

Abstract
Previous attempts to identify neuroprotective targets by studying the ischemic cascade and devising ways to suppress it have failed to translate to efficacious therapies for acute ischemic stroke1. We hypothesized that studying the molecular determinants of endogenous neuroprotection in two well-established paradigms, the resistance of CA3 hippocampal neurons to global ischemia2 and the tolerance conferred by ischemic preconditioning (IPC)3, would reveal new neuroprotective targets. We found that the product of the tuberous sclerosis complex 1 gene (TSC1), hamartin, is selectively induced by ischemia in hippocampal CA3 neurons. In CA1 neurons, hamartin was unaffected by ischemia but was upregulated by IPC preceding ischemia, which protects the otherwise vulnerable CA1 cells. Suppression of hamartin expression with TSC1 shRNA viral vectors both in vitro and in vivo increased the vulnerability of neurons to cell death following oxygen glucose deprivation (OGD) and ischemia. In vivo suppression of TSC1 expression increased locomotor activity and decreased habituation in a hippocampal-dependent task. Overexpression of hamartin increased resistance to OGD by inducing productive autophagy through an mTORC1-dependent mechanism.
Keywords: endogenous neuroprotection, hamartin, global cerebral ischemia, CA3, proteomics, preconditioning, hippocampus, autophagy
Transient global ischemia can be caused by cardiac arrest or open heart surgery, resulting in selective and delayed cell death of CA1 hippocampal neurons4. Remarkably, neighbouring CA3 neurons are resistant to such an ischemic insult2. This differential response remains unexplained, despite extensive research focused on understanding the vulnerability of CA1 neurons to ischemia5-7.
To identifythe mechanisms responsible for the resistance of CA3 neurons to ischemia, we performed label-free proteomic analysis of CA1 and CA3 hippocampal regions from rats subjected to either sham ischemia (n=5) or to 10 min severe forebrain ischemia (n=5), induced by bilateral occlusion of both vertebral and common carotid arteries, followed by 24 h of reperfusion (Supplementary Fig. 1). Subcellular fractionation of CA1 and CA3 regions generated membrane and cytoplasmic fractions, allowing us to enrich for membrane proteins and increase the number of proteins identified (Supplementary Fig. 2). We pooled data sets from the membrane and cytoplasmic fractions from each experimental group to increase the power of identifying relevant pathways.
We carried out a detailed comparison and ontological analyses between the different data sets (Supplementary Figs. 3–5, Supplementary Tables 1–6, Supplementary Results and Discussion). We also conducted Ingenuity Pathway Analysis (IPA) to identify proteins and pathways of interest in a nonbiased manner. We compared the pathways significantly altered by ischemia within CA1 and CA3 regions, and identified those proteins selectively induced in CA3. In CA3 neurons, the PI3k/Akt intracellular signalling pathway was most significantly associated with the protein expression changes induced by ischemia compared to sham CA3 (p=0.00032; Fig. 1a and Supplementary Table 7). The expression of several proteins within this pathway, including hamartin and 14-3-3 (gamma, epsilon and theta) was higher following ischemia as compared to sham CA3. The neuroprotective properties of this pathway are diverse and well-documented for global8 and focal ischemia9, as well as for subarachnoid hemorrhage10.
Figure 1. Hamartin expression is selectively induced in the CA3 hippocampal area following ischemia.

(a) Illustration of the PI3k/Akt pathway, which was significantly associated with and selectively induced in the CA3 region following ischemia (p=0.000319, right-tailed Benjamini-Hochberg multiple testing correction). Proteins are described in Supplementary Table 7. (b) Diagram of a protein network showing changes in expression levels significantly associated with ischemia in the CA3 region. In (a) and (b), proteins in pink and yellow were upregulated and downregulated respectively, by ischemia. Proteins in grey were detected by the proteomic analysis, but their expression was unaffected. Proteins in white participate in the pathway or network but were not detected in the proteomic study. Images were created using IPA software. The relationships between the displayed proteins are indicated. Proteins are described in Supplementary Table 8. (c–h) Representative immunoblots (c,e,g) of hamartin expression and densitometric quantification of the immunoblotting data in membrane fractions (d,f,h) from sham CA1 (n=11) and sham CA3 (n=12) (c,d) and following 10 min of ischemia (n=5) and 12 h (n=5) and 24 h (n=10) of reperfusion in CA1 (e,f) and CA3 (g,h) regions. Values were corrected for total protein content, determined by Sypro staining, normalized such that the expression levels from sham ischemia samples were 1 and presented as mean ± S.E.M. (one-way analysis of variance (ANOVA), Bonferonni’s post hoc test, ***p<0.001, ****p<0.0001, compared to sham ischemia). (i) Representative immunofluorescence images (n=4) of hamartin (red), glial fibrillary acidic protein (GFAP; green) and DAPI (blue) staining in sham ischemic and ischemic hippocampal sections, following 24 h of reperfusion. Merged images are shown. Scale bars: 20 μm.
In addition to canonical pathways, IPA identifies networks of interacting proteins. Hamartin was part of the network of proteins most significantly associated with expression changes induced by ischemia in the CA3 region compared to sham CA3 (p<0.05; Fig. 1b and Supplementary Table 8).
Our proteomic datasets were corroborated by immunoblotting experiments (Supplementary Fig. 6) and are consistent with previous studies11-13 (Supplementary Results and Discussion). The identification of hamartin at all levels of our analysis (pathways, networks and individual proteins; Supplementary Results and Discussion, Supplementary Fig. 7 and Supplementary Table 9) led us to investigate its temporal and spatial expression profile in response to ischemia. We investigated the temporal profile of hamartin expression following ischemia within CA1 and CA3 membrane fractions. In sham rats, hamartin expression was similar between CA1 and CA3 fractions (Fig. 1c,d). Within CA1, hamartin expression was unaffected for the first 24 h following ischemia (Fig. 1e,f). Within CA3, hamartin expression was significantly induced after 10 min of ischemia, peaking at 12 h of reperfusion and remaining significantly elevated after 24 h of reperfusion compared to sham ischemia (Fig. 1g,h).
Hamartin showed a punctate nuclear and peri-nuclear expression pattern in the CA1 and CA3 regions, limited to pyramidal cells, with no co-localization with astrocytic markers (Fig. 1i). Importantly, 24 h following ischemia there was a qualitative increase of hamartin expression selectively in the nuclear and peri-nuclear region of CA3 cells, which is in agreement with the proteomic and immunoblotting results. As CA1 neurons subjected to ischemia are morphologically viable following 24 h of reperfusion, with cell death detected histologically at 48 h14, we hypothesized that their inability to upregulate hamartin contributes to their vulnerability.
