

Abstract
The sulfate-reducing bacterium Desulfococcus multivorans uses various aromatic compounds as sources of cell carbon and energy. In this work, we studied the initial steps in the aromatic metabolism of this strictly anaerobic model organism. An ATP-dependent benzoate coenzyme A (CoA) ligase (AMP plus PPi forming) composed of a single 59-kDa subunit was purified from extracts of cells grown on benzoate. Specific activity was highest with benzoate and some benzoate derivatives, whereas aliphatic carboxylic acids were virtually unconverted. The N-terminal amino acid sequence showed high similarities with benzoate CoA ligases from Thauera aromatica and Azoarcus evansii. When cultivated on benzoate, cells strictly required selenium and molybdenum, whereas growth on nonaromatic compounds, such as cyclohexanecarboxylate or lactate, did not depend on the presence of the two trace elements. The growth rate on benzoate was half maximal with 1 nM selenite present in the growth medium. In molybdenum- and/or selenium-depleted cultures, growth on benzoate could be induced by addition of the missing trace elements. In extracts of cells grown on benzoate in the presence of [75Se]selenite, three radioactively labeled proteins with molecular masses of ∼100, 30, and 27 kDa were detected by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis. The 100- and 30-kDa selenoproteins were 5- to 10-fold induced in cells grown on benzoate compared to cells grown on lactate. These results suggest that the dearomatization process in D. multivorans is not catalyzed by the ATP-dependent Fe-S enzyme benzoyl-CoA reductase as in facultative anaerobes but rather involves unknown molybdenum- and selenocysteine-containing proteins.
In recent years, a growing number of anaerobic bacteria from nearly all physiological groups (denitrifying, sulfate-reducing, iron-reducing, phototrophic, and fermenting bacteria) have been found to metabolize a multitude of aromatic compounds as their sole sources of energy and cell carbon in the absence of oxygen. Thus, many novel enzyme reactions have been discovered in the aromatic metabolisms of anaerobic bacteria which provide alternatives to oxygen-dependent activation and dearomatization processes in aerobic metabolism (for reviews, see references 7, 20, 24, 25, and 36). Both aerobic and anaerobic bacteria convert aromatic compounds into a few central intermediates, among which benzoyl-coenzyme A (CoA) plays an important role in anaerobic metabolism. Benzoyl-CoA is formed from benzoate by a specific CoA ligase. In Thauera aromatica and other facultative anaerobes, benzoyl-CoA is then reductively dearomatized by benzoyl-CoA reductase to a nonaromatic cyclic diene. The product is further metabolized by a modified β oxidation in the so-called benzoyl-CoA pathway (24). Overall, benzoyl-CoA is oxidized to three acetyl-CoA and one CO2.
So far, benzoyl-CoA reductase activity has been detected in cell extracts of the denitrifying bacterium T. aromatica (30), the closely related Azoarcus evansii (15) (both members of the β subdivison of Proteobacteria), and the phototrophic Rhodopseudomonas palustris (α subdivision of Proteobacteria) (30), all facultative anaerobic organisms. The primary sequences of benzoyl-CoA reductases from several facultative anaerobes show a high level of similarity to each other (14, 16). This extremely oxygen-sensitive enzyme has been purified from extracts of T. aromatica (6). The enzyme contains three [4Fe-4S] clusters (8) and couples the two-electron reduction of the aromatic moiety of benzoyl-CoA to a stoichiometric hydrolysis of two ATP (10). The product of the two-electron reduction is cyclohexa-1,5-diene-1-carbonyl-CoA (5).
Widdel (44) was the first to isolate strictly anaerobic bacteria capable of utilizing aromatic compounds and sulfate as their sole energy and carbon sources. He found that some sulfate-reducing bacteria, including Desulfococcus multivorans, require the trace elements molybdenum and selenium for growth on benzoate. To date, utilization of aromatic compounds has been demonstrated for several sulfate-reducing bacteria from different phylogenetic groups, such as D. multivorans, Desulfosarcina variabilis, Desulfonema magnum, and Desulfobacula toluolica, as well as several Desulfobacterium and Desulfotomaculum strains (34). However, little is known about the metabolism of aromatic compounds in strictly anaerobic bacteria. The free-energy change of complete benzoate oxidation with sulfate (−203 kJ mol−1) is 1 order of magnitude lower than with nitrate (∼−3,000 kJ mol−1). Complete oxidation of benzoic acid by sulfate-reducing bacteria proceeds via the following equation: C7H6O2 + 4 H2O + 3.75 SO42−→7 HCO3− + 3.75 HS− + 3.25 H+ (ΔG° = −203 kJ mol−1) (42).
In strictly anaerobic syntrophic consortia utilizing aromatic compounds, the energy situation is even less favorable (42). Considering these energy limitations, it was surprising that an AMP-forming benzoate CoA ligase activity was found in syntrophic consortia (1, 18, 39) and in a sulfate-reducing bacterium (37). It is unknown whether the energy-consuming activations represent a general feature of aromatic acid metabolism in obligate anaerobic bacteria. Even if aromatic metabolism could proceed close to the thermodynamic equilibrium (23), the presence of an additional ATP-consuming benzoyl-CoA reductase, as in T. aromatica (consuming two ATP/benzoyl-CoA reduced) is questionable for strictly anaerobic bacteria.
Schöcke and Schink (39) provided evidence that in the syntrophic Syntrophus gentiana a two-electron reduction of benzoyl-CoA, as reported for T. aromatica, is thermodynamically impossible; they suggested that at least a four-electron reduction of the aromatic ring to a cyclohexene-1-carbonyl-CoA derivative would be required. Further evidence for this notion was obtained from experiments with Syntrophus aciditrophicus strain SB. This fermenting bacterium formed [13C]cyclohexanecarboxylate from [13C]benzoate, indicating a six-electron reduction of the benzene ring (17). Very recently, it has been demonstrated that this Syntrophus strain was even able to use benzoate as a sole carbon and energy source in the absence of an H2-consuming coculture (18).
