

Abstract
We report the complete genome sequence of enterobacteriophage SP6, which infects Salmonella enterica serovar Typhimurium. The genome contains 43,769 bp, including a 174-bp direct terminal repeat. The gene content and organization clearly place SP6 in the coliphage T7 group of phages, but there is ∼5 kb at the right end of the genome that is not present in other members of the group, and the homologues of T7 genes 1.3 through 3 appear to have undergone an unusual reorganization. Sequence analysis identified 10 putative promoters for the SP6-encoded RNA polymerase and seven putative rho-independent terminators. The terminator following the gene encoding the major capsid subunit has a termination efficiency of about 50% with the SP6-encoded RNA polymerase. Phylogenetic analysis of phages related to SP6 provided clear evidence for horizontal exchange of sequences in the ancestry of these phages and clearly demarcated exchange boundaries; one of the recombination joints lies within the coding region for a phage exonuclease. Bioinformatic analysis of the SP6 sequence strongly suggested that DNA replication occurs in large part through a bidirectional mechanism, possibly with circular intermediates.
Bacteriophage SP6 is a small double-stranded DNA tailed phage that infects Salmonella enterica serovar Typhimurium LT2. It shares many features with coliphage T7; however, no significant DNA sequence homology was detected between these phages by DNA hybridization (26). Previous studies of SP6 have elucidated its virion structure and the function and regulation of its early genes. Many of these previous studies examined the phage-encoded SP6 RNA polymerase (SP6RP) (7, 9, 26, 27, 30, 41). SP6RP has been used to produce synthetic RNA for a wide variety of applications (29, 30, 33, 37, 43).
As with phage T7, the SP6 genome is transcribed in a temporally ordered manner (26); transcription by SP6RP gives rise to 10 discrete RNA species (26). This finding suggests that discrete sites present in the SP6 genome serve as specific initiation and termination signals. One terminator sequence for the SP6 species IX RNA transcript has been identified, cloned, and sequenced (5). This sequence appears to be analogous to the stem-loop structure found at a comparable position in the T7 genome and to other rho-independent terminators (14). Preliminary studies have provided evidence for the presence of this termination sequence in the SP6 genome (5), but the termination efficiency has not been fully characterized.
Bacteriophage SP6 has been reported to be closely related to phages K1-5, K5, and K1E (51). In addition, Scholl et al. reported that SP6 encodes a tail protein that is in the same family as the tail spike protein of the otherwise apparently unrelated phage P22 (51). However, information about SP6 phage genetics, SP6 molecular biology, and the relationship of the sequence of this phage to other phage and prophage sequences is still limited. We completed the 43,769-bp sequence of the phage SP6 genome, identified the positions of individual genes and genetic signals, and analyzed the genetic relationships among SP6 and some closely related phages, including previously characterized members of the T7 group of phages and a recently identified (42) apparent prophage of Pseudomonas putida KT2440. Here we present findings on the transcription termination efficiency of a previously identified SP6RP terminator sequence, and we describe seven additional potential terminator sites within the SP6 genome. Finally, we describe bioinformatic analyses in which we identified sites of intergenome recombination in the evolutionary history of SP6 and illuminate features of SP6 DNA replication.
(This work was carried out by A. T. Dobbins in partial fulfillment of the requirements for a Ph.D. from Howard University, Washington, D.C.)
MATERIALS AND METHODS
Bacteria and phages.
Bacteriophage SP6 was kindly provided by Michael Chamberlin, University of California, Berkeley. S. enterica serovar Typhimurium LT2 (= ATCC 19585) was purchased from the American Type Culture Collection.
Bacteriophage propagation and purification.
Bacteriophage SP6 particles were isolated from infected cultures of S. enterica serovar Typhimurium by using a modification of the standard large-scale lambda phage isolation protocol, as described previously (49). Briefly, 10 ml of an overnight S. enterica serovar Typhimurium culture was used to seed 1 liter of Luria-Bertani medium. This preparation was incubated at 37°C until an optical density at 595 nm of ∼0.3 to 0.4 was reached. SP6 particles were added at a multiplicity of infection of 0.1 PFU/bacterium. Then the culture was incubated until complete lysis was observed. Residual bacterial cells were removed by centrifugation (5,500 × g for 20 min at 4°C). SP6 particles were concentrated overnight by addition of 5 M NaCl (10%, vol/vol) and polyethylene glycol 8000 (10%, wt/vol), followed by centrifugation as described above. The phage-containing pellet was resuspended in 28 ml of phage buffer (8 g of NaCl per liter, 0.2 g of KCl per liter, 1.44 g of Na2HPO4 · 7H2O per liter, 0.24 g of KH2PO4 per liter; pH 7.4), and the virions were purified by CsCl density gradient centrifugation at 135,000 × g and 18°C for 17 h in a Beckman Ti70.1 rotor (49). DNA was isolated from purified phage particles by sequential phenol-chloroform-isoamyl alcohol (25:24:1, vol/vol/vol) extractions, ethanol precipitation, and resuspension in TE buffer (10 mM Tris [pH 7.5], 1 mM EDTA).
SP6 DNA preparation and sequencing.
Approximately 10 μg of purified phage genomic DNA was sheared hydrodynamically and repaired by using T4 DNA polymerase to produce blunt ends. The 1- to 3-kb fragments of sheared DNA were purified and ligated into the EcoRV site of the pBluescript II KS+ vector. The ligation mixture was used to electrotransform Escherichia coli XLI-Blue cells. Individual recombinant clones were purified by using QiaPrep plasmid purification kits (Qiagen, Santa Clarita, Calif.), and these plasmids were sequenced from both ends of the inserted DNA by using the Applied Biosystems BigDye v3.0 dye terminator chemistry and universal sequencing primers. The labeled reaction mixtures were separated and analyzed by using an ABI Prism 3100 DNA analyzer. Oligonucleotide primers were synthesized and used to prime sequencing reactions with whole-genome templates to provide sequence coverage of underrepresented regions of the genome and to fill the gaps in the sequence assembly. Approximately eightfold sequence coverage was achieved.
Sequence assembly and analysis.
Sequence chromatograms were assembled and analyzed by using the Phred, Phrap, and Consed sequence analysis software (22). Weak areas in the sequence were identified and corrected by using additional primer-directed sequences. The SP6 terminal redundancies were identified by analogy with previously published sequence data, and the hypothesized structure was verified by primer extension sequencing by using primers SP6.67 (coordinates 416 to 395), SP6.69 (coordinates 43560 to 43580), and SP6.22 (coordinates 50 to 64 and 43645 to 43659) (14, 44, 45). The completed genome sequence was assembled in its proper orientation from the Consed assembly.
Open reading frames (ORFs) were identified by using Glimmer (13, 48), GeneMark (6), and DNA Master (J. G. Lawrence) (http://cobamide2.bio.pitt.edu) software and visual inspection. The translated ORF products were compared with known protein sequences by using BLAST software (1). Terminators were identified by using TransTerm (15). Skew analysis was done with DNA Master; phylogenies were constructed by the neighbor-joining method and by the maximum-likelihood method using ClustalW (10) and PHYLIP (19).
Electron microscopy.
Purified phage were applied to carbon-shadowed Parlodion-coated grids and stained with 1% uranyl acetate. Electron micrographs of the phage were taken with a Zeiss EM902 transmission electron microscope operating at 80 kV.
Construction of plasmid pCSM101.
