

Abstract
The spore wall of Saccharomyces cerevisiae is a multilaminar extracellular structure that is formed de novo in the course of sporulation. The outer layers of the spore wall provide spores with resistance to a wide variety of environmental stresses. The major components of the outer spore wall are the polysaccharide chitosan and a polymer formed from the di-amino acid dityrosine. Though the synthesis and export pathways for dityrosine have been described, genes directly involved in dityrosine polymerization and incorporation into the spore wall have not been identified. A synthetic gene array approach to identify new genes involved in outer spore wall synthesis revealed an interconnected network influencing dityrosine assembly. This network is highly redundant both for genes of different activities that compensate for the loss of each other and for related genes of overlapping activity. Several of the genes in this network have paralogs in the yeast genome and deletion of entire paralog sets is sufficient to severely reduce dityrosine fluorescence. Solid-state NMR analysis of partially purified outer spore walls identifies a novel component in spore walls from wild type that is absent in some of the paralog set mutants. Localization of gene products identified in the screen reveals an unexpected role for lipid droplets in outer spore wall formation.
Author Summary
The cell wall of fungi is a complex extracellular matrix and an important target for antifungal drugs. Assembly of the wall during spore formation in baker's yeast is a useful model for fungal wall development. The outermost layers of the spore wall are composed of a polymer of dityrosine connected to an underlying polysaccharide layer. The assembly pathway of this dityrosine polymer is not known. Using a genetic approach we reveal a network of genes that function redundantly to control dityrosine layer synthesis. Solid state NMR analysis of spore walls from wild-type and mutant cells reveals a novel constituent of the spore wall that may link the dityrosine to the underlying polysaccharides and a role for lipid droplets in the incorporation of this new component into the spore wall.
Introduction
All cells surround themselves with some form of extracellular matrix that provides structural integrity to the cell and protection from the environment. While the composition of these extracellular matrices varies, they present all cells with a common problem – how to assemble a complex macromolecular structure in an extracellular milieu. In fungi, the extracellular matrix is referred to as the cell wall and serves as the interface between a fungal cell and its environment. The composition and structure of the wall can determine the ability of cells to survive under different conditions. As a result, cell wall synthesis is an important target for antifungal drugs for the treatment of fungal infections [1].
The cell wall of f`ungi is composed primarily of long chain polysaccharides and heavily glycosylated proteins. In addition, many fungi also contain polyphenolic compounds such as melanin in their walls, though relatively little is known about how these polyphenols are incorporated into the wall [2]. The vegetative cell wall of Saccharomyces cerevisiae has been used extensively as a model for studies of fungal cell walls, though it lacks components such as the polyphenols that are found in other fungi [3]. These constituents are found, however, in the wall of S. cerevisiae spores.
Starvation can induce diploid cells of S. cerevisiae to undergo meiosis and differentiate to form haploid spores [4]. Spores are formed by an unusual cell division in which four daughter cells are generated within the cytoplasm of the mother cell. Intracellular membranes termed prospore membranes engulf and eventually enclose each of the four nuclei generated by meiosis, giving rise to the daughter cells (Figure 1). Closure of a prospore membrane envelops a nucleus within two membranes; the plasma membrane of the spore and an outer membrane that separates the spore plasma membrane from the mother cell cytoplasm. After prospore membrane closure a spore wall is formed de novo around each spore (Figure 1).
Figure 1. Overview of spore wall formation.
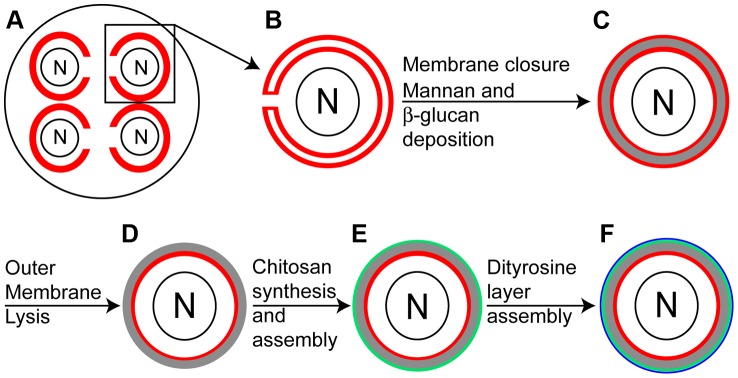
A) Each of the four nuclei (N) in a sporulating cell are engulfed by a prospore membrane (red). B) A single prospore membrane prior to closure. C) After closure of the prospore membrane, mannans and β-glucans (gray) are deposited in the lumen between the spore plasma membrane and outer membrane derived from the prospore membrane (both in red). D) The outer membrane disappears, exposing spore wall material to the ascal cytoplasm. E) Chitosan is synthesized and assembled as a discrete layer (green) on the outside of the β-glucan. F) The dityrosine layer (blue) is formed on the outside of the chitosan layer.
The completed spore wall consists of four distinct layers [5]. The first two layers are composed of mannan and β-glucan and are similar in composition to the vegetative cell wall [6]. These two layers are formed in the lumen between the spore plasma membrane and the outer membrane (Figure 1C). After the β-glucan layer is completed, the outer membrane is lost exposing the spore wall directly to the cytoplasm of the surrounding ascus (Figure 1D) [7]. The two outer layers of the spore wall are made from components unique to the spore [8], [9]. First, a layer of chitosan is formed on top of the β-glucan layer after outer membrane lysis (Figure 1E) [7]. Chitosan is a polymer of β-1,4 linked glucosamine moieties and is generated by the combined action of the chitin synthase Chs3 and the sporulation-specific chitin deacetylases Cda1 and Cda2. Chs3 generates and extrudes from the spore chitin, a β-1,4 linked N-acetylglucosamine polymer and the Cda1/2 enzymes then convert the N-acetylglucosamine subunits in the nascent chitin chains to glucosamine [10], [11]. Once assembled, the chitosan layer forms a surface on which the outermost layer of the spore wall assembles. The major component of this outer layer is the cross-linked amino acid N,N-bis-formyldityrosine (hereafter, dityrosine) (Figure 1F) [8].
Dityrosine is formed in the spore cytoplasm by the Dit1 and Dit2 enzymes [12]–[15]. These dityrosine molecules are then moved to the spore wall by the action of transporters localized in the spore plasma membrane. The primary transporter is encoded by DTR1; however, dtr1Δ cells still display dityrosine fluorescence indicating that alternative transporters exist [16]. Once exported, dityrosine is incorporated into a large, insoluble polymer, the chemical structure of which remains to be determined [9].
Together, the outer spore wall chitosan and dityrosine layers confer enhanced resistance to environmental stresses on the spore, including the ability to pass through the digestive tracts of insects, permitting dispersal in the environment [7], [10], [12]. The composition of the spore wall and its construction in a constrained developmental window make it an excellent model system for the study of fungal cell wall assembly.
Dityrosine in the spore wall is fluorescent and screens for mutants that lack this fluorescence have identified a variety of genes involved in sporulation, including DIT1 and DIT2 [12]. However, no genes specifically involved in assembly of the dityrosine polymer have been reported. To search for such genes, a synthetic genetic array approach was used to identify mutants that display reduced dityrosine fluorescence in combination with a mutant in the dityrosine transporter DTR1. Double mutant analysis of the genes identified in the primary screen reveal a highly interconnected network of genes contributing to dityrosine assembly. Strikingly, many of the major nodes in this network were found to have paralogs in the yeast genome. For several of these paralogous gene sets, deletion of all the paralogs leads to loss of dityrosine from the spore wall. These results reveal a highly redundant network of genes that regulate the assembly of the dityrosine polymer.
Results
A sensitized screen identifies new genes important for dityrosine fluorescence
The transporter Dtr1 functions in the export of dityrosine from the spore cytoplasm, but deletion of DTR1 results in only a modest reduction in dityrosine incorporation into the spore wall, presumably due to the presence of alternative transporters [16]. We reasoned that extracellular dityrosine might nonetheless be limiting in dtr1Δ mutants and so this mutant might be more sensitive to additional perturbations in outer spore wall assembly. Thus, we performed a screen to identify mutants that result in loss of dityrosine fluorescence in combination with dtr1Δ.
Using a modified synthetic genetic array protocol a dtr1Δ strain was crossed to a collection of knockout mutants [17]. As high sporulation frequency is required, we used a set of 301 knockouts of sporulation-induced genes that was previously constructed in the efficiently sporulating SK1 background [18]. Diploids homozygous for dtr1Δ and mutXΔ were tested for fluorescence after sporulation on plates by a patch assay. Fluorescence was monitored by sporulating cells on nitrocellulose filters and then exposing the filters to shortwave UV light. For each gene, homozygous mutXΔ DTR1 diploids were also examined using the same assay. Mutants that displayed wild-type fluorescence in the DTR1 background but reduced fluorescence in combination with dtr1Δ in at least two of three replicates, were considered to have a genetic interaction with dtr1Δ.