In this work, the initial enzymatic step of benzoate metabolism was studied in the strictly anaerobic model organism D. multivorans (δ group of Proteobacteria). Because no molybdenum or selenium enzymes are known to be involved in the anaerobic aromatic metabolism in denitrifying bacteria, the dependence of D. multivorans on these trace elements during growth on benzoate was reinvestigated. The results provide evidence that the initial step in benzoate metabolism is catalyzed by an AMP-forming benzoate CoA ligase and is thus identical to those in facultative anaerobes. In contrast, the strict dependence on the trace elements selenium and molybdenum and the induction of selenocysteine-containing proteins suggest that the subsequent step of benzoyl-CoA dearomatization in D. multivorans differs from that in facultative anaerobic bacteria.
MATERIALS AND METHODS
Growth of D. multivorans and preparation of cell extracts.
D. multivorans (DSM 2059) was grown in a bicarbonate-buffered (pH 7 to 7.3) mineral salt medium under strictly anaerobic conditions in gas-tight stoppered glass bottles at 30°C (44). Accumulated H2S was removed by degassing with N2-CO2 (80:20 [vol/vol]) every 2 days, which also lowered the increasing pH to the initial value. Cultivation on a larger scale was carried out in 50-liter plastic bottles. Cells were harvested by centrifugation (20,000 × g) and stored in liquid nitrogen. Benzoate (5 mM), cyclohexanecarboxylate (5 mM), or lactate (20 mM) was added as an electron donor and carbon source from anaerobically prepared stock solutions; sodium sulfate (30 mM) served as an electron acceptor. The reducing conditions were adjusted by addition of sodium sulfide, using resazurin as a redox indicator. Growth was monitored by absorbance measurement at 578 nm (1-cm light path). At an optical density at 578 nm (OD578) of >0.5, the medium was resupplemented with the electron donor and acceptor. Trace elements were added from anaerobically prepared sodium selenite (20 μM) or sodium molybdate (150 μM) stock solution. If the dependence on selenium and molybdenum was tested, both trace elements were omitted from the medium. Unless otherwise stated, preparation of cell extracts was as follows. Frozen cells were suspended in 20 mM triethanolamine hydrochloride- KOH buffer, pH 7.8 (1 g of cells in 1.5 ml of buffer), 4 mM MgCl2, 10% glycerol, 1 mM dithioerythritol (DTE), and 0.1 mg of DNase I. Cell lysates were obtained by passage through a French press cell at 137 MPa. After centrifugation at 100,000 × g (1 h; 4°C), the supernatant was used for further studies.
Assay of benzoate CoA ligase.
Benzoate CoA ligase activity, as well as the stoichiometry of AMP formation, substrate specificity, and Km values for benzoate, ATP, and CoA, were determined at 37°C using a coupled continuous spectrophotometric assay, as described previously (47). Briefly, the formation of AMP (reaction 1) was coupled enzymatically to myokinase (reaction 2), pyruvate kinase (reaction 3), and lactate dehydrogenase (reaction 4); oxidation of NADH was monitored spectrophotometrically at a λ of 365 nm (ɛNADH = 3.4 × 103 M−1 cm−1):
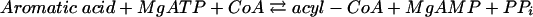 |
(1) |
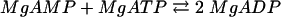 |
(2) |
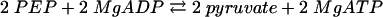 |
(3) |
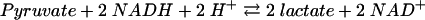 |
(4) |
The assay mixture contained 100 mM Tris-HCl, pH 7.8, 10 mM MgCl2, 2 mM ATP, 0.45 mM NADH, 2 mM phosphoenolpyruvate, 0.4 mM CoA, and 0.5 mM benzoate; the activities of the enzymes were 8 nkat myokinase, 8 nkat pyruvate kinase, and 20 nkat lactate dehydrogenase (each per 10-ml enzyme assay).
For determination of the substrate preference of purified benzoate CoA ligase, the reaction was started by the addition of various carboxylic acids (2 to 4 mM [see Table 3]). For the determination of the apparent Km values of benzoate, MgATP, and CoA, the reaction was started with various concentrations of the respective substrates (0.2 to 10 times the Km value). The other components of the assay were present at the following concentrations: 0.5 mM benzoate, 0.5 mM CoA, 2 mM ATP, and 10 mM MgCl2. The Km values were determined by fitting the data to Michaelis-Menten curves using the Prism GraphPad software package (Graphpad Software Inc., San Diego, Calif.). Km values were derived from three different measurements, with a minimum of four different substrate concentrations. The stoichiometry between benzoate consumption and NADH oxidation was determined by adding benzoate at limiting concentrations; the amount of NADH consumed was estimated from ΔA365. The protein concentration was determined by the method of Bradford (13) using bovine serum albumin as a standard. Purity control was by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (31).
TABLE 3.
Substrate preference of benzoate CoA ligase from D. multivorans
| Substrate (mM) | Relative activity (%)a |
|---|---|
| Benzoate (0.5) | 100 |
| 2-Fluorobenzoate (2) | 100 |
| 3-Fluorobenzoate (2) | 104 |
| 4-Fluorobenzoate (2) | 120 |
| 2-Chlorobenzoate (2) | <1 |
| 3-Chlorobenzoate (2) | <1 |
| 4-Chlorobenzoate (2) | <1 |
| 2-Methylbenzoate (2) | <1 |
| 3-Methylbenzoate (2) | 2 |
| 4-Methylbenzoate (2) | <1 |
| 2-Hydroxybenzoate (4) | <1 |
| 3-Hydroxybenzoate (4) | 36 |
| 4-Hydroxybenzoate (2) | <1 |
| 2-Aminobenzoate (4) | 34 |
| 3-Aminobenzoate (4) | 28 |
| 4-Aminobenzoate (4) | 60 |
| Phenylacetate (4) | 2 |
| Cyclohex-1-enecarboxylate (2) | 56 |
| Cyclohex-3-enecarboxylate (2) | 84 |
| Cyclohexanecarboxylate (2) | 6 |
| Glutarate (2) | <1 |
| Succinate (2) | <1 |
| Acetate (2) | <1 |
Mean value of triple determinations; standard deviations were <10%. One hundred percent corresponds to 8.2 μmol min−1 mg−1.