Hamartin was also upregulated in the CA1 region following IPC, where a short duration of ischemia protects these otherwise vulnerable cells from neuronal death induced by severe forebrain ischemia3. Following ischemia, hamartin protein expression was significantly higher (p<0.01) in the CA1 region of rats subjected to 2 min IPC compared to sham IPC (Supplementary Fig. 8).
To confirm whether expression alters neuronal vulnerability to ischemia, we carried out OGD studies in rat hippocampal neurons. We silenced hamartin expression using a lentiviral vector expressing an shRNA targeting TSC1 (TSC1 shRNA; Supplementary Fig. 9), and subjected cultures treated with a TSC1 shRNA vector to 3 h OGD and 24 h reperfusion (Fig. 2a). Viability assays revealed that TSC1 shRNA-transduced cultures exhibited 34±6.7% higher cell death relative to control shRNA-transduced cultures (p<0.0001; Fig. 2b,c). We observed a similar effect with rat cortical neurons (Supplementary Fig. 10). To control for TSC1 shRNA off-target effects we rescued hamartin expression in TSC1 shRNA-treated hippocampal cultures with a lentiviral vector expressing human TSC1 (Supplementary Fig. 11) and found that overexpression of hamartin reduced cell death after OGD to control levels, suggesting the knockdown is specific (Fig. 2d,e). Our results are consistent with studies on Tsc1−/− mouse embryonic fibroblasts exhibiting increased apoptosis after glucose deprivation15. Additionally, Tsc1−/−hippocampal neurons show high susceptibility to oxidative and endoplasmic reticulum stress16 and have high sensitivity to spontaneous glutamate release17. As oxidative stress and glutamate-induced excitotoxicity are major components of ischemic cell death, our findings suggest Tsc1 conditional-knock-out mice may be very sensitive to ischemia.
Figure 2. Hamartin regulates neuronal susceptibility to OGD-induced cell death.
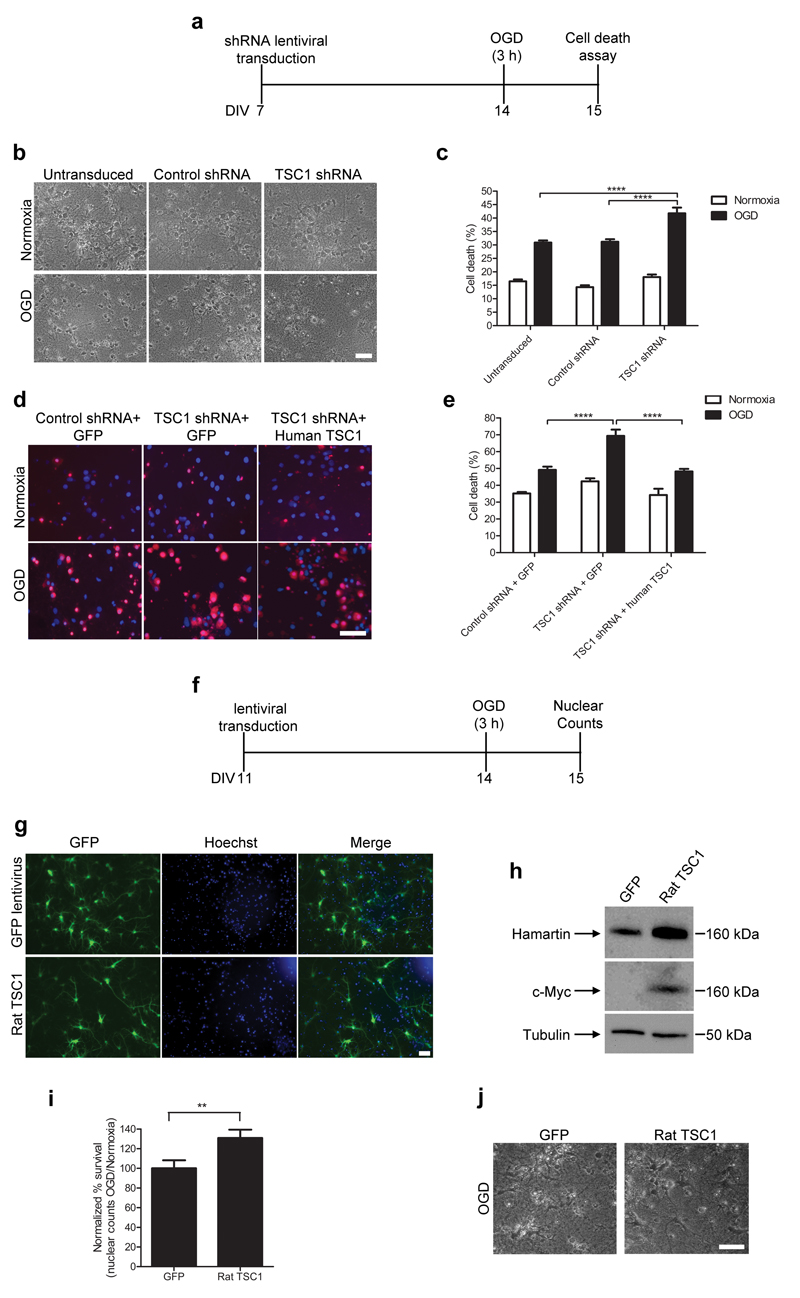
(a) Schematic of in vitro experiments on hippocampal cultures transduced with shRNA vectors and exposed to OGD. DIV, days in vitro. (b,c) Representative phase photomicrographs (b) and quantification of cell death (c) following OGD and normoxia by lactate dehydrogenase (LDH). Cells were untransduced or treated with either TSC1 or control shRNA vectors (n=8–13; one-way ANOVA with Bonferroni’s post hoc test, ****p<0.0001). (d) Merged fluorescent images from cultures treated as shown, stained with ethidium homodimer III (red) and Hoechst 33342 (blue). (e) Quantification of necrotic neurons in (d) as a percentage of total cells (n=4–8 one-way ANOVA with Bonferroni’s post hoc test, ****p<0.0001). (f) Experimental design of in vitro experiments for hippocampal cultures transduced with rat TSC1 vectors and exposed to OGD. (g) Immunofluorescent images of cultures transduced with GFP or rat TSC1 (left). Hoechst 33342 was used for nuclear stain (middle). Merged images are shown on the right. (h) Representative immunoblots of cultures transduced with Myc-tagged rat TSC1 (n=3). (i) Quantification of neurons surviving OGD and 24 h of reperfusion, normalized to intact nuclei counts of OGD to normoxia for GFP-transduced cultures (n=12; two-tailed t-test, **p<0.01). (j) Representative phase photomicrographs of hippocampal cultures from the experiments quantified in (i). Scale bars are 50 μm. All data are expressed as mean ± S.E.M.
