

Abstract
DNA polymerase V, composed of a heterotrimer of the DNA damage-inducible UmuC and UmuD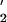 proteins, working in conjunction with RecA, single-stranded DNA (ssDNA)-binding protein (SSB), β sliding clamp, and γ clamp loading complex, are responsible for most SOS lesion-targeted mutations in Escherichia coli, by catalyzing translesion synthesis (TLS). DNA polymerase II, the product of the damage-inducible polB (dinA ) gene plays a pivotal role in replication-restart, a process that bypasses DNA damage in an error-free manner. Replication-restart takes place almost immediately after the DNA is damaged (≈2 min post-UV irradiation), whereas TLS occurs after pol V is induced ≈50 min later. We discuss recent data for pol V-catalyzed TLS and pol II-catalyzed replication-restart. Specific roles during TLS for pol V and each of its accessory factors have been recently determined. Although the precise molecular mechanism of pol II-dependent replication-restart remains to be elucidated, it has recently been shown to operate in conjunction with RecFOR and PriA proteins.
proteins, working in conjunction with RecA, single-stranded DNA (ssDNA)-binding protein (SSB), β sliding clamp, and γ clamp loading complex, are responsible for most SOS lesion-targeted mutations in Escherichia coli, by catalyzing translesion synthesis (TLS). DNA polymerase II, the product of the damage-inducible polB (dinA ) gene plays a pivotal role in replication-restart, a process that bypasses DNA damage in an error-free manner. Replication-restart takes place almost immediately after the DNA is damaged (≈2 min post-UV irradiation), whereas TLS occurs after pol V is induced ≈50 min later. We discuss recent data for pol V-catalyzed TLS and pol II-catalyzed replication-restart. Specific roles during TLS for pol V and each of its accessory factors have been recently determined. Although the precise molecular mechanism of pol II-dependent replication-restart remains to be elucidated, it has recently been shown to operate in conjunction with RecFOR and PriA proteins.
Two seemingly unconnected questions arising during the early and mid 1970s were to decipher the biochemical basis of SOS mutagenesis in Escherichia coli, often referred to as SOS error-prone repair (1), and to determine a cellular role for E. coli DNA polymerase II. A tentative link between the two was established when it was determined that pol II was induced as part of the LexA-regulon (2). pol II was subsequently shown to be encoded by the DNA damage-inducible polB (dinA) gene (3–5). A ΔpolB strain shows no measurable UV sensitivity, and SOS-induced mutagenesis occurs at normal levels (6, 7). However, a ΔpolB ΔumuDC double mutant strain is more sensitive to killing by UV light than either of the single mutant strains, implying that the two SOS-induced polymerases might play compensatory roles in vivo (8).
The ability of pols II and V to complement each other does not mean that these activities are functionally redundant, and indeed they are not. pol V is able to copy UV-damaged DNA in a process referred to as error-prone translesion synthesis (TLS). TLS generates mutations targeted specifically to DNA template damage sites (9–12). In contrast, pol II copies chromosomal DNA during error-free replication-restart (8). Although both polymerases are induced by DNA damage, they appear to function on widely disparate time frames—pol II-catalyzed replication-restart occurs 2 min post-UV irradiation whereas pol V-catalyzed TLS begins roughly 50 min later (8). In this paper, we discuss current models for the roles of pol V in TLS and pol II in replication-restart.
Coping with DNA Damage in E. coli
There are over 40 genes induced on DNA damage in E. coli that have been identified recently by using microarray chip technology (13), of which at least 31 are known to be negatively regulated at the transcriptional level by the LexA protein (14). Many of these genes encode proteins required to repair DNA damage (15). The overriding importance of DNA repair is apparent from the observation that a single pyrimidine dimer is lethal in E. coli strains defective for excision and recombinational repair (16). These experiments were among the first to demonstrate the essential contribution of DNA repair to cell survival. Excision and recombination repair pathways are referred to as “error-free” because they do not result in an increase in mutation rate above spontaneous background levels (1).
In contrast to error-free repair, damage-inducible TLS generates a significant mutational load (17). Most TLS depends on the damage inducible UmuD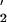 and UmuC proteins, which heterotrimerize to form E. coli pol V (UmuD
and UmuC proteins, which heterotrimerize to form E. coli pol V (UmuD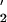 C; refs. 18–20). By copying lesions that block normal replication fork progression, pol V-induced mutations are primarily targeted directly opposite DNA template damage sites; however, pol V is also responsible for causing untargeted mutations at undamaged template sites (21).