A dual-promoter (SP6-T7)-bearing plasmid, pSP6/T7-19, was modified to carry a 210-bp SP6 fragment that contained a terminator sequence for the species IX RNA transcript of SP6 (5). The SP6RP terminator-bearing sequence was first identified in a HindIII digest of the SP6 genome (HindIII fragment E) that contained the RNA species IX transcript by Southern blotting and in vitro transcription analyses. HindIII fragment E was digested with BglII to obtain the 210-bp fragment. The pSP6/T7 plasmid was digested with BamHI and HindIII and ligated to the BglII-HindIII 210-bp fragment to produce a new construct designated pCSM101 (5).
In vitro transcription assays.
Transcription was performed in vitro by using both circular and linear molecules of pCSM101 as the DNA templates. Plasmid pCSM101 was linearized by digestion with PvuII or HindIII. All assay mixtures contained 100 μg of plasmid DNA per ml, [α-32P]CTP (300 cpm/pmol), and 10 U of SP6RP. Transcription was allowed to proceed for 30 min at 37°C. The reaction was stopped by addition of EDTA (pH 8.0) to a final concentration of 20 mM. Samples were fractionated on a 12% polyacrylamide-7 M urea denaturing gel, which was subsequently analyzed by autoradiography.
Gene nomenclature.
Annotations of previously sequenced genomes of T7-like phages have used the T7 gene numbering system, so that homologous genes have the same gene numbers. This has advantages for performing comparisons among members of the group. For SP6 there are enough differences in gene organization and content that using this system would be potentially confusing. SP6 has been independently sequenced by another research group (52), and we have agreed with the workers who performed that study on a common gene numbering system in which gene numbers increase from left to right on the map. Below, we indicate the T7 gene number in parentheses where appropriate. Thus, for example, the gene encoding the SP6RP, which is a homologue of T7 gene 1, is designated gene 7 and is usually indicated below as gene 7 (T7-1).
Nucleotide sequence accession numbers.
The DNA sequence of the SP6 bacteriophage genome reported here has been deposited in the GenBank database under accession number AY288927. The accession number for the SP6RP terminator sequence is L25625, and the accession number for the P. putida genome is NC 002947.
RESULTS
Characterization of bacteriophage SP6 virions.
Figure 1 shows negatively stained electron micrographs of SP6 virions. The heads are hexagonal and therefore apparently icosahedral; they are isometric and have a diameter of ∼50 to 60 nm. At this level of resolution the heads are indistinguishable from those of T7 or any of a large number of phages with ∼40- to 50-kbp genomes. The tails lack any extended tail shaft and have an irregular bushy structure, presumably the phage's tail fibers. While the fine structure of the fibers was not revealed by the images, we believe that the appearance of this part of the SP6 virion and therefore presumably the underlying structure is distinguishable from the appearance of the corresponding parts of either T7 or P22 virions (2, 54).
FIG. 1.
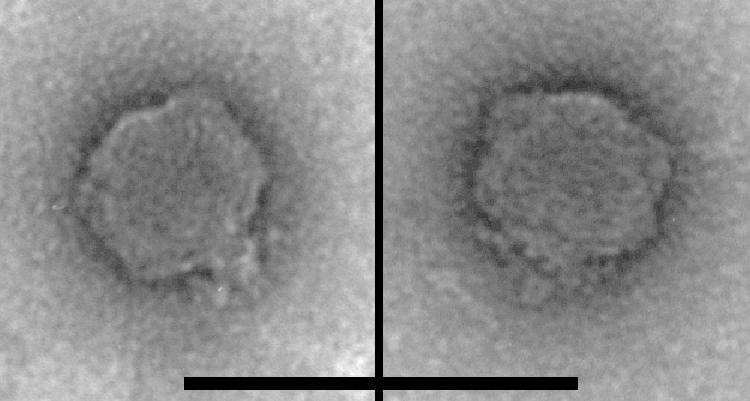
Electron micrographs of two SP6 virions, negatively stained with uranyl acetate. Scale bar = 100 nm.
Determination of the SP6 genomic sequence.
The complete DNA sequence of the SP6 genome was determined by using a shotgun sequencing approach. A total of 480 clones were sequenced in this manner; gaps and areas of sparse coverage were addressed by direct sequencing of virion DNA with custom primers. Final assembly yielded a circular sequence containing 43,595 bp. Because the DNA extracted from the virions was linear, this result suggests either that the virion DNA is circularly permuted and terminally redundant (like that of phage T4) or that it is terminally redundant but not permuted (like that of phage T7). Assuming the latter, we designed primers to anneal interior to the presumed terminal duplication, which primed synthesis in the direction of the presumed ends of the virion DNA (primers SP6.67 and SP6.69). Sequencing reactions with these primers and with virion DNA as the template terminated abruptly at what we assumed are the unique ends of the virion DNA. Comparison of these two sequences indicated that there are 174-bp exact direct repeats at the ends of the virion DNA, resulting in a total genome length of 43,769 bp. Support for this structure was obtained by synthesizing an additional primer (SP6.22) to anneal within the terminal repeat. In this case synthesis did not stop abruptly at the end; this was predicted because the primer should have annealed to both copies of the terminal repeat and therefore encountered a molecular end and terminated only half the time. The overall G+C content of the SP6 genome is 47.2%. The accuracy of the sequence assembly was confirmed by comparing the predicted restriction enzyme map with restriction enzyme maps determined by us (data not shown) and by other workers (26). Fortuitously, the SP6 sequence has been determined independently by a second research group (52), and the sequences agree exactly, indicating strongly that the sequences are error free.
Assignment of probable SP6 genes.
We identified 52 ORFs with good coding potential and plausible translation start sequences; all were transcribed in the rightward direction on the SP6 genome (Fig. 2 and Table 1). The translated ORF products were compared with known protein sequences by using BLAST. The coordinates and best matches are shown in Table 1. The majority of plausible database matches occurred with members of the T7 group (T7, T3, φYeO3-12, gh-1), and with the exceptions noted below, the functional orders of the genes on the genomes are the same. Thus, our results confirm that SP6 is properly described as a phage that is related to T7 and its allies, despite the presence of a P22-like tail spike gene and the lack of DNA hybridization between SP6 and T7. The sequences of three genes were reported previously: the SP6RP gene (27), the SP6 DNA primase gene (55), and the SP6 tail spike gene (51). The majority of the predicted SP6 proteins (28 of 52 proteins [54%]) have no similarity to proteins in the GenBank database. Consequently, we assigned putative functions only to those predicted proteins with clear similarity to T7-group proteins that have experimentally determined functions or that have had their functions in SP6 verified experimentally.
FIG. 2.

Map of the SP6 genome. Rectangles represent genes, and the different colors indicate different classes of genes, as indicated in the key below the map. All transcription is from left to right. The gene numbering scheme is described in Materials and Methods; the designations in parentheses are the names of the homologous T7 genes. Promoters and terminators are indicated above the line by designations that begin with P and t, respectively, and below the line by symbols. Gene functions, either determined experimentally or inferred by homology, are indicated where they are known. The scale is in kilobase pairs.
TABLE 1.