As hamartin suppression in vitro rendered neurons more vulnerable to ischemia, we investigated whether hamartin is sufficient to protect neurons from ischemic insults (Fig. 2f). Rat hippocampal neurons transduced with a lentiviral vector expressing rat Tsc1–eGFP (rat TSC1) overexpressed hamartin compared to neurons treated with a control vector expressing eGFP alone (GFP) (Fig. 2g,h). To detect exogenous expression of full-length hamartin, the rat TSC1 lentiviral vector contained a c-Myc tag at the 3′ end of the Tsc1 sequence (Fig. 2h). Transduction efficiency assessed by eGFP expression was ~50% (Fig. 2g). Hippocampal neurons transduced with rat TSC1 exhibited significantly higher resistance to OGD and reperfusion compared to GFP-transduced neurons. The number of cells surviving OGD versus normoxia was 31±8.6% higher in rat TSC1- compared to GFP-transduced cells, (p=0.0066; Fig. 2i,j). These results provide direct evidence that hamartin is neuroprotective.
To translate our findings to an in vivo paradigm, we used the same shRNA lentiviral vectors to suppress hamartin expression in rat CA3 neurons. Hamartin expression was significantly reduced (p<0.01) in CA3 neurons 14 d following unilateral administration of the shRNA vector targeting TSC1 (Supplementary Fig. 12). The magnitude of suppression was less than that achieved in vitro, perhaps as a result of there being both transduced and untransduced cells in the microdissected tissue. Fourteen days after administration of TSC1 shRNA and control shRNA into the ipsilateral and contralateral hippocampus, respectively, we subjected rats to ischemia or sham ischemia (Fig. 3a). Because global ischemia produces a uniform bilateral insult, we used the contralateral side as control. Following ischemia, the ipsilateral TSC1 shRNA-treated side had 105.9±4.0 neurons per mm in the CA3 region compared to 139.2±3.2 neurons per mm on the contralateral control shRNA-treated side (p<0.01; Fig. 3b,c). Activation of compensatory mechanisms in some CA3 cells with suppressed Tsc1 expression, as well as a non-linear relationship between shRNA knockdown efficiency and loss of CA3 neuronal resistance, could explain the modest effect of TSC1 shRNA. Tsc1 knockdown did not alter neuronal cell number in the CA3 region of sham operated rats (Fig. 3b,c). These data indicate that the resistance of CA3 neurons to ischemia is mediated by upregulation of hamartin.
Figure 3. Resistance of CA3 neurons to ischemia is mediated by upregulation of hamartin in vivo.

(a) Design of experiments in which rats were stereotaxically injected unilaterally with a TSC1 shRNA vector in the dorsal CA3 pyramidal layer and subjected to either ischemia or sham ischemia (n=5). (b) Quantification of the number of neurons per mm in the dorsal CA3 pyramidal layer (one-way ANOVA with Bonferroni’s post hoc test, **p<0.01) from rats treated as in (a). (c) Representative hematoxylin and eosin stained hippocampal sections from rats treated as described in (a). Arrowheads show the needle tract. Scale bars: 1.00 mm (top row), 0.01 mm (middle and bottom rows). Boxed areas indicate the magnified CA3 regions shown in the bottom images. (d,e) Quantification of boxes crossed (d) and rears performed (e) in an open field test on rats bilaterally injected with TSC1 shRNA (n=6) or control shRNA (n=4) and subjected to ischemia (two-way ANOVA with Bonferroni’s post hoc test, *p<0.05, ***p<0.001). Open field test was carried out at three time-points: baseline, immediately before ischemia (second test) and 7 d after ischemia (third test). Data are mean ± S.E.M.
We also examined whether hippocampal function was affected by Tsc1 suppression in CA3 neurons using an open field test18, as these neurons participate in acquisition and encoding of spatial information19. We quantified horizontal and vertical locomotor activity by measuring the number of boxes crossed and rears performed. Naive rats subjected to sham ischemia exhibited the expected pattern of habituation after repeated testing, with a significant decrease in both the number of boxes crossed and rears performed (Fig. 3d,e). Ischemia results in loss of habituation manifested by increased locomotor activity, which is consistent with the extent of neuronal loss in the pyramidal layer of the hippocampus20. Rats injected with either TSC1 shRNA or control shRNA bilaterally in the CA3 region showed a significant increase in locomotor activity after ischemia (Fig. 3d,e). Importantly, TSC1 shRNA-treated rats had a significantly higher increase in both parameters compared to control shRNA-treated rats (p<0.05). Quantification of CA3 neurons after ischemia (Supplementary Fig. 13) revealed similar results to the unilateral experiments, indicating that the increased locomotor activity and loss of habituation were associated with a loss of CA3 neurons.
An important downstream function of hamartin and its partner tuberin is mTORC1 suppression via direct inhibition of small GTPase Rheb21. To examine the role of mTORC1 in the vulnerability of hippocampal neurons to OGD following hamartin suppression, we monitored mTORC1 activity by assaying phosphorylation levels of S6 Ribosomal Protein (S6RP). S6RP phosphorylation was higher in TSC1 shRNA-treated rat hippocampal cells compared to control shRNA-transduced cells, suggesting increased mTORC1 activity (Fig. 4a). Treatment of TSC1 shRNA-transduced cultures with the mTORC1 inhibitor rapamycin prevented S6RP phosphorylation and increased neuronal survival in response to OGD to 70.9±4.71% from 45.9±3.19% for cultures treated with TSC1 shRNA alone (Fig. 4a,b). Therefore, silencing of hamartin exacerbates OGD-induced injury by activating mTORC1. Importantly, rapamycin has been a promising candidate for treatment in Parkinson’s disease22, cardioprotection23 and focal cerebral ischemia24.
Figure 4. Hamartin promotes neuronal survival by inhibiting mTORC1 and inducing productive autophagy.

(a) Representative immunoblot of phosphorylated S6RP expression in control shRNA or TSC1 shRNA-transduced rat hippocampal neuronal cultures (under normoxia or OGD) in the presence or absence of 10 nM rapamycin (n=3). (b) Quantification of percentage survival (OGD to normoxia nuclear counts ratio) from control shRNA- or TSC1 shRNA-transduced cultures, with or without 10 nM rapamycin (n=3–9). (c,d) Representative immunoblots depicting LC3-I, LC3-II and p62 expression (c) and quantification of LC3-II and p62 levels (d) in the hippocampal cultures described in (a) (n=3). (e) Quantification of macroautophagy-dependent (3MA-sensitive) lysosomal degradation in control-transduced and TSC1 shRNA-transduced cultures (n=3). (f,g) Representative immunoblots of LC3-I, LC3-II and p62 expression (f) and quantification of LC3-II and p62 expression (g) from GFP- and rat TSC1-transduced cultures exposed to normoxia or OGD (n=3). (h) Quantification of macroautophagy-dependent lysosomal degradation in GFP- and rat TSC1-transduced cultures (n=3). (i) Quantification of percentage survival (OGD to normoxia nuclear count ratio) of GFP- and rat TSC1-transduced cultures, described in Figure 2i with or without 3MA (10 mM). For (d) and (g), data are normalized to control shRNA treated levels. Sypro staining was used as loading control. For (e) and (h), data are normalized to 3MA-sensitive degradation from control shRNA-transduced cultures following normoxia. Values are mean ± S.E.M. of n=3 independent experiments, and within each experiment at least triplicate samples per condition were assessed (two-way ANOVA with Bonferroni’s post hoc test (b, d, e, h, i) or two tailed t-test (g), *p<0.05, **p<0.01, ***p<0.001).
