

Background: Plasma membrane calcium ATPases interact dynamically with the submembrane actin cytoskeleton.
Results: Biophysical and functional assays show that purified plasma membrane calcium ATPase binds to G-actin and is activated by short actin oligomers.
Conclusion: Plasma membrane calcium ATPases are regulated by polymerizing actin independently of regulation by calmodulin.
Significance: Dynamic actin participates in cytosolic Ca2+ homeostasis by regulating plasma membrane calcium ATPase activity.
Keywords: Actin, ATPases, Calcium, Calmodulin, Cytoskeleton, Transport
Abstract
As recently described by our group, plasma membrane calcium ATPase (PMCA) activity can be regulated by the actin cytoskeleton. In this study, we characterize the interaction of purified G-actin with isolated PMCA and examine the effect of G-actin during the first polymerization steps. As measured by surface plasmon resonance, G-actin directly interacts with PMCA with an apparent 1:1 stoichiometry in the presence of Ca2+ with an apparent affinity in the micromolar range. As assessed by the photoactivatable probe 1-O-hexadecanoyl-2-O-[9-[[[2-[125I]iodo-4-(trifluoromethyl-3H-diazirin-3-yl)benzyl]oxy]carbonyl]nonanoyl]-sn-glycero-3-phosphocholine, the association of PMCA to actin produced a shift in the distribution of the conformers of the pump toward a calmodulin-activated conformation. G-actin stimulates Ca2+-ATPase activity of the enzyme when incubated under polymerizing conditions, displaying a cooperative behavior. The increase in the Ca2+-ATPase activity was related to an increase in the apparent affinity for Ca2+ and an increase in the phosphoenzyme levels at steady state. Although surface plasmon resonance experiments revealed only one binding site for G-actin, results clearly indicate that more than one molecule of G-actin was needed for a regulatory effect on the pump. Polymerization studies showed that the experimental conditions are compatible with the presence of actin in the first stages of assembly. Altogether, these observations suggest that the stimulatory effect is exerted by short oligomers of actin. The functional interaction between actin oligomers and PMCA represents a novel regulatory pathway by which the cortical actin cytoskeleton participates in the regulation of cytosolic Ca2+ homeostasis.
Introduction
Plasma membrane calcium pumps (PMCAs)3 expel Ca2+ from all eukaryotic cells by using the energy of ATP. They play an important role in the maintenance of low intracellular Ca2+ concentrations, being responsible not only for long term regulation of resting Ca2+ levels but also determining the peak of Ca2+ concentration achieved after some cellular stimuli and counteracting transient increases that occur during Ca2+ signaling (1).
The activity of PMCAs is regulated by calmodulin (2, 3), acidic phospholipids (4, 5), phosphorylation by kinases A and C (6, 7), proteolysis by calpain (8), and oligomerization (9). Most of the activation mechanisms implicate the C-terminal region of the pump containing the high affinity calmodulin binding domain, which is involved in the autoinhibition of the pump (10). Stimulation of the pump is a key feature because this enzyme generally shows low basal activity; in most cases, stimulation is related to an increase in the apparent affinity for Ca2+ but can also be accompanied by an increase in the maximum activity (Vmax). Our group has recently reported a novel regulatory mechanism on PMCA activity that involves the actin cytoskeleton (11, 12).
Actin is an abundant component of the cortical cytoskeleton, and it forms a large submembrane filamentous network. Although stable, it is also a highly dynamic structure. Reorganization of the actin cytoskeleton in response to various stimuli is important in many cellular processes, including motility, adhesion, phagocytosis, cytokinesis, secretion, and ion transport across the plasma membrane (13–19). Several transporters are known to be directly associated with and modulated by the actin cytoskeleton, such as epithelial sodium channels (20), channels of cystic fibrosis receptor (21), the Na+/H+ exchanger (22), and the Na+,K+-ATPase (23), among others.
We have previously shown that purified actin in its filamentous form (F-actin) inhibits the activity of PMCA by ∼50% with an apparent affinity in the micromolar range (12). Interestingly, we observed that not only was actin depolymerization associated with a loss in the inhibitory effect (11) but incubation of PMCA with G-actin caused the stimulation of the activity of the pump showing a continuous decrease in the magnitude of activation as polymerization progressed (12). Based on these data, we proposed that G-actin and/or short actin oligomers may be responsible for PMCA activation (12).
The monomer pool of actin in non-muscle animal cells is around 50% of total cellular actin (24). This unpolymerized pool is maintained by binding G-actin to a number of different monomer-binding proteins (25, 26). Data on the distribution and properties of the monomer pool is scarce, but some studies have indicated that it is associated with dynamic regions of the cell, and foci of unassembled actin were found to be concentrated in the cell periphery (27, 28). Moreover, G-actin is known to be recruited to the plasma membrane in response to a variety of cell receptor activation signals (29–32). Short actin filaments are formed by severing proteins and debranching from the actin submembranous network (33), which can be followed by further depolymerization. From these observations, we can hypothesize that monomeric actin and short oligomers would be available in the cell cortex to interact with and potentially regulate PMCA. This is supported by the finding of nonfilamentous actin complexes in resting platelets that contain PMCA and other cytoskeletal proteins (34). Thus, elucidating the role of nonfilamentous actin in PMCA regulation will be key to understanding the impact of dynamic changes of the membrane-proximal actin cytoskeleton in the regulation of Ca2+ transport.
To this aim, we assessed the direct interaction between G-actin and PMCA by surface plasmon resonance analysis. Using this technology, we were able to determine the stoichiometry and affinity of the interaction. We also investigated the calmodulin binding domain of the pump as a putative binding site for the association with actin. We therefore measured the Ca2+-ATPase activity and the levels of phosphorylated intermediates of PMCA to functionally describe the effect of G-actin and short oligomers. We also investigated whether actin stimulation was associated with changes in the affinity of the pump for Ca2+ and/or in the Vmax. Finally, we assessed if the direct interaction with actin oligomers results in a conformational change in the PMCA similar to that observed for other activators of the pump.
MATERIALS AND METHODS
Reagents
All chemicals used in this work were of analytical grade and purchased from Sigma. Pyrene-actin was purchased from Cytoskeleton Inc., and calmodulin was from Calbiochem. Sensor Chips CM5 and kit reagents for the immobilization process were purchased from Biacore-GE, Sweden. Recently drawn human blood for the isolation of PMCA was obtained from the Hematology Section of Fundación Fundosol (Argentina). Blood donation in Argentina is voluntary, and therefore the donor provides informed consent for the donation of blood and for the subsequent legitimate use of the blood by the transfusion service.
Purification of Human Erythrocyte PMCA
PMCA4 is the predominant isoform of human erythrocytes, which contain about 80% of this isoform and 20% of PMCA1 (35). PMCA was isolated from calmodulin-depleted erythrocyte membranes by the calmodulin-affinity chromatography procedure (36). Briefly, membrane proteins were solubilized in a 0.5% C12E10-containing buffer. After centrifugation, the supernatant was loaded into a Sepharose-calmodulin column in the presence of 1 mm Ca2+. The column was thoroughly washed with 0.05% C12E10-containing buffer. PMCA was eluted in 20% (w/v) glycerol, 0.005% C12E10, 120 mm KCl, 1 mm MgCl2, 10 mm MOPS-K (pH 7.4 at 4 °C), 2 mm EGTA, 2 mm dithiothreitol. Purified PMCA was assayed for protein concentration and homogeneity (concentrations 350–800 nm; single band at Mr 134,000) by SDS-PAGE and stored in liquid nitrogen until use. It has been demonstrated previously that the activity and the regulatory properties of the protein are preserved in either solubilized or reconstituted purified preparations compared with that located in the erythrocyte (37).
Actin Purification
Acetone powder of rabbit skeletal muscle was prepared according to the method of Straub (38) and stored at −20 °C for 0–3 years. Actin was purified from the acetone powder following the method of Spudich and Watt (39) with minor modifications. Protein concentration was determined by measuring the absorbance at 290 nm using an extinction coefficient of 0.63 (mg/ml)−1 cm−1. SDS-PAGE was performed to assess the purity of the preparation (typically ∼98%). Purified monomeric actin was stored frozen at −80 °C after freezing in liquid nitrogen at high concentrations (≥5 mg/ml) in a small volume (≤100 μl). Under this storage condition, the sample was stable for 6 months (40). At the time of use, frozen samples were thawed rapidly at 37 °C and clarified. The clarification step consists of ×20 dilution of the sample in fresh Buffer G (2 mm Tris-HCl, 0.2 mm Na2ATP, 0.5 mm dithiothreitol, 0.2 mm CaCl2, 0.005% azide, pH 8.0 at 25 °C); after leaving the preparation at 4 °C for 1 h to depolymerize actin oligomers that could have been formed during the freeze-thaw steps, the sample was centrifuged at 14,000 rpm at 4 °C for 30 min to remove residual nucleating centers.
SPR Experiments
Biomolecular interaction analyses were carried out using a BIAcore T100 instrument (Biacore). All the experiments were carried out at 25 °C.
For coupling of G-actin, PMCA, and C28 peptide on CM-dextran sensor chip (CM5), G-actin was coupled to the carboxymethyl-dextran matrix of CM5 sensor chips (Biacore) using the amine coupling kit (Biacore) as described previously (41). The activation and immobilization periods were set between 3 and 7 min to couple the desired amount of protein yielding between 600 and 1200 resonance units (RU). Prior to immobilization, G-actin in Buffer G was diluted to 10 μg/ml in 10 mm sodium acetate buffer (pH 4.0 at 25 °C). After immobilization, the sensor surface was extensively washed by several injections of a buffer containing 2 mm Tris-HCl (pH 8.0 at 25 °C) and 0.2 mm CaCl2.
PMCA was coupled using the same amine coupling procedure. Prior to immobilization, PMCA at a concentration of 50 nm was dialyzed at 4 °C against a buffer containing 10 mm sodium acetate (pH 5.0 at 25 °C), 50 mm KCl, 1 mm MgCl2, 70 μm CaCl2, 0.005% C12E10, and 20% glycerol. PMCA was injected for ∼7 min to obtain an immobilization level between 400 and 800 RU. Immediately after the immobilization procedure, the sensor surface was washed with a solution containing 15 mm MOPS-KOH (pH 7.2 at 25 °C), 1 mm MgCl2, 70 μm CaCl2, 0.005% C12E10 to have the immobilized PMCA reconstituted into detergent micelles. C28 peptide, which corresponds to the calmodulin binding domain of PMCA, was dissolved in 10 mm sodium acetate (pH 5.0 at 25 °C) at a concentration of 0.15 mg/ml and immobilized following the same procedure as described for PMCA. In all cases, an identical procedure was carried out on the surface of a second flow cell except for the protein injection, constituting the control cell for nonspecific binding.
For binding analysis on PMCA and C28 peptide-immobilized surfaces, calmodulin was diluted 2-fold in a buffer composed of 20 mm MOPS-KOH (pH 7.2 at 25 °C), 120 mm KCl, 1 mm MgCl2, 0.005% C12E10, and 70 μm CaCl2, in a range between 0.4 and 25 nm and injected at a flow rate of 30 μl/min for 60 s. 10 μm G-actin was dialyzed at 4 °C against a buffer composed of 2 mm Tris-HCl (pH 7.7 at 25 °C), 70 μm CaCl2, and 0.005% C12E10 and injected in a range of concentrations between 0.6 to 5 μm (obtained by 2-fold dilution) at a flow rate of 30 μl/min for 60 s. In both cases, dissociation and surface regeneration were carried out with the running buffer.
