of TGR5, a plasma membrane receptor for BAs. Our results reveal several unusual aspects of TGR5 signaling, trafficking, and regulation. First, diverse natural (DCA, TLCA, and OA) and synthetic (CCDC) agonists that robustly activate TGR5 did not induce TGR5 trafficking from the plasma membrane to endosomes. Second, except for very high concentrations of CCDC, a potent TGR5 agonist, these agonists did not induce alterations in the subcellular distribution of β-arrestins or promote detectable interactions of β-arrestin1/2 and GRK2/5/6 with TGR5. Third, TGR5 agonists stimulated interaction between TGR5 and Gαs and the formation of cAMP at the plasma membrane and in the cytosol. However, cAMP generation did not exhibit tachyphylaxis or a diminished response to repeated stimulation, which are both indicative of a lack of desensitization. Fourth, TGR5 associates with and transactivates the EGFR within lipid rafts at the plasma membrane, which leads to activation of ERK1/2. These findings provide insight into the biological actions of BAs that are mediated by TGR5 and are of relevance to the properties of TGR5 agonists/antagonists that are under development for the treatment of metabolic, inflammatory, digestive, and sensory disorders.
Agonist-induced Trafficking of TGR5
Many activated receptors, including the β2 adrenergic receptors, PAR2, neurokinin 1 receptor, and μ- and δ-opioid receptors, traffic from the plasma membrane to endosomes (22, 23). Endosomal sorting then targets the internalized receptors back to the plasma membrane, where recycling mediates resensitization (e.g. β2 adrenergic receptor, neurokinin 1 receptor, μ-opioid receptor), or to lysosomes, where degradation mediates receptor down-regulation (e.g. PAR2, δ-opioid receptor). Our results show that TGR5 is prominently localized to the plasma membrane when expressed in HEK293 cells and in NCM460 colonocytes, which express endogenous TGR5. Surprisingly, stimulation of these cells with diverse agonists of TGR5, including endogenous agonists (DCA, TLCA), a naturally occurring selective agonist (OA), and a highly potent synthetic agonist (CCDA), did not induce detectable trafficking of TGR5 to endosomes, as determined by immunofluorescence and confocal microscopy and by using BRET. All of these agents activated TGR5, as assessed by measurement of cAMP formation. In marked contrast, agonists of PAR2, which was coexpressed in HEK cells with TGR5, induced robust endocytosis of PAR2. These results confirm our earlier reports of agonist-stimulated endocytosis of PAR2 (30) and indicate that the HEK cell line was fully capable of supporting GPCR endocytosis. Moreover, other GPCRs, including the neurotensin 1 receptor (45) and neurokinin 1 receptor4, undergo agonist-stimulated endocytosis in NCM460 colonocytes.
β-arrestins are key regulators of GPCR signaling and trafficking. β-arrestins interact with agonist-occupied and GRK-phosphorylated receptors with graded affinities, depending upon the extent of receptor phosphorylation. “Class A” GPCRs (e.g. β2 adrenergic receptor, bradykinin B2 receptor, μ-opioid receptor, neurokinin 3 receptor) have few GRK phosphorylation sites, exhibit selective, low affinity and transient interactions with β-arrestin2, and recycle rapidly (52–54). “Class B” GPCRs (e.g. neurokinin 1 receptor, neurotensin 1 receptor, vasopressin V2 receptor) are highly phosphorylated, interact with both β-arrestin1 and 2 with high affinity for prolonged periods in endosomes, and recycle slowly. TGR5 is unusual, belonging to neither category of receptor. None of the tested agonists induced detectable redistribution of β-arrestin1 or β-arrestin2 expressed in HEK293 or NCM460 cells, as assessed by confocal microscopy. Analysis of TGR5 and β-arrestin interactions by BRET, a sensitive and quantitative assessment of GPCR and β-arrestin interactions, generally confirmed these results. Thus, DCA, TLCA, and OA did not induce BRET signals between TGR5 and β-arrestin1/2 or GRK2/5/6, whereas in the same cells, agonists of PAR2 stimulated strong BRET interactions between PAR2 and these proteins, in line with other reports (55). CCDC alone caused a small BRET signal between TGR5, β-arrestin2, and GRK2, but this was only apparent at high CCDC concentrations, and there was no signal between TGR5 and β-arrestin1 or GRK5/6. Our results contradict an earlier report that TGR5-GFP traffics to endosomes in CHO cells (9). The reason for this difference could be related to differences in cell types, although, in this study, we confirmed the lack of TGR5 endocytosis in a cell line that naturally expresses this receptor (NCM460 colonocytes).