Mutants in 38 genes that displayed reduced dityrosine fluorescence only in combination with dtr1Δ were identified (Table 1). Most of these genes were not identified in previous screens of this same collection of knockouts in SK1 for other phenotypes associated with outer spore wall defects including ether sensitivity, spore wall permeability, and sensitivity to digestion by Drosophila indicating that the synthetic screen worked to uncover new genes with possible roles in outer spore wall assembly [7], [19], [20]. Several of the mutants identified were in uncharacterized ORFs with no gene designation. For reasons explained below, we have named some of these ORFs. The acronym for OSW6 (IRC18/YJL038w) and OSW7 (YFR039c) stands for Outer Spore Wall), while the acronym for LDS1 (YAL018w), and LDS2 (YOL047c) stands for Lipid Droplet in Sporulation.
Table 1. Mutants displaying synthetic dityrosine defects with dtr1Δ.
| ORF | Gene | Found in earlier spore wall screensa | Commentsb |
| YAL018c | LDS1 | paralog of LDS2 | |
| YBR134w | Ether | dubious ORF/overlaps HSL7 | |
| YBR250w | SPO23 | ||
| YCR010c | ADY2 | acetate transporter | |
| YDL076c | RXT3 | HDAC complex subunit | |
| YDR108w | TRS85 | TRAPP subunit | |
| YDR273w | DON1 | leading edge complex protein | |
| YDR480w | DIG2 | ||
| YDR506c | GMC1 | synaptonemal complex assembly | |
| YDR525w | API2 | dubious ORF | |
| YEL016c | NPP2 | nucleotide pyrophosphorylase | |
| YER106w | MAM1 | Permeable | kinetochore protein |
| YER182w | FMP10 | mitochondrial protein | |
| YFR023w | PES4 | RNA binding motif | |
| YFR039c | OSW7 | Fly | signal peptide |
| YGL096w | TOS8 | putative transcription factor | |
| YGL144c | ROG1 | putative lipase | |
| YHR153c | SPO16 | synaptonemal complex assembly | |
| YIR013c | GAT4 | putative transcription factor YJL037w | |
| YJL037w | IRC18/OSW6 | Fly | paralog of OSW4 |
| YJL038c | LOH1/OSW4 | Permeable | paralog of OSW6 |
| YJL162c | JJJ2 | DNAJ domain | |
| YJR036c | HUL4 | E3 ligase | |
| YJR099w | YUH1 | ubiquitin hydrolase | |
| YKL121w | DGR2 | ||
| YLL030c | RRT7 | dubious ORF | |
| YLL054c | |||
| YLR030w | |||
| YLR238w | FAR10 | FAR complex subunit | |
| YLR373c | VID22 | Ether | |
| YML119w | |||
| YMR147w | |||
| YMR148w | OSW5 | Permeable | |
| YMR262w | |||
| YOL047c | LDS2 | paralog of LDS1 | |
| YOR350c | MNE1 | mitochondrial protein | |
| YPL033c | SRL4 | Permeable | |
| YPL184c | MRN1 | RNA binding |
Whether a mutation in the gene was identified in earlier screens of the same collection for ether sensitive spores [7], spores with permeable spore walls [20], or spores with lowered resistance to digestion by Drosophila [19].
Information from the SGD: www.yeastgenome.org.
A highly redundant network of genes is involved in dityrosine layer assembly
A significant fraction of the mutants examined in the screen (13%) displayed fluorescence defects in the sensitized dtr1Δ background. This suggests that the process is somehow buffered so that individual mutants have only modest effects. One possible explanation for the failure of individual mutants to produce strong effects on dityrosine would be the presence of redundant pathways leading to dityrosine incorporation. If so, combining mutants in genes involved in two separate pathways might lead to decreased dityrosine fluorescence even in the presence of DTR1. This hypothesis was tested by constructing double mutants between genes identified in our screen and examination of dityrosine fluorescence.
As gene products directly involved in assembly of polymerized dityrosine are likely localized to the spore wall, we focused on those with predicted signal peptides or transmembrane domains, or with predicted enzymatic activities. In addition, we also included the genes OSW3 and YEL023c since these genes have been implicated in outer spore wall formation in other studies [our unpublished results and [20]]. Most double mutant combinations were constructed twice and each strain was tested in triplicate for dityrosine fluorescence. Double mutants exhibiting a reduction in dityrosine fluorescence in at least two of the three replicates were scored as a genetic interaction (Table S1).
Reduced dityrosine fluorescence was observed for many different mutant combinations. Most of the mutants tested displayed interactions with at least seven other mutants. The extent of these interactions can be seen in graphic representation of the interaction network which shows a highly interconnected set of genes with relatively few outliers having limited synthetic interactions (Figure 2). This architecture suggests that assembly of the dityrosine layer is accomplished through a number of alternative processes with overlapping function. Major hubs of the network, such as OSW6 or LDS2, show interactions with most of the other genes and might represent particularly important functions in assembly of the outer spore wall.
Figure 2. Genetic interactions between genes involved in dityrosine layer formation.

Cytoscape was used to represent the genetic interactions listed in Table S1. Lines between the circles indicate reduction of dityrosine fluorescence in the double mutant. Double mutants were constructed from two independent pairs of parental strains and tested in three biological replicates (see Methods). Arrowheads indicate whether the interaction was seen in one (one arrowhead) or both (two arrowheads) double mutant strains. Circles in darker purple indicate genes with >10 interacting partners. The size of the circles increases with the number of genetic interactions seen for each gene.
Paralogous genes are redundant for dityrosine assembly
The double mutant analysis suggests that different processes can compensate for each other in spore wall assembly. Another type of redundancy results when different genes encode proteins with the same function. Of the 20 genes shown in the network in Figure 2, 11 are genes with potential paralogs in the S. cerevisiae genome (Table 2). If the paralogous genes are functionally redundant, then mutation of all the genes within a set should lead to a more severe phenotype. This idea was tested by construction of diploids homozygous for deletions of all of the genes within the first six paralog sets listed in Table 2 (attempts to construct deletions for the other three sets were unsuccessful). These diploids were then examined for dityrosine fluorescence both by patch assay and quantitatively using fluorescence microscopy [20] (Figure 3). Multiple mutants in all six of the paralog sets displayed reduced dityrosine fluorescence relative to single mutants, supporting the idea that these genes encode functionally redundant proteins. Quantitatively, dityrosine fluorescence was reduced by at least 45%, in deletions of all the paralog sets. The most severe defects were seen in the lds1Δ lds2Δ rrt8Δ and osw4Δ osw6Δ mutant strains where the fluorescence signal was reduced to the levels of a dit1Δ, which lacks dityrosine.
Table 2. Paralogous gene sets implicated in outer spore wall formation.
| ORF | Gene | Sporulation-induceda | % Similarityb |
| Lds set: | |||
| YAL018c | LDS1 | Y | 53% w/RRT8 |
| YOL047c | LDS2 | Y | 38% w/LDS1 |
| YOL048c | RRT8 | N | 58% w/LDS2 |
| Osw4/6 set: | |||
| YJL038c | OSW4/LOH1 | Y | 50% w/OSW6 |
| YJL037w | OSW6/IRC18 | Y | |
| Dtr set: | |||
| YBR180w | DTR1 | Y | 59% w/QDR3 |
| YIL120w | QDR1 | N | 48% w/DTR1 |
| YBR043c | QDR3 | N | 48% w/QDR1 |
| Gat set: | |||
| YLR013w | GAT3 | Y | 70% w/GAT4 |
| YIR013c | GAT4 | Y | |
| Osw/She set: | |||
| YFR039c | OSW7 | Y | 48% w/SHE10 |
| YGL228w | SHE10 | N | |
| Npp set: | |||
| YCR026c | NPP1 | N | 57% w/NPP2 |
| YEL016c | NPP2 | Y | |
| Fet set: | |||
| YDR506c | GMC1 | Y | 52% w/FET5 |
| YMR058w | FET3 | N | 50% w/GMC1 |
| YFL041w | FET5 | Y | 65% w/FET3 |
| Rog set: | |||
| YGL144c | ROG1 | Y | 63% w/ROG1 |
| YDL109c | N | ||
| Pes set: | |||
| YFR023w | PES4 | Y | 59% w/MIP6 |
| YHR015w | MIP6 | Y | |
Figure 3. The effect of deletion of paralog sets on dityrosine fluorescence.