For binding analysis on the G-actin immobilized surface, PMCA was dialyzed at 4 °C against a buffer containing 20 mm MOPS-KOH (pH 7.2 at 25 °C), 120 mm KCl, 1 mm MgCl2, 70 μm CaCl2, and 0.005% C12E10. Depending on the initial concentration, which varies according to the purification yield, PMCA was concentrated to 2.0 μm and diluted 2-fold in a range of concentrations between 0.01 nm to 2.0 μm. PMCA was injected at a flow rate of 30 μl/min for 30 and 60 s. Pulses of a nonpolymerizing buffer (2 mm Tris-HCl, pH 8.0 at 25 °C, 200 μm CaCl2) were injected to regenerate the surface.
For calculation of the affinity constants, dissociation constants (Kd) were determined under equilibrium conditions or by kinetics analysis after correction for nonspecific binding, for which proteins were passed over a reference surface constituted by a blocked empty flow cell, as described previously (42, 43).
Actin Polymerization Assay
Prior to use, pyrene-actin (Cytoskeleton) was diluted from stock solution (20 mg/ml) and clarified as described above. Pyrene-actin in the supernatant was added to nonlabeled actin to obtain 6% of label. Actin polymerization was monitored by registering the change in the fluorescence signal in a thermostated spectrofluorimeter Jasco FP-6500 using a quartz cuvette of 3 × 3 mm. The excitation and emission wavelengths were 365 and 407 nm, respectively, with a bandwidth of 3 and 10 nm, respectively (44). Measurements were done at 25 °C. To avoid bleaching the fluorophore, the sample was exposed to the lamp intermittently. Polymerization was initiated by addition of a small aliquot of a mixture of concentrated KCl, MgCl2, and ATP to final concentrations of 120 mm, 3.75 mm, and 30 μm, respectively, or buffer G as a control for the stability test. To evaluate the extent of actin polymerization in PMCA activity assays, polymerization was initiated under identical conditions as those used for PMCA activity measurements.
Ca2+-ATPase-specific Activity of PMCA
ATP hydrolysis was assayed either by measuring the [32P]Pi released from [γ-32P]ATP as described by Richards et al. (45) or alternatively performed according to the method of Webb (46) using a commercial kit (EnzChek Phosphate Assay kit from Molecular Probes). In both cases, the reaction medium contained 120 mm KCl, 30 mm MOPS-K (pH 7.4 at 25 °C), 3.75 mm MgCl2, 70 μg/ml C12E10, 10 μg/ml phosphatidylcholine, 1 mm EGTA, and enough CaCl2 to give the desired final [Ca2+]free. The reaction was started by the addition of ATP (final concentration of 30 μm for radioactive assays and 2 mm for the nonradioactive measurements). Enzyme concentration was 0.8 μg/ml. Blanks were carried out in the same medium without free Ca2+, in the presence of 1 mm EGTA; and control experiments for actin ATPase activity were done in the same medium in the absence of PMCA. Measurements were carried out at 25 °C.
Determination of PMCA-phosphorylated Intermediates
The phosphorylated intermediates (EP) were measured as the amount of acid-stable 32P incorporated in the enzyme from [γ-32P]ATP after stopping the reaction with an ice-cold solution containing 10% trichloroacetic acid. Phosphorylated PMCA was measured 1 min after initiation of the reaction in a medium containing 120 mm KCl, 30 mm MOPS-K (pH 7.4 at 25 °C), 3.75 mm MgCl2, 70 μg/ml C12E10, 10 μg/ml phosphatidylcholine, 1 mm EGTA, and enough CaCl2 to give a final [Ca2+]free of 70 μm. PMCA concentration was 0.8 μg/ml. The reaction was initiated by the addition of 30 μm ATP concomitant with the addition of actin or buffer for control experiments. Measurements were carried out at 25 °C. The isolation and quantification of the EP intermediates were performed according to the method described by Echarte et al. (47). Briefly, samples were spun down at 7000 × g for 3.5 min at 4 °C, and pellets were washed once with 7% TCA and 150 mm H3PO4 and once with distilled water and processed for SDS-PAGE. Electrophoresis was performed at pH 6.3 (14 °C) in 7.5% polyacrylamide gels. Gels were stained, dried, and exposed to a Storage Phosphor Screen of Molecular Devices (Amersham Biosciences). Autoradiograms and stained gels were scanned (HP Scanjet G2410 scanner), and images were analyzed using the software GelPro.
Measurement of [Ca2+]
The free calcium concentration in the reaction medium was measured using a selective Ca2+ electrode (93-20, Orion Research, Inc.) as described by Kratje et al. (48).
PMCA Labeling with [125I]TID-PC/16
[125I]TID-PC/16 was prepared as described in Mangialavori et al. (49). A dried film of the photoactivatable reagent was suspended in DMPC/C12E10 (10:70 μg/ml)-mixed micelles containing 1 μg/ml PMCA, 120 mm KCl, 30 mm MOPS-K (pH 7.4 at 25 °C), 3.75 mm MgCl2, 1 mm EGTA, and enough CaCl2 to give 70 μm [Ca2+]free. The PMCA preparation was incubated for 20 min at 25 °C before the treatment. G-actin was added at different final concentrations, and after 1 min, samples were irradiated for 15 min with light from a filtered UV source (λ ∼360 nm). Quantification of total and labeled protein was carried out as described previously (49). Briefly, after protein separation by SDS-PAGE, polypeptides were stained with Coomassie Blue R, and bands corresponding to the PMCA molecular weight were excised from the gel. The incorporation of radioactivity was directly measured on a γ-counter, and the amount of protein was determined by measuring the eluted stain from each band. Specific incorporation was calculated as the ratio between measured radioactivity and amount of protein present in each band.
Data Analysis
All measurements were performed in triplicate to quintuplicate unless specified otherwise in the figures. SPR data were analyzed using BIAEvaluation T 100 software. Equations were fitted to the results by nonlinear regression based on the Gauss-Newton algorithm using commercial programs (Excel and Sigma-Plot for Windows, the latter being able to provide not only the best fitting values of the parameters but also their standard errors).
RESULTS
Characterization of the Direct Binding of G-actin to PMCA
The interaction between PMCA and G-actin was studied using SPR technology. This technique was based on the immobilization of one of the binding partners on a sensor surface, although the other constitutes the flowing analyte, enabling a label-free, real time analysis of biomolecular interactions. The binding phenomenon is monitored as a change in SPR angle, which is the result of a change in mass on the sensor chip surface. Scheme 1 describes the experimental approach used in this study to characterize the direct binding of PMCA to G-actin.
SCHEME 1.

Binding analysis between PMCA and G-actin using the SPR technique. A, binding of solubilized PMCA to immobilized G-actin. G-actin is covalently attached to the CM5 sensor by reactive NH2 groups using the conventional protocol for activating the carboxyl groups of the surface. Solubilized PMCAs constitute the flowing analyte. C12E10 monomers are in equilibrium with micelles. B, binding of G-actin to immobilized PMCA. Covalently attached PMCA is stabilized on the sensor surface through continuous addition of C12E10. Immobilization is carried out using the same amine coupling procedure described above. G-actin constitutes the flowing analyte and is maintained in the monomeric state by using a running buffer of low ionic strength without Mg2+.
PMCA Binding to Immobilized G-actin
As a first experimental approach, we immobilized G-actin on the sensor surface at a level of 800–1200 RU, while PMCA constituted the flowing analyte. A typical sensorgram obtained for this experimental configuration in the presence of 70 μm Ca2+ is shown in Fig. 1A. The experimental data were well described by a kinetic global fit considering a 1:1 interaction (A + B ↔ AB). The binding equilibrium condition could not be attained experimentally, and therefore a steady state analysis could not be performed. Results from the kinetic analysis show a kon = (2.8 ± 0.3) 103 m−1 s−1 and a koff = (2.2 ± 0.1) 10−3 s−1. The Kd value was 786 ± 91 nm, obtained according to Equation 1,
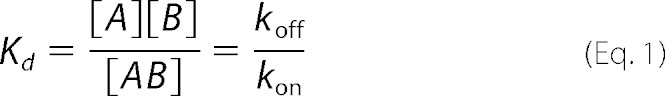 |
where A and B represent PMCA and G-actin; kon and koff are the association and dissociation rate constants of the interaction between PMCA and immobilized G-actin, and Kd is the equilibrium dissociation constant.
FIGURE 1.
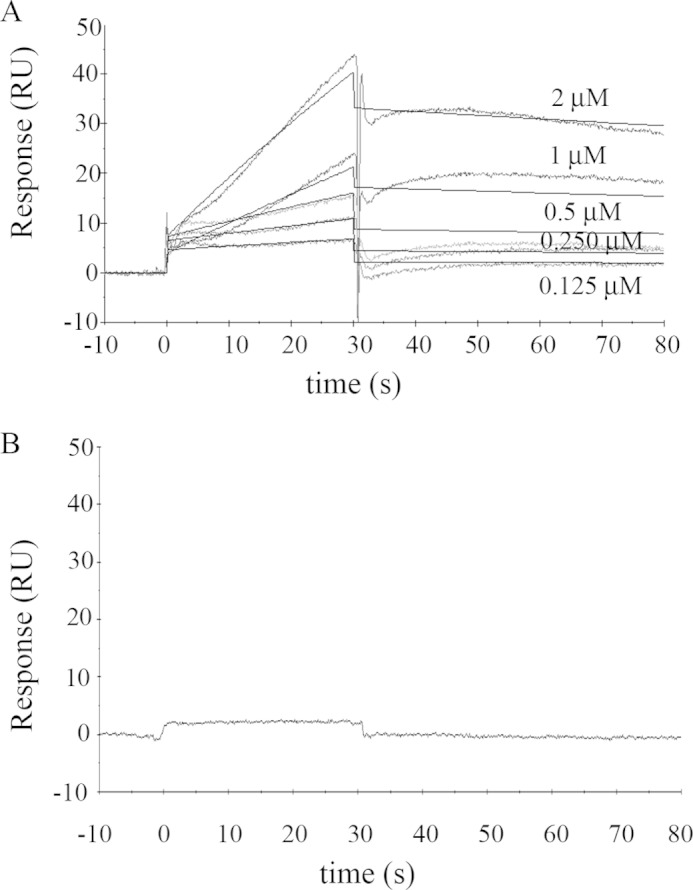
Binding of solubilized human erythrocyte PMCA to immobilized G-actin. A, representative sensorgram of PMCA binding to immobilized G-actin obtained for different PMCA concentrations in the range of 0.125 to 2 μm. G-actin immobilization level was ∼ 800 RU. The running buffer consisted of a low ionic strength Tris-HCl buffer with 70 μm Ca2+ and 0.005% C12E10 to maintain PMCA solubilized in micelles. The time for the association phase was set to 30 s. Steady state for binding interaction was not attained, and therefore a kinetic analysis was carried out. Curves were corrected for bulk effects by simple subtraction of the corresponding control sensorgrams. The dark gray lines represent the experimental curves, and the continuous black lines are the corresponding fits. B, representative sensorgram of PMCA (0.75 μm) injected in a running buffer devoid of Ca2+ by the addition of 2 mm EGTA shows no binding interaction. All other running conditions were identical to those in A. Note that human erythrocyte PMCA corresponds mostly to isoform PMCA4b (see under “Materials and Methods”).