ORFs identifieda
| Gene | CDS start position | CDS stop position | CDS length (nucleotides) | Protein length (amino acids) | Start codon | Stop codon | Comments | Best homolog | Best E value |
|---|---|---|---|---|---|---|---|---|---|
| 0.2 | 711 | 884 | 174 | 57 | ATG | TAA | |||
| 0.6 | 998 | 1099 | 102 | 33 | ATG | TGA | |||
| 1 | 1099 | 1275 | 177 | 58 | ATG | TAA | |||
| 2 | 1268 | 1477 | 210 | 69 | ATG | TAA | |||
| 3 (T7-0.3) | 1637 | 1996 | 360 | 119 | ATG | TAA | Inhibits EcoB and EcoK host restriction | T7 gp0.3 | e-43 |
| 5 | 2181 | 2372 | 192 | 63 | ATG | TAA | |||
| 6 | 2435 | 3319 | 885 | 294 | ATG | TAA | |||
| 7 (T7-1) | 3392 | 6016 | 2,625 | 874 | ATG | TAA | SP6 DNA-directed RNA polymerase | 0 | |
| 8 (T7-1.1) | 6271 | 6468 | 198 | 65 | GTG | TAA | |||
| 9 (T7-4) | 6472 | 8457 | 1,986 | 661 | ATG | TGA | SP6 DNA primase | 0 | |
| 10 | 8601 | 9212 | 612 | 203 | ATG | TGA | |||
| 11 | 9199 | 9369 | 171 | 56 | ATG | TAA | |||
| 12 | 9431 | 9592 | 162 | 53 | ATG | TAA | |||
| 13 (T7-5) | 9579 | 12128 | 2,550 | 849 | ATG | TAA | Phage putative DNA polymerase | Pseudomonas aeruginosa phage PaP3 | e-31 |
| 14 | 12128 | 12226 | 99 | 32 | ATG | TGA | |||
| 14.5 | 12396 | 12542 | 147 | 48 | ATG | TGA | |||
| 15 | 12629 | 13006 | 378 | 125 | ATG | TAA | |||
| 16 | 13216 | 14022 | 807 | 268 | ATG | TAA | |||
| 17 | 14036 | 14254 | 219 | 72 | ATG | TAA | |||
| 18 | 14359 | 14727 | 369 | 122 | ATG | TAA | |||
| 20 (T7-6) | 15010 | 16038 | 1,029 | 342 | ATG | TGA | Similarity to phage exonuclease | Pseudomonas aeruginosa phage PaP3 | e-15 |
| 21 (T7-3) | 16023 | 16433 | 411 | 136 | ATG | TAA | Packaging and recombination endonuclease VII | T4 gp49 | e-6 |
| 22 | 16519 | 17433 | 915 | 304 | ATG | TAA | |||
| 23 (T7-1.7) | 17534 | 18004 | 471 | 156 | ATG | TAA | T7 gp1.7 (C-terminal only) | e-20 | |
| 24 (T7-1.3) | 18004 | 18951 | 948 | 315 | ATG | TGA | |||
| 25 | 18941 | 19135 | 195 | 64 | ATG | TGA | |||
| 26 | 19146 | 19274 | 129 | 42 | ATG | TGA | |||
| 27 | 19271 | 19732 | 462 | 153 | ATG | TAA | Roseophage SIOI gp26.2 | e-7 | |
| 28 | 19742 | 19951 | 210 | 69 | ATG | TGA | |||
| 29 (T7-8) | 19953 | 21500 | 1,548 | 515 | ATG | TAA | Head portal protein | Pseudomonas putida KT2440 | e-59 |
| 30 (T7-9) | 21504 | 22406 | 903 | 300 | GTG | TAA | Scaffolding protein | Pseudomonas putida KT2440 | e-7 |
| 31 (T7-10) | 22486 | 23691 | 1,206 | 401 | ATG | TAA | Major capsid protein | ||
| 32 (T7-11) | 23744 | 24484 | 741 | 246 | ATG | TAA | Tail protein | Pseudomonas putida KT2440 | e-10 |
| 33 (T7-12) | 24484 | 26895 | 2,412 | 803 | ATG | TAA | Tail protein | Pseudomonas putida KT2440 | e-70 |
| 34 (T7-14) | 26896 | 27615 | 720 | 239 | ATG | TAA | Internal virion protein | Pseudomonas phage gh-1 | e-4 |
| 35 | 27616 | 30552 | 2,937 | 978 | ATG | TAG | Lysozyme motif at C terminus | ||
| 36 (T7-16) | 30619 | 34431 | 3,813 | 1,270 | ATG | TGA | Internal virion protein | Pseudomonas putida KT2440 | e-7 |
| 37 (T7-17) | 34431 | 35390 | 960 | 319 | ATG | TAG | Tail fiber (partial) | ||
| 38 (T7-17.5) | 35399 | 35596 | 198 | 65 | ATG | TAA | Putative holin | ||
| 39 (T7-18) | 35580 | 35879 | 300 | 99 | GTG | TAA | Small terminase subunit | ||
| 40 (T7-19) | 35879 | 37774 | 1,896 | 631 | ATG | TAA | Large terminase subunit | Pseudomonas putida KT2440 | e-81 |
| 41 | 37921 | 38220 | 300 | 99 | ATG | TAA | |||
| 42 | 38235 | 38534 | 300 | 99 | ATG | TAA | |||
| 42.3 | 38544 | 38789 | 246 | 81 | ATG | TAA | |||
| 42.7 | 38812 | 38979 | 168 | 55 | GTG | TGA | |||
| 43 | 38976 | 39332 | 357 | 118 | GTG | TAA | Hypothetical bacterial protein | Desulfovibrio desulfuricans G20 | e-16 |
| 44 | 39423 | 39593 | 171 | 56 | ATG | TAG | |||
| 45 | 39671 | 39880 | 210 | 69 | ATG | TAA | |||
| 46 | 40011 | 41663 | 1,653 | 550 | ATG | TAA | P22-like tail spike (partial) | P22 gp9 | 0 |
| 47 | 41774 | 43261 | 1,488 | 495 | GTG | TGA | |||
| 48 | 43269 | 43412 | 144 | 47 | ATG | TAG | |||
| 49 | 43466 | 43543 | 78 | 25 | ATG | TAA |
CDS, coding sequence.
The SP6 genes are generally tightly spaced, and in many cases the stop codon of one gene overlaps the start codon of the following gene. There are several relatively large (∼100- to 300-bp) apparently noncoding regions within the genome (e.g., between genes 7 and 8 and between genes 45 and 46). Although no tRNA genes were identified in these or other locations by using a tRNA scanning program (36), several putative transcription promoters and terminators were identified in these regions (see below).
SP6 gene products with similarity to known proteins were classified into three major groups: proteins involved in nucleic acid and other metabolism, virion structure and assembly proteins, and proteins that match database proteins with unknown functions. The genes for proteins involved in metabolism and the structure and assembly functional groups are generally clustered on the phage chromosome. Genes 3 (T7-0.3), 7 (T7-1), 9 (T7-4), 13 (T7-5), 20 (T7-6), 21 (T7-3), and 24 (T7-1.3) fall in the metabolism group; the products of two of these genes, gp7 (T7-gp1) and gp9 (T7-gp4), are known from previous work to function as a DNA-directed RNA polymerase and a DNA primase, respectively (27, 55). The structure and assembly genes are clustered and span from gene 29 (T7-8) through gene 37 (T7-17), which encode virion components (with gene 35 not assigned because of a lack of a database match), plus genes 39 (T7-18) and 40 (T7-19) encoding putative DNA packaging proteins and gene 46 encoding the P22 tail spike homologue (51). The majority of predicted SP6 gene products (33 of 52 products [63%]) do not match proteins whose functions are known. These products include three predicted proteins (gp8, gp23, and gp27) that exhibit similarity to other phage proteins (T7 gp1.1, T7 gp1.7, and Roseobacter phage SIO1 gp26.2, respectively), while another two products (gp43 and gp46) match nonphage proteins whose functions are not known. The remaining 28 genes are currently unique to SP6.