β-arrestins couple GPCRs to clathrin and AP2 and, thereby, mediate endocytosis of many receptors, including PAR2 (23, 44). Our observation that agonists do not recruit GRK2/5/6 or β-arrestin1/2 to TGR5 is thus consistent with the lack of agonist-stimulated endocytosis of TGR5. The structural basis for the inability of TGR5 to recruit β-arrestins and traffic to endosomes is unknown. One possibility is that TGR5 is resistant to GRK-mediated phosphorylation. However, although TGR5 did not interact robustly with GRK2/5/6, TGR5 does contain a series of serines within the intracellular carboxyl-terminal domain that are predicted sites for phosphorylation by GRKs (56). Further studies are required to assess whether TGR5 agonists cause phosphorylation of this receptor.
Although uncommon, agonists of some other GPCRs also fail to induce β-arrestin recruitment and receptor endocytosis. For example, enkephalins, endorphin, and etorphine recruit β-arrestins to the μ-opioid receptor and stimulate robust receptor endocytosis, but morphine is generally less effective, and herkinorin does not cause β-arrestin2 recruitment or receptor endocytosis (57–61).
Although TGR5 did not traffic to endosomes, DCA did induce the redistribution of TGR5 to plasma membrane domains, which were detected by immunogold labeling of TGR5 and electron microscopy. The identity of these compartments is not known, although they likely correspond to lipid rafts because treatment of cells with methyl-β-cyclodextrin, which depletes the plasma membrane of cholesterol and blocks formation of lipid rafts, prevented TGR5 trafficking to these domains. Further studies are required to define the nature and composition of these domains. Other GPCRs are also present in lipid rafts, both basally and after agonist stimulation, including neurokinin, opioid, and bradykinin receptors (24). Although further studies are required to define the nature of these TGR5-containing lipid domains, the concentration of GPCRs and related signaling proteins within such microdomains can enhance the efficiency and fidelity of signal transduction (24).
The Spatiotemporal Control of TGR5 Signaling
GPCR signaling must be under tight physiological control because dysregulated signaling can cause disease. Regulation at the level of the receptor can include processes that control the duration of receptor interaction with heterotrimeric G proteins or that regulate the subcellular location of the receptor and, thus, its proximity to signaling proteins. By using FRET sensors on the basis of Epac2 that allow quantitative assessment of the subcellular location and time course of cAMP formation (38), we observed that DCA evoked the generation of cAMP at the plasma membrane and in the cytosol. Remarkably, cAMP formation was completely sustained in the continued presence of DCA for at least 100 min. There was no evidence of tachyphylaxis of this signal, although cAMP levels quickly declined when the cells were washed. In contrast, isoprenaline-stimulated formation of cAMP underwent tachyphylaxis. This lack of desensitization of DCA-evoked formation of cAMP is consistent with the inability of DCA to induce the recruitment of β-arrestins to TGR5 because β-arrestins play a central role in receptor desensitization. Although compartmentalized signaling is a key determinant of the specific actions of GPCRs, further studies are required to define the functional importance of TGR5-mediated generation of cAMP at the plasma membrane and throughout the cell. The generation of cAMP at the plasma membrane may allow TGR5 to efficiently activate ion channels. Thus, BAs activate TGR5 on primary spinal afferent neurons to induce enhanced excitability and scratching behavior (21) by a mechanism that involves activation of transient receptor potential ankyrin 1 at the plasma membrane by a cAMP and protein kinase A-dependent process4.