PMCA is known to oligomerize (dimerize) into a fully active calmodulin-independent form at an enzyme concentration of 75 nm (9, 50, 51). Because all PMCA concentrations tested here (125–2000 nm) were above this value, it is very likely that the observed binding phenomena occurred between G-actin and the dimeric species of PMCA.
Because the affinity of PMCA for G-actin is lower than the affinity for PMCA itself, G-actin binding to monomeric PMCA cannot be studied using this system, but as shown below, it is possible when PMCA is immobilized on the sensor surface.
Requirement of Ca2+ in the Medium
Because changes in intracellular calcium concentrations regulate the activity of PMCA, we decided to explore whether the presence of calcium affects the binding of G-actin to PMCA. Ca2+ depletion was achieved by adding 2 mm EGTA to the running buffer. All other buffers contained 70 μm Ca2+. Because the running buffer also contains 1 mm Mg2+, the immobilized G-actin can replace Ca2+ by Mg2+ (52). The presence of Mg2+ prevents actin denaturation because it is known that in the absence of a divalent cation G-actin is unstable and tends to denature. However, the fact that G-actin is immobilized prevents it from polymerizing due to the presence of Mg2+. Fig. 1B shows that in the absence of Ca2+ no binding of G-actin to the PMCA can be detected, leading to the conclusion that Ca2+ is necessary for the interaction to occur.
To explain this observation, we propose two alternative hypotheses as follows: (i) G-actin does not bind to the monomeric species of PMCA given that pump oligomerization has been demonstrated to require Ca2+ (50), or (ii) G-actin does not bind to PMCA in the absence of Ca2+ regardless of the PMCA oligomerization state.
G-actin Binding to Immobilized PMCA
To address the first possibility, i.e. whether G-actin can also bind the monomeric species of the pump, PMCA was immobilized on the sensor surface (400–800 RU). To obtain monomeric PMCA covalently attached to the surface, the protein was injected at a low concentration (50 nm) with subsequent washing steps after the immobilization procedure to promote dimer dissociation. Performing SPR experiments with PMCA immobilized on a sensor chip surface represented a more challenging task than the experiment reported in Fig. 1, mainly for the following two reasons. (i) PMCA is a large membrane protein that requires a hydrophobic environment to maintain proper folding. (ii) Actin tends to polymerize. Regarding PMCA instability, the strategy consisted in immobilizing the protein stabilized in micelles of detergent. This same stabilization approach was successfully applied to G protein-coupled receptors (53) where, as in our case, the reconstitution detergent was the same as used for the extraction and isolation of the protein. Thus, the detergent was present in all the buffers used (immobilization, injection, dissociation, and regeneration buffers) and is thought to be in equilibrium with the protein micelles attached to the surface (as shown in Scheme 1). To overcome the G-actin aggregation/polymerization tendency, we dialyzed a preparation of pre-clarified G-actin against a buffer of low ionic strength and no Mg2+ to stabilize the monomeric state of actin; this buffer also constituted the running buffer.
Calmodulin Binding to Immobilized PMCA as Evidence of the Native State of Immobilized PMCA
To verify the accessibility of the flowing analyte to the cytosolic face of immobilized PMCA and to confirm the preservation of its native conformation, binding analyses of CaM to immobilized PMCA were carried out. Because the C-terminal domain of the PMCA is particularly sensitive to denaturation and proteolysis, CaM is a highly suitable candidate to probe the integrity of the pump.
Fig. 2A shows a typical sensorgram obtained for these experiments. In Fig. 2B it can be observed that curve of the analysis of the binding response is a function of the CaM concentration, which displays a hyperbolic behavior with a Kd of 1.6 ± 0.3 nm. This is in agreement with the reported apparent affinity of monomeric PMCA for CaM (9) and reveals that the PMCA intracellular face is accessible to the flowing analyte and preserves its native conformation after immobilization and during the SPR assays. After each measurement, PMCA was washed with a 1 mm EGTA solution, and no binding of CaM was detected in the absence of Ca2+.
FIGURE 2.

Calmodulin binding to immobilized PMCA. A, human erythrocyte PMCA at an immobilization level of ∼1000 RU was stabilized on the sensor surface in C12E10 micelles. Immediately after immobilization, a buffer containing 0.005% C12E10 was injected. Calmodulin was injected in the range 1.8–15 nm in a buffer composed of 20 mm MOPS-KOH (pH 7.2 at 25 °C), 120 mm KCl, 1 mm MgCl2, 0.005% C12E10, and 70 μm Ca2+ at a flow rate of 30 μl/min for 60 s. B, nonlinear analysis of the binding response obtained in the SPR assays was performed using a 1:1 interaction fit. The Kd values that best fit the experimental data were 1.6 ± 0.3 nm.
G-actin Remains Monomeric during SPR Assays
To perform the binding experiment using G-actin as the flowing analyte, the sample was prepared as described above to ensure the presence of monomeric actin. We further tested that G-actin did not polymerize or aggregate during SPR experiments by using a pyrene-labeled actin assay. This probe is used to monitor the actin assembly process as the labeled monomer increases its fluorescence signal when it is incorporated into a filament (44). As shown in Fig. 3, G-actin remains in its monomeric form as no enhancement in the fluorescence signal could be detected. When Polymerization Buffer (10× mixture of KCl, MgCl2, and ATP to final concentrations of 120, 3.75, and 2 mm, respectively) was added to a different aliquot of the same actin preparation, G-actin rapidly polymerized. This experiment indicates that actin is preserved in its native state after sample preparation, and it also constitutes a positive control for the stability test.
FIGURE 3.
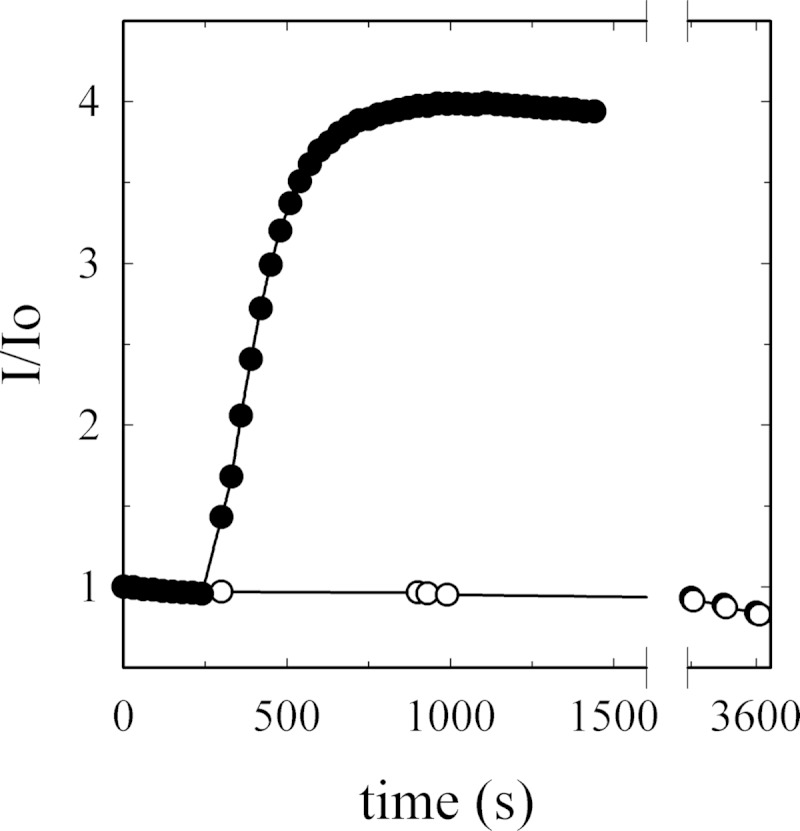
G-actin monomer stability during surface plasmon resonance analysis. G-actin tendency to aggregate or polymerize was assessed by measuring the fluorescence signal associated with pyrene-actin during incorporation into a filament (λex = 365 ± 3; λem = 407 ± 10, in nm). The fluorescence signal of 10 μm G-actin, containing 6% pyrene-label (○), was measured in a medium composed of 2 mm Tris-HCl (pH 7.7 at 25 °C), 70 μm CaCl2, and 0.005% C12E10. No changes in the intensity of fluorescence could be detected during 1 h. The slightly negative slope represents pyrene bleaching. As a positive control of actin polymerization, a 10× polymerization buffer was added to another aliquot of the preparation (●). The data are displayed as I/I0 (fluorescence signal at time t divided by initial fluorescence).
G-actin Binds Immobilized Monomeric PMCA
We next analyzed G-actin binding to the immobilized PMCA. A typical sensorgram is shown in Fig. 4A. Following correction for bulk effects by simple subtraction of the corresponding control sensorgrams (response of G-actin on control flow cell), the sensorgrams display saturating binding curves. The steady state for binding interaction was attained under these conditions. From the maximum response, the apparent Kd value was derived by applying a simple hyperbolic fit (Fig. 4B). This result suggests that G-actin can also bind to monomeric PMCA. The apparent affinity of the interaction is 3.8 ± 1.2 μm.
FIGURE 4.

Binding of G-actin to immobilized PMCA isolated from human erythrocytes. A, representative sensorgram of G-actin binding to immobilized PMCA at an immobilization level of ∼1000 RU is shown. PMCA was stabilized on the sensor surface in a micellar environment provided by the extraction detergent C12E10. Immediately after immobilization, a buffer containing 0.005% C12E10 was injected. G-actin was assayed in the range 0.6–5 μm in a modified Buffer G supplemented with 0.005% C12E10. The time for the association phase was set to 60 s. Curves were corrected for bulk effects by simple subtraction of the corresponding control sensorgrams. B, binding interaction analyzed from the kinetic global fit assuming a 1:1 interaction fit. The Kd values that best fit the experimental data were 3.8 ± 1.2 μm. Dark gray lines represent the experimental curves, and continuous black lines are the corresponding fits.
Taken together, these results suggest that G-actin can bind both forms of the pump, monomeric and oligomeric PMCA. Based on the results in Fig. 1B, Ca2+ may thus be essential for the interaction independently of the pump oligomerization state. However, this assertion cannot be directly tested. In contrast to the reciprocal experiment, in this case the running buffer had to be devoid of Mg2+ to prevent actin polymerization. Thus, it is not possible to assess the binding phenomena in the absence of Ca2+, because actin needs the presence of a divalent cation (Mg2+ or Ca2+) to maintain its native conformation. However, an additional observation from this experimental configuration is that binding occurs despite the absence of Mg2+; this could suggest that Mg2+, in contrast to Ca2+, is not a requirement for the interaction to occur.
Binding of G-actin to Immobilized C28 Peptide
The PMCA C terminus is involved in the regulation by and interaction with cellular signaling molecules (54, 55). Located inside this domain is the calmodulin-binding region (2, 56, 57), which is also known to interact with acidic phospholipids (58) and mediate the oligomerization process of the pump (59–61).
To determine whether this domain was also involved in G-actin binding, we performed SPR experiments using C28 peptide, which corresponds to the sequence of the calmodulin binding domain of hPMCA4b (2, 3). C28 rather than G-actin was immobilized on the sensor surface (at a level of 400–800 RU) due to the peptide's strong tendency to aggregate in solution (62). Fig. 5 shows that no specific interaction was observed between immobilized C28 and G-actin, suggesting that G-actin binds PMCA in a region different from the CaM binding domain.