mTORC1 is an inhibitor of autophagy, a catabolic process implicated in ischemic pathophysiology25. Autophagy degrades damaged organelles and protein aggregates, enclosing them inside autophagosomes and digesting them with hydrolases after autophagosome fusion with lysosomes. Efficient completion of this cascade is termed productive autophagy25. Because hamartin indirectly inhibits mTORC1 activity, we deciphered its neuroprotective mechanism by assessing autophagy. Upon induction of autophagy, microtubule-associated protein 1 light chain 3 (LC3) is processed from a 16 kDa form (LC3-I) to a 14kDa form (LC3-II), which is recruited to autophagosomes and is an indicator of autophagosome formation26. LC3-II expression was significantly higher in TSC1 shRNA-transduced compared to control shRNA-transduced rat hippocampal cultures following both normoxia and OGD (Fig. 4c,d). As suppression of hamartin expression activated mTORC1, these results suggested that autophagosome accumulation resulted from impaired autophagic flux (fusion and degradation of autophagosomes with lysosomes). Expression of sequestosome-1 (p62) protein, which is degraded by lysosomes and accumulates following impairments in autophagy, was significantly increased by suppression of hamartin compared to control shRNA-transduced rat hippocampal cultures following both normoxia or OGD (Fig. 4c,d). We found further evidence for impaired autophagy by measuring lysosomal degradation of long-lived proteins, during macroautophagy. We used the the autophagy inhibitor 3-methyladenine (3MA) to dissect the macroautophagy component of lysosomal activity. During normoxia, TSC1 shRNA-transduced neurons exhibited significantly less macroautophagy-dependent degradation (down 53±10% as compared to control shRNA-transduced cultures), which was completely diminished following OGD (Fig. 4e). Therefore, suppression of hamartin expression results in the accumulation of autophagosomes and impaired autophagic flux both in normoxia and OGD in hippocampal neurons.
Tsc1 overexpression in rat hippocampal neurons upregulated LC3-II expression, suppressed p62 expression by 44±7% and increased 3MA-sensitive degradation 340±40% compared to GFP-transduced cultures after OGD (Fig. 4f–h). Inhibition of autophagy in rat TSC1-transduced cultures with 3MA abolished the protection conferred by overexpression of rat TSC1, reducing neuronal survival to 23±2% from 47±3% for untreated cultures overexpressing hamartin (p<0.001; Fig. 4i). Taken together, these findings suggest that overexpression of hamartin conferred protection to hippocampal neurons to OGD by inducing efficient autophagic flux.
The role of autophagy in cerebral ischemia is controversial, with some studies showing that activation of autophagy is detrimental27, 28, while others supporting a neuroprotective role29, 30. In keeping with our findings, it was previously31 reported that global ischemia increased the number of autophagosomes in CA1 due to decreased autophagosome degradation, which indicates impaired autophagy. Moreover, autophagy was implicated in mediating neuroprotection in response to IPC32, which is consistent with our finding of hamartin upregulation in CA1 neurons following IPC. All these observations highlight the key role of both the extent and time point of autophagy induction in determining the outcome following ischemia.
Hamartin has been studied in the context of cancer, epilepsy and more specifically tuberous sclerosis (which results from TSC1 mutations)33. This study shows the importance of examining endogenous neuroprotection in identifying new targets and suggests that hamartin confers resistance against ischemia by inducing productive autophagy. Although the resistive properties of CA3 neurons to ischemia are absent in other paradigms, such as traumatic brain injury, our finding that hamartin also alters susceptibility of cortical neurons conventionally affected by focal ischemia highlights the translational potential of hamartin as a target for neuroprotection in stroke.
Online Methods
Rats
All procedures were conducted in accordance with regulations of the Animal Care Committee at the University of Calgary or with the 1986 Animals Act (Scientific Procedures) under project licence from the United Kingdom Home Office or with the Institutional Animal Care and Use Committee of the Biomedical Research Foundation Academy of Athens. Experiments were approved by the clinical medicine ethical review committee of the University of Oxford. Male adult Wistar rats (200g±10%) were obtained from Charles River Laboratories (Calgary) or from Harlow, UK.
Global forebrain ischemia
We subjected rats either to 10 min severe ischemia or to 2 min IPC, followed by 72 h of reperfusion and 10 min ischemia using a modified four-vessel occlusion (4-VO) method34. Rats were anaesthetized with isofluorane (1% to 1.5% (v/v) maintenance) in 30% (v/v) O2 and 70% (v/v) N2O. Common carotid arteries were dissected, exposed and a 3-O silk suture was looped around them. The vertebral arteries were electrocauterized. Rats were fastened overnight, with free access to water, to ensure low serum glucose levels. Twenty-four hours after the preparatory surgery, rats were briefly anesthetized and both common carotid arteries were temporarily occluded with aneurysm clips for 2 min (IPC) or 10 min (ischemia). A 2-O silk suture, previously inserted through the neck, posterior to the trachea, esophagus, external jugular veins and common carotid arteries, but anterior to the cervical and paravertebral muscles, was tightened to prevent any collateral blood flow. We further used rats if they showed complete loss of consciousness, loss of tail and foot pad response and loss of righting and corneal reflex. We regulated core temperature at 37.0±0.5°C in all rats during surgery, using a rectal thermometer connected to a feedback-controlled heating pad, and for 24 h following ischemia, by a telemetry probe previously inserted in the peritoneal space controlling an infrared lamp.
For sham ischemia and sham IPC, the vertebral arteries were electrocauterized and the carotid arteries were manipulated but not occluded. We killed rats either immediately after 10 min ischemia or 12 h, 24 h or 7 d following reperfusion.
Core temperature telemetry probe implantation and regulation
We implanted sterilized core temperature telemetry probes (TA-F40, Data Sciences International) into the peritoneal cavity 7 d prior to any further surgery under isofluorane anaesthesia (3% (v/v) induction, 1% (v/v) to 1.5% (v/v) maintenance) in 30% (v/v) O2 and 70% (v/v) N2O. Following ischemia, rats were placed on receivers (RLA-1020, Data Sciences International) connected to infrared lamps. Temperature was sampled in the freely moving rats every 30 sec and regulated (37.0±0.5°C) using the computerized temperature control system ART-2.2.