In addition to stimulating the formation of cAMP at the plasma membrane, TGR5 at the plasma membrane also stimulated activation of ERK1/2 by a mechanism that involved activation of a membrane metalloprotease and transactivation of the EGFR because DCA-stimulated ERK1/2 activation was prevented by a metalloprotease inhibitor (batimastat) and an inhibitor of the EGFR tyrosine kinase (AG1478). These findings support other reports that TGR5 transactivates the EGFR (25, 27). We observed that methyl-β-cyclodextrin and filipin, which disrupt lipid rafts and caveolae by depleting cholesterol, also blocked DCA-stimulated ERK1/2 activation. Our results suggest that TGR5-mediated transactivation of the EGFR occurs within lipid rafts. By using BRET to assess interactions between TGR5-RLuc8 and EGFR-Venus, we observed the existence of preassembled heterodimers between TGR5 and EGFR. Filipin and methyl-β-cyclodextrin prevented the assembly of these heterodimers, suggesting that they assemble efficiently in lipid rafts. Although we do not know whether TGR5 interacts directly with EGFR or whether they are part of a multiprotein complex, other GPCRs can interact with the EGFR in lipid rafts, which can enhance the efficiency of signaling. For example, agonists promote the formation of a multiprotein complex containing the angiotensin 1 receptor and EGFR that is necessary for efficient signaling in hepatic C9 cells (50, 51). Similarly, isoprenaline stimulates the assembly of complexes containing the β2 adrenergic receptor and EGFR in COS-7 cells (49).
Considered together, our results suggest that TGR5 and EGFR form heterodimers within lipid rafts that promote the efficient BA-induced transactivation of the EGFR by a mechanism that involves activation of a membrane metalloprotease, release of an unidentified EGFR agonist, and autocrine activation of EGFR. This mechanism operates in HEK cells and in NCM460 colonocytes that express endogenous TGR5 and ensures efficient ERK activation. Although lipid rafts are essential for TGR5 interaction with EGFR, transactivation of the EGFR, and ERK signaling, they are not required for direct EGF activation of EGFR and ERK signaling or for TGR5-mediated cAMP generation, which were unaffected by filipin and methyl-β-cyclodextrin. Whether TGR5-mediated transactivation of the EGFR causes EGFR endocytosis and down-regulation requires further investigation.
Implications for the Functional Effects of TGR5 Agonists
TGR5 has been implicated in several important biological processes, including the control of energy balance (11), insulin and glucose homeostasis (12, 13), inflammation (4, 9, 10, 18), bile formation and secretion (14, 15, 17, 62), intestinal motility and secretion (19, 20), and BA-evoked itch and analgesia (21). The circulating levels of BAs normally wax and wane during feeding and fasting, and during cholestatic diseases, cells would be continuously exposed to high levels of BAs. Our results suggest that, in contrast to many other GPCRs that desensitize and traffic to endosomes soon after activation, activated TGR5 is retained in plasma membrane lipid rafts, from where it can transmit sustained signals. Thus, the mechanisms that control TGR5 signaling in the short term are presently unknown, and whether long-term exposure to agonists regulates TGR5 at the transcriptional level remains to be determined.
TGR5 has emerged as a promising target for metabolic, inflammatory, and digestive diseases, where agonists may promote energy expenditure and insulin release, dampen inflammation, and promote colon transit (4). Therefore, it will be important to determine how these agonists regulate TGR5. Our results show that one synthetic agonist, CCDC, can induce association of TGR5 with β-arrestin2 and GRK2, in contrast to the actions of endogenous BAs. Although CCDC could be a biased agonist of TGR5, the ability of this agonist to stimulate β-arrestin2 and GRK2 recruitment could simply reflect its increased potency. Whether other TGR5 agonists differentially affect TGR5 signaling and trafficking and also function as biased agonists remains to be determined. TGR5 has recently been implicated in the pruritus that is associated with cholestatic disease, raising the possibility that receptor antagonists could provide a novel treatment for itch (21). However, the effects of antagonists on TGR5 signaling are unexplored. In summary, the BA receptor TGR5 transmits signals from plasma membrane lipid rafts that are sustained, β-arrestin-independent, and depend on interaction with the EGFR.
Acknowledgments
We thank Drs. Michael Crouch and Ron Osmond of TGR Biosciences, Adelaide for the AlphaScreen SureFire kits.
This work was supported, in whole or in part, by National Institutes of Health Grant P30-DK026743 from NIDDK (to C. U. C.). This work was also supported by the Northern California Institute for Research and Education, the Veterans Health Administration, National Health and Medical Research Council (NHMRC) Grants 63303 and 103188, and Monash University (to N. W. B.); by NHMRC Grant 454858 (to D. P. P.); and by British Heart Foundation Grant FS/08/017/25027 (to G. S. C.).