C; refs. 18–20). By copying lesions that block normal replication fork progression, pol V-induced mutations are primarily targeted directly opposite DNA template damage sites; however, pol V is also responsible for causing untargeted mutations at undamaged template sites (21).
An in Vitro Model System for SOS Mutagenesis.
Three commonly occurring DNA lesions that have been used as models to study SOS mutagenesis in vivo and in vitro are TT cis-syn cyclobutane dimers, TT (6–4) photoproducts, and abasic (apurinic/apyrimidinic) moieties. TT dimers and (6–4) photoproducts arise from UV irradiation (1), and abasic sites occur either spontaneously or from the action of DNA glycosylases (22). TT (6–4) photoproducts and abasic moieties are strongly mutagenic (11, 23), but TT dimers are much less so (9). However, each of the lesions presents a strong block to DNA replication in vivo (9, 11, 23) and in in vitro model systems using purified DNA polymerases and polymerase accessory proteins (12).
A model biochemical system devised by H. Echols and coworkers (24) has facilitated reconstitution of SOS mutagenesis in vitro (Fig. 1a). The proteins involved in copying blocking template lesions are pol V, RecA, SSB, and β sliding clamp and γ clamp loading complex (18). In accordance with Echols' original suggestion, we have continued to refer to the group of proteins including pol V, RecA, SSB, and β clamp/γ clamp-loading complex by the term “pol V Mut,” where the designation “Mut” refers to a mutasomal complex (25). Although there is strong biochemical evidence that these proteins mutually interact proximal to a DNA template permitting bypass of the lesion (12, 26), there are currently no data reporting protein–protein interactions in the absence of DNA to form a stable macromolecular structure. It is therefore possible that the protein–protein interactions required for TLS lesion (12, 26) are mediated by the primer/template DNA.
Figure 1.

Primer-template DNA constructs used to reconstitute SOS mutagenesis in vitro. (a) When pol V, incubated in the presence of RecA, SSB, β sliding clamp, and γ clamp loading complex is used to copy a 30-mer/M13 primer-template DNA, synthesis proceeds past the lesion X to the end of the template strand (18). TLS requires the presence of pol V and RecA proteins for the case of TT cis-syn photodimers, TT (6–4) photoproducts, and abasic sites (19). (b) RecA, SSB, and pol V bind to regions of ssDNA far from the lesion site. The use of a shorter p/t DNA (30-mer/240-mer) reduces nonessential ssDNA, enabling a measurement of the stoichiometries and the effects of each mutasomal component on TLS (26).
Echols had originally suggested that pol III core was a component of the mutasome, based primarily on genetic evidence indicating a requirement for pol III in SOS mutagenesis (27, 28). We have found, however, that the presence of pol III core is not required for TLS (18, 19), and in its presence pol V-catalyzed TLS is inhibited (19). However, it cannot be ruled out that pol V is part of a macromolecular replication-repair complex, i.e., a DNA synthesis “factory” (29, 30), that might include the presence of pol III core.
pol V Mut copies TT (6–4) photoproducts, TT cis-syn photodimers, and abasic lesions at least 50-fold more efficiently than they are copied by either pol III or pol IV (12). Furthermore, pol V Mut-catalyzed nucleotide misincorporation frequencies are in excellent agreement with mutation frequencies determined in vivo (12). The most important example is the use of pol V to copy the TT (6–4) photoproduct. Unlike TT cis-syn photodimers, which impede replication but are only weakly mutagenic, TT (6–4) photoproducts both are inhibitory and are highly mutagenic, causing T → C transitions opposite the 3′-T of the (6–4) photoproduct. In agreement with mutational data, the incorporation of G by pol V occurs with a 6:1 preference compared with incorporation of A opposite a 3′-T (11, 12, 31). In contrast, pol III and pol IV favor the non-mutagenic incorporation of A (12).
Compared with the 3′-T of the (6–4) photoproduct, the 5′-T is a much less mutagenic site, with transition frequencies typically less than 2%. In agreement, pol V Mut incorporates A in preference to G opposite a 5′-T by factors ranging from about 25- to 50-fold (12). The specificity for incorporation of A opposite a 5′-T is retained when extending from either a G or A situated opposite the 3′-T site. pol III and pol IV have little ability to copy beyond the 3′-T when compared with pol V (12). Thus, the steady state kinetic measurements using E. coli DNA polymerases to copy TT cis-syn photodimers and abasic sites showed that recapitulation of the in vivo data is achieved by using pol V (12).
Effects of RecA, SSB, and β,γ Processivity Complex on pol V Activity.