FIGURE 5.

Binding of G-actin to immobilized C28 peptide. A, representative sensorgram of G-actin binding to immobilized C28 peptide at an immobilization level of ∼650 RU is shown. G-actin was assayed in the range 0.5–16 μm in a modified Buffer G (2 mm Tris-HCl, 70 μm Ca2+, pH 7.7 at 25 °C). The time for the association phase was set to 60 s. Curves were corrected for bulk effects by simple subtraction of the corresponding control sensorgrams. B, nonspecific binding of G-actin to the C28 peptide.
Characterization of the Effect of Actin on PMCA Ca2+-ATPase Activity and Phosphoenzyme Levels
Actin stimulation of PMCA has been observed to decrease as actin polymerization progresses (12). Thus, to characterize the activation phenomenon, we first needed to establish the conditions under which actin polymerization was minimized when measuring PMCA Ca2+-ATPase activity. We studied the polymerization kinetics under the experimental conditions used for Ca2+-ATPase determination using pyrene-labeled actin. Because the polymerization kinetics is highly dependent on actin concentration, we assayed different concentrations within the range used for the functional experiments. Based on the results shown in Fig. 6, we decided to perform PMCA activity experiments during 1 min after initiating the reaction, because we found that during this period of time all actin concentrations tested were not yet beyond the first stages of polymerization, i.e. actin is found in its monomeric and/or short oligomer form.
FIGURE 6.
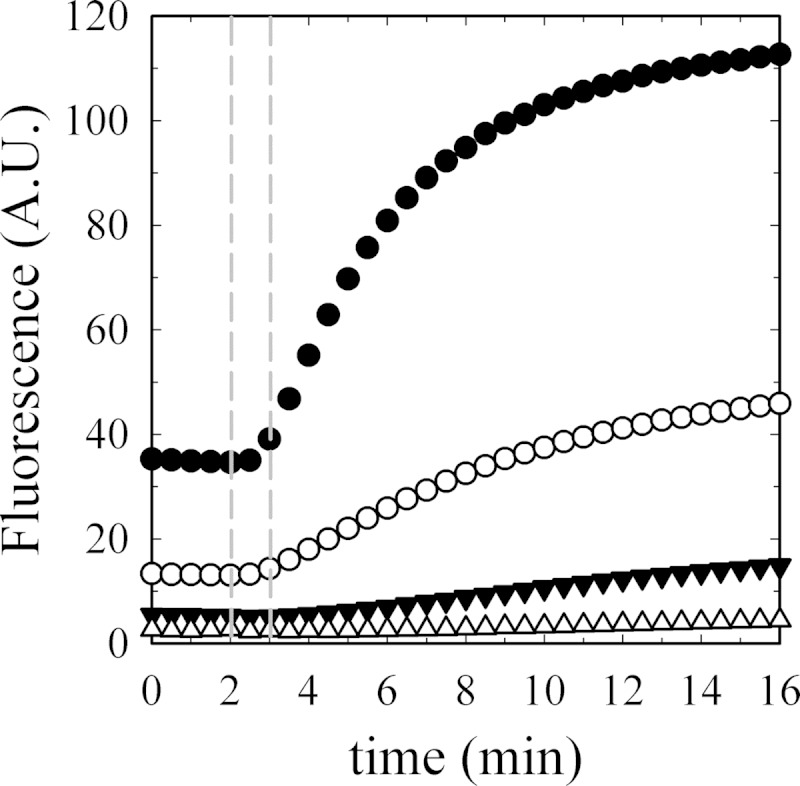
Time course of actin polymerization in Ca2+-ATPase and EP experiments. Actin polymerization (6% pyrene-label) was measured as a function of G-actin concentration as follows: ●, 5.00; ○, 2.50; ▾, 1.25; and Δ, 0.62 μm final concentrations. The first 120 s show the base-line signals that are different for each actin concentration because the percentage of label is constant and therefore not the total amount. Polymerization was initiated as described under “Materials and Methods” at 120 s. Data were collected every 30 s. The range between the two vertical dashed lines shows the time window used to perform Ca2+-ATPase activity experiments and phosphoenzyme level determinations.
When purified PMCA was incubated with increasing concentrations of G-actin, an increase in the rate of ATP hydrolysis was observed (Fig. 7A, inset) as measured by the [32P]Pi released from [γ-32P]ATP. As can be observed in the inset in Fig. 7A, ATP hydrolysis is linear under these conditions ensuring that initial velocity conditions are met. ATPase activity due to actin was not detected at any of the concentrations used (data not shown). This can be explained by the fact that the ATP bound to the assembling monomers is hydrolyzed not during the assembly step but subsequent to incorporation within the filament (63). Moreover, the release of the phosphate from actin-ADP-Pi has an even lower probability than hydrolysis with a measured t½ of ∼2 min (64, 65). A stimulatory effect of actin on PMCA due to Ca2+ released from actin, as actin exchanges bound Ca2+ by Mg2+ in the nucleation step in vitro (66, 67), was ruled out because the experiment was carried out in the presence of EGTA, which buffers small fluctuations in [Ca2+].
FIGURE 7.

Effect of actin on the Ca2+-ATPase activity and phosphorylated intermediates in isolated human erythrocyte PMCA during early stages of actin polymerization. A, Ca2+-ATPase activity was measured in PMCA reconstituted in DMPC/C12E10-mixed micelles in the presence of increasing amounts of purified G-actin. The reaction was started by the addition of [32P]ATP to a final concentration of 30 μm concomitant with the addition of G-actin. Each point represents the slope ± S.E. of the Pi released (inset) during 1 min after initiation of the reaction. The continuous line corresponds to the plot of Equation 2 as the best fit to the experimental data. Inset, Ca2+-dependent Pi release as a function of time for increasing concentrations of G-actin; data are from three independent experiments. B, effect of actin on the phosphoenzyme levels. Intermediate levels of PMCA reconstituted in DMPC/C12E10-mixed micelles as a function of G-actin concentration were measured 1 min after initiating the reaction in a medium of identical composition as for the experiment in A. The reaction was started by the addition of [32P]ATP to a final concentration of 30 μm concomitant with the addition of G-actin. The continuous line corresponds to the plot of Equation 2 as the best fit to the experimental data. C, turnover (kpEP) of the phosphoenzyme was calculated as the ratio between the Ca2+-ATPase activity taken from A and the EP values obtained in B at the different G-actin concentrations. The continuous line corresponds to the plot of Equation 2 as the best fit to the experimental data.
From the slope of each curve corresponding to the different G-actin concentrations (Fig. 7A, inset), the PMCA-specific activity was calculated. Equation 2 shows the best fit of the dependence of Ca2+-ATPase activity on G-actin concentration,
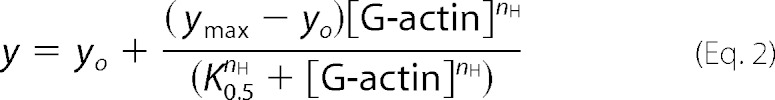 |
where yo is the Ca2+-ATPase activity in the absence of actin; ymax is the maximum Ca2+-ATPase activity; K0.5 corresponds to G-actin concentration at which a value of (ymax − yo)/2 in the activity is obtained, and nH represents the Hill coefficient.
The best fitting value of K0.5 for actin activation of the enzyme was 1.73 ± 0.06 μm with a maximum effect of more than 300% the value in the absence of G-actin. The Hill coefficient was 3.64 ± 0.39, which suggests that more than one molecule of G-actin is needed to increase PMCA activity. These results, together with the finding of a single binding site for G-actin and the evidence of the presence of short oligomers under the conditions of ATPase determination, suggest that short oligomers are responsible for the activating effect on PMCA.
PMCA forms an acid-stable phosphorylated intermediate (EP) during its reaction cycle (68, 69). Measurement of EP is of great relevance for the characterization of partial reactions of the enzyme cycle and has provided valuable information for the elucidation of the mechanism of its modulators (70, 71). To further characterize the stimulatory effect of actin on PMCA, we studied the dependence of the levels of PMCA-phosphorylated intermediates on G-actin concentration (Fig. 7B). For this purpose, we measured the total phosphoenzyme formation from [γ-32P]ATP as a function of G-actin concentration under identical experimental conditions as those used for the Ca2+-ATPase activity assay. We previously determined that steady state conditions were attained (data not shown). The levels of PMCA-phosphorylated intermediates increase with G-actin concentration (Fig. 7B). Applying Equation 2 yields a K0.5 = 1.57 ± 0.05 μm and nH = 4.59 ± 0.65. These results show that actin, in its short oligomeric form, is able to regulate the steps of the reaction cycle of the enzyme involving the phosphorylated species of the pump. The increase in the levels of EP can be due to an increase in the rate of formation or alternatively due to a decrease in the breakdown of EP. Because the rate of the full cycle of the pump is increased by actin (increase in the specific activity; Fig. 7A), it is likely that the actin effect on the EP is due to an increase in the rate of formation of EP.
Fig. 7C shows the turnover of EP (kpEP), which was calculated as the ratio between the Ca2+-ATPase activity from Fig. 7A and the EP values obtained in Fig. 7B at the different G-actin concentrations. The continuous line corresponds to the plot of Equation 2 as the best fit to the experimental data with K0.5 = 1.61 ± 0.05 μm and nH = 3.02 ± 0.23.
Taken together, these results suggest that actin oligomers enhance the Ca2+-ATPase activity of PMCA by favoring the steps of the reaction cycle that lead to the formation of the phosphorylated intermediates of the pump (increase in EP levels) and decreasing the transit time for this step (increase in kpEP).
Effect of Actin on the Ca2+ Dependence of PMCA ATPase Activity and Calmodulin Activation
The effect of G-actin on PMCA Ca2+-ATPase activity was also determined as a function of Ca2+ at saturating concentrations of ATP (2 mm). We measured Ca2+-ATPase activity by continuously monitoring the release of inorganic phosphate from ATP using the method described by Webb (46). The increase in ATP concentration did not dramatically affect actin polymerization kinetics during the 1st min (data not shown). Reactions were initiated by adding the mixture of the activating agent (5 μm G-actin, 100 nm CaM, or 5 μm G-actin plus 100 nm CaM) and 2 mm ATP. PMCA Ca2+-ATPase-specific activity was determined from the slope of the absorbance at 365 nm as a function of time during the 1st min after the start of the reaction. In all cases, the response as a function of time was linear (data not shown) indicating the initial velocity condition. The experimental results obtained for the specific activity as a function of [Ca2+] for all the treatments were fitted to a simple hyperbola.
As can be seen in Fig. 8, actin, likely in its short oligomeric form, increases the apparent affinity of PMCA for Ca2+ by more than 3-fold compared with the basal conditions where actin was replaced by Buffer G. As before, to rule out that the exchange of the cation (Ca2+ by Mg2+) by actin during the nucleation step leads to artifacts, an EGTA-Ca2+ buffer was used to maintain the [Ca2+] constant.
FIGURE 8.