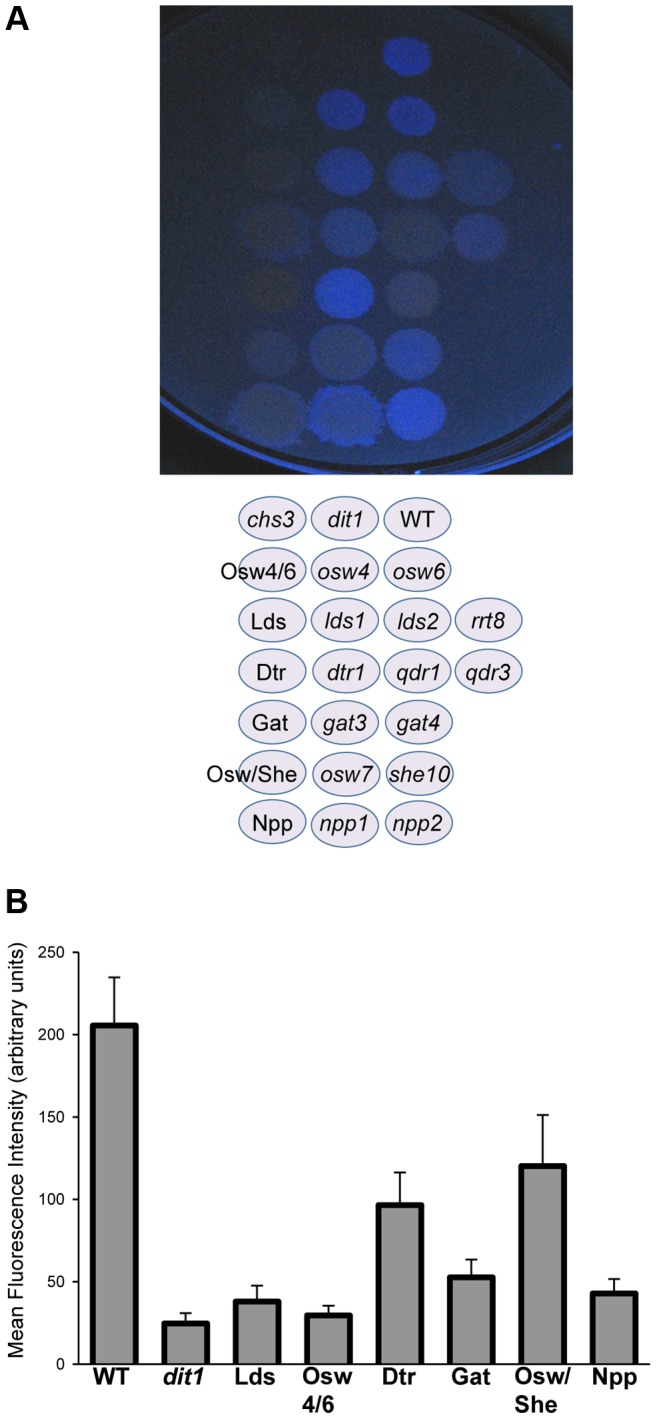
A) Patch assay of dityrosine fluorescence. Single and multiple mutant diploid cells of the indicated genotypes were sporulated on filters and the filter was exposed to UV light to detect dityrosine fluorescence. B) Quantitation of dityrosine fluorescence in the paralog mutant strains. Dityrosine fluorescence from the spore wall was quantified for strains of the indicated genotypes using a fluorescence microscopy assay. For each strain, the fluorescence value represents the average of twenty individual cells. Error bars indicate one standard deviation. Multiple mutant diploids are indicated by the name of the set of genes deleted as listed in Table 2.
Paralogous genes are not required for the presence of a chitosan layer
The deletion of the paralogous pairs (and triples) is distinct from the combination with dtr1Δ or other mutants in that these mutations likely remove a single function (e.g., Osw4/6 function) rather than weakening two different aspects of assembly (e.g., Osw4 function and Lds1 function). For this reason, the strains carrying deletions of all the genes for a particular paralog set were phenotypically characterized to determine what role each paralogous group plays in outer spore wall assembly. For simplicity, each multiple mutant strain will be indicated by the name of the paralog set given in Table 2, e.g. the lds1Δ lds2Δ rrt8Δ strain will be referred to as the Lds mutant.
Assembly of the dityrosine layer requires the underlying chitosan layer, so loss of dityrosine fluorescence could be an indirect effect of loss of chitosan [10]. Each paralog set mutant strain was stained with the chitin/chitosan binding dye Calcofluor White as well as the chitosan specific dye Eosin Y and examined in the fluorescence microscope. In wild-type cells, spores stain weakly with Calcofluor White because the dityrosine layer blocks the dye from binding the underlying chitosan [21]. By contrast, Eosin Y binds efficiently to the chitosan in the wild-type spores [22] (Figure 4, top row). Deletion of the CDA1 and CDA2 genes encoding chitin deacetylases results in spore walls that contain chitin instead of chitosan, resulting in the absence of a dityrosine layer [11]. Consistent with this fact, cda1Δ cda2Δ spores stain with Calcofluor White but not with Eosin Y (Figure 4, 2nd row). A chs3Δ mutant lacks both chitin and chitosan and as a result stains with neither dye (Figure 4, 3rd row). A dit1Δ mutant contains chitosan but lacks dityrosine and binding of both dyes is seen (Figure 4, 4th row).
Figure 4. The effect of deletion of paralog sets on chitosan.
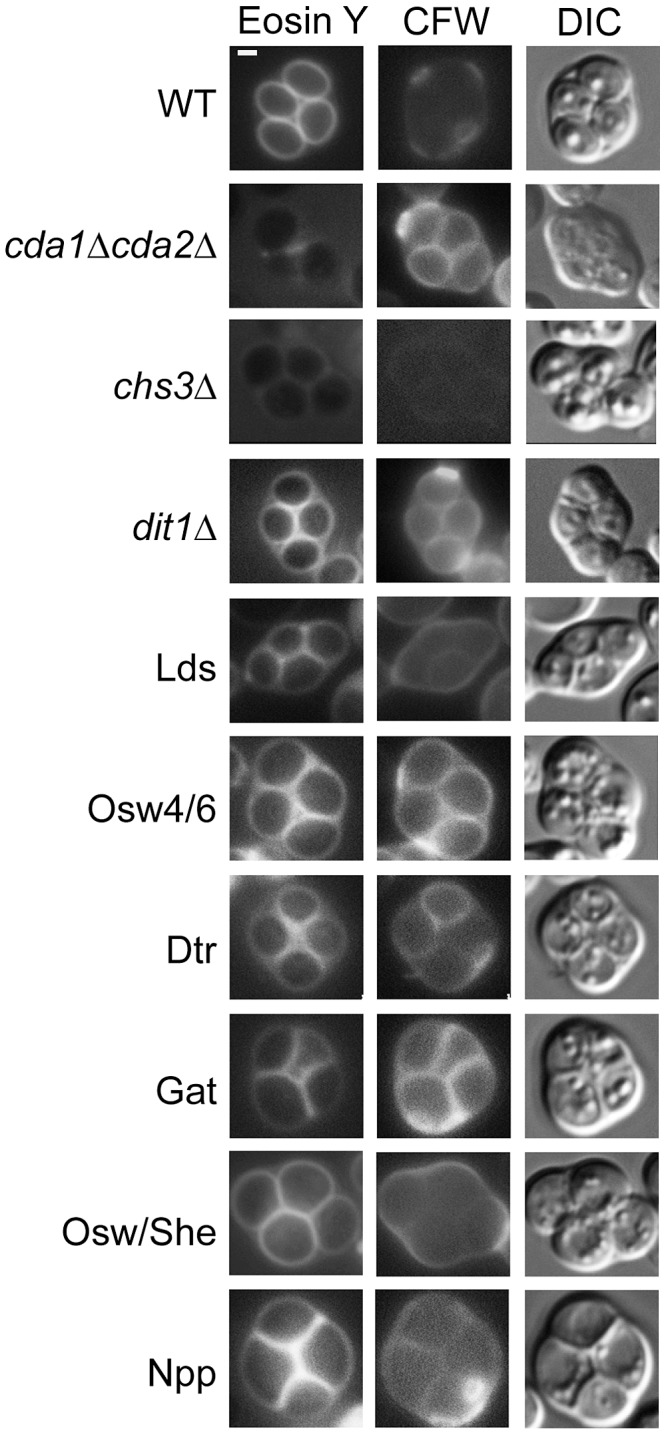
Strains of the indicated genotype were sporulated and stained with both Eosin Y and Calcofluor White (CFW) to visualize the chitosan layer. The cda1Δ cda2Δ, chs3Δ, and dit1Δ strains provide standards for the conditions chitin/no chitosan/no dityrosine, no chitin/no chitosan/no dityrosine, and chitin/chitosan/no dityrosine, respectively. For all strains >30 cells were examined and >60% display the staining patterns shown here. Quantitation for each strain is given in Table S2. DIC = differential interference contrast. Scale bar = 1 micron. Multiple mutant diploids are indicated by the name of the set of genes deleted as listed in Table 2.