RecA and SSB essentially bind indiscriminately, but with high affinity to regions of single-stranded DNA (ssDNA) (32). pol V also binds to ssDNA, although with considerably weaker affinity (33, 34). Given the propensity of these proteins to coat any stretch of ssDNA, it is necessary to minimize “nonessential” primer-template (p/t) DNA to investigate a specific role for each mutasomal component in copying localized DNA damage sites. To accomplish this goal, we have recently refined H. Echols' in vitro model system (24) by reducing the length of the p/t DNA to a 30-mer/240-mer (Fig. 1b). This “minimal” p/t DNA is nevertheless long enough to allow formation of a RecA filament (26). The presence of a RecA filament proximal to a template damage site appears to be a requirement for pol V-catalyzed TLS lesion (12, 26, 35).
pol V carries out distributive synthesis in the absence of mutasomal accessory proteins. An estimate of its ability to copy undamaged DNA templates indicates that 1 nt is incorporated for roughly 100 pol V–p/t DNA encounters. Thus, pol V tends to dissociate rapidly before adding a nucleotide when it binds at a 3′-OH primer end.
The addition of either RecA or SSB separately to the reaction results in a strong stimulation of pol V activity on undamaged DNA—about 340-fold for RecA and about 1,040-fold for SSB (26). Stimulation by RecA occurs only in the presence of ATP, necessary for assembling a RecA-ssDNA filament (32). The presence of an intact RecA filament proximal to a lesion appears as an absolute requirement for TLS—there is no detectable incorporation opposite a damaged template base in the absence of RecA (26). The concentration of RecA resulting in maximum pol V activity on undamaged DNA as well as maximum pol V-catalyzed TLS efficiency occurs at 1 RecA per 3 to 4 nt (26), which is consistent with requirements for optimized nucleoprotein filament assembly (32).
The strong stimulation in pol V activity in the presence of RecA is accompanied by a 340-fold reduction in the steady-state apparent Michaelis constant (Km,dNTP characterizing the dNTP substrate concentration required to attain one-half the maximum nucleotide incorporation velocity, Vmax), with no change in the apparent Vmax value. The 1,040-fold stimulation of pol V by SSB is brought about by a 150-fold decrease in Km,dNTP and 7-fold increase in Vmax. A possible interpretation of the data showing a marked reduction in Km,dNTP is that the affinity of pol V for p/t 3′-ends is enhanced in the presence of both RecA and SSB.
The activity of pol V is stimulated 3-fold in the presence of β,γ processivity complex, caused by a reduction in its apparent Km,dNTP value (26). The presence of β,γ complex confers a marked increase in pol V processivity when copying natural or damaged DNA templates. Although this result is consistent with a reduction in Km,dNTP corresponding to an increase pol V–p/t DNA binding affinity, a pre-steady state kinetic analysis will be performed to determine whether RecA and SSB have a similar effect on the binding of pol V to primer-3′-ends.
Even though RecA, SSB, and β,γ complex stimulate pol V activity (12, 18, 26), the key question is, “What are the specific roles played by RecA, SSB, and β,γ complex during TLS?” Roles for RecA both in catalyzing generalized recombination between homologous DNA sequences and serving as a coprotease during cleavage of LexA and UmuD proteins are well known (1). However, RecA plays yet another, absolutely essential role during TLS. SOS mutagenesis does not occur in a recA1730 mutant strain that retains recombination and coprotease (i.e., SOS induction) functions (36–38).
Despite the effect of the mutasomal proteins in stimulating pol V activity at normal, undamaged template sites, there is no observable TLS unless RecA is also present (26). TLS is absent when RecA1730 replaces wild-type RecA in the in vitro assay (18), which agrees with in vivo data (36–38). The mutually supportive biochemical and genetic data imply that specific interactions between RecA and pol V in the vicinity of a template lesion are required for pol V-catalyzed TLS. R. Devoret and colleagues (37, 38) have suggested that lesion bypass can occur only if pol V maintains contact with the 3′-filament tip. Support for this model comes from electron microscopic studies that reveal that, at subsaturating concentrations, UmuD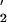 C preferentially binds to one end of a RecA nucleoprotein filament (34).
C preferentially binds to one end of a RecA nucleoprotein filament (34).
A “Cowcatcher” Model for pol V Mut-Catalyzed TLS.
The key to understanding the biochemical mechanism of TLS requires a detailed analysis of how pol V, SSB, and RecA interact in the vicinity of a damaged template base. RecA filaments formed in the presence of ATP assemble and disassemble in a 5′ to 3′ direction on ssDNA. Disassembly requires ATP hydrolysis whereas assembly does not (32). The assembly and disassembly reactions are in a dynamic equilibrium. In contrast, a RecA filament formed with ATPγS (a poorly hydrolyzable analog of ATP) is considerably more stable (32). We designate filaments formed in the presence of ATPγS as “stabilized” RecA filaments.