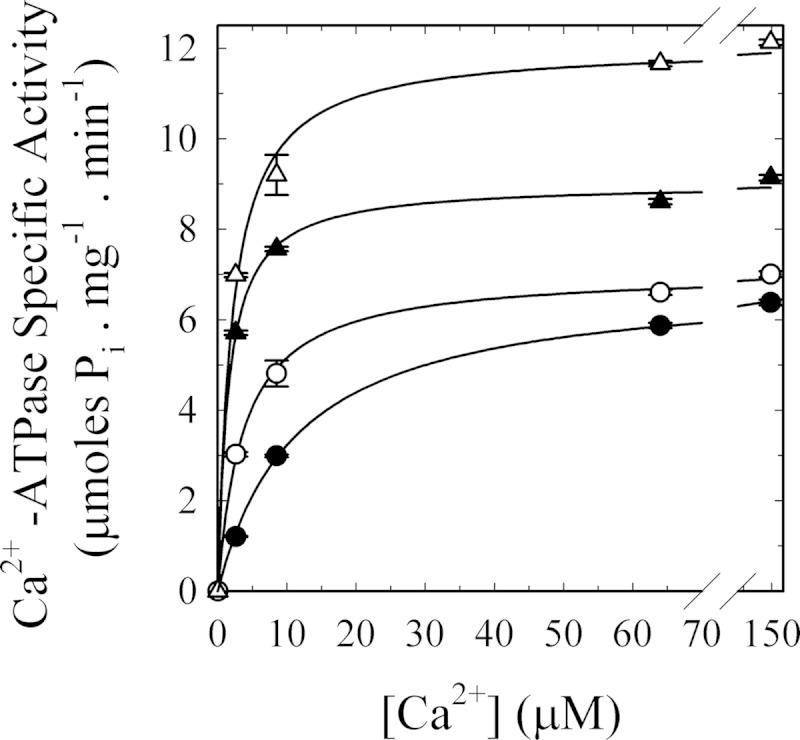
[Ca2+] dependence of G-actin effect on human erythrocyte PMCA Ca2+-ATPase activity. Ca2+-ATPase activity was measured at different [Ca2+] in the presence of the following: 100 nm calmodulin (▴) as a positive control for PMCA activation; 5 μm G-actin (○); 100 nm calmodulin plus 5 μm G-actin (Δ), or buffer (●) as the basal condition. The reaction was started by the addition of 2 mm ATP concomitant with the addition of the effector/buffer. The reaction medium contained 120 mm KCl, 30 mm MOPS-K (pH 7.4 at 25 °C), 3.75 mm MgCl2, 70 μg/ml C12E10, 10 μg/ml phosphatidylcholine, 1 mm EGTA, and enough CaCl2 to give the desired final [Ca2+]free. PMCA concentration was 0.8 μg/ml. Pi release was determined by the continuous method of Webb (46). Values are the mean ± S.E. from three different experiments. When not apparent, error bars are within the symbols. The values of K0.5 and Vmax for the different treatments are as follows: basal conditions 11.4 ± 0.4 μm and 6.9 ± 0.1 μmol of Pi·mg−1·min−1, respectively; addition of 100 nm CaM 1.5 ± 0.1 μm and 9.0 ± 0.1 μmol of Pi·mg−1·min−1, respectively; addition of 5 μm G-actin 3.7 ± 0.2 μm and 7.1 ± 0.1 μmol of Pi·mg−1·min−1, respectively; addition of 100 nm CaM plus 5 μm G-actin 2.1 ± 0.3 μm and 12.1 ± 0.3 μmol of Pi·mg−1·min−1, respectively.
In contrast to what was observed in the radioactive Pi release assay, at high Ca2+ concentrations no significant increase in PMCA activity was observed, thus leading to the conclusion that in the presence of saturating concentrations of both substrates (Ca2+ and ATP), the effect of actin is barely noticeable indicating that it does not affect the Vmax of the pump.
When simultaneously added with CaM, G-actin did not further increase the apparent affinity for Ca2+. The ratio Vmax/K0.5 is not significantly different in these two conditions indicating that CaM and actin exert their regulation on intermediates of the reaction cycle that are separated by irreversible steps.
Effect of Actin on the Conformational State of PMCA
We have previously reported that treatments that lead to an activation of PMCA are related to a decrease in the accessible area of the membrane domain of the pump to the surrounding lipids, strongly suggesting the involvement of a conformational change in the transmembrane segments (49, 72, 73).
To assess whether the direct binding of actin to PMCA would also induce a conformational change typical of the known activators of the pump, we measured the specific incorporation of [125I]TID-PC/16 to PMCA as a function of G-actin concentration (Fig. 9A). [125I]TID-PC/16 is a photoactivatable phosphatidylcholine analog that partitions in the phospholipid milieu and, upon photolysis, reacts with the surrounding molecules in its immediate vicinity. This reagent can thus be used to study the interaction between the PMCA (or any membrane protein) with the lipids in its immediate environment. Changes in the specific incorporation of [125I]TID-PC/16 into the PMCA reflect conformational changes of the membrane domain of the pump characteristic for its different reaction states (74).
FIGURE 9.

PMCA conformational change induced by G-actin measured as the relative incorporation of [125I]TID-PC/16 to PMCA. A, purified human erythrocyte PMCA was incubated with [125I]TID-PC/16 as described under “Materials and Methods” after which purified G-actin was added at different concentrations. The reaction medium contained 70 μm Ca2+. After 1 min of actin addition, the reaction was stopped by exposing the samples to a UV light source. Data are the mean ± S.E. of two independent experiments. The continuous line corresponds to the best fitted parameters of Equation 3 to the experimental data. B, effect of saturating concentrations of 120 nm CaM, 10 μm phosphatidic acid, and 5 μm actin on the incorporation of [125I]TID-PC/16 to purified PMCA. As expected for a calmodulin-like activator, the specific incorporation of [125I]TID-PC/16 decreased with actin from near 150% to a value of 95.1 ± 2.4%, although CaM and phosphatidic acid (PA) decreased the specific incorporation to 76 ± 1 and 89 ± 2%, respectively.
The reaction medium was identical to that used in activity experiments except for the absence of ATP. Actin polymerization kinetics under these conditions were not significantly different from those in Fig. 6. The probe was preincubated with the PMCA and then for 1 min with different G-actin concentrations at 25 °C. As standardized previously, the level of [125I]TID-PC/16 incorporation in the absence of Ca2+ (2 mm EGTA) was taken as 100%. The presence of saturating Ca2+ concentrations (70 μm) in the absence of actin (replaced by Buffer G) gave a specific incorporation of ∼150%, as described previously (49). The experimental data in the presence of G-actin were best fitted to Equation 3 with a Kd = 1.10 ± 0.08 μm and nH = 2.78 ± 0.56.
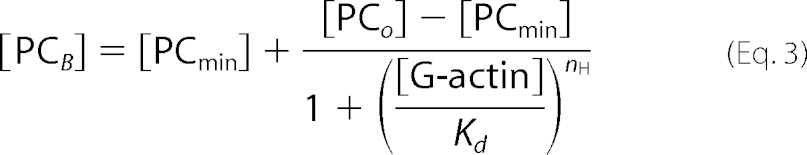 |
Fig. 9B shows the effect of saturating concentrations of other “calmodulin-like” activators on the incorporation of the probe to PMCA. As expected for an activator, actin, most likely short oligomers, decreased the specific incorporation of [125I]TID-PC/16 from 150.0 ± 2.7 to a value of 95.1 ± 2.4%.
DISCUSSION
Previous studies from our laboratory showed that the actin cytoskeleton differentially regulates the activity of PMCA depending on its polymerization state (11, 12). Although the inhibitory role of actin filaments has been well described, the characterization of the interaction between G-actin and the pump as well as the stimulatory effect during the first stages of polymerization have not been addressed.
In this study, we demonstrate a direct and specific binding between PMCA and G-actin, and we characterize the stimulatory effect providing strong evidence in favor of short actin oligomers as the actin entity responsible for the stimulatory effect.
Binding experiments, based on surface plasmon resonance technology, were carried out with the two full-length native proteins. This represented a challenging task because one of them is an integral membrane protein and the other nucleates and polymerizes under physiological conditions. For this reason, and as our first choice, the experimental configuration consisted in immobilizing the soluble protein (G-actin) on the sensor surface composed of a hydrogel matrix, whereas the membrane protein (PMCA) solubilized in detergent micelles was injected as the flowing analyte. This experimental configuration has the advantage of preventing the aggregation/polymerization of G-actin, as it was covalently attached to the surface. This allowed us to work under polymerizing conditions without actin assembly. By this approach, we obtained an apparent affinity constant of the PMCA for G-actin of ∼800 nm. However, to obtain measurable binding responses, PMCA had to be injected in high concentrations, all of them above the Kd value of the dimerization process of the pump. PMCA is known to oligomerize, and it has been demonstrated that self-association also occurs when the enzyme is isolated and reconstituted in micelles (75); hence, we should assume that all PMCA injected was in the dimeric/oligomeric forms. Because the SPR signal depends on the mass of protein bound to the surface, it is critical to use homogenous and nonaggregated protein samples. Thus, the obtained apparent affinity value is difficult to interpret due to the possible heterogeneity in the composition of the analyte. However, the data clearly indicate a specific interaction between G-actin and oligomeric PMCA, which may be of relevance at the cellular level. Oligomeric PMCA has been reported to be present in caveolae where the pump molecules are 18–25-fold concentrated compared with the non-caveolae portion of the plasma membrane (76) and where its biologically most relevant activators, acidic phospholipids and calmodulin, are enriched (77, 78). These regions of the membrane are tightly connected with the underlying actin cytoskeleton (79). Thus, PMCA interaction with and regulation by actin in these regions could be of physiological interest as these structures are involved in calcium homeostasis among other diverse functions (80).
To quantitatively characterize the binding interaction of G-actin and PMCA, the monomeric species of the pump had to be assayed, which required inverting the experimental configuration to obtain monomeric PMCA immobilized on the sensor surface. However, performing the experiment in the mirror configuration of the one described above presented some limitations as follows: (i) the medium had to be of low ionic strength and devoid of Mg2+ because, in this case, G-actin was the flowing analyte and was able to assemble under polymerizing conditions; hence, the mirror experiment could not be performed under identical conditions, and (ii) PMCA had to be maintained on the surface in a stable micellar environment, for which detergent had to be in a permanent equilibrium between the surface and the bulk solution. Despite these difficulties, we successfully achieved the immobilization of a stabilized pump and were able to perform the assays with native PMCA with its cytosolic face available for the flowing analyte, as evidenced by the CaM binding experiments.
Data obtained from these experiments indicate that G-actin can also interact with monomeric PMCA. The binding interaction is well described by a 1:1 interaction model with an apparent affinity in the low micromolar range (Kd = 3.8 ± 1.2 μm). Similar affinity values have been reported for the direct interaction between actin and other transporters such as the mitochondrial channel voltage-dependent anion channel and aquaporin-2 (81, 82). Despite not being considered strong interactions, it is important to note that ion transporters bind to G-actin with similar affinity compared with actin monomer-binding proteins such as profilin (Kd = 1.5 × 10−7 m), actin depolymerization factor (ADF)/cofilin (Kd = 4 × 10−6 m), twinfilin (Kd = 5 × 10−7 m), and Rac1 (Kd = 1.7 × 10−6 m). Low affinity interactions between G-actin and many of its binding partners may be indicative of the highly dynamic role it plays in cell physiology, where such interactions may be preferred over the high affinity stable ones.