Purification of benzoate CoA ligase.
Purification of benzoate CoA ligase was carried out under aerobic conditions and started from 10 g (wet mass) of cells; the preparation of cell extract and ultracentrifugation were as described above.
Ammonium sulfate precipitation and dialysis.
The soluble protein fraction obtained after ultracentrifugation was precipitated with a saturated ammonium sulfate solution, pH 7.8, containing 1 mM Na2EDTA to 33% saturation. After centrifugation (12,000 × g for 15 min), the ammonium sulfate in the supernatant was increased to 55% saturation. The resulting precipitate was dissolved in 7.6 ml of basal buffer (10 mM Tris-HCl, pH 7.8, 2 mM MgCl2, 2 mM DTE) and dialyzed twice for 12 h each time against basal buffer (exclusion mass, 12 to 14 kDa; Medicell International Ltd., London, England).
DEAE-Sepharose chromatography.
The dialyzed protein solution was applied at a flow rate of 1 ml min−1 to a DEAE-Sepharose column (Fast Flow; diameter, 10 mm; volume, 10 ml; Amersham Biosciences), which had been equilibrated with basal buffer. The column was washed with 30 ml of basal buffer and then with 20 ml of 50 mM KCl in basal buffer. The ligase was eluted with a linear gradient of 50 to 200 mM KCl in basal buffer (100 ml). Fractions (5 ml) were collected and tested for benzoate CoA ligase activity.
Q-Sepharose chromatography.
Pooled fractions with benzoate CoA ligase activity were diluted with an equal volume of basal buffer and applied to a fast protein liquid chromatography Hi Load Q-Sepharose column (diameter, 16 mm; volume, 14 ml; Amersham Biosciences) at a flow rate of 2 ml min−1. The column was equilibrated with basal buffer containing 150 mM KCl and was subsequently washed with 20 ml of the same buffer. The ligase was eluted in a linear gradient from 150 to 400 mM KCl in basal buffer (120 ml); the CoA ligase activity eluted at ∼170 mM KCl.
Affinity chromatography.
The protein fraction containing benzoate CoA ligase activity obtained by Q-Sepharose chromatography was diluted with an equal volume of basal buffer and applied at a flow rate of 1 ml min−1 to a Reactive Green cross-linked agarose column (Reactive Green 19-agarose; diameter, 15 mm; volume, 10 ml; Sigma-Aldrich), which had been equilibrated with 50 ml of basal buffer (1 ml min−1). After being washed with 40 ml of basal buffer, the ligase was eluted in a linear gradient of 0.1 to 1 M KCl in basal buffer (120 ml); the benzoate CoA ligase activity eluted at ∼650 mM KCl.
Mono Q chromatography.
The benzoate CoA ligase activity-containing pool obtained from affinity chromatography was concentrated to ∼2 ml by ultrafiltration (exclusion mass, 30 kDa; Amicon, Beverly, Mass.) and dialyzed twice against 1 liter of basal buffer (see above). The enzyme was then applied to a Mono Q anion-exchange column (volume, 1 ml; diameter, 0.5 cm; Amersham Biosciences), which had been equilibrated with basal buffer (flow rate, 1 ml min−1). The column was washed with 5 ml of 150 mM KCl in basal buffer; benzoate CoA ligase was eluted in a 30-ml linear gradient from 150 to 300 ml of KCl. The ligase activity eluted at ∼180 mM KCl. The ligase was again concentrated to 1.5 ml and frozen after addition of 20% glycerol (final volume). In this form, benzoate CoA ligase was stored at −20°C for several weeks without significant loss of activity.
75Se-labeling studies.
An exponentially growing D. multivorans culture supplemented with 20 nM sodium selenite was diluted 1:10 in fresh medium supplemented with 1.2 μM sodium [75Se]selenite (6.4 kBq; addition was made from a 535-MBq mmol−1 stock solution). Benzoate, lactate, or cyclohexanecarboxylate (5 mM each) served as a growth substrate. To compensate for the increasing pH, aliquots of sodium hydrogen sulfate were added to the medium (1 to 5 mM). After growth for 6 days at 30°C (OD578, 0.5 to 0.6), cells were harvested by centrifugation. After being washed with buffer containing 50 mM HEPES, pH 7.0, 10 mM KCl, and 5 mM MgCl2, the cells were disrupted by ultrasonification in the presence of DNase I (0.5 μg ml−1). After centrifugation, the supernatant was subjected to SDS-PAGE analysis (31). In control experiments, the supernatant was incubated with 2 μg of RNase A ml−1 or 1.5 μg of proteinase K for 30 min at 37°C prior to SDS-PAGE. Proteins were stained with Coomassie brilliant blue (46). Phosphorylase b (94 kDa), bovine serum albumin (67 kDa), ovalbumin (43 kDa), carbonic acid anhydrase (30 kDa), and lysozyme (14 kDa) were used as molecular mass standards. Labeled macromolecules were visualized by phosphorimaging of dried gels.
Two-dimensional gel electrophoresis.
Two-dimensional gel electrophoresis (Immobiline DryStrip gels, 18 cm; linear pH gradient from pH 3 to 10; Amersham Pharmacia Biotech) was performed using an IPGphor isoelectric focusing system (Amersham Biosciences) in the first dimension, according to the manufacturer's protocol. Before application to the first dimension, cell extracts were dialyzed against 5 mM Tris-HCl, pH 7.8 (4°C; 3 h). Protein (120 to 400 μg per separation) was applied to the IPG strips. Separation in the second dimension was by SDS-10% PAGE (31). For detection of 75Se-labeled proteins on two-dimensional gels, D. multivorans was cultivated in the presence of 1.2 μM [75Se]selenite as described above. In these experiments, a higher specific radioactivity (5.3 MBq/10-ml culture volume) was used than for one-dimensional gels.
N-terminal amino acid sequencing.