N. W. Bunnett, unpublished data.
- BA
- bile acid
- DCA
- deoxycholic acid
- TLCA
- taurolithocholic acid
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- EGFR
- epidermal growth factor receptor
- OA
- oleanolic acid
- CCDC
- 3-(2-chlorophenyl)-N-(4-chlorophenyl)-N, 5-dimethylisoxazole-4-carboxamide
- PAR
- protease-activated receptor
- BRET
- bioluminescence resonance energy transfer
- HBSS
- Hanks' balanced salt solution
- CAMYEL
- cAMP sensor using YFP-Epac-RLuc
- YFP
- yellow fluorescent protein.
REFERENCES
- 1. Bajor A., Gillberg P. G., Abrahamsson H. (2010) Bile acids. Short and long term effects in the intestine. Scand. J. Gastroenterol. 45, 645–664 [DOI] [PubMed] [Google Scholar]
- 2. Hofmann A. F., Hagey L. R. (2008) Bile acids. Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol. Life Sci. 65, 2461–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lefebvre P., Cariou B., Lien F., Kuipers F., Staels B. (2009) Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 89, 147–191 [DOI] [PubMed] [Google Scholar]
- 4. Pols T. W., Noriega L. G., Nomura M., Auwerx J., Schoonjans K. (2011) The bile acid membrane receptor TGR5. A valuable metabolic target. Dig. Dis. 29, 37–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Claudel T., Staels B., Kuipers F. (2005) The Farnesoid X receptor. A molecular link between bile acid and lipid and glucose metabolism. Arterioscler. Thromb. Vasc. Biol. 25, 2020–2030 [DOI] [PubMed] [Google Scholar]
- 6. Wang H., Chen J., Hollister K., Sowers L. C., Forman B. M. (1999) Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 3, 543–553 [DOI] [PubMed] [Google Scholar]
- 7. Staudinger J. L., Goodwin B., Jones S. A., Hawkins-Brown D., MacKenzie K. I., LaTour A., Liu Y., Klaassen C. D., Brown K. K., Reinhard J., Willson T. M., Koller B. H., Kliewer S. A. (2001) The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. U.S.A. 98, 3369–3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Makishima M., Lu T. T., Xie W., Whitfield G. K., Domoto H., Evans R. M., Haussler M. R., Mangelsdorf D. J. (2002) Vitamin D receptor as an intestinal bile acid sensor. Science 296, 1313–1316 [DOI] [PubMed] [Google Scholar]
- 9. Kawamata Y., Fujii R., Hosoya M., Harada M., Yoshida H., Miwa M., Fukusumi S., Habata Y., Itoh T., Shintani Y., Hinuma S., Fujisawa Y., Fujino M. (2003) A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 278, 9435–9440 [DOI] [PubMed] [Google Scholar]
- 10. Maruyama T., Miyamoto Y., Nakamura T., Tamai Y., Okada H., Sugiyama E., Nakamura T., Itadani H., Tanaka K. (2002) Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 298, 714–719 [DOI] [PubMed] [Google Scholar]
- 11. Watanabe M., Houten S. M., Mataki C., Christoffolete M. A., Kim B. W., Sato H., Messaddeq N., Harney J. W., Ezaki O., Kodama T., Schoonjans K., Bianco A. C., Auwerx J. (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439, 484–489 [DOI] [PubMed] [Google Scholar]
- 12. Katsuma S., Hirasawa A., Tsujimoto G. (2005) Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 329, 386–390 [DOI] [PubMed] [Google Scholar]
- 13. Thomas C., Gioiello A., Noriega L., Strehle A., Oury J., Rizzo G., Macchiarulo A., Yamamoto H., Mataki C., Pruzanski M., Pellicciari R., Auwerx J., Schoonjans K. (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 10, 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keitel V., Cupisti K., Ullmer C., Knoefel W. T., Kubitz R., Häussinger D. (2009) The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology 50, 861–870 [DOI] [PubMed] [Google Scholar]
- 15. Lavoie B., Balemba O. B., Godfrey C., Watson C. A., Vassileva G., Corvera C. U., Nelson M. T., Mawe G. M. (2010) Hydrophobic bile salts inhibit gallbladder smooth muscle function via stimulation of GPBAR1 receptors and activation of KATP channels. J. Physiol. 588, 3295–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Keitel V., Häussinger D. (2011) TGR5 in the biliary tree. Dig. Dis. 29, 45–47 [DOI] [PubMed] [Google Scholar]