None of the paralog sets is required for formation of the chitosan layer, as all of the paralogous mutant strains stained with Eosin Y. However, staining of the Lds mutant strain was noticeably less intense than in dit1Δ, suggesting that this mutant may have some loss of chitosan in addition to dityrosine. Compared to wild type, the Lds, Osw4/6, Dtr, and Gat mutants all displayed increased Calcofluor White staining consistent with reduced levels of dityrosine in the wall. The Osw/She and Npp strains did not show clear increases in Calcofluor White staining. For Osw/She this is perhaps consistent with the quantitatively modest reduction in dityrosine fluorescence in the double mutant (Figure 3B).
Paralogous genes are required for functional spore walls
Spore walls function to protect cells form various exogenous stresses. One assay that is commonly used for spore wall function is sensitivity to ether vapor. Ether is toxic to vegetative cells but the outer layers of the spore wall confer resistance to ether exposure [23]. To test whether the various paralog sets are required not just for wild-type levels of dityrosine fluorescence but also for making functional spore walls, diploids containing deletions of each set were tested for ether sensitivity. The outer layers of the spore wall contribute to ether resistance, but to different extents so that dit1Δ mutants, which lack dityrosine, are sensitive but less so than chs3Δ mutants, which lack both chitosan and dityrosine (Figure 5). All of the paralog set mutants showed increased sensitivity to ether. In fact, all of the combinations appeared more sensitive to ether than a dit1Δ strain, suggesting that the spore wall defect in these mutants may be more significant than just the loss of dityrosine. In the case of the Osw4/6 and Osw/She mutants, deletion of both paralogs is necessary to reveal the sensitive phenotype. In the other paralog sets, however, deletion of one of the genes is sufficient to account for much of the sensitivity. This is most obvious in the Lds set where the rrt8Δ single mutant is as ether sensitive as the lds1Δ lds2Δ rrt8Δ triple mutant. The patterns of dityrosine fluorescence in the individual mutants (Figure 3A) do not necessarily reflect the patterns of ether sensitivity. Thus, the mechanistic link between ether resistance and outer spore wall structure is unclear. However, the ether tests reveal that the spore walls in all of the paralog mutants are compromised in their ability to confer stress resistance to the spore.
Figure 5. The effect of deletion of paralog sets on ether resistance of spores.
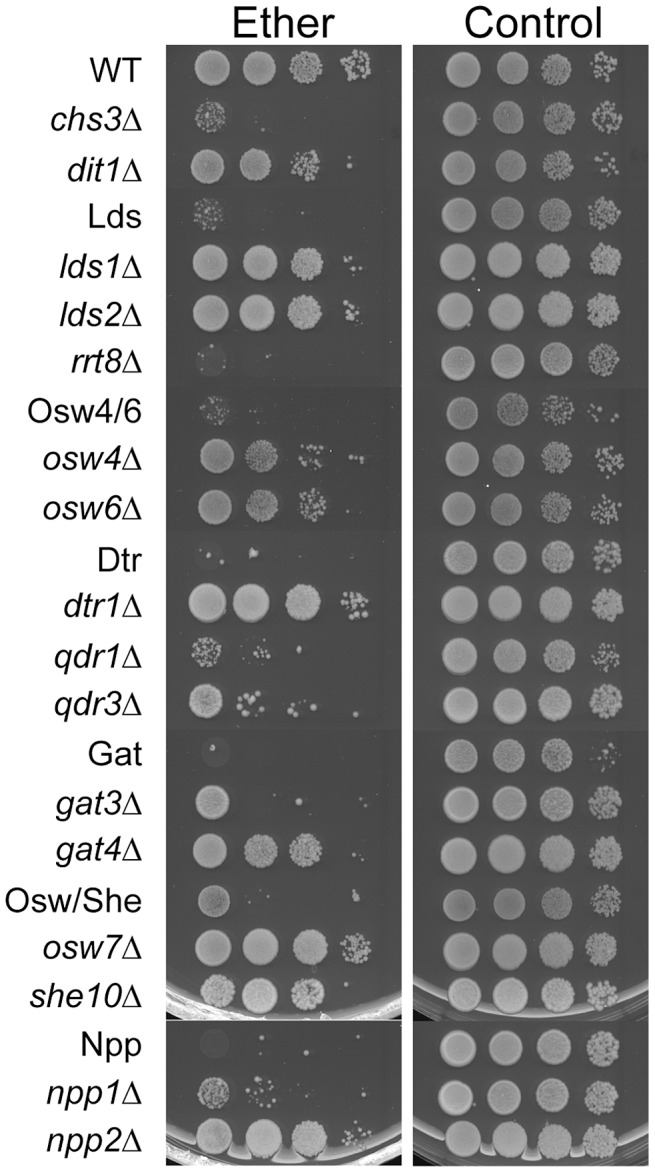
Strains of the indicated genotypes were sporulated in liquid to >70% asci and then 10-fold serial dilutions were spotted onto YPD plates. Left panels, plates exposed to ether vapor for 45 minutes before incubation at 30°C. Right panels, no ether control. Multiple mutant diploids are indicated by the name of the set of genes deleted as listed in Table 2.
Solid state NMR identifies a novel component of the spore wall
The hypothesis that multiple assembly routes are used for dityrosine incorporation into the spore wall suggests that different mutants might accumulate different intermediates in the biosynthetic pathway. Solid-state nuclear magnetic resonance (NMR) has been proven to be an effective tool for the analysis of intact bacterial cell walls [24], [25] and we sought to apply this technology to analyze the spore wall. An established protocol for purification of spore walls was adapted to isolate large quantities of spore wall fragments enriched for just the outer spore wall layers. These purified outer spore walls from wild type spores were analyzed using 13C solid state NMR spectroscopy. The samples were not isotopically enriched but relied on the natural abundance of 13C in the wall components. The 13C spectrum from a wild-type spore wall prepared in this way is shown in Figure 6. The identities of many of the chemical shifts were confirmed by comparison to spectra prepared using purified chitosan and tyrosine (Figure S1). The strongest resonances are from the six carbons of the glucosamine ring of chitosan (labeled in red in Figure 6). The conversion of chitin to chitosan is incomplete during spore wall formation as shown by the unique resonance at 22 ppm that corresponds to the methyl carbon of the acetyl moiety on N-acetyl-glucosamine groups present in the polymer (indicated by –CH3 in Figure 6). The carbonyl resonance associated with the N-acetyl-glucosamine groups is also observed at ∼174 ppm, but overlaps with the carbonyl resonance of dityrosine. A similar incomplete conversion of chitin to chitosan has been reported in cell walls of other fungi [26]. Small resonances at chemical shifts between 130 ppm and 155 ppm (labeled in green) correspond to the carbons of the dityrosine rings as confirmed by the loss of these signals when the spore wall of a dit1Δ mutant was examined (Figure 6, lower spectrum). As expected, the resonances corresponding to chitosan (and the residual N-acetyl-glucosamine groups of chitin) are still observed in the dit1Δ spore walls. In the spectrum of the dit1Δ mutant, the integrated intensities of carbonyl and methyl resonances of N-acetyl-glucosamine groups is close to 1∶1, indicating that the shoulder on the C = O resonance in the wild-type spectrum is primarily due to dityrosine. In addition, a number of significant resonances in the range of 25 to 35 ppm were observed that are not from chitosan or dityrosine and, thus, represent an unknown component of the outer spore wall, which we designate as component χ (labeled in blue) (Figure 6).
Figure 6. NMR analysis of the outer spore wall.

Outer spore walls were purified from wild type (AN120) or dit1Δ cells and then analyzed by solid state 13C NMR. Resonances assigned to chitosan carbons are designated by red labels and resonances from dityrosine are indicated by green labels. Unassigned chemical shifts from component χ are indicated in blue. Upper spectrum, spore wall from wild type. Lower spectrum, spore wall from dit1Δ. For comparison, spectra are scaled so the C5/3 resonance of chitosan is of constant height.
Solid state NMR reveals distinct outer spore wall defects in the paralog mutants
Spore walls prepared from six different paralog mutant strains were similarly analyzed by solid state NMR. Complete spectra for each strain are shown in Figure S2. The dityrosine and component χ regions of the spectra are shown in Figure 7. The relative heights of the carbonyl and Cζ resonances can be used to assess the amount of dityrosine in the spore wall (Figure 7A). For component χ, the height of the resonances relative to the -CH3 peak provides an indicator of its abundance (Figure 7B). The phenotypes of the various mutants fell on a spectrum with respect to their severity: the Npp mutant showed no strong effect on dityrosine or component χ incorporation; the Osw4/6 mutant had reduced dityrosine but not component χ incorporation; the Osw/She mutant showed reduced dityrosine and a modest reduction component χ incorporation; the Gat and Dtr mutant strains had strong reductions in both dityrosine and component χ; and the Lds mutant appeared to completely lack resonances for both dityrosine and component χ This NMR analysis confirms the effect of the paralog set mutants on dityrosine incorporation. In addition it identifies a new component of the outer spore wall, and shows that the Lds family proteins are essential for incorporation of both dityrosine and component χ into the wall.