Purified benzoate CoA ligase was blotted from SDS-polyacrylamide gels to a polyvinylidene difluoride membrane. N-terminal amino acid sequencing was carried out by Michael Kiess (TobLab Co., Munich, Germany), applying a modified Edman degradation protocol (27). Phenylthiohydantoin derivatives of the amino acids were detected by a Procise 492 amino acid sequenator (Applied Biosystems, Weiterstedt, Germany). Amino acid sequence comparisons were performed using the Blast search tool (http://www.ncbi.nlm.nih.gov/BLAST/) with an expected value threshold of 10,000.
RESULTS
Purification of benzoate CoA ligase.
Benzoate CoA ligase was purified from extracts of D. multivorans grown on benzoate and sulfate. Purification followed the procedure established for the purification of benzoate CoA ligases from T. aromatica (41) and A. evansii (19), followed by Mono Q chromatography, to obtain a pure enzyme preparation. Due to high NADH-oxidizing background activities, benzoate CoA ligase could not be measured accurately in the 100,000 × g supernatant and in the resuspended pellet after 55% ammonium sulfate precipitation. Therefore, enrichment of the benzoate-dependent NADH oxidation activity in the coupled spectrophotometric assay is reported relative to the enzyme fraction obtained after DEAE-Sepharose chromatography (Table 1). The enrichment of benzoate CoA ligase was 6.8-fold, with a yield of 34%. The preparation yielded a single protein species with a molecular mass of 59 kDa, as judged from SDS-PAGE analysis (Fig. 1); the purity was estimated to be >95%.
TABLE 1.
Purification of benzoate CoA ligase from D. multivoransa
| Purification step | Amt of protein (mg) | Total activity (U) | Sp act (U mg−1) | Enrich- ment (n-fold) | Recovery (%) |
|---|---|---|---|---|---|
| 100,000 × g supernatant | 470 | ND | |||
| (NH4)2SO4 precipitation | 125 | ND | |||
| DEAE-Sepharose | 6.3 | 7.8 | 1.2 | 1 | 94 |
| Q-Sepharose | 4.5 | 8.3 | 1.8 | 1.5 | 100 |
| Reactive Green | 2.4 | 6.9 | 2.9 | 2.4 | 82 |
| Mono Q | 0.3 | 2.4 | 8.2 | 6.8 | 34 |
The enzyme was purified from 10 g (wet mass) of cells of D. multivorans grown on benzoate under sulfate-reducing conditions. One unit corresponds to 1 μmol of benzoyl-CoA formed per min. Due to high background reaction, benzoate CoA ligase could not be measured after the first two purification steps (indicated as ND [not determined]).
FIG. 1.

SDS-polyacrylamide gel of purified benzoate CoA ligase. Lane 1, molecular mass standard; lane 2, benzoate CoA ligase (3 μg) obtained after the last purification step (Mono Q chromatography). Staining was with Coomassie blue.
Properties of benzoate CoA ligase.
To assess the identity of the 59-kDa protein, its N-terminal amino acid sequence was determined. It showed significant similarity to N-terminal amino acid sequences of benzoate CoA ligases from T. aromatica and A. evansii; all of the enzymes consist of single subunits with similar molecular masses (Table 2). Notably, the N-terminal sequence showed no similarity to any nonaromatic carboxylic acid CoA ligase. The specific activity of purified benzoate CoA ligase was 8.2 μmol mg−1 min−1, which is similar to that of the enzyme from T. aromatica (13.3 μmol mg−1 min−1 [41]). For testing the substrate preference, benzoate was replaced with other aromatic and nonaromatic carboxylic acids in the spectrophotometric assay (Table 3). The activity was highest with benzoate (considered 100% activity) and fluorinated analogues (100 to 120%). Considerable activity was also obtained with aminobenzoate derivatives and 3-hydroxybenzoate, albeit only at concentrations higher than 2 mM, indicating poor binding of these substrates. Partially reduced alicyclic carboxylic acids also served as substrates at high rates, whereas aliphatic carboxylic acids were virtually unconverted (<1% of the rate obtained with benzoate). The apparent Km values (mean value ± standard deviation) for benzoate (25 ± 5 μM), CoA (58 ± 7 μM), and ATP (145 ± 33 μM) are in the physiological range. The substrate preference pattern, as well as the low Km values, strongly suggests that this CoA ligase activity is specific for benzoate and some analogues.
TABLE 2.
Similarities of the N-terminal amino acid sequences of purified benzoate-CoA ligase from D. multivorans and other characterized benzoate CoA ligases
The product of ATP hydrolysis was identified using the coupled spectrophotometric assay by adding limiting amounts of benzoate (Table 4). The ratio of benzoate added to NAD+ formed was between 2.0 and 2.2. This value indicates that AMP plus PPi, rather than ADP plus Pi, is formed by the ligase, given that one NAD+-ADP is formed in the coupled assay. Note that one AMP is converted to two ADP by myokinase (with ATP) in the coupled assay (see Materials and Methods).
TABLE 4.
Stoichiometry of benzoate CoA ligase reaction as determined in the coupled spectrophotometric assaya
| NAD+ formed (μmol) | Benzoate consumed (μmol) | NAD+/benzoate molar ratio |
|---|---|---|
| 8.2 | 4 | 2.1 |
| 15.9 | 8 | 2.0 |
| 43.5 | 20 | 2.2 |
| 81.5 | 40 | 2.0 |
Benzoate-dependent hydrolysis of ATP is coupled to the oxidation of NADH.
Selenium- and molybdenum-dependent growth.