Although G-actin was able to bind PMCA, as evidenced in SPR experiments, the monomer had no functional effect on the pump (Fig. 7A). Actin stimulation of PMCA Ca2+-ATPase activity shows a sigmoidal behavior (nH = 3.64 ± 0.40) suggesting that more than one molecule of G-actin is needed to exert its effect, which means the following: (a) G-actin needs to assemble into short oligomers, most likely trimers or tetramers to exert its action on PMCA catalytic activity, or (b) there is more than one site of interaction for G-actin. From the results of the binding experiments, we ruled out the second possibility as the interaction between G-actin and PMCA was well described by a 1:1 binding model.
The effect of actin on PMCA function was tested under polymerizing conditions, as the reaction medium for PMCA Ca2+-ATPase activity determinations strongly favors actin assembly (Fig. 6). Medium conditions during the experiments were compatible with the initial polymerization steps. The assembly process was observed to occur for higher actin concentrations, although little or no polymerization was detected for the lower concentrations (determined 1 min after initiation of the reaction). G-actin at concentrations showing polymerization exerted a stimulatory effect on PMCA activity, while lower concentrations, which showed no detectable assembly, had no effect on the activity of the pump. Thus, the actin assembly event seems to correlate with the activation of the pump, and the oligomer hypothesis is the most plausible explanation for the stimulatory effect observed. Moreover, all nH values obtained in the experiments of this work (Figs. 7, A and B, and 9A) are in the range of 3–4. These values are compatible with the reported number of monomeric subunits that constitute the nucleus (83, 84), i.e. the thermodynamically favored association of actin monomers from which the elongation of filaments takes place. This further supports the need of an assembly structure to stimulate the pump as the minimum number of actin monomers needed to exert the stimulatory effect corresponds to the minimum actin monomers that are capable of starting the polymerization process.
As polymerization kinetics are highly dependent on actin concentration, faster assembly rates are achieved at higher actin concentrations. However, as pyrene fluorescence intensity is insensitive to the actin filament length distribution, it cannot be determined if an increase in the fluorescence signal is due to an increase in the number of filaments or in their length. It thus still remains unknown if higher concentrations of actin have larger effects because the actin oligomers reach an optimum length or because there are more oligomer units being assembled. Some indirect evidence would suggest that the length is playing a role because incubation for 1 min with 10 μm G-actin led to an increase of only 75% of the basal activity (data not shown). At this concentration, actin rapidly assembles under the experimental conditions, further supporting the idea that “short” actin oligomers formed in the very first stages of polymerization are responsible for the maximum stimulatory effect achieved. Regarding the affinity of the effect, it is important to note that it has been expressed in terms of the molar concentration of initial G-actin; however, if short oligomers are the entities exerting the effect, this value would be much higher (lower Kd).
To further characterize the regulation of the PMCA by actin, we studied the dependence of the level of PMCA phosphorylated intermediates on G-actin concentration under identical conditions. Yet, here again, it appears that short oligomers are responsible for the observed effect. Oligomeric actin produces an increase in the levels and in the turnover of the phosphoenzyme; this can explain, at least in part, the increase in the Ca2+-ATPase activity of the pump. It is likely that the increase in the levels of EP is due to an increase in the rate of any of the steps that lead to EP formation because a decrease in EP breakdown would have resulted in a decrease in the rate of the whole cycle of the pump.
Moreover, actin oligomers increase the Ca2+-ATPase activity of the pump by increasing its apparent affinity for Ca2+. In eukaryotic cells, the rise in cytosolic calcium activates proteins that sever or nucleate actin filaments, yielding actin oligomers with their barbed ends blocked. Thus, these events promote actin disassembly (85). As an example, gelsolin and villin shorten actin filaments in the presence of calcium in the micromolar range and are inactive when the calcium concentration is below this range (86, 87). In addition, recent studies showed that in the presence of actin-binding proteins, filaments disassemble in large and rapid bursts (88). It is thus possible that PMCA stimulation by short oligomers may be one of the physiological feedback responses of the actin cytoskeleton to an intracellular calcium increase. Importantly, actin oligomers not only affect the Ca2+ affinity of the pump but Ca2+ itself may regulate both the formation of this activating agent and its interaction with the pump; and as we observed, in the absence of Ca2+ no binding interaction occurred (Fig. 1B). This is analogous to the regulation by CaM, which also increases the apparent affinity of the pump for Ca2+, and where Ca2+ is involved in the formation and interaction of the “activating agent,” i.e. the complex Ca2+-CaM. The lack of a specific interaction between G-actin and (oligomeric) PMCA in the absence of Ca2+ (Fig. 1B) together with the observation of a specific binding between G-actin and monomeric PMCA in the presence of Ca2+ (Fig. 4) would indicate that Ca2+ is essential for the interaction to occur, independently of the oligomerization state of the pump. In contrast to the need for Ca2+, Mg2+ is not a requirement for the interaction to occur.
Because the evidence presented here indicates that short oligomers are the stimulatory entity of actin, it would be of interest to study their direct binding to PMCA. By measuring the specific incorporation of [125I]TID-PC/16 to PMCA, it was possible to study both the apparent affinity of short oligomers for PMCA and the change in the pump conformation upon the interaction with them. The incorporation of the probe to PMCA as a function of G-actin concentration was performed under identical conditions as were used for activity experiments except for the absence of ATP. Under these conditions, PMCA was considered to be in equilibrium (no ATP), and actin was considered to polymerize as shown in Fig. 6.
Oligomeric actin induced a conformational change in the PMCA characterized by a decrease in the surface accessible to the lipids as a result of rearrangements in the transmembrane segments of the pump (49). This effect is analogous to the behavior observed for other activators of the pump such as CaM and phosphatidic acid (72, 73).
When we measured the equilibrium constant for the dissociation of short oligomers as the change of the transmembrane conformation of the pump and expressed it in terms of initial G-actin concentration, the parameters were comparable with the ones obtained in the SPR binding studies, which were also carried out under equilibrium conditions. Importantly, the apparent affinity of the pump for G-actin (Kd = 3.8 ± 1.2 μm) is lower than that obtained in the [125I]TID-PC/16 incorporation experiment (Kd = 1.10 ± 0.08 μm), which likely reflects the affinity for actin short oligomers, although it is expressed in terms of the initial concentration for G-actin. As discussed below, this value would be even lower in terms of actin oligomers.
Altogether, these results suggest that PMCA can bind both G-actin and short oligomers and that short oligomers but not G-actin are the entity that exerts the stimulatory effect on the pump (Scheme 2). A direct comparison between the apparent affinities of actin for PMCA measured in equilibrium (SPR) and in steady state (Ca2+-ATPase activity and phosphorylation) is not valid, but it seems that both are within the micromolar range (between 1 and 3 μm) expressed as initial G-actin. This appears to be the most suitable form to express the results because the exact molecular entity (or entities) of actin, which exert the effect, and the amount of each of these entities present under our experimental conditions are not known. Thus, the values of the constants expressed in this form represent the concentration of initial G-actin that under a defined polymerizing condition and during the 1st min after initiation of the reaction produces a population of oligomers capable of displaying the half-maximal effect.
SCHEME 2.

Summary of G-actin-PMCA interactions determined by SPR and activity measurements. Top, interaction of immobilized G-actin (immob-G-Act) with soluble PMCA dimers in micelles ((PMCA)2mic(sol)), determined by SPR in the presence of Ca2+ and Mg2+. The calculated Kd was ∼0.8 μm. Middle, interaction of immobilized micellar PMCA with soluble G-actin in the presence of Ca2+ but absence of Mg2+. The Kd determined by SPR was ∼3.8 μm. Bottom, stimulation of the basal ATPase activity of the micellar PMCA (PMCAmicbasal) by G-actin in the presence of Ca2+ and Mg2+. The K0.5 is ∼1.7 μm and n = 3–4 G-actin monomers/PMCA are required for activation. For details, see the text.
Whether preformed actin oligomers bind PMCA or one monomer first binds and then successive subunits are added to the complex remains unknown. In the latter case, PMCA could act as a nucleator agent. When polymerization assays were performed under activity conditions using 5 μm G-actin and 6 nm PMCA (identical to those in the experiments of Figs. 7 and 8), no difference in the polymerization kinetics was observed in the presence or absence of PMCA (data not shown). This suggests that PMCA does not act as a nucleation center for actin polymerization.
Unlike CaM, G-actin did not bind the CaM binding domain as evidenced by the unspecific interaction with C28. Oligomers also do not bind this domain because CaM and actin had additive effects on PMCA catalytic activity (Fig. 8). A possible candidate for the binding site may be the positively charged acidic phospholipid site in the PMCA, because actin is negatively charged at physiological pH. Lysine-rich clusters in actin-binding proteins have been identified to be involved in actin binding, e.g. the DAIKKK sequence in actin depolymerization factor, cofilin, and tropomyosin (89); the KKGGKKKG sequence of myosin (90); the KSKLKKT sequence in thymosin β4 (91), and the sequence KKEK in villin (92). Although there is no conserved sequence for actin binding between the different actin-binding proteins, a consistent motif rich in basic residues exists, which is also present in the PMCA sequence at the cytosolic loop between the transmembrane segments 2 and 3. This region contains the acidic phospholipid binding domain in which the KKEK motif is found (Fig. 10). Moreover, it has been observed that phosphatidylinositol 4,5-bisphosphate, an acidic phospholipid, displaces bound actin from actin-binding proteins (94–96). As can be observed in Fig. 10, the acidic phospholipid-binding site is a highly conserved sequence present in all human PMCAs. From this observation, we propose this sequence as the putative binding site for actin and expect all isoforms of the pump to potentially interact with actin. Experimental work will be required to confirm the involvement of this domain in PMCA interactions with actin.
FIGURE 10.

PMCA sequences corresponding to the acidic phospholipid binding domains. Amino acid number: a, 342 for hPMCA2xb, 373 for hPMCA2wb, and 328 for hPMCA2zb; b, 347 for hPMCA3xb and 333 for hPMCA3zb; c, 339 for hPMCA4xb and 327 for hPMCA4zb.
Acknowledgment
We thank Dr. Rolando Rossi for helpful comments on the manuscript.
This work was supported by Agencia Nacional de Promoción Cientifica y Tecnológica, Consejo Nacional de Investigaciones Cientificas y Técnicas, and Universidad de Buenos Aires Ciencia y Técnica (Argentina).
- PMCA
- plasma membrane calcium pump
- CaM
- calmodulin
- DMPC/PC
- 1,2-dimyristoyl-sn-glycero-3-phosphocholine
- C12E10
- polyoxyethylene(10) dodecyl ether
- SPR
- surface plasmon resonance
- RU
- resonance unit
- [125I]TID-PC/16
- 1-O-hexadecanoyl-2-O-[9-[[[2-[125I]iodo-4-(trifluoromethyl-3H-diazirin-3-yl)benzyl]oxy]carbonyl]nonanoyl]-sn-glycero-3-phosphocholine.