Figure 7. NMR analysis of outer spore walls in the paralog mutant strains.

A) The dityrosine region of the 13C NMR spectrum is shown for spore walls from wild type, dit1Δ and the indicated paralog mutant strains. Positions of the dityrosine chemical shifts and chitin carbonyl chemical shifts are indicated above the wild type spectrum. The spectra have been scaled so the height of the carbonyl resonance is constant. B) The component χ region of the 13C NMR spectrum is shown for spore walls from wild type, dit1Δ and the indicated paralog mutant strains. Positions of the component χ chemical shifts and chitin methyl group shifts are indicated above the wild type spectrum. The spectra have been scaled to the C5/3 resonance as in Figure 6. Multiple mutant diploids are indicated by the name of the set of genes deleted as listed in Table 2.
The Lds proteins are localized to a specific population of lipid droplets
The possible role of the Lds proteins in component χ synthesis led us to examine the localization of these proteins. In sporulating cells, Lds1-GFP, Lds2-GFP, and Rrt8-GFP displayed similar localizations, concentrating in discrete patches or puncta along the ascal sides of the growing prospore membranes in cells in mid-Meiosis II. In post-meiotic cells, the proteins localized more uniformly around the outside of spores, consistent with localization to the spore wall (Figure 8A). To assess the functionality of the fusion protein, an RRT8-GFP strain was mated to an rrt8Δ, the resulting diploid was sporulated, and the ether sensitivity of the spores was examined. RRT8-GFP partially complemented the ether sensitivity of the rrt8Δ mutant, indicating that the fusion protein is at least partially functional and suggesting that the localization is relevant to Lds function (C. Lin, unpublished observations).
Figure 8. Localization of the Lds proteins involved in outer spore wall synthesis.

A) Diploids expressing GFP fusions to the indicated protein as well as mTag-BFP-Spo2051–91 as a prospore membrane (PrM) marker were sporulated and examined by fluorescence microscopy. Examples of a cell in the middle of Meiosis II and a post-meiotic cell are shown. B) Sporulating wild-type cells expressing mTag-BFP-Spo2051–91 were stained with the lipid droplet marker Bodipy TR. Arrowheads indicate lipid droplets associated with the ascal side of the prospore membrane. Arrows indicate lipid droplets within the prospore membrane C) Cells expressing both Lds2-GFP and mTag-BFP-Spo2051–91 were stained with Bodipy TR. Arrowheads indicate localization of Lds2-GFP to a lipid droplet outside of the prospore membrane. For all experiments at least 20 cells at the indicated stage were scored and 100% of the cells displayed patterns similar to those shown here. Quantitation for each experiment is given in Table S2. Scale bars = 1 micron.
An Rrt8-GFP fusion protein has previously been reported to localize to lipid droplets in vegetative cells, and an association of lipid droplets with the prospore membrane has been previously reported in electron microscopy studies [27]. To determine if the foci observed for Lds set proteins during sporulation are lipid droplets, the localization of this organelle in sporulating cells was investigated. BODIPY 493/503, which has a green fluorescence, can be used to stain lipid droplets in yeast [28]. However, because the Lds proteins are fused to GFP that also fluoresces green, an alternative method for visualizing lipid droplets was required for co-localization experiments. Red fluorescent BODIPY TR displayed an identical staining pattern to BODIPY 493/503 in vegetative yeast cells making it a good alternative to BODIPY 493/503 (Figure S3). When cells in Meiosis II were stained with BODIPY TR, bright staining lipid droplets were seen inside of the prospore membrane with smaller but clear staining of droplets outside of the prospore membrane as well (Figure 8B). In post-meiotic cells, lipid droplets are seen inside of the spores, but the BODIPY TR staining outside of the prospore membrane is lost. As BODIPY stains lipid droplets by partitioning into the hydrophobic core of the droplet, this loss of staining suggests that the lipid constituents of the droplets outside of the prospore membrane are consumed during the process of spore wall development.
To determine whether the Lds proteins are present in lipid droplets, cells expressing both a blue fluorescent marker for the prospore membrane and Lds2-GFP were sporulated and stained with BODIPY TR. In mid-Meiosis II cells, Lds2-GFP puncta overlapped with or were directly adjacent to a subset of the lipid droplets that appear to be outside of the prospore membrane. No localization of Lds2-GFP to lipid droplets inside of the prospore membrane was observed (Figure 8C). In post-meiotic cells, no overlap of GFP with BODIPY TR staining was seen, indicating that the lipid droplets to which Lds2-GFP localizes are the ones consumed during spore wall formation. Thus, the Lds proteins localize to a specific subset of developmentally regulated lipid droplets.
The fluorescence images suggest that the specific lipid droplets to which the Lds proteins localize are found on or outside of the prospore membrane. However, the resolution of the fluorescence images is not sufficient to clearly determine the position of the lipid droplets relative to the prospore membrane. Transmission electron microscopy was used to examine the behavior of lipid droplets in sporulating wild type cells at higher resolution (Figure 9). Consistent with the fluorescence experiments, in mid-Meiosis II cells, lipid droplets can bee seen in close contact with the side of the prospore membrane facing the presumptive ascal cytoplasm (Figure 9A, B). These droplets are often irregularly shaped and their surface stains more darkly with permanganate than droplets inside of the prospore membrane. After prospore membrane closure, lipid droplets remain associated with the outer membrane (Figure 9C). In more mature asci, where spore wall assembly is nearly complete, lipid droplets in the ascal cytoplasm are less numerous, but can still be found associated with the outer spore wall after outer membrane lysis (Figure 9D). Thus, there is a sub-population of lipid droplets that remain associated with the exterior surface of the spore throughout spore wall formation. The localization of the Lds proteins to these lipid droplets positions the proteins so that they may contribute directly to outer spore wall morphogenesis.
Figure 9. Electron microscopy of lipid droplets in sporulating cells.

Electron micrographs of wild type cells. A) A cell in Meiosis II showing two prospore membranes (green arrows) engulfing nuclear lobes (N). Lipid droplets can be seen associated with the prospore membrane (yellow arrowheads). Scale bar = 500 nm. B) Higher magnification of one of the prospore membranes in A. Scale bar = 100 nm. C) Post meiotic cell showing dark-stained lipid droplets (yellow arrowhead) associated with the outer membrane (green arrow). Lipid droplets inside of the spore (yellow arrow) do not stain as darkly with permanganate. N = nucleus. Scale bar = 500 nm. D) Lipid droplets in the ascus (yellow arrowhead) associated with the outer spore wall (red arrow) of mature spores. Yellow arrow indicated a lipid droplet within the spore cytoplasm. N = nucleus. Scale bar = 500 nm.
Discussion
Many screens for sporulation defective mutants in S. cerevisiae have been published and yet very few genes have been reported to affect the incorporation of dityrosine into the spore wall, which has been a significant obstacle to studies of wall development [18], [29]–[32]. This work shows that two levels of genetic redundancy are utilized in dityrosine assembly, which provides an explanation for the failure of earlier mutant screens to reveal the genes involved. The first level of redundancy is a redundancy of process in which mutations in two genes not related by sequence is required to reveal a phenotype. In these instances, the gene products, e.g., the secreted protease Osw3 and the ubiquitin ligase Hul4, have unrelated functions and yet the presence of one somehow buffers the loss of the other. How this is achieved is unclear. One possibility is that dityrosine might be incorporated into the wall via multiple mechanisms, for example different chemical linkages, and that loss of any individual linkage is insufficient to significantly reduce the fluorescence of the wall.
The second level is redundancy of function; many of the genes involved have paralogs of overlapping activities present in the genome and so deletion of multiple paralogs is required to produce a strong phenotype. Paralogous genes are relatively common in S. cerevisiae due to a whole genome duplication event that occurred during the evolution of Saccharomyces [33]. Of the paralogs listed in Table 2, five pairs (LDS1/LDS2, ROG1/YDL109c, NPP1/NPP2, OSW7/SHE10, PES4/MIP6) can be assigned to the whole genome duplication [34].
Paralogs with overlapping function are predicted to be lost from the genome without selective pressures for retention of both copies [35]. Several explanations have been advanced to account for redundant paralogs, including compensatory regulation, dosage effects, and incomplete specialization [35]. In particular, redundant genes involved in development in metazoans have been suggested to serve as a buffering system to protect against error [36], [37]. A similar logic may account for the redundancy in spore wall development. Many of the genes involved in spore wall development function only during this phase of the yeast life cycle and, therefore, cannot be selected for during mitotic growth. Redundancy of these activities may provide “insurance” against loss-of-function mutations arising during vegetative growth. By this reasoning, one might expect increased redundancy amongst genes involved in sporulation. It is notable in this regard that targeted deletion of sporulation-induced genes in S. cerevisiae revealed obvious phenotypes for only 11% of the mutants [18]. Similar studies in S. pombe reveal a somewhat higher percentage (30%), but again the majority of sporulation-induced genes display no deletion phenotype, suggesting that redundancy might be a common feature of developmental processes in yeast [38], [39]. Synthetic mutant analysis is well suited to address the genetics of redundant systems. The application of SGA analysis, as presented here, should be a valuable tool for dissecting other aspects of sporulation.