Since benzoate CoA ligase (AMP forming) catalyzes an energy-consuming reaction in benzoate catabolism in D. multivorans, the presence of an ATP-dependent T. aromatica-type benzoyl-CoA reductase (iron-sulfur enzyme) was questionable. Preliminary studies by Widdel (44) indicated that molybdenum and selenium play a role in the growth of some sulfate reducers on benzoate. To test the presence of molybdo- and selenoenzymes involved in benzoate metabolism, the dependence on both trace elements was quantitatively reinvestigated. The doubling time of D. multivorans was determined within the first 6 to 8 days of growth (up to an OD578 of 0.3), as growth at an OD578 of >0.4 was no longer exponential, probably due to accumulating products (e.g., hydrogen sulfide). Growth of D. multivorans on lactate or cyclohexanecarboxylate showed no significant molybdate (150 nM) or selenite (20 nM) dependence (Fig. 2A and B). In contrast, growth on benzoate occurred only in the presence of both trace elements in the medium (Fig. 2C). In the absence of molybdate and/or selenite, growth of D. multivorans on benzoate could be induced with a short lag phase by adding the missing trace elements (Fig. 2D). The doubling times of D. multivorans on aromatic and nonaromatic substrates were similar (≤40 h).
FIG. 2.

Effects of Mo (150 nM) and Se (20 nM) on growth of D. multivorans with different energy and carbon sources. At an OD578 of >0.5, the growth rate leveled off, probably due to accumulating sulfide. (A to C) Growth on lactate (20 mM) (A), cyclohexanecarboxylate (B), and benzoate (C) (5 mM each). The media contained Mo (150 nM) and Se (20 nM) (•), Mo but no Se (▿), Se but no Mo (◊), and neither Se nor Mo (▪). (D) Growth on benzoate with Mo but no Se (▾) and with Se but no Mo (▴). The arrows indicate addition of the missing trace elements.
To test the effectiveness of selenium uptake and incorporation, a selenite-depleted D. multivorans culture was incubated with selenite concentrations varying from 0.01 nM to 100 μM (Fig. 3). The OD578 values reached after 8 days indicate that 10 nM selenite is optimal for growth. A concentration of 1 nM selenite supported growth to half the optimal OD578, whereas lower concentrations did not support growth; concentrations of ≥10 μM selenite inhibited growth.
FIG. 3.

Effects of selenite concentrations on growth of D. multivorans with benzoate. Growth was determined as the OD578 reached after 8 days.
Benzoate-induced 75Se-labeled proteins.
The molybdenum and selenium dependence of growth on benzoate suggested a requirement for molybdenum- and selenium-containing proteins in the benzoate metabolism of D. multivorans. To test this, we attempted to label these proteins radioactively. The low growth rate of D. multivorans allowed only 75Se-labeling studies (half-life of 75Se, 120 days; half-life of 99Mo, ∼60 h). Moreover, only selenium in the form of selenocysteine, and not molybdenum, is known to be covalently incorporated in proteins.
Figure 4 shows SDS-polyacrylamide gels and autoradiographs of dried gels after electrophoretic separation of 75Se-labeled cell extracts. When cells were grown on benzoate, three 75Se-labeled bands with molecular masses of ∼100, 30, and 27 kDa were detected (Fig. 4A, lane 1). Since the radioactivity was not released from the protein during sample preparation for PAGE (80°C heating), selenium is most probably present in the form of selenocysteine. The absence of any background labeling suggests specific incorporation of 75Se into these proteins. After pretreatment of cell extracts with proteinase K, all labeled bands disappeared on SDS-polyacrylamide gels, whereas RNase treatment had no effect (Fig. 4A, lanes 2 and 3), indicating that all labeled bands are selenoproteins. To examine differential expression of selenium-containing proteins, D. multivorans was grown with [75Se]selenite on either cyclohexanecarboxylate or lactate. The amounts of the 100-kDa selenoprotein were 5- to 10-fold lower in extracts of cells grown on lactate or cyclohexanecarboxylate than for benzoate-grown cells, and the 30-kDa selenoprotein was hardly detectable in cells grown on lactate or cyclohexanecarboxylate (Fig. 4B; only the results with benzoate and lactate are shown). The 27-kDa selenoprotein from lactate- or cyclohexanecarboxylate-grown cells had approximately half the intensity observed in benzoate-grown cells. The differential expression of the 100-kDa band was also detected on Coomassie-stained SDS-polyacrylamide gels (Fig. 4C). The results indicate that production of selenoproteins is induced during growth on benzoate compared to growth on lactate or cyclohexanecarboxylate.
FIG. 4.

SDS-polyacrylamide gel of cell extracts from D. multivorans grown on [75Se]selenite-containing mineral medium. (A and B) Autoradiographs of an SDS-10% polyacrylamide gel (A) and an SDS-12.5% polyacrylamide gel (B). (C) 100-kDa section of the Coomassie blue-stained SDS-12.5% polyacrylamide gel. Lanes 1 to 4 and 7, crude extract of cells grown on benzoate (lane 2, as lane 1, but after treatment of cell extract with proteinase K; lane 3, crude extract after treatment with RNase A); lanes 5 and 8, crude extract of cells grown on lactate; lane 6, 97-kDa molecular mass standard. The protein migration front in panel B is marked by an arrow. The arrow in panel C points to an ∼100-kDa protein band which is differentially expressed in cells grown on benzoate and lactate.
For further analysis of benzoate-induced 75Se-labeled proteins, extracts from cells grown on benzoate were subjected to two-dimensional SDS-PAGE analysis, with isoelectric focusing from pH 3 to 10 as the additional dimension (Fig. 5). As in the one-dimensional gels, three radioactively labeled proteins with masses of 27, 30, and ∼100 kDa were identified (Fig. 5B). However, only the 100-kDa 75Se-labeled protein could be visualized by Coomassie staining on two-dimensional gels (Fig. 5A). It could not be blotted in sufficient amounts for N-terminal amino acid sequence analysis. Therefore, the N-terminal amino acid sequences of the three 75Se-labeled protein bands could not be determined.
FIG. 5.

Two-dimensional SDS-PAGE of proteins from cell extracts of D. multivorans. (A) Coomassie blue-stained two-dimensional SDS-polyacrylamide gel of extracts from cells grown on benzoate. The rectangles mark the positions of the radioactively labeled protein bands. (B) Autoradiograph of a two-dimensional SDS-polyacrylamide gel of extracts from cells grown on benzoate and [75Se]selenite. The vertical dimension is an SDS-10% polyacrylamide gel; the horizontal dimension is isoelectric focusing using a pH gradient as indicated.