Three key observations lead to a model for TLS: (i) pol V Mut, which synthesizes DNA distributively on “dynamic” RecA filaments (formed with ATP), copies stabilized RecA filaments processively (19, 26); (ii) pol V cannot copy a stabilized RecA filament in the absence of SSB (26); (iii) synthesis on a stabilized RecA filament is processive only when β,γ complex is present (26). This last result is especially significant, suggesting that bound RecA must be stripped from the template strand during replication because a 100-Å diameter RecA filament (32) cannot pass through a 30-Å β diameter dimer clamp opening (39). A model for TLS is shown in Fig. 2.
Figure 2.

A “cowcatcher” model describing DNA polymerase V mutasome-catalyzed TLS. (a) A DNA polymerase III holoenzyme replication complex (HE; yellow) stalls when encountering a template lesion (X). The continued unwinding action of DnaB helicase (not shown) opens up a stretch of ssDNA template downstream of X. (b) pol III core dissociates from the 3′-primer end proximal to the lesion, and an activated RecA nucleoprotein filament (RecA*; light blue) is assembled in a 5′ to 3′ direction on the ssDNA in a reaction requiring ATP, but not ATP hydrolysis. (c) The RecA nucleoprotein filament continues to advance to reach the site of DNA damage. (d) pol V binds to the 3′-primer end vacated by pol III core. The activity and binding affinity of pol V are strongly stimulated by the presence of RecA, SSB, and β sliding clamp (12, 26). (e) The key feature of the model is that pol V (red) + SSB (green), operating jointly as a locomotive cowcatcher, strip RecA from the DNA template in a 3′ to 5′ direction immediately ahead of an advancing pol V molecule. The cowcatcher stripping reaction does not require ATP hydrolysis and takes place concurrently with the “standard” 5′ to 3′ RecA filament disassembly reaction requiring ATP hydrolysis. (f) After TLS, pol V dissociates from DNA when contact with the tip of a RecA filament is lost. Thus, bidirectional RecA filament disassembly helps to confine mutations to DNA damage sites by ensuring that undamaged DNA template bases are not copied by the low fidelity pol V. The pol III HE replication complex reassembles after dissociation of pol V. The sketch in e has been enlarged relative to the other parts of the figure to emphasize the cowcatcher aspects of the TLS model. Data supporting this model are contained in ref. 26.
The essential feature of the model is that stripping of the RecA filament takes place in a 3′ to 5′ direction in a reaction requiring pol V + SSB (ref. 26; Fig. 2e). Although the β sliding clamp is not required to strip RecA from the template strand, it nevertheless plays an important role by allowing pol V to retain contact with the 3′-tip of the receding RecA filament, thereby preventing its dissociation from the DNA at least until the lesion has been copied. The stripping mechanism is akin to a locomotive cowcatcher§ composed of pol V + SSB.
The 3′ → 5′ RecA stripping reaction was verified by cleavage of the primer and stabilized RecA template filament strands with a combination of endo- and exonucleases (26). A sketch depicting the nuclease protection assay (Fig. 3) contains a summary of the experimental data (26). The primer/template, which was refractory to cleavage by HinfI after a short incubation with pol V Mut (Fig. 3a), is cleaved once the HinfI restriction site is copied (Fig. 3b). After treatment with HinfI, the cleaved p/t DNA is digested by using a combination of RecJ and λ exonucleases (Fig. 3c) that selectively degrade single- and double-stranded DNA, respectively, in a 5′ to 3′ direction. In the absence of restriction with HinfI, the template strand is completely refractory to digestion with RecJ (Fig. 3c), thereby proving that the RecA filament remains intact at the 5′-end. However, the DNA is hydrolyzed by λ exonuclease once the entire template is copied, demonstrating that RecA is no longer bound to the nascent duplex DNA (Fig. 3c). Control reactions demonstrated that the HinfI, λ, and RecJ nucleases were inactive on an intact RecA filament formed on nonreplicated p/t DNA (26), giving direct evidence that RecA is removed by pol V + SSB in 3′ → 5′ direction, in a reaction that does not use ATP hydrolysis.
Figure 3.