Functions of the paralog sets
What specific roles do these different gene products play in assembly of the outer spore wall? For several of the genes identified, the function of the proteins is likely indirect. For example, Dtr1 and related transporters are required for delivery of precursors for assembly. Similarly, the Gat3 and Gat4 proteins are DNA-binding proteins and, thus, are likely to be required for transcription of other genes whose products act on assembly. Very similar consensus binding sites have been defined for both Gat3 and Gat4 [40]. A search of yeast promoter sequences with these consensus sequences failed to identify any of the genes implicated in outer spore wall assembly (A. M. N., unpublished observations). Identification of the target genes for this pair of transcription factors may, therefore, identify additional genes involved in spore wall development.
Of the genes identified by the screen, OSW4/OSW6 and OSW7/SHE10 are the best candidates to encode proteins directly involved in assembly of the dityrosine layer. These genes encode proteins with predicted signal peptides and GFP fusions to Osw4 localized to the prospore membrane consistent with spore wall localization (C. Lin and A. M. Neiman, unpublished observations). Moreover, the 13C NMR spectra of both of the Osw4/6 and Osw/She mutants are similar to that of dit1Δ, showing reduced dityrosine, but significant levels of both chitosan and component χ. Thus, these mutants appear somewhat specific in their effects on the dityrosine. It remains to be determined if these proteins play an enzymatic or structural role in the wall.
A role for lipid droplets in spore wall assembly
In vegetative cells, lipid droplets are seen as multiple puncta near the nuclear envelope and ER [28]. This distribution is changed in sporulating cells where a subset of lipid droplets is associated specifically with the ascal side of the prospore membrane. The Lds proteins are found on this class of lipid droplets and are essential for outer spore wall assembly, revealing a function for lipid droplets in this process.
These results also suggest the existence of distinct sub-populations of lipid droplets, as defined both by localization and protein composition, during sporulation. Analysis of different lipid droplet proteins in mammalian cells has also revealed discrete sub-populations of lipid droplets in some cell types [41], [42]. Recruitment of distinct classes of droplets for specific functions may be a conserved behavior for this organelle.
The role of the Lds proteins in spore wall assembly is unclear, though the localization of lipid droplets to the surface of the spore wall (after outer membrane lysis) means that the Lds proteins could play a direct role in assembly. In addition to lacking dityrosine, the Lds mutant completely lacks component χ and has reduced Eosin Y and Calcofluor White staining, suggesting that the levels of chitosan may be reduced as well. Thus, the effect on dityrosine may be secondary to these other deficiencies, for instance, incorporation of component χ may be necessary for subsequent dityrosine assembly. Perhaps, some of the reactions in spore wall assembly actually occur on the surface of the lipid droplet, aided by the Lds proteins, before final incorporation of components into the wall. Additionally, both the EM studies and BODIPY staining suggest that the lipid constituents of the prospore membrane-associated lipid droplets are consumed during the course of spore wall assembly. Whether these lipids are used as an energy source for the spore, to expand the prospore membrane, or in some other way contribute to spore wall morphogenesis remains to be determined.
NMR analysis reveals a new component of the spore wall
Our results demonstrate that solid state NMR is an effective assay to examine the composition of the outer spore wall. The 13C spectrum of the outer spore wall is not overly complex, containing ∼20 distinct carbon resonances. This relative simplicity suggests that more sensitive 2-dimensional NMR assays could be effective in defining the structural organization of the spore wall, including how the different components are linked to each other.
In addition, our analysis reveals a previously unknown component of the outer spore wall, which we designate χ. Based on peak height of the NMR resonances, component χ is less abundant in the wall than chitosan, but more abundant than dityrosine. From the analysis of the dit1Δ spore wall it is clear that incorporation of component χ into the wall does not require dityrosine. Moreover, the Lds mutant spectrum shows that the chitosan layer is still formed in the absence of component χ. These data raise the possibility that component χ might act as a linker between the chitosan and dityrosine components of the spore wall.
The chemical nature of component χ remains to be determined, though based on previous biochemical characterizations of purified outer spore walls, it seems unlikely to be either a polysaccharide or a protein [9]. The lack of a carbonyl resonance associated with component χ in the NMR data also indicates that it does not contain amino acids. The positions of the component χ chemical shifts are consistent with reduced carbons such as in alkanes. Given that the Lds proteins required for component χ incorporation are localized to lipid droplets adjacent to the developing spore wall, perhaps generation of component χ requires material that is delivered from the lipid droplets to the spore wall.
It is also noteworthy that levels in the spore wall of component χ are reduced in the dtr1Δ qdr1Δ qdr3Δ mutant strain. This strain lacks dityrosine in the wall because of a failure to export monomeric dityrosine from the spore cytoplasm [16]. However, the reduction of component χ in this strain cannot be an effect of reduced dityrosine because incorporation of component χ is unaffected in dit1Δ cells that lack dityrosine. One straightforward possibility is that like dityrosine, some precursor to component χ is synthesized in the spore cytoplasm and exported to the wall by Dtr1 and related transporters.
Implications for other fungi
Genomic sequencing has revealed that many pathogenic fungi possess the enzymes for the production of chitosan and dityrosine. This is true even of fungi that do not form spores, such as Candida albicans. In fact, dityrosine has been found in the vegetative cell walls of C. albicans and mutants in dityrosine synthesis show drug sensitivity consistent with cell wall defects [43], [44]. Chitosan is also an important component of the wall of the pathogen Cryptococcus neoformans, where it is required for cell wall integrity and for virulence [22], [45]. While Cryptococcus does not synthesize dityrosine, it incorporates an analogous polyphenolic compound, melanin, into the cell wall and melanization is important for resistance of the fungus to various environmental stresses and may play an important role in the evasion of host immune responses during infection [2], [46]. Interestingly, C. neoformans mutants lacking chitosan display a “leaky melanin” phenotype, suggesting that chitosan is required for proper incorporation of melanin into the cell wall [22]. The nature of the connection between the carbohydrate and polyphenol components of the spore wall is, therefore, an important general issue in understanding the structure of the fungal cell wall. Assembly of the outer spore wall in S. cerevisiae provides a tractable model to address this issue. Our results identify several new genes intimately involved in construction of the outer spore wall. Further analysis of these gene products should provide insights into the structure of the spore wall and the mechanisms of its assembly.
Methods
Strains and media
Yeast strains used in this study are listed in Table S3. Unless otherwise indicated, standard yeast media and growth conditions were used [47]. For synthetic medium containing Geneticin (G418), monosodium glutamate (Sigma) was added as a nitrogen source instead of ammonium sulfate. Drug concentrations used were 200 mg/L G418, 3 mg/L cycloheximide, 1 g/L 5-FOA, and 300 mg/L Hygromycin B. Strain CL62 was derived from the ybr180wΔ strain from the systematic yeast knockout collection [48] by selection for growth on plates containing cycloheximide. To construct the paralog deletion strains (CL6, CL7, CL15, CL26, and CL57) lds1Δ, dtr1Δ, gat4Δ, osw7Δ, and npp2Δ strains from the SK1 knockout collection [18] were crossed to rrt8Δ, qdr3Δ, gat3Δ, she10Δ, and npp1Δ strains from the BY4741 knockout collection [48], respectively. After sporulation and dissection of the resulting diploids, double mutant haploids were identified by marker segregation, confirmed by PCR, and CL15, CL26 and CL57 were constructed by mating of appropriate segregants. The single gene deletion strains CL38, CL43, CL44, CL47, and CL59 were also obtained as segregants from these crosses. All knockout alleles were confirmed by PCR. For construction of CL6 and CL7, deletion of the third paralog was achieved by PCR-mediated gene disruption in double mutant haploids [49]. For CL6, primers LDS2-KO-F/LDS2-KO-R (for primer sequences see Table S4) and pAG32 [50] as template were used to generate deletion of LDS2. For CL7, primers QDR1-KO-F and QDR1-KO-R were used to delete QDR1. Single deletions of OSW4 and OSW6 (strains CL52 and CL54) were generated using the primer sets ANO262-A/ANO262-B for OSW4 and ANO263-A/ANO263-B for OSW6 and pFA6a-HIS3MX6 as the template to generate deletions in strains AN117-4B and AN117-16D followed by mating of the haploids. Simultaneous deletion of both OSW4 and OSW6 (strain CL35) was achieved using the same strategy but ANO262 and ANO263 were used as primers. CL50 was constructed by using the primers CDA1&2-KO-F and CDA1&2-KO-R to simultaneously delete both CDA1 and CDA2 in AN117-4B and AN117-16D.