Subcellular fractionation
We microdissected rat brains on ice, sectioned them at 500 μm, and punched-separated the CA1 and CA3 regions based on their visual boundaries under a surgical microscope. We carried out subcellular fractionation using a modified version of Guillemin35. We conducted all steps at 4°C and all buffers were supplemented with 1 mM PMSF, 10 mM NaVO4 and a protease inhibitor cocktail (Roche Diagnostics). We homogenized tissues in 1ml CLB buffer (10 mM HEPES, 10 mM NaCl, 1 mM KH2PO4, 5 mM NaHCO3, 5 mM EDTA, 1 mM CaCl2, 0.5 mM MgCl2, 250 mM sucrose, pH 7.4) with a glass/glass homogenizer. We centrifuged the homogenates at 1,000×g for 10 min. The pellet was resuspended in 1 ml CLB buffer and centrifuged at 1,000×g for 10 min. We combined the resulting supernatants to derive a mixed cytoplasmic and membrane fraction. Following sedimentation at 107,000×g for 30 min, the resulting supernatant (cytoplasmic fraction), was stored at −80 °C. We resuspended the pellet in 300 μl solubilisation buffer (20 mM Tris, 7 M urea, 2 M thiourea, 4% (w/v) CHAPS, pH 7.4) and incubated for 1 h at 4 °C. Samples were centrifuged at 120,000×g for 3 h and the supernatant (membrane proteins) was stored at −80 °C.
Sample preparation and analysis by tandem mass spectrometry
We analyzed digested, desalted and concentrated protein samples (75 μg) by nanoUPLC-MSE as described previously36. We determined protein content for the cytoplasmic fraction using the Dc Protein assay (BioRad), and for the membrane fraction using the EZQ protein assay (Invitrogen). We precipitated protein (75 μg) using the methanol/chloroform method, evaporated, resuspended in 100 mM Tris-pH 7.8, 6 M urea buffer, reduced for 30 min (100 mM Tris-pH 7.8, 195 mM DTT), alkylated for 30 min (100 mM Tris-pH 7.8, 195 mM iodoacetamide), diluted with 100 mM Tris-pH 7.8 (1:5) and digested with sequence grade modified trypsin (Promega) (200 ng μl−1) at 37°C overnight. Digested peptides were desalted and concentrated using Sep Pak C18 column cartridges (Waters) according to the manufacturer’s instructions. We performed sample analysis using a 75 μm I.D.x 25cm C18 nanoAcquity™ UPLC™ column (Waters) and a 90 min gradient: 2% (v/v) to 45% (v/v) solvent B (solvent A: 99.9% (v/v) H2O, 0.1% (v/v) formic acid; solvent B: 99.9% (v/v) acetonitrile, 0.1% (v/v) formic acid) on a Waters nanoAcquity™ UPLC™ system (final flow rate 250nl min−1, 7000 psi) coupled to a Waters QTOFpremier™ tandem mass spectrometer (Waters)36. Data were acquired in high-definition MSE mode and processed with ProteinLynx Global Server (PLGS version 2.2.5, Waters) to rebuild MS/MS spectra by combining all masses with identical retention times. We calibrated the raw data using glu-fibrinopeptide (200fmol μl−1, 700 nl min−1 flow rate, 785.8426 Da [M+2H]2+) as a lock mass. We processed the raw data sets including deisotoping, deconvolution, and peaklists generated on the basis of assigning precursor ions and fragments based on similar retention times. We used a SwissProt database (release 51.0, 10/2006, 241,242 entries) to identify the protein identities with the following parameters: peptide tolerance 15ppm, fragment tolerance 0.015 Da, trypsin missed cleavages 1, variable modifications: carbamidomethylation, M oxidation.
Quantitative analysis of the proteomic dataset
We analyzed quantitative changes in protein expression, based on mass spectrometry peptide ion peak intensities using the Waters Expression Analysis Software (WEPS™). For normalization, the “auto-normalization” function was used. Included protein hits were identified with a confidence of >95%. Identical peptides across samples were clustered based on mass precision (<15 ppm, typically 5ppm) and a retention time tolerance of <0.25 min, using the clustering software included in PLGS 2.2.5. In order to avoid potential errors, if two or more distinct proteins shared an identical peptide but were found to be regulated differently, then the quantitation algorithm did not include the peptide in question. Only proteins with a score higher than 45 were included for further analysis. The associated p value of protein level changes between samples was calculated by the PLGS 2.2.5 software using a Monte Carlo algorithm. Proteins that were detected in all experimental groups were selected for further analysis. Quantitative differences were used in subsequent analysis only for proteins with a p value for the change less than 0.05, and with a minimum 10% difference in their expression levels between the corresponding groups.
Ingenuity pathway analysis
We analyzed identified proteins together with the quantitative data using the IPA software and database (IPA, Ingenuity Systems). Pathway analysis identified the pathways from the IPA library of canonical pathways that were most significant to the data set. Proteins from the data set that met the expression change cut off between conditions of 10% and were associated with a canonical pathway in the Ingenuity Knowledge Base were considered for the analysis. The significance of the association between the data set and the canonical pathway was measured using the right-tailed Benjamini-Hochberg multiple testing correction. The calculated p value determined the probability that the association between the proteins in the data set and the canonical pathway is explained by chance alone.
For protein network generation, the data set containing protein identifiers and corresponding expression values was uploaded into the IPA application. Each protein was mapped to its corresponding object in the Ingenuity Knowledge Base. A protein expression change cut off of 10% between conditions was set to identify proteins whose expression was significantly differentially regulated. These proteins, called network-eligible proteins, were overlaid onto a global molecular network developed from information contained in the Ingenuity Knowledge Base. Networks of network-eligible proteins were then algorithmically generated based on their connectivity.
Ontological analysis
In addition to IPA, we analyzed individual datasets using the online panther classification system (www.pantherdb.org), which classifies proteins ontologically according to their molecular function or their biological process.
Primary hippocampal cultures and lentiviral transduction
We prepared hippocampal and cortical cultures from E18 rat embryos , as previously described37. We plated dissociated hippocampal or cortical cells onto 12– or 24–well plates at a density of 200,000 cells cm−2. We maintained cells in Neurobasal medium (Invitrogen), with 2% B27 supplement (Invitrogen). We replaced half the medium with fresh medium every three days. We supplemented hippocampal cells for the first 3 d with 25 μM L-glutamate to promote cell sprouting and improve neuronal viability. We transduced cells with lentiviral vectors at a multiplicity of infection of 10–20, 7 days in vitro (DIV) for shRNA studies and at 11 DIV for overexpression studies. We added rapamycin (10 nM) or 3MA (10 mM) or vehicle 24 h prior to normoxia or OGD.