Sketch of a nuclease protection analysis used to demonstrate the 3′ → 5′ disassembly of a RecA nucleoprotein filament by pol V + SSB. (a) A “stabilized” RecA nucleoprotein filament (blue circles) has been formed with ATPγS. The RecA filament cannot disassemble in the absence of ATP hydrolysis and is therefore refractory to cleavage by HinfI and by λ 5′ → 3′ double-stranded or RecJ 5′ → 3′ single-stranded exonucleases. Blocked cleavage reactions are depicted by crossed-out arrows. A <1-min reaction with pol V Mut is sufficient to incorporate C opposite G and to extend the primer past the template lesion (X); however, the extended primer does not yet reach the HinfI restriction site located 10 nt downstream from X. Therefore, a HinfI restriction enzyme fails to cut the DNA. (b) A 3-min reaction with pol V Mut is sufficient to copy past the HinfI restriction site, enabling cleavage by the restriction enzyme and subsequent digestion of the 32P-labeled template strand using a combination of RecJ + λ exonucleases (bottom portion of sketch). When the HinfI restriction enzyme is omitted from the reaction, digestion of the template strand does not occur because pol V Mut has not yet copied to the 5′-end of the template strand (top portion of sketch). (c) A 10-min reaction with pol V Mut is sufficient to reach the end of the template strand, enabling digestion with λ exonuclease. The experimental data documenting the pol V + SSB-catalyzed RecA stripping reaction are contained in ref. 26.
Bidirectional Filament Disassembly, a Mechanism for Localized Mutagenesis.
The pol V + SSB-catalyzed 3′ → 5′ disassembly reaction occurs equally well on damaged and undamaged DNA (26), but the fidelity of pol V is extremely low when copying undamaged DNA with typical error rates of about 10−2 to 10−3 (12). To avoid nontargeted mutations, i.e., at template sites other than directly opposite lesions, it is important to have a mechanism to facilitate pol V dissociation subsequent to TLS. A mechanism for dissociation of pol V is provided by 3′ → 5′ disassembly of RecA occurring concomitantly with the “normal” ATP hydrolysis-driven disassembly of the RecA filament, occurring 5′ → 3′ (Fig. 2e).
Bidirectional disassembly of the filament ensures the removal of RecA from the template strand shortly beyond the lesion. pol V dissociates rapidly from the DNA in the absence of RecA, thereby confining mutations to the template damage sites (26). pol V is unlikely to dissociate by itself, because it synthesizes DNA with relatively high processivity on a stabilized RecA filament when SSB and β,γ complex are also present in the reaction (12, 26).
pol II Plays a Pivotal Role in Error-Free Replication-Restart.
Although DNA replication is strongly inhibited almost immediately after irradiation with UV light, it resumes about 30 s later in wild-type cells (8, 40, 41). We proposed that pol II plays a pivotal role in alleviating stalled DNA replication, based on the observation that UV-irradiated cells lacking pol II exhibit about a 50-min delay in the resumption of DNA synthesis (8). We have recently observed a similar 40- to 50-min delay in recovering replication in cells lacking the primosomal protein PriA (S.R., R.W., and M.F.G., unpublished results), suggesting that pol II and PriA operate in the same pathway. In addition, we also observed that recovery of DNA synthesis in recF, recO, or recR strains was limited unless the strain carried a compensatory “coprotease” constitutive mutation in recA (recA730). These observations suggest that RecFOR proteins normally play an important function in generating an active RecA nucleoprotein filament necessary for replication restart (32).
Future Perspectives
An important challenge is to determine roles for each SOS-induced polymerase. pol II is induced immediately after DNA damage, ≈30 s post-UV irradiation (8), whereas pol V induction occurs roughly 50 min later (42). The earlier appearance of pol II makes teleological sense because it is preferable for E. coli to try to rescue its DNA without introducing mutations. Failing that, the cell can still survive by copying its damaged chromosome by using pol V, but at a cost of a substantial mutational load.
The role of pol V seems simplest to analyze. This mutator enzyme exhibits weak polymerase activity in the absence of RecA, SSB, and β/γ complex (12, 19, 26). It might be present primarily, if not solely, to copy damaged DNA, although pol V could also play a role during evolution by generating untargeted mutations on undamaged DNA (21, 43). pol V copies TT cis-syn photodimers, TT (6–4) photoproducts, and abasic moieties efficiently and with the same nucleotide incorporation specificity observed in vivo (19). However, there is a paucity of information on the spectrum of lesions copied by pol V either alone or in conjunction with the other two SOS-induced polymerases. It has recently been shown that pol V is probably not involved in generating adaptive mutations (44) occurring in nondividing E. coli; such functions are ascribed to pols III and IV (DinB; refs. 45 and 46).