Plasmids
Prospore membranes were visualized using pRS426-Spo2051–91-mTagBFP, which expresses a fusion of amino acids 51 to 91 of Spo20 to the N-terminus of mTagBFP under the control of the TEF2 promoter. To construct this plasmid, a yeast codon-optimized version of mTagBFP [51] flanked by PacI and AscI sites was synthesized (GeneWiz Inc., New Jersey) and cloned into pUC57. A PacI-AscI fragment was then used to replace the mCherry sequence in pRS426-Spo2051–91-mCherry.
Screen for synthetic interactions with dtr1Δ
A 48-pin replicator was used to transfer a collection of 301 deletions of sporulation-induced genes in the SK1 background [18] onto 90 mm YPD plates. All strains were arrayed in triplicate. Because the strains in this collection are HO, the arrayed patches were replica plated to SPO plates to generate haploid spores. CL62 cells (MAT a dtr1Δ cyh2) were crossed to these haploid spores by replica plating the sporulated patches to YPD plates spread with CL62. Diploids from this cross were selected on SD-Ura -Trp -His plates. To obtain double deletion mutants, the patches were then transferred to SPO medium and then to SD(MSG) -His +G418 +cycloheximide +5-FOA plates. On this medium, -His and G418 select for the presence of the two knockouts while the cycloheximide and 5-FOA select against any unsporulated diploid cells. Patches containing the double deletion cells were allowed to grow and spontaneously diploidize (all patches should contain both mating types due to the presence of the HO gene) and then replica plated onto nitrocellulose membranes on SPO plates for dityrosine fluorescence detection under UV302.
Construction of double mutants for pairwise interactions
To examine synthetic interactions between different genes implicated in outer spore wall assembly, individual strains carrying deletions of each gene examined (listed in Table S1) were taken from the SK1 knockout collection and crossed to a set of strains from the BY4741 knockout collection carrying deletions of all the remaining genes analyzed. Each double mutant combination was constructed twice, once with each gene as the MAT a parent in the initial mating (i.e. from the yeast knockout collection) and once with each gene as the MATα parent (from the SK1 knockouts). Construction of the initial diploids, isolation of the double mutants, and scoring of dityrosine fluorescence was performed similarly to the dtr1Δ synthetic screen described above except that no selection for cycloheximide resistance was used. Three separate isolates for each SK1 knockout were crossed to the other deletions and reduction of dityrosine fluorescence intensity in two of the three replicates was considered a synthetic interaction.
Visualization for pairwise genetic interaction networks
The network in Figure 2 was created using Cytoscape (version 2.8.3) [52].
Analysis of dityrosine fluorescence by patch assay
Cells to be assayed were grown as patches on YPD plates for 1 day and then replica-plated onto nitrocellulose membranes (Whatman Optitran BA-S 85) on YPD plates. After two days growth, the membranes were transferred to SPO plates and incubated for 3 days at 30°C. The membranes were then transferred to petri dishes containing 200 µl of water, 50 µl of 10 mg/ml zymolyase and 15 µl of β-mercaptoethanol at 30°C for 5 hrs. Finally, the membranes were wetted with 0.1 M NaOH solution to raise the pH and exposed to short wave UV light for the detection of dityrosine fluorescence.
Microscopy and image processing
All images were collected on a Zeiss Axio-Observer Z1 microscope using a Hamamatsu ER-G camera. Images were collected and fluorescence intensity was measured using Zeiss Axiovision software (version 4.7).
Quantitative analysis of dityrosine fluorescence
Dityrosine fluorescence from the spore wall was quantified as described [20]. Briefly, cells were sporulated in liquid SPO medium for three days, centrifuged, washed once with water, re-suspended in 5% ammonia solution to raise the pH, and finally transferred onto microscope slides. Images were collected with a dityrosine fluorescence-specific filter set (Ex. 320 nm/Em. 410 nm) at fixed exposure times. The dityrosine fluorescence intensity (in arbitrary units) was calculated for each cell by taking the average intensity at two points in the spore wall and subtracting the background fluorescence outside of the spore.
Calcofluor White and Eosin Y staining
Sporulated cells were harvested and washed with 1 ml McIlvaine's buffer (0.2 M Na2HPO4/0.1 M citric acid [pH 6.0]) followed by staining with 30 µl Eosin Y disodium salt (Sigma) (5 mg/ml) in 500 µl McIlvaine's buffer for 10 min at room temperature in the dark. Cells were then washed twice in McIlvaine's buffer to remove residual dye and resuspended in 200 µl McIlvaine's buffer. One microliter of a 1 mg/ml Calcofluor White solution (Sigma) was then added to the Eosin Y-stained cells before transfer to microscope slides. Fluorescence of Calcofluor White and Eosin Y stains was examined using DAPI and FITC filter sets, respectively.
BODIPY TR methyl ester staining
Sporulating cells were stained with 5 µM of BODIPY TR methyl ester (Life Technologies) in PBS buffer at room temperature for 10 min and washed twice with PBS buffer. BODIPY TR fluorescence was visualized using a Rhodamine filter set.
Electron microscopy
Sporulating yeast cells were collected and processed for electron microscopy as described in [53].
Test of ether resistance
Cells were sporulated in liquid SPO medium at 30°C for 2 days. All cultures displayed at least 75% sporulation. Serial dilutions (1, 10−1, 10−2, 10−3) of sporulated cells were spotted on two YPD plates. One plate served as an ether-negative control. The other plate was inverted over ether-soaked filter paper (Whatman #3, 1003-090) for 45 min. Plates were incubated at 30°C for one to two days and photographed.
NMR methods
Spore wall sample preparation
Isolation of spore walls was performed similarly to that previously described [54]. ∼25 g (wet weight) of sporulated wild type (AN120) cells were collected (2.5 L of SPO medium at 75% sporulation frequency), resuspended and washed with 0.1 M sodium phosphate buffer (pH 7.2) and then resuspended in 60 ml of 0.1 M sodium phosphate buffer (pH 7.2) containing 20 mg of Zymolyase (US Biologicals, Salem, MA) as well as 0.04% of ß-mercaptoethanol. The suspension was incubated with shaking at 30°C for 2 hr until spores were released from asci. To remove ascal debris, spore pellets were washed three times at 4,000 rpm for 10 min in a swinging bucket rotor with 0.5% Triton X-100 solution and then resuspended in a small volume (around 10 ml) of 0.5% TX-100, layered onto a 30 ml Percoll (MP Biomedicals, Santa Ana, CA) step gradient [Percoll/TX-100/2.5 M sucrose; from bottom to top 0.8/0.0005/0.1→0.7/0.001/0.1→0.6/0.0015/0.1→0.5/0.002/0.1] and spun at 10,000 rpm for 45 min at 4°C in a Sorvall SS34 rotor to separate spores from vegetative cells. Spores were collected from the bottom layer and washed three times with 0.5% TX-100 at 6,000 rpm for 5 min. The collected spores were then mixed with 20 ml of 0.5% Triton X-100 solution and 20 ml of 0.5 mm zirconium/silica beads (BioSpec, Bartlesville, OK) for lysis by bead beating. Spores were disrupted in a BeadBeater (BioSpec, Bartlesville, OK) with pulses of 20 sec ON/40 sec OFF until 80% of spores were disrupted (as determined by light microscopy). The spore wall fragments were separated from intact spores and spore cytoplasm by layering onto a cushion of 60% Percoll/2% TX-100/H2O and centrifugation at 23,000 rpm in a Beckman SW41 rotor at 4°C for 1 hr. Spore wall fragments form a visible band within the Percoll cushion and this band was collected, the Percoll diluted by addition of 35 ml of 0.5% Triton TX-100 and the fragments were pelleted by centrifugation at 6,000 rpm in a Sorvall SS-34 rotor for 5 min. The wall pellets were then washed three times with distilled H2O. Spore walls from mutant strains appear to have a somewhat lower density and so the concentration of Percoll used in the purifications had to be adjusted. For AN264, CL35, CL7, CL15, and CL26, the steps in the Percoll gradient at the first stage were 0.5→0.4→0.3→0.2. The cushion used in the second stage was 40% Percoll/2% TX-100/H2O. For CL6, the Percoll gradient was 0.35→0.25→0.15→0.05 and the Percoll cushion was 35% Percoll/2% TX-100/H2O.