REFERENCES
- 1. Strehler E. E., Zacharias D. A. (2001) Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 81, 21–50 [DOI] [PubMed] [Google Scholar]
- 2. James P., Maeda M., Fischer R., Verma A. K., Krebs J., Penniston J. T., Carafoli E. (1988) Identification and primary structure of a calmodulin binding domain of the Ca2+ pump of human erythrocytes. J. Biol. Chem. 263, 2905–2910 [PubMed] [Google Scholar]
- 3. Enyedi A., Vorherr T., James P., McCormick D. J., Filoteo A. G., Carafoli E., Penniston J. T. (1989) The calmodulin binding domain of the plasma membrane Ca2+ pump interacts both with calmodulin and with another part of the pump. J. Biol. Chem. 264, 12313–12321 [PubMed] [Google Scholar]
- 4. Niggli V., Adunyah E. S., Carafoli E. (1981) Acidic phospholipids, unsaturated fatty acids, and limited proteolysis mimic the effect of calmodulin on the purified erythrocyte Ca2+-ATPase. J. Biol. Chem. 256, 8588–8592 [PubMed] [Google Scholar]
- 5. Niggli V., Adunyah E. S., Penniston J. T., Carafoli E. (1981) Purified (Ca2+-Mg2+)-ATPase of the erythrocyte membrane. Reconstitution and effect of calmodulin and phospholipids. J. Biol. Chem. 256, 395–401 [PubMed] [Google Scholar]
- 6. Neyses L., Reinlib L., Carafoli E. (1985) Phosphorylation of the Ca2+-pumping ATPase of heart sarcolemma and erythrocyte plasma membrane by the cAMP-dependent protein kinase. J. Biol. Chem. 260, 10283–10287 [PubMed] [Google Scholar]
- 7. Zylinska L., Guerini D., Gromadzinska E., Lachowicz L. (1998) Protein kinases A and C phosphorylate purified Ca2+-ATPase from rat cortex, cerebellum, and hippocampus. Biochim. Biophys. Acta 1448, 99–108 [DOI] [PubMed] [Google Scholar]
- 8. James P., Vorherr T., Krebs J., Morelli A., Castello G., McCormick D. J., Penniston J. T., De Flora A., Carafoli E. (1989) Modulation of erythrocyte Ca2+-ATPase by selective calpain cleavage of the calmodulin-binding domain. J. Biol. Chem. 15, 8289–8296 [PubMed] [Google Scholar]
- 9. Kosk-Kosicka D., Bzdega T. (1988) Activation of the erythrocyte Ca2+-ATPase by either self-association or interaction with calmodulin. J. Biol. Chem. 263, 18184–18189 [PubMed] [Google Scholar]
- 10. Sarkadi B., Szász I., Gárdos G. (1980) Characteristics and regulation of active calcium transport in inside-out red cell membrane vesicles. Biochim. Biophys. Acta 598, 326–338 [DOI] [PubMed] [Google Scholar]
- 11. Vanagas L., Rossi R. C., Caride A. J., Filoteo A. G., Strehler E. E., Rossi J. P. (2007) Plasma membrane calcium pump activity is affected by the membrane protein concentration: evidence for the involvement of the actin cytoskeleton. Biochim. Biophys. Acta 1768, 1641–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vanagas L., de La Fuente M. C., Dalghi M., Ferreira-Gomes M., Rossi R. C., Strehler E. E., Mangialavori I. C., Rossi J. P. (2013) Differential effects of G- and F-actin on the plasma membrane calcium pump activity. Cell Biochem. Biophys. 66, 187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mitchison T. J., Cramer L. P. (1996) Actin-based cell motility and cell locomotion. Cell 84, 371–379 [DOI] [PubMed] [Google Scholar]
- 14. Ridley A. (2000) Rho GTPases. Integrating integrin signaling. J. Cell Biol. 150, F107–F109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qualmann B., Kessels M. M., Kelly R. B. (2000) Molecular links between endocytosis and the actin cytoskeleton. J. Cell Biol. 150, F111–F116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chimini G., Chavrier P. (2000) Function of Rho family proteins in actin dynamics during phagocytosis and engulfment. Nat. Cell Biol. 2, E191–E196 [DOI] [PubMed] [Google Scholar]
- 17. Mandato C. A., Benink H. A., Bement W. M. (2000) Microtubule-actomyosin interactions in cortical flow and cytokinesis. Cell Motil. Cytoskeleton 45, 87–92 [DOI] [PubMed] [Google Scholar]
- 18. Valentijn J. A., Valentijn K., Pastore L. M., Jamieson J. D. (2000) Actin coating of secretory granules during regulated exocytosis correlates with the release of rab3D. Proc. Natl. Acad. Sci. U.S.A. 97, 1091–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khurana S. (2000) Role of actin cytoskeleton in regulation of ion transport: examples from epithelial cells. J. Membr. Biol. 178, 73–87 [DOI] [PubMed] [Google Scholar]
- 20. Berdiev B. K., Prat A. G., Cantiello H. F., Ausiello D. A., Fuller C. M., Jovov B., Benos D. J., Ismailov I. I. (1996) Regulation of epithelial sodium channels by short actin filaments. J. Biol. Chem. 271, 17704–17710 [DOI] [PubMed] [Google Scholar]
- 21. Ismailov I. I., Berdiev B. K., Shlyonsky V. G., Fuller C. M., Prat A. G., Jovov B., Cantiello H. F., Ausiello D. A., Benos D. J. (1997) Role of actin in regulation of epithelial sodium channels by CFTR. Am. J. Physiol. 272, C1077–C1086 [DOI] [PubMed] [Google Scholar]
- 22. Kurashima K., D'Souza S., Szászi K., Ramjeesingh R., Orlowski J., Grinstein S. (1999) The apical Na+/H+exchanger isoform NHE3 is regulated by the actin cytoskeleton. J. Biol. Chem. 274, 29843–29849 [DOI] [PubMed] [Google Scholar]
- 23. Cantiello H. F. (1995) Actin filaments stimulate the Na+-K+-ATPase. Am. J. Physiol. 269, F637–F643 [DOI] [PubMed] [Google Scholar]
- 24. Sheterline P., Clayton J., Sparrow J. C. (1998) Actin. 4th Ed., Oxford University Press, New York [Google Scholar]
- 25. dos Remedios C. G., Chhabra D., Kekic M., Dedova I. V., Tsubakihara M., Berry D. A., Nosworthy N. J. (2003) Actin binding proteins: regulation of cytoskeletal microfilaments. Physiol. Rev. 83, 433–473 [DOI] [PubMed] [Google Scholar]
- 26. Fechheimer M., Zigmond S. (1993) Focusing on unpolymerized actin. J. Cell Biol. 123, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spudich A., Wrenn J. T., Wessells N. K. (1988) Unfertilized sea urchin eggs contain a discrete cortical shell of actin that is subdivided into two organizational states. Cell Motil. Cytoskeleton 9, 85–96 [DOI] [PubMed] [Google Scholar]
- 28. Bonder E. M., Fishkind D. J., Cotran N. M., Begg D. A. (1989) The cortical actin-membrane cytoskeleton of unfertilized sea urchin eggs: analysis of the spatial organization and relationship of filamentous actin, nonfilamentous actin, and egg spectrin. Dev. Biol. 134, 327–341 [DOI] [PubMed] [Google Scholar]
- 29. Brozinick J. T., Jr., Berkemeier B. A., Elmendorf J. S. (2007) “Actin” g on GLUT4: membrane & cytoskeletal components of insulin secretion. Curr. Diabetes Rev. 3, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scita G., Confalonieri S., Lappalainen P., Suetsugu S. (2008) IRSp53: crossing the road of membrane and actin dynamics in the formation of membrane protusions. Trends Cell Biol. 18, 52–60 [DOI] [PubMed] [Google Scholar]
- 31. Weed S. A., Parsons J. T. (2001) Cortactin:coupling membrane dynamics to cortical actin assembly. Oncogene 20, 6418–6434 [DOI] [PubMed] [Google Scholar]
- 32. Huang Y., Burkhardt J. K. (2007) T-cell receptor-dependent actin regulatory mechanisms. J. Cell Sci. 120, 723–730 [DOI] [PubMed] [Google Scholar]
- 33. Billadeau D. D., Burkhardt J. K. (2006) Regulation of cytoskeletal dynamics at the immune synapse: new stars join the actin troupe. Traffic 11, 1451–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bozulic L. D., Malik M. T., Powell D. W., Nanez A., Link A. J., Ramos K. S., Dean W. L. (2007) Plasma membrane Ca2+-ATPase associates with CLP36, α-actinin, and actin in human platelets. Thromb. Haemost. 97, 587–597 [PubMed] [Google Scholar]
- 35. Guerini D., Pan B., Carafoli E. (2003) Expression, purification, and characterization of isoform 1 of the plasma membrane Ca2+ pump: focus on calpain sensitivity. J. Biol. Chem. 278, 38141–38148 [DOI] [PubMed] [Google Scholar]
- 36. Niggli V., Penniston J. T., Carafoli E. (1979) Purification of the (Ca2+-Mg2+)-ATPase from human erythrocyte membranes using a calmodulin affinity column. J. Biol. Chem. 254, 9955–9958 [PubMed] [Google Scholar]
- 37. Filomatori C. V., Rega A. F. (2003) On the mechanism of activation of the plasma membrane Ca2+-ATPase by ATP and acidic phospholipids. J. Biol. Chem. 278, 22265–22271 [DOI] [PubMed] [Google Scholar]
- 38. Straub F. B. (1943) Actin II. Studies from the Inst. Med. Chem. Univ. Szeged 3, 23–37 [Google Scholar]
- 39. Spudich J. A., Watt S. (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246, 4866–4871 [PubMed] [Google Scholar]
- 40. Xu J., Schwarz W. H., Käs J. A., Stossel T. P., Janmey P. A., Pollard T. D. (1998) Mechanical properties of actin filament networks depend on preparation, polymerization conditions, and storage of actin monomers. Biophys. J. 74, 2731–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Johnsson B., Löfås S., Lindquist G. (1991) Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Anal. Biochem. 198, 268–277 [DOI] [PubMed] [Google Scholar]
- 42. Malchiodi E. L., Eisenstein E., Fields B. A., Ohlendorf D. H., Schlievert P. M., Karjalainen K., Mariuzza R. A. (1995) Superantigen binding to a T cell receptor β chain of known three-dimensional structure. J. Exp. Med. 182, 1833–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fernández M. M., De Marzi M. C., Berguer P., Burzyn D., Langley R. J., Piazzon I., Mariuzza R. A., Malchiodi E. L. (2006) Binding of natural variants of staphylococcal superantigens SEG and SEI to TCR and MHC class II molecule. Mol. Immunol. 43, 927–938 [DOI] [PubMed] [Google Scholar]
- 44. Cooper J. A., Walker S. B., Pollard T. D. (1983) Pyrene actin: documentation of the validity of a sensitive assay for actin polymerization. J. Muscle Res. Cell Motil. 4, 253–262 [DOI] [PubMed] [Google Scholar]
- 45. Richards D. E., Rega A. F., Garrahan P. J. (1978) Two classes of site for ATP in the Ca2+-ATPase from human red cell membranes. Biochim. Biophys. Acta 511, 194–201 [DOI] [PubMed] [Google Scholar]
- 46. Webb M. R. (1992) A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc. Natl. Acad. Sci. U.S.A. 89, 4884–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Echarte M. M., Levi V., Villamil A. M., Rossi R. C., Rossi J. P. (2001) Quantitation of plasma membrane calcium pump phosphorylated intermediates by electrophoresis. Anal. Biochem. 289, 267–273 [DOI] [PubMed] [Google Scholar]