SP6 gene 38 (T7-17.5) is a homolog of the T7 holin gene, encoding a lysis function, and it is located in a comparable part of the gene order. In contrast, SP6 has no candidate for a homologue of T7 gp18.5. T7 gp18.5 is a homologue of the phage λ Rz protein, which has been shown to be required for cell lysis (in the case of λ) only when divalent cation concentrations are high. For the other well-conserved genes of T7, SP6 is missing obvious homologues of genes 0.7 (encoding protein kinase), 2.5 (SSB protein), 3.5 (amidase), 5.7, 13 (virion assembly factor), and 15 (interior virion protein), although in all of these cases except genes 3.5 and 13 there are genes without assigned functions in the corresponding genome positions that may have equivalent roles.
A well-established unusual feature of the gene encoding the major capsid subunit in phages T7, T3, and φYeO3-12 (but not gh-1) is that there is a translational frameshift signal near the end of the coding region which causes about 15% of the ribosomes translating the mRNA to undergo a −1 frameshift, resulting in a protein (gp10B) that is (in T7) 53 amino acids larger than the conventionally translated gp10A protein due to the C-terminal extension (11, 12). The functional significance of this frameshift is not well understood, but it has been suggested that in the case of a Listeria phage with such an extension (57) the longer protein could occupy the five-fold corners of the icosahedral capsid. Examination of the SP6 capsid gene (gene 31) sequence failed to reveal any obvious shifty sequence, there is no significant ORF available downstream from the stop codon and the small space between genes 31 and 32 is almost entirely occupied by the transcription terminator described below. Thus, in this regard SP6 appears to be unlike T7 and T3 and similar to phages like the T7 family member phage gh-1 (28), as well as phages λ and P22, and many other phages which have only a single form of the major capsid protein.
There are three other special features of the SP6 proteins that can be inferred from their sequences. First, gp35 has a small domain near its C terminus with weak sequence similarity to cell wall lysozymes. Previously, this protein was shown experimentally to have lysozyme activity (39). Several phages are known to have a lysin activity encoded in virion proteins that is distinct from the endolysin that releases phages from the cell following lytic growth (3, 39); these proteins may facilitate DNA entry during infection. However, in contrast to the lysin domain at the C terminus of SP6 gp35, most phages belonging to the T7 family, including T7, T3, φYe03-12, gh-1, the P. putida prophage, and Yersinia pestis phage φA1122 (21), have instead a lysin domain at the N terminus of the protein encoded by the downstream gene, corresponding to SP6 gene 36 (T7-16). The recently sequenced Pseudomonas aeruginosa phage φKMV (31) has the SP6 module arrangement. The difference between the locations of the lysin domains in SP6 and φKMV and the locations in the other phages belonging to the T7 family is probably not simply a reflection of an ancestral transfer of a lysin-encoding domain from one gene to the adjacent gene, inasmuch as the SP6 and φKMV lysin domains belong to the true lysozyme sequence family, while the lysin domains of the other phages belong to the transglycosylase sequence family.
Second, gp37 (T7-gp17) matches over its N-terminal ∼160 amino acids the corresponding parts in T7-like phages. The remainder of the SP6 protein, roughly another 160 amino acids, has no detectable sequence matches. In T7, gp17 is the tail fiber; the N-terminal domain (homologous to a portion of the SP6 protein) is thought to bind the fiber to the virion, while the remainder of the protein constitutes the body of the fiber, including the cell binding function (54). Thus, SP6 gp37 (T7-gp17) may be capable of binding to the fiber binding site on the virion, but it probably does not have the additional sequences necessary to form a distinct tail fiber.
Third, gp46 is missing the region corresponding to the first ∼115 amino acids of the homologous P22 tail spike protein. In the P22 protein, this domain attaches the tail spike to the virion (38); we conjecture that the SP6 protein may make the part of the tail spike needed to interact with the surface of the cell and that it attaches to the virion by some other mechanism. This curious feature of the SP6 tail spike protein has been noted previously (20); Freiberg et al. also showed that these apparently truncated tail spikes are nonetheless components of the virion. A hypothesis to reconcile these observations is that the two tail-fiber-like proteins (gp37 and gp46) function as a complex of both types of subunits, with gp37 (T7-gp17) forming a linkage between the virion and the receptor-binding gp46 protein.
SP6RP promoter sequences.
The SP6 genome was searched for sequences that correspond to the SP6RP core promoter sequence (5′-ATTTAGGTGACACTATAG [53]). Thirteen related sequences were identified that have three or fewer mismatches; a degenerate consensus sequence, KAWTTARGKGACACTATAG, was derived based on the promoter sequences with two or fewer mismatches; and the search was repeated. No additional putative promoters were identified. Of the 13 sequences, 3 (+1 positions at bp 2121, 6304, and 29410) are unlikely to function since they depart from the core sequence at positions 1, −1, and −15, respectively, which would dramatically decrease promoter activity (53). The remaining 10 sites are excellent candidates for SP6 promoters; based on saturation mutagenesis experiments, the departures of these sequences from the core consensus sequence should have little effect on promoter activity (Fig. 3) (53). The sequence with the greatest departure from the core consensus is P3, which has substitutions at positions −12, −16, and −17, although mutational studies (53) indicated that these substitutions do not have a large impact on promoter activity (Fig. 3). Most of the putative promoters lie in intergenic regions; the only exceptions are P2 and P3, which are located approximately 100 bp from the 3′ ends of genes 9 and 10. We do not know how these 10 putative promoters correspond to the 11 SP6 promoters described previously (34).
FIG. 3.

Putative phage-specific promoters of SP6. Ten potential SP6 promoters were identified by searching for sequences closely related to three previously identified promoters (7), shown here as P6, P8, and P9. Each promoter designation is indicated on the left and is followed by the nucleotide position at +1. The letters at the top indicate the effects of mutations at positions on promoter activity in vivo, as reported by Shin et al. (53). The four letters indicate how many of the three possible substitutions lead to a reduction in activity to a level that is less than one-third the consensus level (i.e., P8); A indicates that all three substitutions reduced activity to this extent, B indicates that two substitutions reduced activity to this extent, C indicates that one substitution reduced activity to this extent, and D indicates that no substitution reduced activity to this extent (53). We do not know how these putative promoters are related to those described previously (34).
The number of putative phage-specific promoters is less in SP6 than in either T3 (14 promoters) or T7 (16 promoters), although several promoters occupy related locations in these genomes. For example, all three phages have promoters that are located in the vicinity of homologues of T7 gene 1 (immediately downstream), gene 4 (downstream of gene 1.7 near the 5′ end), and between genes 9 and 10. Several promoters located within common genes in phages T3 and T7 are notably absent from SP6. For example, T3 and T7 both have a promoter between genes 8 and 9, T3 has a promoter between genes 10 and 11, and a T7 promoter is present between genes 16 and 17. No promoters are evident in corresponding positions in SP6.
In phages T3 and T7 there are five E. coli promoters at the left end of the genome that are required for expression of the phage RNA polymerase and other very early functions (Fig. 2). Although SP6 has only four E. coli promoters in this region, they likely serve similar functions and are located in similar positions. The positions of the predicted start sites are as follows: Pc1, position 608; Pc2, position 1520; Pc3, position 2075; and Pc4, position 3354 (Fig. 2). The main difference is that there appears to be just a single promoter upstream of gene 1, whereas there are three promoters at similar positions in both T3 and T7.