DISCUSSION
In this work, benzoate CoA ligase from D. multivorans was purified and characterized. The molecular and kinetic properties of the enzyme were highly similar to those of benzoate CoA ligases from denitrifying bacteria. This finding supports the notion that benzoyl-CoA is the common intermediate in benzoate metabolism in both facultative and obligate anaerobic bacteria. The formation of benzoyl-CoA by benzoate CoA ligase is coupled to the cleavage of at least one energy-rich phosphoanhydride linkage benzoate. Even if hydrolysis of the formed PPi would be coupled to energy conservation (e.g., by a membrane-bound proton translocating pyrophosphatase, as reported for S. gentianae (38), the question arises whether sulfate-reducing bacteria can afford a second ATP-dependent activation reaction for breaking the aromaticity of the benzene ring. In denitrifying bacteria, and probably in phototrophic bacteria, iron-sulfur proteins are involved in the dearomatization of benzoyl-CoA (including benzoyl-CoA reductase, the electron donor ferredoxin, and the ferredoxin-regenerating 2-oxoglutarate-ferredoxin oxidoreductase [7]). Therefore, the requirement for the trace elements molybdenum and selenium for growth on benzoate and the induction of selenocysteine-containing proteins suggest that key processes in the benzoate metabolism of D. multivorans (and probably other sulfate reducers) proceed via a different, probably ATP-independent mechanism.
Due to their low expression, the nature of the selenoproteins could not be identified by N-terminal sequencing. This finding suggests that the selenoproteins have high specific activities, as reported for several other selenocysteine-containing enzymes (2, 4). In addition, the growth rates were similar on benzoate and nonaromatic compounds (Fig. 2), indicating that the enzymatic steps involved in dearomatization of the benzene nucleus are not rate limiting for growth of D. multivorans. However, the possibility cannot be ruled out that in addition to selenocysteine-containing proteins D. multivorans contains enzymes with a noncovalently bound Se cofactor, which would escape detection by SDS-PAGE analysis. The 30-kDa selenoprotein exhibits the most significant induction during growth on benzoate. The only known bacterial selenoprotein with a similar molecular mass is selenophosphate synthetase (SelD; 35 ± 2 kDa); it catalyzes the formation of selenophosphate from ATP and selenide (45). However, SelD is involved in anabolic selenocysteine synthesis and is usually expressed only at very low levels. Thus, the 30-kDa protein might be a component of a novel type of bacterial selenoprotein with a specific function in aromatic metabolism.
The simultaneous dependence on molybdenum and selenium for growth on benzoate could be due to the presence of a protein belonging to the family of mononuclear molybdopterin enzymes, in which a selenocysteine of the polypeptide backbone is coordinated with the molybdenum atom at the active site (usually in a 90- to 100-kDa subunit). Characterized examples of such enzymes are involved in formate oxidation belonging to the dimethyl sulfoxide family of molybdenum enzymes, e.g., the formate dehydrogenases of Escherichia coli (FDH-H/N/O) (3, 11, 28) or strict anaerobes (22, 33) and the related tungsten- and selenocysteine-containing formyl-methanofuran dehydrogenases (43). However, a possible specific function of a molybdenum-selenocysteine enzyme in benzoate metabolism could be in benzene ring hydroxylation instead. Many molybdenum enzymes of the xanthine oxidase family hydroxylate heteroaromatic compounds at the carbon atom adjacent to the nitrogen atom (for recent reviews, see references 26 and 32). Among these, nicotinic acid hydroxylase (21) and some xanthine dehydrogenases (40) contain selenium, albeit not as selenocysteine but as an inorganic selenium cofactor. Notably, a putative hydroxylation of the homoaromatic ring of benzoyl-CoA can be considered the reverse of the molybdo-enzyme 4-hydroxybenzoyl-CoA reductase (dehydroxylating) reaction. This member of the xanthine oxidase family catalyzes the reductive removal of the phenolic hydroxyl group from 4-hydroxybenzoyl-CoA, yielding benzoyl-CoA and water, using reduced ferredoxin as an electron donor (9, 12). Reversibility of aromatic ring dehydroxylation could be accomplished with a high-potential electron acceptor (>−100 mV) (9). The adenosine phosphosulfate-sulfite redox couple is in this range; therefore, a putative ring hydroxylation is thermodynamically feasible in sulfate-reducing bacteria. Hydroxylation reactions could activate benzoyl-CoA for a subsequent reductive dearomatization due to the substantial weakening of the aromatic character of a dihydroxylated benzene ring, as in the case of meta-positioned hydroxyl groups. Resorcinol (29) or hydroxyhydroquinone (35), both carrying two meta-positioned hydroxyl groups, are reductively dearomatized by electron donors, such as reduced ferredoxin, without coupling to an exergonic process.
Taken together with the mechanistic and energetic considerations discussed above, the results indicate that novel molybdo- and selenoproteins are involved in benzoyl-CoA metabolism of the strictly anaerobic model organism D. multivorans. These enzymes may represent an alternative to the known ATP-dependent dearomatizing enzymes of facultative anaerobes.
Acknowledgments
We are grateful to C. Ebenau-Jehle for help with the cultivation of bacteria and with two-dimensional gel electrophoresis and to P. Schlörit for initial benzoate CoA ligase activity measurements. We thank A. Böck (Munich, Germany), G. Fuchs and H. Heider (Freiburg, Germany), and A. Guss (Urbana, Ill.) for helpful suggestions. We are grateful to J. Andreesen (Halle, Germany) for providing [75Se]selenite.
This work was funded by the Deutsche Forschungsgemeinschaft (BO 1565/4-1).