- 17. Maruyama T., Tanaka K., Suzuki J., Miyoshi H., Harada N., Nakamura T., Miyamoto Y., Kanatani A., Tamai Y. (2006) Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J. Endocrinol. 191, 197–205 [DOI] [PubMed] [Google Scholar]
- 18. Wang Y. D., Chen W. D., Yu D., Forman B. M., Huang W. (2011) The G protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor κ light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology 54, 1421–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alemi F., Poole D. P., Chiu J., Schoonjans K., Cattaruzza F., Grider J. R., Bunnett N. W., Corvera C. U. (2013) The receptor TGR5 mediates the prokinetic actions of intestinal bile acids and is required for normal defecation in mice. Gastroenterology 144, 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poole D. P., Godfrey C., Cattaruzza F., Cottrell G. S., Kirkland J. G., Pelayo J. C., Bunnett N. W., Corvera C. U. (2010) Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastro. Motil. 22, 814–825, e227–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alemi F., Kwon E., Poole D. P., Lieu T., Lyo V., Cattaruzza F., Cevikbas F., Steinhoff M., Nassini R., Materazzi S., Guerrero-Alba R., Valdez-Morales E., Cottrell G. S., Schoonjans K., Geppetti P., Vanner S. J., Bunnett N. W., Corvera C. U. (2013) The TGR5 receptor mediates bile acid-induced itch and analgesia. J. Clin. Invest. 123, 1513–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanyaloglu A. C., von Zastrow M. (2008) Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 48, 537–568 [DOI] [PubMed] [Google Scholar]
- 23. Murphy J. E., Padilla B. E., Hasdemir B., Cottrell G. S., Bunnett N. W. (2009) Endosomes. A legitimate platform for the signaling train. Proc. Natl. Acad. Sci. U.S.A. 106, 17615–17622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patel H. H., Murray F., Insel P. A. (2008) G-protein-coupled receptor-signaling components in membrane raft and caveolae microdomains. Handb. Exp. Pharmacol. 186, 167–184 [DOI] [PubMed] [Google Scholar]
- 25. Werneburg N. W., Yoon J. H., Higuchi H., Gores G. J. (2003) Bile acids activate EGF receptor via a TGF-α-dependent mechanism in human cholangiocyte cell lines. Am. J. Physiol. 285, G31–36 [DOI] [PubMed] [Google Scholar]
- 26. Yang J. I., Yoon J. H., Myung S. J., Gwak G. Y., Kim W., Chung G. E., Lee S. H., Lee S. M., Kim C. Y., Lee H. S. (2007) Bile acid-induced TGR5-dependent c-Jun-N terminal kinase activation leads to enhanced caspase 8 activation in hepatocytes. Biochem. Biophys. Res. Commun. 361, 156–161 [DOI] [PubMed] [Google Scholar]
- 27. Yasuda H., Hirata S., Inoue K., Mashima H., Ohnishi H., Yoshiba M. (2007) Involvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cells. Biochem. Biophys. Res. Commun. 354, 154–159 [DOI] [PubMed] [Google Scholar]
- 28. Sato H., Macchiarulo A., Thomas C., Gioiello A., Une M., Hofmann A. F., Saladin R., Schoonjans K., Pellicciari R., Auwerx J. (2008) Novel potent and selective bile acid derivatives as TGR5 agonists. Biological screening, structure-activity relationships, and molecular modeling studies. J. Med. Chem. 51, 1831–1841 [DOI] [PubMed] [Google Scholar]
- 29. Evans K. A., Budzik B. W., Ross S. A., Wisnoski D. D., Jin J., Rivero R. A., Vimal M., Szewczyk G. R., Jayawickreme C., Moncol D. L., Rimele T. J., Armour S. L., Weaver S. P., Griffin R. J., Tadepalli S. M., Jeune M. R., Shearer T. W., Chen Z. B., Chen L., Anderson D. L., Becherer J. D., De Los Frailes M., Colilla F. J. (2009) Discovery of 3-aryl-4-isoxazolecarboxamides as TGR5 receptor agonists. J. Med. Chem. 52, 7962–7965 [DOI] [PubMed] [Google Scholar]
- 30. Jacob C., Cottrell G. S., Gehringer D., Schmidlin F., Grady E. F., Bunnett N. W. (2005) c-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J. Biol. Chem. 280, 16076–16087 [DOI] [PubMed] [Google Scholar]