SP6RP terminator sequences and termination efficiency.
The SP6 sequence was also examined for the presence of factor-independent transcriptional terminators. A number of sequences capable of forming stem-loop mRNA structures were identified, but few of these are followed by the characteristic run of U bases. Seven potential terminators were identified. One (t7) is located between genes 46 and 47 (the first base of the stem is at position 41669) and is followed by 3 U residues. This putative terminator was identified previously (51), although the sequence reported previously differs from that described here, perhaps due to secondary-structure elements confounding the previous study. A second terminator (t5) is located in the short intergenic region between genes 31 and 32 (the stem starts at position 23698) immediately following the gene encoding the major capsid subunit; phages T3 and T7 also have terminators downstream of their capsid genes. The capsid protein is likely the most abundantly synthesized protein encoded by the phage, and its gene is also preceded by a phage-specific promoter. This should allow a higher level of transcription for the capsid protein gene; furthermore, the efficiency of the terminator should determine the level of transcription for the downstream structural genes. Likewise, the RNA polymerase gene 7 (T7-1) is followed by a terminator (t1; the start of the stem is at position 6036) and is preceded by an E. coli promoter (Pc4). Putative terminators were also identified between genes 35 and 36 (t6; the stem starts at position 30566) and downstream of genes 14, 15, and 18 (t2, t3, and t4, with stems starting at positions 12238, 13137, and 14733, respectively). Two potential stem-loop structures are present between genes 3 and 5 and between genes 45 and 46; these structures might function as RNase III sites.
Figure 4 shows the results of characterization of the t5 terminator located downstream of the capsid gene. The terminator was cloned into a plasmid downstream from an SP6 promoter, and [32P]RNA was made by in vitro transcription with purified SP6RNAP and was separated by gel electrophoresis. Figure 4, lane 1, shows the results of transcription when the circular plasmid template was used. The discrete band is the size expected for termination at the terminator; readthrough products are visible only as a high-molecular-weight smear. Lanes 2 and 3 show the results of the same reaction performed with templates that had been cut at the unique HindIII and PvuII sites, respectively, which were located downstream from the terminator. In these cases the readthrough products ended uniquely at the cut end of the template. The ratio of the two bands in lane 2 or 3 provides a measure of termination efficiency in this in vitro reaction; in multiple determinations the termination efficiencies ranged from 40 to 50%.
FIG. 4.

Transcription termination: denaturing polyacrylamide gel analysis of in vitro transcription products synthesized from linearized and circular pCSMl0l templates with SP6RP. The numbers on the left indicate the lengths of the transcripts (in nucleotides). Lanes 1, 2, and 3, transcription products of closed circular, HindIII-digested, and PvuII-digested pCSM101 templates, respectively; lanes 4, 5, and 6, transcription products of closed circular, HindIII-digested, and PvuII-digested pSP6/T7-19 (parent plasmid) templates, respectively.
Genome comparisons.
Figure 5 shows a comparison of the genomes of SP6 and of four related phages. Genes that have homologues in other phages, as judged by significant sequence similarity, are indicated; for convenience, the genes of SP6 were given the numbers of the corresponding T7 homologues. The large-scale features of the comparison show that there are two major differences in the overall genome organization between SP6 and the other phages analyzed. First, SP6 has an extra group of genes at the right end of the genome, which extend roughly 5 kb beyond the position corresponding to the right end for the other phages in the group. These extra genes include the gene encoding the P22-like tail spike protein. It is possible that these genes are an evolutionarily recent addition to the SP6 genome; if so, we note that the recombination event(s) that brought them into the genome preserved the terminal repeat at the right end of the genome.
FIG. 5.

Comparison of five T7-like phages. All genes in SP6 with identifiable homologues in the other taxa are colored; in addition, gene 49 (similar to P22 tail spike protein gene 9) is shown in green. Genes in black or in blue show phages T3 and φYeO3-12 as most closely related, while those that are in red show phages T3 and T7 as most closely related. The genes indicated in blue appear to have been inverted in the genome (by displacement and inversion of gene order) but have individually reinverted so that they are transcribed in the same direction as all other genes. There appear to have been at least two large recombination events involving large portions of the genome; these domains may involve genes with cooperating functions (RNA transactions, DNA replication, head assembly, and tail assembly). At the bottom, phylogenies are constructed for selected genes using neighbor joining (47). Bootstrap values for each dendrogram are shown. In addition, the results of Felsenstein tests of phylogenetically informative sites (16) are shown below each phylogeny, indicating the numbers of sites supporting (i) T3,φYeo-T7**, (ii) T3,T7-φYeo**, and (iii) T7,φYeo-T3** phylogenies, respectively, where the double asterisk indicates the homologous genes from either phage SP6 (line 1) or gh-1 (line 2).
The second major difference in genome organization is in the region of the genomes containing the homologues of T7 genes 1.3, 1.7, and 2. Compared to the arrangement in the other phages, it appears that these genes in SP6 were either transposed to a different position en masse and had their orientations inverted individually or were independently translocated to adjacent positions downstream of their supposed T7-like ancestral location.
In addition to these major differences in genome organization, there are numerous smaller insertions, deletions, and substitutions in SP6 compared to the other phages in Fig. 5 (as there are in any pairwise comparison for the other phages). An intriguing example is the six genes in SP6 located between the homologues of T7 genes 5 and 6. In addition to being unlike the genes at this position in the other phages, these genes are associated with three putative promoters and three putative transcription terminators. These genes bear some resemblance to a class of genes (morons) found in some other phage groups, particularly the lambdoid group, that have been identified as evolutionarily recent additions to the genome and that are often associated with promoters and terminators (25).
In addition to the sequence matches between SP6 and the other phages shown in Fig. 5, some of the closest matches of the SP6 proteins are matches with proteins encoded in the recently sequenced genome of the bacterium P. putida KT2440 (42). The P. putida genes are parts of an apparent prophage covering 40 kb of the genome (42, 56), and they include homologues of nearly all the SP6 genes that have homologues in the other phages in Fig. 5, arranged in essentially the same order. The existence of the P. putida KT2440 prophage is surprising because other described phages belonging to the T7 family are all strictly virulent. The view that this is a bona fide prophage and not simply the result of chance recombination events is supported by the fact that it includes an integrase gene belonging to the tyrosine recombinase family and the fact that it appears to have integrated into a tRNA gene, reconstituting the 3′ end of the severed tRNA gene with phage DNA, as is the case with many other temperate phages. It is most parsimonious to conclude that the prophage was generated by integrase-mediated recombination between the host chromosome and a circular form of the phage chromosome, the latter of which was generated by recombination between the terminal repeats of the virion DNA. In this context it is interesting that, at least with respect to the genes that can be recognized as homologues of genes in other members of the T7 family, the gene order in the prophage is not permuted with respect to the order on the physical map of the phage genomes.
This argues that the physical ends of the virion DNA that gave rise to the prophage were near the attachment site, most likely in the region between the gene 19 homologue and the right end of the prophage. If this view is correct, then it suggests that a short module of sequence, containing an integrase gene and attachment site and corresponding to the rightmost ∼4 kb of the prophage, may have inserted at the left end of the genome of an ancestral lytic phage, conferring the ability to integrate as a prophage. Such an insertion would be analogous to the insertion of a sequence module at the right end of an ancestor of the SP6 genome that we propose above, which gave the phage a new type of tail fiber.