REFERENCES
- 1.Auburger, G., and J. Winter. 1992. Purification and characterization of benzoyl-CoA ligase from a syntrophic, benzoate-degrading, anaerobic mixed culture. Appl. Microbiol. Biotechnol. 37:789-795. [DOI] [PubMed] [Google Scholar]
- 2.Axley, M. J., A. Böck, and T. C., Stadtman. 1991. Catalytic properties of an Escherichia coli formate dehydrogenase mutant in which sulfur replaces selenium. Proc. Natl. Acad. Sci. USA 88:8450-8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benoit, S., H. Abaibou, and M. A. Mandrand-Berthelot. 1998. Topological analysis of the aerobic membrane-bound formate dehydrogenase of Escherichia coli. J. Bacteriol. 180:6625-6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berry, M. J., J. D. Kieffer, and P. R. Larsen. 1991. Evidence that cysteine, not selenocysteine, is in the catalytic site of type II iodothyronine deiodinase. Endocrinology 129:550-552. [DOI] [PubMed] [Google Scholar]
- 5.Boll, M., D. Laempe, W. Eisenreich, A. Bacher, T. Mittelberger, J. Heinze, and G. Fuchs. 2000. Non-aromatic products from anoxic conversion of benzoyl-CoA with benzoyl-CoA reductase and cyclohexa-1,5-diene-1-carbonyl-CoA hydratase. J. Biol. Chem. 275:21889-21895. [DOI] [PubMed] [Google Scholar]
- 6.Boll, M., and G. Fuchs. 1995. Benzoyl-CoA reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. ATP-dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K 172. Eur. J. Biochem. 234:921-933. [DOI] [PubMed] [Google Scholar]
- 7.Boll, M., G. Fuchs, and H. Heider. 2002. Anaerobic oxidation of aromatic compounds and hydrocarbons. Curr. Opin. Chem. Biol. 6:604-611. [DOI] [PubMed] [Google Scholar]
- 8.Boll, M., G. Fuchs, C. Meier, A. X. Trautwein, and D. J. Lowe. 2000. EPR and Mössbauer studies of benzoyl-CoA reductase. J. Biol. Chem. 275:31857-31868. [DOI] [PubMed] [Google Scholar]
- 9.Boll, M., G. Fuchs, C. Meier, A. X. Trautwein, A. El Kasmi, S. W. Ragsdale, G. Buchanan, and D. J. Lowe. 2001. Redox centers of 4-hydroxybenzoyl-CoA reductase, a member of the xanthine oxidase family of molybdenum-containing enzymes. J. Biol. Chem. 276:47853-47862. [DOI] [PubMed] [Google Scholar]
- 10.Boll, M., S. J. P. Albracht, and G. Fuchs. 1997. Benzoyl-CoA reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. A study of adenosintriphosphatase activity, ATP stoichiometry of the reaction and EPR properties of the enzyme. Eur. J. Biochem. 244:840-851. [DOI] [PubMed] [Google Scholar]
- 11.Boyington, J. C., V. N. Gladyshev, S. V. Khangulov, T. C. Stadtman, and P. Sun. 1997. Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science 275:1305-1308. [DOI] [PubMed] [Google Scholar]
- 12.Brackmann, R., and G. Fuchs. 1993. Enzymes of anaerobic metabolism of phenolic compounds—4-hydroxybenzoyl-CoA reductase (dehydroxylating) from a denitrifying Pseudomonas species. Eur. J. Biochem. 213:563-571. [DOI] [PubMed] [Google Scholar]
- 13.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 14.Breese, K., M. Boll, J. Alt-Mörbe, H. Schägger, and G. Fuchs. 1998. Genes coding for benzoyl-CoA pathway of anaerobic aromatic metabolism in the bacterium Thauera aromatica. Eur. J. Biochem. 256:148-154. [DOI] [PubMed] [Google Scholar]
- 15.Ebenau-Jehle, C., M. Boll, and G. Fuchs. 2003. 2-Oxoglutarate:NADP+ oxidoreductase in Azoarcus evansii: properties and function in electron transfer reactions in aromatic ring reduction. J. Bacteriol. 185:6119-6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egland, P. G., D. A. Pelletier, M. Dispensa, J. Gibson, and C. S. Harwood. 1997. A cluster of bacterial genes for anaerobic benzene ring biodegradation. Proc. Natl. Acad. Sci. USA 94:6484-6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elshahed, M. S., and M. J. McInerney. 2001. Benzoate fermentation by the anaerobic bacterium Syntrophus aciditrophicus in the absence of hydrogen-using microorganisms. Appl. Environ. Microbiol. 67:5520-5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elshahed, M. S., V. K. Bhutpathiraju, N. Q. Wofford, M. A. Nanny, and M. J. McInerney. 2001. Metabolism of benzoate, cyclohex-1-ene carboxylate, and cyclohexane carboxylate by “Synthrophus aciditrophicus” strain SB in syntrophic association with H2-using microorganisms. Appl. Environ. Microbiol. 67:1728-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gescher, J., M. E. Mohamed, and G. Fuchs. 2002. Genes coding for a new pathway of aerobic benzoate metabolism in Azoarcus evansii. J. Bacteriol. 184:6301-6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson, J., and C. S. Harwood. 2002. Metabolic diversity in aromatic compound utilization by anaerobic microbes. Annu. Rev. Microbiol. 56:345-369. [DOI] [PubMed] [Google Scholar]
- 21.Gladyshev, V. N., S. V. Khangulov, and T. C. Stadtman. 1996. Properties of the selenium- and molybdenum-containing nicotinic acid hydroxylasen from Clostridium barkeri. Biochemistry 35:212-223. [DOI] [PubMed] [Google Scholar]
- 22.Graentzdoerffer, A., D. Rauh, A. Pich, and J. R. Andreesen. 2003. Molecular and biochemical characterization of two tungsten- and selenium-containing formate dehydrogenases from Eubacterium acidaminophilum that are associated with components of an iron-only hydrogenase. Arch. Microbiol. 179:116-130. [DOI] [PubMed] [Google Scholar]