Oxygen glucose deprivation
We induced OGD in hippocampal cultures by washing twice and immersing in 500 μl deoxygenated custom Neurobasal media without glucose, aspartate, glutamate, glutamine, or pyruvate (Invitrogen). We removed oxygen by bubbling the immersion solution for 30 min with a premixed gas (85% N2, 10% H2, 5% CO2). We sealed cultures inside a modular chamber (Billups-Rothenberg) flushed for 10 min with the same premixed gas and placed inside an incubator for 3 h. Anoxia was confirmed using disposable anaerobic indicator strips (GasPak, BD Biosciences). We treated control cultures similarly but with normoxic custom Neurobasal media supplemented with 3 mM d-glucose in a normoxic incubator.
We induced OGD in cortical cultures by washing twice and immersing in 500 μl deoxygenated glucose-free balanced salt solution (BSS; 20 mM HEPES, 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 0.03 mM glycine, pH 7.4) in a CellHouse 170 hypoxic incubator (Heto Holten) under 95% N2 and 5% CO2 (<0.1% O2). The immersion solution was pre-equilibrated overnight in the hypoxic incubator, resulting to a pO2 less than 10 mmHg, measured with an ABL-77 gas analyser (Radiometer). We treated control cultures in a similar fashion but with normoxic BSS solution containing 3 mM d-glucose in a normoxic incubator. Following OGD, both hippocampal and cortical cultures were fed with their original medium, which was replaced 1:1 with fresh medium, and incubated for 24 h in a normoxic incubator.
Cell death assays
We assessed cell death in primary neuronal cultures quantitatively using the LDH release Cytotox96® assay (Promega) and the necrotic/healthy cells detection kit (PromoKine).
For both assays, we carried out measurements in triplicate for at least n=3 independent experiments in a blinded fashion. For the LDH assay, the results were expressed as a ratio of the amount of LDH released in the cell culture media to the total LDH content, measured in lysed sister cultures.
For the necrotic/healthy cells detection kit, cells were co-stained with Hoechst 33342 and ethidium homodimer III. We visualised fluorescence using a fluorescent microscope (NIKON) with a 20x lens. At least 500 neurons were counted. We acquired images using the Simple-PCI software (Digital Pixel). The data were expressed as a percentage of the number of neurons stained with ethidium homodimer III to the number stained with Hoechst 33342.
We quantified alternatively viable cells by counting the number of intact nuclei in a haemocytometer, after lysing the cells in a detergent-containing solution38. Cell counts were performed in triplicate in a blinded fahion.
Lentiviral vectors
Lentiviral vectors (Sigma-Aldrich) used were: pLKO.1 TSC1 shRNA (5′- CCG GCC GGG AGC TGT TCC GTA ATA ACT CGA GTT ATT ACG GAA CAG CTC CCG GTT TTT G-3′), which targets nucleotides 2,262-2,282 of the rat TSC1 mRNA (GenBank accession number NM021854)); pLKO.1 control shRNA (5′-CCG GCA ACA AGA TGA AGA GCA CCA ACT CGA GTT GGT GCT CTT CAT CTT GTT GTT TTT- 3′), which contains a scrambled sequence that does not target any known rat genes; pLKO.1 TurboGFP, which contains the TurboGFP gene.
Recombinant lentiviral particles encoding rat Tsc1-eGFP, human TSC1 or a control eGFP plasmid were generated by co-transfection of human embryonic kidney (HEK) 293T cells with each expression plasmid (pEZ-Lv201 vector) together with the GeneCopoeia Lenti-Pac HIV Expression Packaging Kit, according to manufacturer’s instructions (Lifesciences, Source Bioscience). We cultured HEK 293T cells in DMEM (Invitrogen) supplemented with 10% FBS and 1% penicillin/streptomycin. Two days before transfection, we plated 1.5×106 cells onto 10 cm dishes. For transfection, 2.5μg of vector DNA (rat or human TSC1, or control eGFP plasmid) and 2.5μg of Lenti-Pac HIV mix were added in 293T cells in the presence of the Endofectin Lenti transfection reagent (GeneCopoeia). After 16 h, we removed the medium and the cells were washed twice with PBS and returned to the normal culture medium supplemented with 1/500 volume of the Titer Boost reagent. We collected medium containing recombinant lentiviral particles 48 h post-transfection and centrifuged (400×g, 10 min, 4 °C) to remove debris. After filtration through a 0.45 μm filter unit (Millipore, USA) we centrifuged the supernatant at 46,100×g for 4.5 h at 4 °C in SS34 rotor (Sorvall). We resuspended the viral particles in 50μl per tube of PBS supplemented with 0.5% BSA, aliquoted and stored at −80°C. We determined lentiviral titers by seeding HeLa cells in 12–well plates at 5×104 cells per well, 3–4 h before infection with serial dilutions of the concentrated viral stock. After incubation for 2 d, the medium was removed and the eGFP expressing cells were identified by FACS. Titers ranged from 4×108 to 2.5×109 infectious units (IU ml−1). The human TSC1 mRNA sequence contains six nucleotide mismatches compared to the rat Tsc1 mRNA base pairs 2,262–2,282 making it resistant to TSC1 shRNA knock-down.
Stereotaxic administration of lentiviral vectors
We administered lentiviral vectors into the hippocampus of live rats by stereotaxic injection 14 d before any further treatment. A 5.0 μl volume containing 5×109 particles ml−1 was injected into the hippocampus (3.8mm posterior to Bregma, 3.8 mm lateral to Bregma, 3.0 mm below the dura) using a 10 μl Hamilton syringe with a 34–gauge needle at a flow rate of 0.2 μl min−1. The needle was left in place for an additional 3 min and withdrawn at rate of 1 mm min−1. We stereotaxically administered the control pLKO.1 TurboGFP vector to determine the extent of gene expression in the hippocampus. GFP immunoreactivity was detected ± 0.5 mm anterio-posteriorly from the injection site.
Immunoblotting
We precipitated cytoplasmic and membrane fractions or whole cell homogenates (20–50 μg) using methanol/chloroform resolved by SDS-PAGE and transferred to PVDF membranes, stained with Sypro or Ponceau S to determine loading (total density of eight most prominent bands per lane; representative part of lane shown in Figures 1 and 4), and probed with the indicated antibodies indicated in Figures 1, 2 and 4 and Supplementary Figures 2,6,8-12. We developed immunoreactive species using ECL (Amersham), quantified by densitrometry (VisionWorks Analysis Software, UVP Biospectrum AC imaging system) and corrected for loading using the quantified data from loading controls.
Immunofluorescence
We cut coronal sections (6 μm) of paraffin-embedded brains at 3.6 to 4.16 mm posterior to bregma. After deparaffinization, brain sections were rehydrated, treated for antigen retrieval (DAKO) and blocked with 10% (v/v) goat serum in PBS. We incubated sections with the indicated primary antibodies indicated in Figure 1 and Supplementary Figure 12 overnight at 4°C, followed by Cy3 or Cy5 secondary antibodies. Mounting medium contained DAPI (Vector Laboratories) as a counter stain. We detected fluorescence with fluorescent microscopy and images were processed for background subtraction using the NIS elements imaging software (Nikon).