- 31. Kocan M., Dalrymple M. B., Seeber R. M., Feldman B. J., Pfleger K. D. (2010) Enhanced BRET technology for the monitoring of agonist-induced and agonist-independent interactions between GPCRs and β-arrestins. Front. Endocrinol. 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Savage E. E., Wootten D., Christopoulos A., Sexton P. M., Furness S. G. (2013) A simple method to generate stable cell lines for the analysis of transient protein-protein interactions. BioTechniques 54, 217–221 [DOI] [PubMed] [Google Scholar]
- 33. Lan T. H., Liu Q., Li C., Wu G., Lambert N. A. (2012) Sensitive and high resolution localization and tracking of membrane proteins in live cells with BRET. Traffic 13, 1450–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Galés C., Rebois R. V., Hogue M., Trieu P., Breit A., Hébert T. E., Bouvier M. (2005) Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods 2, 177–184 [DOI] [PubMed] [Google Scholar]
- 35. Wachten S., Masada N., Ayling L. J., Ciruela A., Nikolaev V. O., Lohse M. J., Cooper D. M. (2010) Distinct pools of cAMP centre on different isoforms of adenylyl cyclase in pituitary-derived GH3B6 cells. J. Cell Sci. 123, 95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hasdemir B., Bunnett N. W., Cottrell G. S. (2007) Hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) mediates post-endocytic trafficking of protease-activated receptor 2 and calcitonin receptor-like receptor. J. Biol. Chem. 282, 29646–29657 [DOI] [PubMed] [Google Scholar]
- 37. Jiang L. I., Collins J., Davis R., Lin K. M., DeCamp D., Roach T., Hsueh R., Rebres R. A., Ross E. M., Taussig R., Fraser I., Sternweis P. C. (2007) Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 282, 10576–10584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Halls M. L., Cooper D. M. (2010) Sub-picomolar relaxin signalling by a pre-assembled RXFP1, AKAP79, AC2, β-arrestin 2, PDE4D3 complex. EMBO J. 29, 2772–2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ. 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kocan M., See H. B., Sampaio N. G., Eidne K. A., Feldman B. J., Pfleger K. D. (2009) Agonist-independent interactions between β-arrestins and mutant vasopressin type II receptors associated with nephrogenic syndrome of inappropriate antidiuresis. Mol. Endocrinol. 23, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kocan M., See H. B., Seeber R. M., Eidne K. A., Pfleger K. D. (2008) Demonstration of improvements to the bioluminescence resonance energy transfer (BRET) technology for the monitoring of G protein-coupled receptors in live cells. J. Biomol. Screen. 13, 888–898 [DOI] [PubMed] [Google Scholar]
- 42. Mercier J. F., Salahpour A., Angers S., Breit A., Bouvier M. (2002) Quantitative assessment of β 1- and β 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J. Biol. Chem. 277, 44925–44931 [DOI] [PubMed] [Google Scholar]
- 43. Sato H., Genet C., Strehle A., Thomas C., Lobstein A., Wagner A., Mioskowski C., Auwerx J., Saladin R. (2007) Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem. Biophys. Res. Commun. 362, 793–798 [DOI] [PubMed] [Google Scholar]
- 44. Déry O., Thoma M. S., Wong H., Grady E. F., Bunnett N. W. (1999) Trafficking of proteinase-activated receptor-2 and β-arrestin-1 tagged with green fluorescent protein. β-Arrestin-dependent endocytosis of a proteinase receptor. J. Biol. Chem. 274, 18524–18535 [DOI] [PubMed] [Google Scholar]
- 45. Law I. K., Murphy J. E., Bakirtzi K., Bunnett N. W., Pothoulakis C. (2012) Neurotensin-induced proinflammatory signaling in human colonocytes is regulated by β-arrestins and endothelin-converting enzyme-1-dependent endocytosis and resensitization of neurotensin receptor 1. J. Biol. Chem. 287, 15066–15075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kelly E., Bailey C. P., Henderson G. (2008) Agonist-selective mechanisms of GPCR desensitization. Br. J. Pharmacol. 153, S379–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Prenzel N., Zwick E., Daub H., Leserer M., Abraham R., Wallasch C., Ullrich A. (1999) EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402, 884–888 [DOI] [PubMed] [Google Scholar]