Phylogenetic analyses.
Comparative analyses of temperate phage genomes show that these phages are genetic mosaics with respect to each other, evidently as the result of horizontal exchange of genes or blocks of genes in the ancestry of the group (8, 23-25). Recent analyses of phages T3 and A1122 and others of the T7-like group provide qualitative and experimental evidence for horizontal exchange in this group as well (21, 45). We applied phylogenetic analyses to the T7-like group, including SP6, to determine the degree of recombination among this quintessentially virulent group of phages. The genes that have homologues in other phages are shown in Fig. 5. For each set of homologues we derived a phylogenetic tree based on protein sequence alignments, using the neighbor-joining method (47). These trees, shown below the genome maps in Fig. 5, fall into two mutually incongruent classes: for the genes shown in black or blue, the T3 and φYeO3-12 genes are closer to each other than they are to any of the other homologues; for the genes shown in red, the T3 gene clusters with the T7, rather than the φYeO3-12, homologue. These relationships demonstrate that the genes depicted in red have different evolutionary histories than do those depicted in black or blue, reflecting the action of horizontal genetic exchange (32) in their evolutionary history.
We assessed the robustness of these relationships in two ways. First, 1,000 data sets were generated by bootstrapping (17), and the percentage of the resulting phylogenies supporting each node is shown on each phylogeny in Fig. 5. In addition, the numbers of informative sites supporting each of three alternative phylogenies in the Felsenstein test are shown at the bottom of Fig. 5; the results of the Felsenstein tests corroborate the bootstrap values with the exception of gene 20 (T7-6), which is chimeric (Fig. 6).
FIG. 6.

Analysis of phylogenetically informative sites showing support for two distinct phylogenies for the N terminus and C terminus of the gp20 (T7-gp6) protein. This pattern was not seen for any other gene (see Fig. 5). (A) Phylogeny of the gene as a whole. (B) Phylogenetically informative sites extracted from an alignment of the homologues of the protein from the four phages indicated. The numbers indicate positions in the alignment. (C) Phylogeny of the N terminus of the protein. The sequences were divided at aligned residue 180 (C-terminal residue of the SCDKDFKTIP sequence in T7), which was between the two sites defining domains with different phylogenetic histories. This phylogeny is supported robustly (bootstrap value for both nodes, 100%). (D) Distinctly different phylogeny inferred from the C-terminal portion of the gp6 exonuclease.
Figure 6A shows a neighbor-joining tree based on the entirety of gene 20 (T7-6); branch lengths now reflect distances. The phylogenetically informative sites of the 5′ and 3′ ends of this gene clearly support two different relationships (Fig. 6B); the probability of the pattern arising by chance is on the order of 10−12. Figures 6C and D show neighbor-joining trees based on the N-terminal and C-terminal parts of the protein sequences, dividing at position 180 in the alignment. The phylogeny of the N termini is congruent with those of the red genes in Fig. 5, while the phylogeny of the C termini is congruent with those of the black and blue genes. These results imply that the two halves of gene 21 (T7-6) have different evolutionary histories.
Analyses of other phages rarely show mosaic boundaries within coding regions (25), presumably because mosaic proteins are typically not functional. Known examples of such mosaic boundaries appear to fall at domain boundaries within the protein. There is no direct structural information available for the exonuclease (gp6) of T7-like phages, but there is a high-resolution structure for the homologous exonuclease of phage T5 (4). The latter enzyme is a two-domain protein, and when the T7 and T5 sequences are aligned, the inferred site of recombination in the T7 family of proteins falls on a loop that spans the two domains of the T5 protein.
Skew analysis and DNA replication.
DNA replication introduces mutations differentially into leading and lagging strands, resulting in mutational bias. This bias can be exploited to infer differences in the way in which the replication fork passes over different parts of the genome (35). We scanned the SP6 genome for locations where the change in bias was maximal, indicating a switch between leading strand replication and lagging strand replication. Two locations were identified (at bp 10500 and 32500), separated by ∼50% of the genome length; we interpret these positions to correspond to the origin and terminus of theta DNA replication. Figure 7 plots the positions of eight base sequences that show strong strand bias in the proposed replication arms. The magnitude of the octomeric skew is different for the two replichores of the genome (eightfold versus threefold). The defeat in octomeric skew in the central portion of the SP6 genome may reflect (i) strong compositional bias imparted by the unidirectionally transcribed genes in this genome which prevents replication-imparted mutational bias from accumulating or (ii) the action of unidirectional (sigma) replication which reinforces leading strand bias in one replichore but defeats it in the other replichore, where the lagging strand during theta replication is replicated as a leading strand during sigma replication. We have assigned the position in gene 13 (T7-5) as the replication origin for two reasons. First, gene 13 (T7-5) encodes the phage DNA polymerase; there are examples in other phages, such as λ and its relatives, where the replication origin occurs within the coding region of a gene encoding a replication protein. Second, one would predict the pattern of reinforcement and defeat of strand bias that we observed in SP6 if the origins for theta and sigma replication were both in gene 13 (T7-5).
FIG. 7.

Mapping the origin of replication by mutational bias. The genome was analyzed by circular permutation to find breakpoints where changes in strand-specific degenerate-octomeric skew were the strongest. Two sites were identified; positions were refined after fine-scale analysis of the distribution of several hundred octomers showing at least 80% strand bias, as shown in the center. The graph shows the cumulative distribution of the sequences on the Watson-and-Crick strands of DNA for 3% intervals of the genome.
DISCUSSION
Genome features.
Salmonella phage SP6 has long been considered very similar to the well-studied coliphages T7 and T3 and other phages, based on a similar virulent life cycle, the presence of a single-polypeptide virus-encoded RNA polymerase, and similar virion morphology. The genome sequence reported here largely confirms these anticipated relationships, but it also reveals some informative differences in detail, particularly those determined by comparative analyses with the other phages in the group.
Genome mosaicism.
The mosaic patterns in the genomes that resulted from recombination in the ancestry of these phages are reminiscent of the patterns seen in other groups of mostly temperate phages (8, 23-25, 46). This is of some theoretical interest in that strictly virulent phages (which included the T7-SP6 group until now) are expected to have severely reduced opportunities for recombination with similar phages, primarily because the probability of nearly simultaneous infection of one cell by two phages is drastically reduced in natural conditions owing to the low concentration of phage capable of infecting any given host cell. Most recombination in temperate phages is thought to involve at least one prophage, an opportunity not available to strictly virulent phages (32). Growth in the absence of genetic exchange is not a sustainable life style because such an organism is unable to avoid the inevitable accumulation of deleterious mutations, a process termed Muller's ratchet (18, 40). In this sense, it is expected, and reassuring, that the T7-SP6 group of phages shows clear evidence of substantial amounts of genetic exchange. The T7-like prophage in the P. putida genome (42) suggests that perhaps the temperate members of this group act as gene donors and gene recipients (during nonproductive infections), thus driving recombination among virulent phages via their temperate cousins.
Transcriptional control.
SP6 appears to employ a scheme for transcriptional regulation similar to that employed by T7, and we identified 10 putative promoters and seven putative terminators. Most of these promoters and terminators are located in spaces between genes, as is typical for such elements, and they include the three promoters that have been characterized biochemically (7). In a previous study (5) the phage-specific transcripts in an SP6-infected cell were characterized and mapped on the genome map. Although the precision of that mapping was limited by the restriction fragments that were used for hybridization, the results of the experiments are compatible with the positions of the promoters and terminators which we found here through bioinformatic analysis.