Antibody reagents for immunoblotting (IB) and immunofluorescence (IF) experiments
We used antibody to 14-3-3 theta (sc732, Santa Cruz), 1:2000 dilution (IB), antibody to hamartin (ab32936, Abcam), 1:1000 and 1:100 for IB and IF respectively, antibody to β-tubulin (ab7792, Abcam), 1:9,000 (IB), antibody to GFP (AB3080, Millipore), 1:100 (IF), antibody to GFAP (ab7260, Abcam), 1:1000 (IF), Cy3-conjugated goat anti-chicken IgG (ab97145, Abcam) at 1:100 (IF), Cy5-conjugated goat anti-rabbit IgG (AP187S, Millipore) at 1:200 (IF), anti-protein disulfide isomerase (ab3672, Abcam) at 1:250 (IB), antibody to calnexin (ab22595, Abcam), 1:1000, antibody to Na+, K+ ATPase (sc58631, Santa Cruz), 1:5000 (IB), antibody to histone H3 (ab61251, Abcam), 1:5000 (IB), antibody to p-S6RP ser 235/236 (4857S, Cell signaling technology), 1:100 at (IB), antibody to p62 (MABC32, Millipore), 1:100 (IB), antibody to LC3 (5F10, Nanotools Antikoeper technik), 1:200 (IB)
Perfusion-fixation
Rats were killed by transcardiac perfusion with chilled (4 °C) PBS solution and 4% (v/v) formalaldehyde in PBS. Brains were removed and incubated overnight in chilled 10% (v/v) formaldehyde in PBS, before dehydration and embedding in paraffin wax.
Hematoxylin and eosin staining
We cut coronal sections (10μm) of paraffin-embedded brains at 3.6 to 4.16 mm posterior to bregma. After deparaffinization in Clearene and rehydration, sections were stained with hematoxylin and eosin 39.
CA1 and CA3 cell counting
We counted healthy, viable cells in the entire CA3 molecular layer of the hippocampus in hematoxylin and eosin stained coronal sections at 3.6, 3.8 and 4.16 mm posterior to bregma. Three sections per rat were examined. Averaged measurements were expressed as number of neurons mm−1 ± S.E.M. Measurements were done in a blinded fashion.
Preparation of whole cell homogenates
We prepared whole cell homogenates from microdissected CA3 tissue and primary cortical cultures. We harvested hippocampal or cortical cultures in homogenisation buffer (50 mM Tris-HCl, 150 mM NaCl, 1mM EDTA, 1% (v/v) Triton X-100, 0.1% (w/v) SDS, pH 7.4, 4 °C) supplemented with protease inhibitors, 1 mM PMSF and phosphatase inhibitors. We centrifuged homogenates at 1,000×g for 10 min. The pellet was resuspended in homogenisation buffer and centrifuged at 1,000×g for 10 min. The resulting supernatants were combined. For tissue, the same procedure was followed, but homogenisation was done using a polytron homogenizer (Omni International).
Protein degradation assay
We measured total protein degradation by radioactive labeling as previously described37, determined as the percentage of the initial total acid precipitable radioactivity (protein) in the cell lysates transformed to acid soluble radioactivity (amino acids and small peptides) in the medium during the incubation. We quantified total lysosomal degradation as the NH4Cl-inhibited degradation, while macroautophagic degradation was quantified using the 3MA- inhibited proteolysis.
Open field test
The open field test was conducted by an investigator blinded to treatment, as previously described40. Rats used in open field testing were grouped according to the boxes crossed and rears performed at baseline and randomly assigned to different experimental treatments, so there was no significant difference between groups prior to further treatments. The open field apparatus was a 60×100 cm black wooden box with walls 18 cm high. The floor was divided into fifteen 20×20 cm identical squares. We recorded the activity of each rat for a 3 min session. We manually counted and recorded in a blinded fashion the numbers of squares crossed and rears performed.
Statistical analyses
Data are shown as mean ± S.E.M. Unless stated otherwise, we carried out statistical analysis with GraphPad Prism 5 using a two-tailed Student's t-test if two groups were compared, one-way ANOVA with Bonferroni’s multiple comparisons post-test for comparisons of more than two groups and two-way ANOVA with Bonferroni post-test when two independent variables were assessed. Differences were considered significant for p<0.05.
Supplementary Material
Acknowledgments
This work was supported by the UK Medical Research Council grant G0500495 and by the Dunhill Medical Trust. AMB is a senior investigator of the UK National Institute for Health Research (NIHR) and received funding from Fondation Leducq for neurovascular coupling. GH was funded through the NIHR Integrated Academic Training Programme and Oxford University Clinical Academic Graduate School. B.K. and C.W.D. were supported by the NIHR Biomedical Research Centre, Oxford, UK. S.N. was supported by the Deutsche Forschungsgemeinschaft. G.T. and S.M.W. received funding from NHS Blood and Transplant (NHSBT) and the National Institute for Health Research (NIHR) under its Programme Grants Scheme (NIHR Programmes RP-PG-0310-10001 and -10003). We would also like to thank R. Deacon from the Department of Experimental Psychology, University of Oxford, for providing us the open field apparatus and for his guidance with the behavioral experiments. We thank E. Martin Rendon for her input concerning the lentiviral vectors and J. Peeling for his evaluation of the manuscript. We dedicate this paper, to the memory of our colleague and mentor, Dr John P. MacManus (1943-2012).
Abbreviations
- 3MA
3 methyladenine
- 4-VO
four-vessel occlusion
- DAPI
4′,6-diamidino-2-phenylindole
- DIV
days in vitro
- GFAP
glial fibrillary acidic protein
- IB
immunoblotting
- IPA
ingenuity pathways analysis
- IPC
ischemic preconditioning
- LC3
Microtubule-associated protein 1 light chain 3
- MOI
multiplicity of infection
- mTORC1
mammalian target of rapamycin complex 1
- p62
sequestosome-1
- S6RP
S6 Ribosomal Protein
- shRNA
small interfering RNA hairpin sequence
- OGD
oxygen glucose deprivation
- TSC1
tuberous sclerosis complex 1
Footnotes
Competing financial interests The authors declare no competing financial interests.