There remains much to be learned regarding the mechanism of TLS. The cowcatcher model (Fig. 2) describes the biochemical mechanism for TLS, but only after pol V has been bound to the p/t DNA. The model does not address the lesion-targeting mechanism that enables the replacement of a stalled pol III replication complex with pol V. A simple mass action “search and copy” seems improbable given the limited number of pol V molecules with the cell (47). Perhaps pol V might be part of a multiprotein replication-repair “factory” (48) that can switch one polymerase for another, depending on the specific nature of the impediment. However, there are currently no experimental data to support such an exotic mechanism although interactions between pol V and pol III have been reported (19, 49). Bidirectional filament disassembly may keep pol V from extensively copying undamaged DNA (Fig. 2e). However, pol V-catalyzed mutations on undamaged DNA, i.e., untargeted mutations, do occur (21). Thus, on the other side of the coin, the replacement of pol V with pol III once TLS is completed also remains to be determined.
Might the specific interactions observed between pol V and RecA, SSB, and β clamp help to explain the lesion targeting mechanism (12, 26)? If, as seems likely, replication fork progression ceases just in front of a template lesion, our data suggest that pol V exhibits much higher affinity for p/t DNA when present as part of a mutasomal complex (12, 26). Binding experiments on a pre-steady state time scale could prove instrumental in addressing the mechanism of lesion targeting.
Although we are beginning to understand the role of pol V in E. coli, considerably less is known about the role of pol II. It is clear that the induction of pol II is instrumental in initiating replication restart (8), yet the specific role of pol II during replication restart remains uncertain. A 50-min delay in the DNA synthesis recovery after UV radiation for cells mutated in pol II (8) or PriA (S.R., R.W., and M.F.G., unpublished results) implies that these enzymes are likely to act in concert. However, the precise step at which pol II contributes to replication restart is, at the moment, simply a “best guess.” The fact that a coprotease constitutive allele of recA is able to suppress the genetic requirements for the RecFOR complex suggests that RecFOR may play an important, but indirect, role in replication restart by helping “activate” RecA protein for its many functions. Clearly, testing such hypotheses awaits a reconstituted biochemical system using purified proteins.
Although pol II is induced on DNA damage, the LexA binding site within the polB operator is one of the weakest in the entire LexA regulon (14). Consequently, constitutive levels of pol II are high, in a range of 40 to 60 molecules (5). Aside from its role in replication restart, genetic data suggest that pol II is involved in partially replicating chromosomal and episomal DNA (50), copying specific DNA lesions (51, 52), and contributing to episomal DNA repair in stationary phase E. coli (53). Indeed, a 5-fold increase in adaptive mutation is observed when pol II is absent in nondividing cells (53), and such a phenotype is apparently caused by swapping the high fidelity pol II enzyme (54) with the much lower fidelity pol IV (45, 46). Thus, much still remains to be learned about the cellular functions of enigmatic pol II.
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grants GM42554 and GM21422. P.P. was supported by a NIH-National Institute on Aging postdoctoral training grant.
Abbreviations
- TLS
translesion synthesis
- p/t
primer-template
- ssDNA
single-stranded DNA
- SSB
ssDNA-binding protein
Footnotes
This paper results from the National Academy of Sciences colloquium, “Links Between Recombination and Replication: Vital Roles of Recombination,” held November 10–12, 2000, in Irvine, CA.
A locomotive cowcatcher is a pointed device attached to the front of locomotives designed to push obstacles off the track ahead of an advancing train.