We further characterized the terminator located immediately following the major capsid gene. We believe that this gene is expressed at a high level from its own promoter and that the terminator reduces the expression of the downstream genes by 40 to 50% relative to the expression of the capsid gene (Fig. 4). The reduced transcription of the nine downstream genes located prior to the next phage promoter presumably results in a lower abundance of their products in the virion. However, we note that that transcription is not necessarily the sole point of regulation, since, for example, in phage λ the efficiency of translating mRNA encoding different genes in the late operon varies over a nearly 1,000-fold range (50).
DNA replication.
Octomer skew analysis makes a good case for bidirectional replication in SP6 starting from an origin in gene 13 (T7-5). A bidirectional origin has been proposed for phage T7 downstream of gene 1; genomes are thought to recombine via their terminal direct repeats to form linear concatemers, in which the invasion of recombination forks begins unidirectional replication forks. A similar scenario for SP6 would produce the skew plot shown in Fig. 7, which shows that the 3′ end of the genome was replicated from a bidirectional origin on the adjacent, concatamerized phage genome.
However, the skew plot is also consistent with circularization of the SP6 genome, whereby the 3′ end of the genome is replicated by its own bidirectional origin. We favor this model since (i) the concentration of DNA ends would favor the formation of monomeric circles until there are very large numbers of SP6 genomes present and (ii) recombination forks in any linear concatemer would yield circular products of one or more phage genomes. In addition, since we argue that the P. putida prophage most likely entered the chromosome through a circular intermediate (see above), we believe that such a mechanism would also be available for DNA replication. Further work is required to resolve this issue.
Acknowledgments
This work was supported by NIH grant S06 GM08016 to M.G. and by NIH grant GM51975 to R.W.H., G.F.H., and J.G.L.
We thank Eugene T. Butler III for bringing SP6 to the lab, Agnes Day of Howard University for support of the project, and Hugh Nicholas of the Pittsburgh Supercomputing Center for assistance with the transcription terminator searches.
REFERENCES
- 1.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, T. 1960. On the fine structure of the temperate bacteriophages P1, P2 and P22, p. 1008. In Proceedings of the European Regional Conference on Electron Microscopy, vol. 2. Nederlandse Vereniging voor Electronenmicroscopie, Delft, The Netherlands.
- 3.Arisaka, F., S. Kanamaru, P. Leiman, and M. G. Rossmann. 2003. The tail lysozyme complex of bacteriophage T4. Int. J. Biochem. Cell Biol. 35:16-21. [DOI] [PubMed] [Google Scholar]
- 4.Artymiuk, P. J., T. A. Ceska, D. Suck, and J. R. Sayers. 1997. Prokaryotic 5′-3′ exonucleases share a common core structure with gamma-delta resolvase. Nucleic Acids Res. 25:4224-4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basham, D. A. 1989. In vitro analysis of site selective termination of transcription by SP6 RNA polymerase. Ph.D. thesis. Howard University, Washington, D.C.
- 6.Børsheim, K. Y. 1993. Native marine bacteriophages. FEMS Microbiol. Ecol. 102:141-159. [Google Scholar]
- 7.Brown, J. E., J. F. Klement, and W. T. McAllister. 1986. Sequences of three promoters for the bacteriophage SP6 RNA polymerase. Nucleic Acids Res. 14:3521-3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brussow, H., and R. W. Hendrix. 2002. Phage genomics: small is beautiful. Cell 108:13-16. [DOI] [PubMed] [Google Scholar]
- 9.Butler, E. T., and M. J. Chamberlin. 1982. Bacteriophage SP6-specific RNA polymerase. I. Isolation and characterization of the enzyme. J. Biol. Chem. 257:5772-5778. [PubMed] [Google Scholar]
- 10.Chenna, R., H. Sugawara, T. Koike, R. Lopez, T. J. Gibson, D. G. Higgins, and J. D. Thompson. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31:3497-3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Condron, B. G., J. F. Atkins, and R. F. Gesteland. 1991. Frameshifting in gene 10 of bacteriophage T7. J. Bacteriol. 173:6998-7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Condron, B. G., R. F. Gesteland, and J. F. Atkins. 1991. An analysis of sequences stimulating frameshifting in the decoding of gene 10 of bacteriophage T7. Nucleic Acids Res. 19:5607-5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delcher, A. L., D. Harmon, S. Kasif, O. White, and S. L. Salzberg. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27:4636-4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn, J. J., and F. W. Studier. 1983. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 166:477-535. [DOI] [PubMed] [Google Scholar]
- 15.Ermolaeva, M. D., H. G. Khalak, O. White, H. O. Smith, and S. L. Salzberg. 2000. Prediction of transcription terminators in bacterial genomes. J. Mol. Biol. 301:27-33. [DOI] [PubMed] [Google Scholar]
- 16.Felsenstein, J. 1985. Confidence limits on phylogenies with a molecular clock. Syst. Zool. 34:152-161. [Google Scholar]
- 17.Felsenstein, J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783-791. [DOI] [PubMed] [Google Scholar]
- 18.Felsenstein, J. 1974. The evolutionary advantage of recombination. Genetics 78:737-756. [DOI] [PMC free article] [PubMed]
- 19.Felsenstein, J. 1989. PHYLIP—phylogeny inference package (version 3.2). Cladistics 5:164-166.