- 23.Jackson, B. E., and M. J. McInerney. 2002. Anaerobic microbial metabolism can proceed close to thermodynamic limits. Nature 415:454-456. [DOI] [PubMed] [Google Scholar]
- 24.Harwood, C. S., G. Burchhardt, H. Herrmann, and G. Fuchs. 1999. Anaerobic metabolism of aromatic compounds via the benzoyl-CoA pathway. FEMS Microbiol. Rev. 22:439-458. [Google Scholar]
- 25.Heider, J., and G. Fuchs. 1997. Anaerobic degradation of aromatic compounds. Eur. J. Biochem. 243:577-596. [DOI] [PubMed] [Google Scholar]
- 26.Hille, R. 2002. Molybdenum and tungsten in biology. Trends Biochem. Sci. 27:360-367. [DOI] [PubMed] [Google Scholar]
- 27.Hunkapiller, M. W., R. M. Hewick, W. J. Dreyer, and L. E. Hood. 1983. High sensitivity sequencing with a gas phase sequenator. Methods Enzymol. 91:399-413. [DOI] [PubMed] [Google Scholar]
- 28.Jormakka, M., S. Tornroth, B. Byrne, and S. Iwata. 2002. Molecular basis of proton motive force generation: structure of formate dehydrogenase-N. Science 295:1863-1868. [DOI] [PubMed] [Google Scholar]
- 29.Kluge, C., A. Tschech, and G. Fuchs. 1990. Anaerobic metabolism of resorcyclic acids (m-dihydroxybenzoic acids) and resorcinol (1,3-benzenediol) in a fermenting and in a denitrifying bacterium. Arch. Microbiol. 155:68-74. [Google Scholar]
- 30.Koch, J., W. Eisenreich, A. Bacher, and G. Fuchs. 1993. Products of enzymatic reduction of benzoyl-CoA, a key reaction in anaerobic aromatic metabolism. Eur. J. Biochem. 211:649-661. [DOI] [PubMed] [Google Scholar]
- 31.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 32.Lowe, D. J. 2002. Enzymes of the xanthine oxidase family: the role of molybdenum. Met. Ions Biol. Syst. 39:455-479. [PubMed] [Google Scholar]
- 33.Raaijmakers, H., S. Macieira, J. M. Dias, S. Teixeira, S. Bursakov, R. Huber, J. J. Moura, I. Moura, and M. J. Romao. 2002. Gene sequence and the 1.8 Å crystal structure of the tungsten-containing formate dehydrogenase from Desulfovibrio gigas. Structure 10:1261-1272. [DOI] [PubMed] [Google Scholar]
- 34.Rabus, R., T. Hansen, and F. Widdel. 3 August 2001, posting date. Dissimilatory sulfate- and sulfur-reducing prokaryotes. In M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (ed.), The prokaryotes. [Online.] Springer-Verlag, New York, N.Y. http://link.springer-ny.com/link/service/books/10125.
- 35.Reichenbecher, W., B. Philipp, M. J. Suter, and B. Schink. 2000. Hydroxyhydroquinone reductase, the initial enzyme involved in the degradation of hydroxyhydroquinone (1,2,4-trihydroxybenzene) by Desulfovibrio inopinatus. Arch. Microbiol. 173:206-212. [DOI] [PubMed] [Google Scholar]
- 36.Schink, B., B. Philipp, and J. Müller. 2000. Anaerobic degradation of phenolic compounds. Naturwissenschaften 87:12-23. [DOI] [PubMed] [Google Scholar]
- 37.Schnell, S., and B. Schink. 1991. Anaerobic aniline degradation via reductive deamination of 4-aminobenzoyl-CoA in Desulfobacterium anilini. Arch. Microbiol. 155:183-190. [Google Scholar]
- 38.Schöcke, L., and B. Schink. 1998. Membrane-bound proton-translocating pyrophosphatase of Syntrophus gentianae, a syntrophically benzoate-degrading fermenting bacterium. Eur. J. Biochem. 256:589-594. [DOI] [PubMed] [Google Scholar]
- 39.Schöcke, L., and B. Schink. 1999. Energetics and biochemistry of fermentative benzoate degradation by Syntrophus gentianae. Arch. Microbiol. 171:331-337. [DOI] [PubMed] [Google Scholar]
- 40.Schräder, T., A. Rienhöfer, and J. R. Andreesen. 1999. Selenium-containing xanthine dehydrogenase from Eubacterium barkeri. Eur. J. Biochem. 264:862-871. [DOI] [PubMed] [Google Scholar]
- 41.Schühle, K., J. Gescher, U. Feil, M. Paul, M. Jahn, H. Schägger, and G. Fuchs. 2002. Benzoate-coenzyme A ligase from Thauera aromatica: an enzyme acting in anaerobic and aerobic pathways. J. Bacteriol. 185:4920-4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thauer, R. K., K. Jungermann, and K. Decker. 1997. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 41:100-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vorholt, J. A., M. Vaupel, and R. K. Thauer. 1997. A selenium-dependent and a selenium-independent formylmethanofuran dehydrogenase and their transcriptional regulation in the hyperthermophilic Methanopyrus kandleri. Mol. Microbiol. 23:1033-1034. [DOI] [PubMed] [Google Scholar]
- 44.Widdel, F. 1980. Anaerober Abbau von Fettsäuren und Benzoesäure durch neu isolierte Arten. Ph.D. thesis. University of Göttingen, Göttingen, Germany.
- 45.Wilting, R., K. Vamvakidou, and A. Böck. 1998. Functional expression in Escherichia coli of the Haemophilus influenzae gene coding for selenocysteine-containing selenophosphate synthetase. Arch. Microbiol. 169:71-75. [DOI] [PubMed] [Google Scholar]
- 46.Zehr, B. D., T. J. Savin, and R. E. Hall. 1989. A one-step, low background Coomassie staining procedure for polyacrylamide gels. Anal. Biochem. 182:157-159. [DOI] [PubMed] [Google Scholar]
- 47.Ziegler, K., R. Buder, J. Winter, and G. Fuchs. 1989. Activation of aromatic acids and aerobic aromatic metabolism in a denitrifying Pseudomonas strain. Arch. Microbiol. 151:171-176. [Google Scholar]