- 48. DeFea K. A., Zalevsky J., Thoma M. S., Déry O., Mullins R. D., Bunnett N. W. (2000) β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 148, 1267–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maudsley S., Pierce K. L., Zamah A. M., Miller W. E., Ahn S., Daaka Y., Lefkowitz R. J., Luttrell L. M. (2000) The β(2)-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J. Biol. Chem. 275, 9572–9580 [DOI] [PubMed] [Google Scholar]
- 50. Olivares-Reyes J. A., Shah B. H., Hernández-Aranda J., García-Caballero A., Farshori M. P., García-Sáinz J. A., Catt K. J. (2005) Agonist-induced interactions between angiotensin AT1 and epidermal growth factor receptors. Mol. Pharmacol. 68, 356–364 [DOI] [PubMed] [Google Scholar]
- 51. Yin X., Li B., Chen H., Catt K. J. (2008) Differential signaling pathways in angiotensin II- and epidermal growth factor-stimulated hepatic C9 cells. Mol. Pharmacol. 74, 1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oakley R. H., Laporte S. A., Holt J. A., Barak L. S., Caron M. G. (2001) Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-β-arrestin complexes after receptor endocytosis. J. Biol. Chem. 276, 19452–19460 [DOI] [PubMed] [Google Scholar]
- 53. Oakley R. H., Laporte S. A., Holt J. A., Caron M. G., Barak L. S. (2000) Differential affinities of visual arrestin, β arrestin1, and β arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275, 17201–17210 [DOI] [PubMed] [Google Scholar]
- 54. Schmidlin F., Déry O., Bunnett N. W., Grady E. F. (2002) Heterologous regulation of trafficking and signaling of G protein-coupled receptors. β-Arrestin-dependent interactions between neurokinin receptors. Proc. Natl. Acad. Sci. U.S.A. 99, 3324–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ramachandran R., Mihara K., Chung H., Renaux B., Lau C. S., Muruve D. A., DeFea K. A., Bouvier M., Hollenberg M. D. (2011) Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J. Biol. Chem. 286, 24638–24648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Blom N., Gammeltoft S., Brunak S. (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294, 1351–1362 [DOI] [PubMed] [Google Scholar]
- 57. Groer C. E., Tidgewell K., Moyer R. A., Harding W. W., Rothman R. B., Prisinzano T. E., Bohn L. M. (2007) An opioid agonist that does not induce μ-opioid receptor-arrestin interactions or receptor internalization. Mol. Pharmacol. 71, 549–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Haberstock-Debic H., Kim K. A., Yu Y. J., von Zastrow M. (2005) Morphine promotes rapid, arrestin-dependent endocytosis of μ-opioid receptors in striatal neurons. J. Neurosci. 25, 7847–7857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Keith D. E., Anton B., Murray S. R., Zaki P. A., Chu P. C., Lissin D. V., Monteillet-Agius G., Stewart P. L., Evans C. J., von Zastrow M. (1998) μ-Opioid receptor internalization. Opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol. Pharmacol. 53, 377–384 [PubMed] [Google Scholar]
- 60. Sternini C., Spann M., Anton B., Keith D. E., Jr., Bunnett N. W., von Zastrow M., Evans C., Brecha N. C. (1996) Agonist-selective endocytosis of μ opioid receptor by neurons in vivo. Proc. Natl. Acad. Sci. U.S.A. 93, 9241–9246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Whistler J. L., von Zastrow M. (1998) Morphine-activated opioid receptors elude desensitization by β-arrestin. Proc. Natl. Acad. Sci. U.S.A. 95, 9914–9919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li T., Holmstrom S. R., Kir S., Umetani M., Schmidt D. R., Kliewer S. A., Mangelsdorf D. J. (2011) The G protein-coupled bile acid receptor, TGR5, stimulates gallbladder filling. Mol. Endocrinol. 25, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]