References
- 1.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 2.Bonner C A, Randall S K, Rayssiguier C, Radman M, Eritja R, Kaplan B E, McEntee K, Goodman M F. J Biol Chem. 1988;263:18946–18952. [PubMed] [Google Scholar]
- 3.Bonner C A, Hays S, McEntee K, Goodman M F. Proc Natl Acad Sci USA. 1990;87:7663–7667. doi: 10.1073/pnas.87.19.7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwasaki H, Nakata A, Walker G, Shinagawa H. J Bacteriol. 1990;172:6268–6273. doi: 10.1128/jb.172.11.6268-6273.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiu Z, Goodman M F. J Biol Chem. 1997;272:8611–8617. doi: 10.1074/jbc.272.13.8611. [DOI] [PubMed] [Google Scholar]
- 6.Kow Y W, Faundez G, Hays S, Bonner C A, Goodman M F, Wallace S S. J Bacteriol. 1993;175:561–564. doi: 10.1128/jb.175.2.561-564.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Escarcellar M, Hicks J, Gudmundsson G, Trump G, Touati D, Lovett S, Foster P, McEntee K, Goodman M F. J Bacteriol. 1994;176:6221–6228. doi: 10.1128/jb.176.20.6221-6228.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rangarajan S, Woodgate R, Goodman M F. Proc Natl Acad Sci USA. 1999;96:9224–9229. doi: 10.1073/pnas.96.16.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lawrence C W, Banerjee S K, Borden A, LeClerc J E. Mol Gen Genet. 1990;166:166–168. doi: 10.1007/BF00283040. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee S K, Borden A, Christensen R B, LeClerc J E, Lawrence C W. J Bacteriol. 1990;172:2105–2112. doi: 10.1128/jb.172.4.2105-2112.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LeClerc J E, Borden A, Lawrence C W. Proc Natl Acad Sci USA. 1991;88:9685–9689. doi: 10.1073/pnas.88.21.9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang M, Pham P, Shen X, Taylor J-S, O'Donnell M, Woodgate R, Goodman M F. Nature (London) 2000;404:1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- 13.Courcelle J A, Khodursky A, Peter B, Brown P O, Hanawalt P C. Genetics. 2001;158:41–64. doi: 10.1093/genetics/158.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez de Henestrosa A R, Ogi T, Aoyagi S, Chafin D, Hayes J J, Ohmori H, Woodgate R. Mol Microbiol. 2000;35:1560–1572. doi: 10.1046/j.1365-2958.2000.01826.x. [DOI] [PubMed] [Google Scholar]
- 15.Koch W H, Woodgate R. In: DNA Damage and Repair. Nicholoff J A, Hoekstra M F, editors. Totowa, NJ: Humana; 1998. [Google Scholar]
- 16.Rupp W D, Howard-Flanders P. J Mol Biol. 1968;31:291–304. doi: 10.1016/0022-2836(68)90445-2. [DOI] [PubMed] [Google Scholar]
- 17.Koffel-Schwartz N, Coin F, Veaute X, Fuchs R P P. Proc Natl Acad Sci USA. 1996;93:7805–7810. doi: 10.1073/pnas.93.15.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang M, Bruck I, Eritja R, Turner J, Frank E G, Woodgate R, O'Donnell M, Goodman M F. Proc Natl Acad Sci USA. 1998;95:9755–9760. doi: 10.1073/pnas.95.17.9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang M, Shen X, Frank E G, O'Donnell M, Woodgate R, Goodman M F. Proc Natl Acad Sci USA. 1999;96:8919–8924. doi: 10.1073/pnas.96.16.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reuven N B, Arad G, Maor-Shoshani A, Livneh Z. J Biol Chem. 1999;274:31763–31766. doi: 10.1074/jbc.274.45.31763. [DOI] [PubMed] [Google Scholar]
- 21.Fijalkowska I J, Dunn R L, Schaaper R M. J Bacteriol. 1997;179:7435–7445. doi: 10.1128/jb.179.23.7435-7445.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindahl T. Annu Rev Biochem. 1982;51:61–87. doi: 10.1146/annurev.bi.51.070182.000425. [DOI] [PubMed] [Google Scholar]
- 23.Lawrence C W, Borden A, Banerjee S K, LeClerc J E. Nucleic Acids Res. 1990;18:2153–2157. doi: 10.1093/nar/18.8.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajagopalan M, Lu C, Woodgate R, O'Donnell M, Goodman M F, Echols H. Proc Natl Acad Sci USA. 1992;89:10777–10781. doi: 10.1073/pnas.89.22.10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Echols H, Goodman M F. Mutat Res. 1990;236:301–311. doi: 10.1016/0921-8777(90)90013-u. [DOI] [PubMed] [Google Scholar]
- 26.Pham P, Bertram J G, O'Donnell M, Woodgate R, Goodman M F. Nature (London) 2001;409:366–370. doi: 10.1038/35053116. [DOI] [PubMed] [Google Scholar]