- 20.Freiberg, A., R. Morona, L. Van den Bosch, C. Jung, J. Behlke, N. Carlin, R. Seckler, and U. Baxa. 2003. The tail spike protein of Shigella phage Sf6. A structural homolog of Salmonella phage P22 tail spike protein without sequence similarity in the beta-helix domain. J. Biol. Chem. 278:1542-1548. [DOI] [PubMed] [Google Scholar]
- 21.Garcia, E., J. M. Elliott, E. Ramanculov, P. S. Chain, M. C. Chu, and I. J. Molineux. 2003. The genome sequence of Yersinia pestis bacteriophage φA1122 reveals an intimate history with the coliphage T3 and T7 genomes. J. Bacteriol. 185:5248-5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordon, D., C. Abajian, and P. Green. 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8:195-202. [DOI] [PubMed] [Google Scholar]
- 23.Hendrix, R. W. 2002. Bacteriophages: evolution of the majority. Theor. Popul. Biol. 61:471-480. [DOI] [PubMed] [Google Scholar]
- 24.Hendrix, R. W., M. C. Smith, R. N. Burns, M. E. Ford, and G. F. Hatfull. 1999. Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage. Proc. Natl. Acad. Sci. 96:2192-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juhala, R. J., M. E. Ford, R. L. Duda, A. Youlton, G. F. Hatfull, and R. W. Hendrix. 2000. Genomic sequences of bacteriophages HK97 and HK022: pervasive genetic mosaicism in the lambdoid bacteriophages. J. Mol. Biol. 299:27-51. [DOI] [PubMed] [Google Scholar]
- 26.Kassavetis, G. A., E. T. Butler, D. Roulland, and M. J. Chamberlin. 1982. Bacteriophage SP6-specific RNA polymerase. II. Mapping of SP6 DNA and selective in vitro transcription. J. Biol. Chem. 257:5779-5788. [PubMed] [Google Scholar]
- 27.Kotani, H., Y. Ishizaki, N. Hiraoka, and A. Obayashi. 1987. Nucleotide sequence and expression of the cloned gene of bacteriophage SP6 RNA polymerase. Nucleic Acids Res. 15:2653-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kovalyova, I. V., and A. M. Kropinski. 2003. The complete genomic sequence of lytic bacteriophage gh-1 infecting Pseudomonas putida—evidence for close relationship to the T7 group. Virology 311:305-315. [DOI] [PubMed] [Google Scholar]
- 29.Krieg, P. A., and D. A. Melton. 1984. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res. 12:7057-7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krieg, P. A., and D. A. Melton. 1987. In vitro RNA synthesis with SP6 RNA polymerase. Methods Enzymol. 155:397-415. [DOI] [PubMed] [Google Scholar]
- 31.Lavigne, R., M. V. Burkal'tseva, J. Robben, N. N. Sykilinda, L. P. Kurochkina, B. Grymonprez, B. Jonckx, V. N. Krylov, V. V. Mesyanzhinov, and G. Volckaert. 2003. The genome of bacteriophage phiKMV, a T7-like virus infecting Pseudomonas aeruginosa. Virology 312:49-59. [DOI] [PubMed] [Google Scholar]
- 32.Lawrence, J. G., G. F. Hatfull, and R. W. Hendrix. 2002. Imbroglios of viral taxonomy: genetic exchange and failings of phenetic approaches. J. Bacteriol. 184:4891-4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leary, S. L., H. J. Baum, and Z. G. Loewy. 1991. DNA-dependent RNA polymerase from bacteriophage T3 transcribes and amplifies an RNA template in vitro. Gene 106:93-96. [DOI] [PubMed] [Google Scholar]
- 34.Lee, S. S., and C. Kang. 1993. Two base pairs at −9 and −8 distinguish between the bacteriophage T7 and SP6 promoters. J. Biol. Chem. 268:19299-19304. [PubMed] [Google Scholar]
- 35.Lobry, J. R. 1996. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. E vol. 13:660-665. [DOI] [PubMed] [Google Scholar]
- 36.Lowe, T. M., and S. R. Eddy. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melton, D. A., P. A. Krieg, M. R. Rebagliati, T. Maniatis, K. Zinn, and M. R. Green. 1984. Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Res. 12:7035-7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller, S., B. Schuler, and R. Seckler. 1998. Phage P22 tail spike protein: removal of head-binding domain unmasks effects of folding mutations on native-state thermal stability. Protein Sci. 7:2223-2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moak, M., and I. J. Molineux. 2004. Peptidoglycan hydrolytic activities associated with bacteriophage virions. Mol. Microbiol. 51:1169-1183. [DOI] [PubMed]
- 40.Muller, H. 1932. Some genetic aspects of sex. Am. Nat. 66:118-138. [Google Scholar]
- 41.Nam, S. C., and C. W. Kang. 1988. Transcription initiation site selection and abortive initiation cycling of phage SP6 RNA polymerase. J. Biol. Chem. 263:18123-18127. [PubMed] [Google Scholar]
- 42.Nelson, K. E., C. Weinel, I. T. Paulsen, R. J. Dodson, H. Hilbert, V. A. Martins dos Santos, D. E. Fouts, S. R. Gill, M. Pop, M. Holmes, L. Brinkac, M. Beanan, R. T. DeBoy, S. Daugherty, J. Kolonay, R. Madupu, W. Nelson, O. White, J. Peterson, H. Khouri, I. Hance, P. C. Lee, E. Holtzapple, D. Scanlan, K. Tran, A. Moazzez, T. Utterback, M. Rizzo, K. Lee, D. Kosack, D. Moestl, H. Wedler, J. Lauber, D. Stjepandic, J. Hoheisel, M. Straetz, S. Heim, C. Kiewitz, J. A. Eisen, K. N. Timmis, A. Dusterhoft, B. Tummler, and C. M. Fraser. 2002. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 4:799-808. [DOI] [PubMed] [Google Scholar]
- 43.Nielsen, D. A., and D. J. Shapiro. 1986. Preparation of capped RNA transcripts using T7 RNA polymerase. Nucleic Acids Res. 14:5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pajunen, M., S. Kiljunen, and M. Skurnik. 2000. Bacteriophage φYeO3-12, specific for Yersinia enterocolitica serotype O:3, is related to coliphages T3 and T7. J. Bacteriol. 182:5114-5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pajunen, M. I., M. R. Elizondo, M. Skurnik, J. Kieleczawa, and I. J. Molineux. 2002. Complete nucleotide sequence and likely recombinatorial origin of bacteriophage T3. J. Mol. Biol. 319:1115-1132. [DOI] [PubMed] [Google Scholar]
- 46.Pedulla, M. L., M. E. Ford, J. M. Houtz, T. Karthikeyan, C. Wadsworth, J. A. Lewis, D. Jacobs-Sera, J. Falbo, J. Gross, N. R. Pannunzio, W. Brucker, V. Kumar, J. Kandasamy, L. Keenan, S. Bardarov, J. Kriakov, J. G. Lawrence, W. R. Jacobs, R. W. Hendrix, and G. F. Hatfull. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171-182. [DOI] [PubMed] [Google Scholar]
- 47.Saitou, N., and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. E vol. 4:406-425. [DOI] [PubMed] [Google Scholar]
- 48.Salzberg, S. L., A. L. Delcher, S. Kasif, and O. White. 1998. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 26:544-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 50.Sampson, L. L., R. W. Hendrix, W. M. Huang, and S. R. Casjens. 1988. Translation initiation controls the relative rates of expression of the bacteriophage lambda late genes. Proc. Natl. Acad. Sci. 85:5439-5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scholl, D., S. Adhya, and C. R. Merril. 2002. Bacteriophage SP6 is closely related to phages K1-5, K5, and K1E but encodes a tail protein very similar to that of the distantly related P22. J. Bacteriol. 184:2833-2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scholl, D., J. Kieleczawa, P. Kemp, J. Rush, C. C. Richardson, C. R. Merril, S. Adhya, and I. J. Molineux. 2004. Genomic analysis of bacteriophages SP6 and K1-5, an estranged subgroup of the T7 supergroup. J. Mol. Biol. 1151-1171. 335: [DOI] [PubMed]
- 53.Shin, I., J. Kim, C. R. Cantor, and C. Kang. 2000. Effects of saturation mutagenesis of the phage SP6 promoter on transcription activity, presented by activity logos. Proc. Natl. Acad. Sci. 97:3890-3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steven, A. C., B. L. Trus, J. V. Maizel, M. Unser, D. A. Parry, J. S. Wall, J. F. Hainfeld, and F. W. Studier. 1988. Molecular substructure of a viral receptor-recognition protein. The gp17 tail-fiber of bacteriophage T7. J. Mol. Biol. 200:351-365. [DOI] [PubMed] [Google Scholar]
- 55.Tseng, T. Y., D. N. Frick, and C. C. Richardson. 2000. Characterization of a novel DNA primase from the Salmonella typhimurium bacteriophage SP6. Biochemistry 39:1643-1654. [DOI] [PubMed] [Google Scholar]
- 56.Weinel, C., K. E. Nelson, and B. Tummler. 2002. Global features of the Pseudomonas putida KT2440 genome sequence. Environ. Microbiol. 4:809-818. [DOI] [PubMed] [Google Scholar]
- 57.Zimmer, M., E. Sattelberger, R. B. Inman, R. Calendar, and M. J. Loessner. 2003. Genome and proteome of Listeria monocytogenes phage PSA: an unusual case for programmed +1 translational frameshifting in structural protein synthesis. Mol. Microbiol. 50:303-317. [DOI] [PubMed] [Google Scholar]