- 48. Kratje R. B., Garrahan P. J., Rega A. F. (1983) The effects of alkali metal ions on active Ca2+ transport in reconstituted ghosts from human red cells. Biochim. Biophys. Acta 731, 40–46 [DOI] [PubMed] [Google Scholar]
- 49. Mangialavori I., Giraldo A. M., Buslje C. M., Gomes M. F., Caride A. J., Rossi J. P. (2009) A new conformation in sarcoplasmic reticulum calcium pump and plasma membrane Ca2+ pumps revealed by a photoactivatable phospholipidic probe. J. Biol. Chem. 284, 4823–4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kosk-Kosicka D., Bzdega T., Wawrzynow A. (1989) Fluorescence energy transfer studies of purified erythrocyte Ca2+-ATPase. Ca2+-regulated activation by oligomerization. J. Biol. Chem. 264, 19495–19499 [PubMed] [Google Scholar]
- 51. Sackett D. L., Kosk-Kosicka D. (1996) The active species of plasma membrane Ca2+-ATPase are a dimer and a monomer-calmodulin complex. J. Biol. Chem. 271, 9987–9991 [DOI] [PubMed] [Google Scholar]
- 52. Gershman L. C., Selden L. A., Estes J. E. (1991) High affinity divalent cation exchange on actin. Association rate measurements support the simple competitive model. J. Biol. Chem. 266, 76–82 [PubMed] [Google Scholar]
- 53. Navratilova I., Dioszegi M., Myszka D. G. (2006) Analyzing ligand and small molecule binding activity of solubilized GPCRs using biosensor technology. Anal. Biochem. 355, 132–139 [DOI] [PubMed] [Google Scholar]
- 54. Penniston J. T., Enyedi A. (1998) Modulation of the plasma membrane Ca2+ pump. J. Membr. Biol. 165, 101–109 [DOI] [PubMed] [Google Scholar]
- 55. Strehler E. E., Treiman M. (2004) Calcium pumps of plasma membrane and cell interior. Curr. Mol. Med. 4, 323–335 [DOI] [PubMed] [Google Scholar]
- 56. Schumacher M. A., Rivard A. F., Bächinger H. P., Adelman J. P. (2001) Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 410, 1120–1124 [DOI] [PubMed] [Google Scholar]
- 57. Sarkadi B., Enyedi A., Földes-Papp Z., Gárdos G. (1986) Molecular characterization of the in situ red cell membrane calcium pump by limited proteolysis. J. Biol. Chem. 261, 9552–9557 [PubMed] [Google Scholar]
- 58. Filoteo A. G., Enyedi A., Penniston J. T. (1992) The lipid-binding peptide from the plasma membrane Ca2+ pump binds calmodulin, and the primary calmodulin-binding domain interacts with lipid. J. Biol. Chem. 267, 11800–11805 [PubMed] [Google Scholar]
- 59. Kosk-Kosicka D., Bzdega T. (1990) Effects of calmodulin on erythrocyte Ca2+-ATPase activation and oligomerization. Biochemistry 29, 3772–3777 [DOI] [PubMed] [Google Scholar]
- 60. Kosk-Kosicka D., Bzdega T., Johnson J. D. (1990) Fluorescence studies on calmodulin binding to erythrocyte Ca2+-ATPase in different oligomerization states. Biochemistry 29, 1875–1879 [DOI] [PubMed] [Google Scholar]
- 61. Vorherr T., Kessler T., Hofmann F., Carafoli E. (1991) The calmodulin-binding domain mediates the self-association of the plasma membrane Ca2+ pump. J. Biol. Chem. 266, 22–27 [PubMed] [Google Scholar]
- 62. Vorherr T., James P., Krebs J., Enyedi A., McCormick D. J., Penniston J. T., Carafoli E. (1990) Interaction of calmodulin with the calmodulin binding domain of the plasma membrane Ca2+ pump. Biochemistry 29, 355–365 [DOI] [PubMed] [Google Scholar]
- 63. Pollard T. D., Weeds A. G. (1984) The rate constant for ATP hydrolysis by polymerized actin. FEBS Lett. 170, 94–98 [DOI] [PubMed] [Google Scholar]
- 64. Korn E. D., Carlier M. F., Pantaloni D. (1987) Actin polymerization and ATP hydrolysis. Science 238, 638–644 [DOI] [PubMed] [Google Scholar]
- 65. Carlier M. F. (1991) Nucleotide hydrolysis in cytoskeletal assembly. Curr. Opin. Cell Biol. 3, 12–17 [DOI] [PubMed] [Google Scholar]
- 66. Carlier M. F., Pantaloni D., Korn E. D. (1986) Fluorescence measurements of the binding of cations to high-affinity and low-affinity sites on ATP-G-actin. J. Biol. Chem. 261, 10785–10792 [PubMed] [Google Scholar]
- 67. Vorobiev S., Strokopytov B., Drubin D. G., Frieden C., Ono S., Condeelis J., Rubenstein P. A., Almo S. C. (2003) The structure of nonvertebrate actin: implications for the ATP hydrolytic mechanism. Proc. Natl. Acad. Sci. U.S.A. 100, 5760–5765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Katz S., Blostein R. (1975) Ca2+-stimulated membrane phosphorylation and ATPase activity of the human erythrocyte. Biochim. Biophys. Acta 389, 314–324 [DOI] [PubMed] [Google Scholar]
- 69. Lichtner R., Wolf H. U. (1980) Characterization of the phosphorylated intermediate of the isolated high-affinity (Ca2+-Mg2+)-ATPase of human erythrocyte membranes. Biochim. Biophys. Acta 598, 486–493 [DOI] [PubMed] [Google Scholar]
- 70. Rega A. F., Garrahan P. J. (1986) The Ca2+ Pump of Plasma Membranes, pp. 77–90, CRC Press, Boca Raton, FL [Google Scholar]
- 71. Bredeston L. M., Rega A. F. (2002) Pre-steady-state phosphorylation and dephosphorylation of detergent-purified plasma-membrane Ca2+-ATPase. Biochem. J. 361, 355–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mangialavori I., Ferreira-Gomes M., Pignataro M. F., Strehler E. E., Rossi J. P. (2010) Determination of the dissociation constants for Ca2+ and calmodulin from the plasma membrane Ca2+ pump by a lipid probe that senses membrane domain changes. J. Biol. Chem. 285, 123–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mangialavori I., Villamil-Giraldo A. M., Pignataro M. F., Ferreira-Gomes M., Caride A. J., Rossi J. P. (2011) Plasma membrane calcium pump (PMCA) differential exposure of hydrophobic domains after calmodulin and phosphatidic acid activation. J. Biol. Chem. 286, 18397–18404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mangialavori I. C., Caride A. J., Rossi R. C., Rossi J. P., Strehler E. E. (2011) Diving into the lipid bilayer to investigate the transmembrane organization and conformational state transitions of P-type ion ATPases. Curr. Chem. Biol. 5, 118–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kosk-Kosicka D., Inesi G. (1985) Cooperative calcium binding and calmodulin regulation in the calcium-dependent adenosine triphosphatase purified from the erythrocyte membrane. FEBS Lett. 189, 67–71 [DOI] [PubMed] [Google Scholar]
- 76. Fujimoto T. (1993) Calcium pump of the plasma membrane is localized in caveolae. J. Cell Biol. 120, 1147–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pike L. J., Casey L. (1996) Localization and turnover of phosphatidylinositol 4,5-bisphosphate in caveolin-enriched membrane domains. J. Biol. Chem. 271, 26453–26456 [DOI] [PubMed] [Google Scholar]
- 78. Shaul P. W., Smart E. J., Robinson L. J., German Z., Yuhanna I. S., Ying Y., Anderson R. G., Michel T. (1996) Acylation targets endothelial nitric-oxide synthase to plasmalemmal caveolae. J. Biol. Chem. 271, 6518–6522 [DOI] [PubMed] [Google Scholar]
- 79. Head B. P., Patel H. H., Roth D. M., Murray F., Swaney J. S., Niesman I. R., Farquhar M. G., Insel P. A. (2006) Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J. Biol. Chem. 281, 26391–26399 [DOI] [PubMed] [Google Scholar]
- 80. Carafoli E. (1987) Intracellular calcium homeostasis. Annu. Rev. Biochem. 56, 395–433 [DOI] [PubMed] [Google Scholar]
- 81. Roman I., Figys J., Steurs G., Zizi M. (2006) Direct measurement of VDAC-actin interaction by surface plasmon resonance. Biochim. Biophys. Acta 1758, 479–486 [DOI] [PubMed] [Google Scholar]
- 82. Noda Y., Horikawa S., Katayama Y., Sasaki S. (2004) Water channel aquaporin-2 directly binds to actin. Biochem. Biophys. Res. Commun. 322, 740–745 [DOI] [PubMed] [Google Scholar]
- 83. Wegner A., Engel J. (1975) Kinetics of the cooperative association of actin to actin filaments. Biophys. Chem. 3, 215–225 [DOI] [PubMed] [Google Scholar]
- 84. Tobacman L. S., Korn E. D. (1983) The kinetics of actin nucleation and polymerization. J. Biol. Chem. 258, 3207–3214 [PubMed] [Google Scholar]
- 85. Stossel T. P. (1989) From signal to pseudopod. How cells control cytoplasmic actin assembly. J. Biol. Chem. 264, 18261–18264 [PubMed] [Google Scholar]
- 86. Yin H. L., Hartwig J. H., Maruyama K., Stossel T. P. (1981) Ca2+ control of actin filament length. Effects of macrophage gelsolin on actin polymerization. J. Biol. Chem. 256, 9693–9697 [PubMed] [Google Scholar]
- 87. Bretscher A., Weber K. (1980) Villin is a major protein of the microvillus cytoskeleton which binds both G and F actin in a calcium-dependent manner. Cell 20, 839–847 [DOI] [PubMed] [Google Scholar]
- 88. Kueh H. Y., Charras G. T., Mitchison T. J., Brieher W. M. (2008) Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J. Cell Biol. 182, 341–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yonezawa N., Nishida E., Ohba M., Seki M., Kumagai H., Sakai H. (1989) An actin-interacting heptapeptide in the cofilin sequence. Eur. J. Biochem. 183, 235–238 [DOI] [PubMed] [Google Scholar]
- 90. Yamamoto K. (1991) Identification of the site important for the actin-activated MgATPase activity of myosin subfragment-1. J. Mol. Biol. 217, 229–233 [DOI] [PubMed] [Google Scholar]
- 91. Safer D., Elzinga M., Nachmias V. T. (1991) Thymosin β4 and Fx, an actin-sequestering peptide, are indistinguishable. J. Biol. Chem. 266, 4029–4032 [PubMed] [Google Scholar]
- 92. Friederich E., Vancompernolle K., Huet C., Goethals M., Finidori J., Vandekerckhove J., Louvard D. (1992) An actin-binding site containing a conserved motif of charged amino acid residues is essential for the morphogenic effect of villin. Cell 70, 81–92 [DOI] [PubMed] [Google Scholar]
- 93. Deleted in proof. [Google Scholar]
- 94. Kusano K., Abe H., Obinata T. (1999) Detection of a sequence involved in actin-binding and phosphoinositide-binding in the N-terminal side of cofilin. Mol. Cell. Biochem. 190, 133–141 [PubMed] [Google Scholar]
- 95. Goldschmidt-Clermont P. J., Machesky L. M., Baldassare J. J., Pollard T. D. (1990) The actin-binding protein profilin binds to PIP2 and inhibits its hydrolysis by phospholipase C. Science 247, 1575–1578 [DOI] [PubMed] [Google Scholar]
- 96. Xian W., Vegners R., Janmey P. A., Braunlin W. H. (1995) Spectroscopic studies of a phosphoinositide-binding peptide from gelsolin: behavior in solutions of mixed solvent and anionic micelles. Biophys. J. 69, 2695–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]