- 27.Bridges B A, Mottershead R P, Sedgwick S G. Mol Gen Genet. 1976;144:53–58. doi: 10.1007/BF00277304. [DOI] [PubMed] [Google Scholar]
- 28.Hagensee M E, Timme T L, Bryan S K, Moses R E. Proc Natl Acad Sci USA. 1987;84:4195–4199. doi: 10.1073/pnas.84.12.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lemon K P, Grossman A D. Science. 1998;282:1516–1519. doi: 10.1126/science.282.5393.1516. [DOI] [PubMed] [Google Scholar]
- 30.Goodman M F, Tippin B. Nat Rev Mol Cell Biol. 2000;1:101–109. doi: 10.1038/35040051. [DOI] [PubMed] [Google Scholar]
- 31.Smith C A, Wang M, Jiang N, Che L, Zhao X, Taylor J-S. Biochemistry. 1996;35:4146–4154. doi: 10.1021/bi951975c. [DOI] [PubMed] [Google Scholar]
- 32.Kuzminov A. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruck I, Woodgate R, McEntee K, Goodman M F. J Biol Chem. 1996;271:10767–10774. doi: 10.1074/jbc.271.18.10767. [DOI] [PubMed] [Google Scholar]
- 34.Frank E G, Cheng N, Do C, Cerritelli M E, Bruck I, Goodman M F, Egelman E H, Woodgate R, Steven A C. J Mol Biol. 2000;297:585–597. doi: 10.1006/jmbi.2000.3591. [DOI] [PubMed] [Google Scholar]
- 35.Reuven N B, Arad G, Stasiak A Z, Stasiak A, Livneh Z. J Biol Chem. 2001;276:5511–5517. doi: 10.1074/jbc.M006828200. [DOI] [PubMed] [Google Scholar]
- 36.Dutreix M, Moreau P L, Bailone A, Galibert F, Battista J R, Walker G C, Devoret R. J Bacteriol. 1989;171:2415–2423. doi: 10.1128/jb.171.5.2415-2423.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bailone A, Sommer S, Knezevic J, Dutreix M, Devoret R. Biochemie. 1991;73:479–484. doi: 10.1016/0300-9084(91)90115-h. [DOI] [PubMed] [Google Scholar]
- 38.Sommer S, Boudsocq F, Devoret R, Bailone A. Mol Microbiol. 1998;28:281–291. doi: 10.1046/j.1365-2958.1998.00803.x. [DOI] [PubMed] [Google Scholar]
- 39.Kong X-P, Onrust R, O'Donnell M, Kuriyan J. Cell. 1992;69:425–437. doi: 10.1016/0092-8674(92)90445-i. [DOI] [PubMed] [Google Scholar]
- 40.Khidhir A M, Casaregola S, Holland I B. Mol Gen Genet. 1985;199:133–140. doi: 10.1007/BF00327522. [DOI] [PubMed] [Google Scholar]
- 41.Witkin E M, Maniscalco R V, Sweasy J B, McCall J O. Proc Natl Acad Sci USA. 1987;84:6805–6809. doi: 10.1073/pnas.84.19.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boudsocq F, Campbell M, Devoret R, Bailone A. J Mol Biol. 1997;270:201–211. doi: 10.1006/jmbi.1997.1098. [DOI] [PubMed] [Google Scholar]
- 43.Echols H. Cell. 1981;25:1–2. doi: 10.1016/0092-8674(81)90223-3. [DOI] [PubMed] [Google Scholar]
- 44.Cairns J, Foster P L. Genetics. 1991;128:695–701. doi: 10.1093/genetics/128.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foster P L. Cold Spring Harbor Symp Quant Biol. 2000;65:21–29. doi: 10.1101/sqb.2000.65.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKenzie G J, Lee P L, Lombardo M-J, Hastings P J, Rosenberg S M. Mol Cell. 2001;7:571–579. doi: 10.1016/s1097-2765(01)00204-0. [DOI] [PubMed] [Google Scholar]
- 47.Woodgate R, Ennis D G. Mol Gen Genet. 1991;229:10–16. doi: 10.1007/BF00264207. [DOI] [PubMed] [Google Scholar]
- 48.Shapiro L, Losick R. Cell. 2000;100:89–98. doi: 10.1016/s0092-8674(00)81686-4. [DOI] [PubMed] [Google Scholar]
- 49.Sutton M D, Opperman T, Walker G C. Proc Natl Acad Sci USA. 1999;96:12373–12378. doi: 10.1073/pnas.96.22.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rangarajan S, Gudmundsson G, Qiu Z, Foster P L, Goodman M F. Proc Natl Acad Sci USA. 1997;94:946–951. doi: 10.1073/pnas.94.3.946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tessman I, Kennedy M A. Genetics. 1993;136:439–448. doi: 10.1093/genetics/136.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Napolitano R, Janel-Bintz R, Wagner J, Fuchs R P P. EMBO J. 2000;19:6259–6265. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foster P L, Gudmundsson G, Trimarchi J M, Cai H, Goodman M F. Proc Natl Acad Sci USA. 1995;92:7951–7955. doi: 10.1073/pnas.92.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai H, Yu H, McEntee K, Kunkel T A, Goodman M F. J Biol Chem. 1995;270:15327–15335. doi: 10.1074/jbc.270.25.15327. [DOI] [PubMed] [Google Scholar]