Reference List
- 1.O’Collins VE, et al. 1,026 experimental treatments in acute stroke. Ann. Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- 2.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 3.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37:1281–1286. doi: 10.1212/wnl.37.8.1281. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, et al. Expression of the apoptosis-effector gene, Bax, is up-regulated in vulnerable hippocampal CA1 neurons following global ischemia. J. Neurochem. 1996;67:64–71. doi: 10.1046/j.1471-4159.1996.67010064.x. [DOI] [PubMed] [Google Scholar]
- 6.Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J. Neurosci. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun HS, et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat. Neurosci. 2009;12:1300–1307. doi: 10.1038/nn.2395. [DOI] [PubMed] [Google Scholar]
- 8.Zhang QG, et al. Akt inhibits MLK3/JNK3 signaling by inactivating Rac1: a protective mechanism against ischemic brain injury. J. Neurochem. 2006;98:1886–1898. doi: 10.1111/j.1471-4159.2006.04020.x. [DOI] [PubMed] [Google Scholar]
- 9.Shioda N, Han F, Morioka M, Fukunaga K. Bis(1-oxy-2-pyridinethiolato)oxovanadium(IV) enhances neurogenesis via phosphatidylinositol 3-kinase/Akt and extracellular signal regulated kinase activation in the hippocampal subgranular zone after mouse focal cerebral ischemia. Neuroscience. 2008;155:876–887. doi: 10.1016/j.neuroscience.2008.05.056. [DOI] [PubMed] [Google Scholar]
- 10.Endo H, Nito C, Kamada H, Yu F, Chan PH. Akt/GSK3beta survival signaling is involved in acute brain injury after subarachnoid hemorrhage in rats. Stroke. 2006;37:2140–2146. doi: 10.1161/01.STR.0000229888.55078.72. [DOI] [PubMed] [Google Scholar]
- 11.Gozal E, et al. Proteomic analysis of CA1 and CA3 regions of rat hippocampus and differential susceptibility to intermittent hypoxia. J. Neurochem. 2002;83:331–345. doi: 10.1046/j.1471-4159.2002.01134.x. [DOI] [PubMed] [Google Scholar]
- 12.Kawagoe J, et al. Distributions of heat shock protein (HSP) 70 and heat shock cognate protein (HSC) 70 mRNAs after transient focal ischemia in rat brain. Brain Res. 1992;587:195–202. doi: 10.1016/0006-8993(92)90997-n. [DOI] [PubMed] [Google Scholar]
- 13.Kinouchi H, et al. Induction of 70-kDa heat shock protein and hsp70 mRNA following transient focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 1993;13:105–115. doi: 10.1038/jcbfm.1993.13. [DOI] [PubMed] [Google Scholar]
- 14.Colbourne F, Li H, Buchan AM, Clemens JA. Continuing postischemic neuronal death in CA1: influence of ischemia duration and cytoprotective doses of NBQX and SNX-111 in rats. Stroke. 1999;30:662–668. doi: 10.1161/01.str.30.3.662. [DOI] [PubMed] [Google Scholar]
- 15.Choo AY, et al. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol. Cell. 2010;38:487–499. doi: 10.1016/j.molcel.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Nardo A, et al. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J. Neurosci. 2009;29:5926–5937. doi: 10.1523/JNEUROSCI.0778-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- 18.Andersen MB, Zimmer J, Sams-Dodd F. Postischemic hyperactivity in the Mongolian gerbil correlates with loss of hippocampal neurons. Behav. Neurosci. 1997;111:1205–1216. doi: 10.1037//0735-7044.111.6.1205. [DOI] [PubMed] [Google Scholar]
- 19.Kesner RP. Behavioral functions of the CA3 subregion of the hippocampus. Learn. Mem. 2007;14:771–781. doi: 10.1101/lm.688207. [DOI] [PubMed] [Google Scholar]
- 20.Mileson BE, Schwartz RD. The use of locomotor activity as a behavioral screen for neuronal damage following transient forebrain ischemia in gerbils. Neurosci. Lett. 1991;128:71–76. doi: 10.1016/0304-3940(91)90762-i. [DOI] [PubMed] [Google Scholar]
- 21.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 22.Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010;30:1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang SS, et al. Rapamycin protects heart from ischemia/reperfusion injury independent of autophagy by activating PI3 kinase-Akt pathway and mitochondria K(ATP) channel. Pharmazie. 2010;65:760–765. [PubMed] [Google Scholar]
- 24.Chauhan A, Sharma U, Jagannathan NR, Reeta KH, Gupta YK. Rapamycin protects against middle cerebral artery occlusion induced focal cerebral ischemia in rats. Behav. Brain Res. 2011;225:603–609. doi: 10.1016/j.bbr.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 25.Gabryel B, Kost A, Kasprowska D. Neuronal autophagy in cerebral ischemia--a potential target for neuroprotective strategies? Pharmacol. Rep. 2012;64:1–15. doi: 10.1016/s1734-1140(12)70725-9. [DOI] [PubMed] [Google Scholar]
- 26.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng YQ, Liu JX, Li XZ, Xu L, Xu YG. RNA interference-mediated downregulation of Beclin1 attenuates cerebral ischemic injury in rats. Acta Pharmacol. Sin. 2009;30:919–927. doi: 10.1038/aps.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen YD, et al. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- 29.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol. Dis. 2008;32:329–339. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 30.Wang P, et al. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy. 2012;8:77–87. doi: 10.4161/auto.8.1.18274. [DOI] [PubMed] [Google Scholar]
- 31.Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J. Neurochem. 2010;115:68–78. doi: 10.1111/j.1471-4159.2010.06905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheng R, et al. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 2010;6 doi: 10.4161/auto.6.4.11737. [DOI] [PubMed] [Google Scholar]
- 33.van den Ouweland AM, et al. Characterisation of TSC1 promoter deletions in tuberous sclerosis complex patients. Eur. J. Hum. Genet. 2011;19:157–163. doi: 10.1038/ejhg.2010.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Online Methods Reference List
- 34.Pulsinelli WA, Buchan AM. The four-vessel occlusion rat model: method for complete occlusion of vertebral arteries and control of collateral circulation. Stroke. 1988;19:913–914. doi: 10.1161/01.str.19.7.913. [DOI] [PubMed] [Google Scholar]
- 35.Guillemin I, et al. A subcellular prefractionation protocol for minute amounts of mammalian cell cultures and tissue. Proteomics. 2005;5:35–45. doi: 10.1002/pmic.200400892. [DOI] [PubMed] [Google Scholar]
- 36.Xu D, et al. Novel MMP-9 substrates in cancer cells revealed by a label-free quantitative proteomics approach. Mol. Cell Proteomics. 2008;7:2215–2228. doi: 10.1074/mcp.M800095-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS. One. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaur J, Zhao Z, Geransar RM, Papadakis M, Buchan AM. Prior deafferentation confers long term protection to CA1 against transient forebrain ischemia and sustains GluR2 expression. Brain Res. 2006;1075:201–212. doi: 10.1016/j.brainres.2005.12.123. [DOI] [PubMed] [Google Scholar]
- 40.Jiang Y, Deacon R, Anthony DC, Campbell SJ. Inhibition of peripheral TNF can block the malaise associated with CNS inflammatory diseases. Neurobiol. Dis. 2008;32:125–132. doi: 10.1016/j.nbd.2008.06.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.