To remove the inner spore wall layers, collected spore wall fragments were first resuspended in 5 ml of 50 mM Tris (pH 8.0) containing 1% SDS and 5 mM DTT. The sample was heated at 95°C for 1 hr and then 1 mg of protease K (US Biologicals) was added and the solution incubated at 50°C for 24 hr. After the protease K reaction, the samples were again heated to 95°C for 1 hr to inactivate the protease K and then washed 3 times with 0.5% TX-100 followed by 3 washes with 0.1 M sodium phosphate buffer. Samples were then resuspended in 2 ml of 0.1 M sodium phosphate buffer (pH 7.2) containing 0.04% of ß-mercaptoethanol. 1.5 mg of zymolyase (US Biologicals) was added and samples were incubated at 37°C for 24 hr. After zymolyase digestion, samples were washed 3 times with 0.5% TX-100 and then 3 times with H2O before desiccation in a speed vacuum concentrator at low temperature.
Solid-state NMR analysis
Spore wall solid-state NMR experiments were carried out on a 500 MHz Bruker AVANCE spectrometer using an HX magic angle spinning (MAS) probe. For analysis, 50–100 mg of dried spore wall samples were packed into 4 mm rotors. All experiments were performed at room temperature. The MAS speed was between 10 and 13 kHz to eliminate the overlap of relevant resonances with MAS sidebands. A ramped-amplitude cross polarization (CP) sequence was used to record the 1D 13C chemical shift spectra with proton decoupling. The experimental conditions were as follows: a 4 µs 1H 90° pulse was followed by 2 ms simultaneous 13C and 1H contact pulses and an acquisition time of 27 ms. The repetition delay was 3 s. 2000 to 6000 scans were recorded for each spectrum. The data were processed with exponential line broadening of 100 Hz. The carbonyl resonance of powdered glycine at 176.46 ppm relative to tetramethylsilane was used as an external 13C reference.
Supporting Information
Solid state 13C NMR spectra for chitosan, L-tyrosine, and wild-type spore walls. Spore walls were prepared as described in Methods. Chitosan and L-tyrosine are from Sigma (Chicago, Illinois).
(TIF)
Complete NMR spectra for the strains shown in Figure 7. Resonances assigned to chitosan, dityrosine and component χ are indicated above the wild type spectrum. For comparison, all the spectra have been scaled to have the same height of the Chitosan C1 peak.
(TIF)
Two examples of wild type vegetative yeast cells co-stained with BODIPY 495/503 (green) and BODIPY TR (red).
(TIF)
Analysis of double mutant strains. Three biological replicates were scored for each double mutant combination. Any replicate that displayed reduced dityrosine fluorescence is indicated by “−”. “+” indicates no loss of fluorescence observed. Double mutant combinations scored as interactions and used to generate Figure 2 are highlighted in yellow. n.d. = not determined.
(XLS)
Quantitation of fluorescence images.
(DOC)
Strains used in this study.
(DOC)
Oligonucleotides used in this study.
(DOC)
Acknowledgments
The authors are grateful to Martine Ziliox (Stony Brook University) and the Center for Structural Biology for assistance with the NMR analysis and to Nancy Hollingsworth (Stony Brook University) for comments on the manuscript. We are indebted to Liang Jin (Stony Brook University) for plasmids, Jamie Konopka (Stony Brook University) for providing BODIPY TR, and to Maya Schuldiner (Weizmann Institute, Israel) for GFP fusion strains.
Funding Statement
The research was funded by NIH grant R01 GM072540 to AMN. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Denning DW (2003) Echinocandin antifungal drugs. Lancet 362: 1142–1151. [DOI] [PubMed] [Google Scholar]
- 2. Eisenman HC, Casadevall A (2012) Synthesis and assembly of fungal melanin. Appl Microbiol Biotechnol 93: 931–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Orlean P (2012) Architecture and biosynthesis of the Saccharomyces cerevisiae cell wall. Genetics 192: 775–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neiman AM (2011) Sporulation in the Budding Yeast Saccharomyces cerevisiae . Genetics 189: 737–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kreger-Van Rij NJW (1978) Electron microscopy of germinating ascospores of Saccharomyces cerevisiae . Arch Microbiol 117: 73–77. [DOI] [PubMed] [Google Scholar]
- 6. Klis FM, Mol P, Hellingwerf K, Brul S (2002) Dynamics of cell wall structure in Saccharomyces cerevisiae . FEMS Microbiol Rev 26: 239–256. [DOI] [PubMed] [Google Scholar]
- 7. Coluccio A, Bogengruber E, Conrad MN, Dresser ME, Briza P, et al. (2004) Morphogenetic pathway of spore wall assembly in Saccharomyces cerevisiae . Eukaryot Cell 3: 1464–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Briza P, Ellinger A, Winkler G, Breitenbach M (1988) Chemical composition of the yeast ascospore wall. The second outer layer consists of chitosan. J Biol Chem 263: 11569–11574. [PubMed] [Google Scholar]
- 9. Briza P, Ellinger A, Winkler G, Breitenbach M (1990) Characterization of a DL-dityrosine-containing macromolecule from yeast ascospore walls. J Biol Chem 265: 15118–15123. [PubMed] [Google Scholar]
- 10. Pammer M, Briza P, Ellinger A, Schuster T, Stucka R, et al. (1992) DIT101 (CSD2, CAL1), a cell cycle-regulated yeast gene required for synthesis of chitin in cell walls and chitosan in spore walls. Yeast 8: 1089–1099. [DOI] [PubMed] [Google Scholar]
- 11. Christodoulidou A, Briza P, Ellinger A, Bouriotis V (1999) Yeast ascospore wall assembly requires two chitin deacetylase isozymes. FEBS Lett 460: 275–279. [DOI] [PubMed] [Google Scholar]
- 12. Briza P, Breitenbach M, Ellinger A, Segall J (1990) Isolation of two developmentally regulated genes involved in spore wall maturation in Saccharomyces cerevisiae . Genes Dev 4: 1775–1789. [DOI] [PubMed] [Google Scholar]
- 13. Briza P, Winkler G, Kalchhauser H, Breitenbach M (1986) Dityrosine is a prominent component of the yeast ascospore wall. A proof of its structure. J Biol Chem 261: 4288–4294. [PubMed] [Google Scholar]
- 14. Briza P, Eckerstorfer M, Breitenbach M (1994) The sporulation-specific enzymes encoded by the DIT1 and DIT2 genes catalyze a two-step reaction leading to a soluble LL-dityrosine-containing precursor of the yeast spore wall. Proc Natl Acad Sci U S A 91: 4524–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Briza P, Kalchhauser H, Pittenauer E, Allmaier G, Breitenbach M (1996) N,N′-Bisformyl dityrosine is an in vivo precursor of the yeast ascospore wall. Eur J Biochem 239: 124–131. [DOI] [PubMed] [Google Scholar]
- 16. Felder T, Bogengruber E, Tenreiro S, Ellinger A, Sa-Correia I, et al. (2002) Dtrlp, a multidrug resistance transporter of the major facilitator superfamily, plays an essential role in spore wall maturation in Saccharomyces cerevisiae . Eukaryot Cell 1: 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tong AH, Lesage G, Bader GD, Ding H, Xu H, et al. (2004) Global mapping of the yeast genetic interaction network. Science 303: 808–813. [DOI] [PubMed] [Google Scholar]
- 18. Rabitsch KP, Toth A, Galova M, Schleiffer A, Schaffner G, et al. (2001) A screen for genes required for meiosis and spore formation based on whole-genome expression. Curr Biol 11: 1001–1009. [DOI] [PubMed] [Google Scholar]
- 19. Coluccio AE, Rodriguez RK, Kernan MJ, Neiman AM (2008) The yeast spore wall enables spores to survive passage through the digestive tract of Drosophila. PLoS ONE 3: e2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suda Y, Rodriguez RK, Coluccio AE, Neiman AM (2009) A screen for spore wall permeability mutants identifies a secreted protease required for proper spore wall assembly. PLoS One 4: e7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tachikawa H, Bloecher A, Tatchell K, Neiman AM (2001) A Gip1p-Glc7p phosphatase complex regulates septin organization and spore wall formation. J Cell Biol 155: 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baker LG, Specht CA, Donlin MJ, Lodge JK (2007) Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot Cell 6: 855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dawes IW, Hardie ID (1974) Selective killing of vegetative cells in sporulated yeast cultures by exposure to diethyl ether. Mol Gen Genet 131: 281–289. [DOI] [PubMed] [Google Scholar]
- 24. Kern T, Hediger S, Muller P, Giustini C, Joris B, et al. (2008) Toward the characterization of peptidoglycan structure and protein-peptidoglycan interactions by solid-state NMR spectroscopy. J Am Chem Soc 130: 5618–5619. [DOI] [PubMed] [Google Scholar]
- 25. Shenouda NS, Pan Y, Schaefer J, Wilson GE (1996) A simple solid-state NMR method for determining peptidoglycan crosslinking in Bacillus subtilis. Biochim Biophys Acta 1289: 217–220. [DOI] [PubMed] [Google Scholar]
- 26. Fukamizo T, Sonoda K, Toyoda H, Ouchi S, Goto S (1990) Solid State 13C-NMR Analysis of Cell Wall Components of Fusarium oxysporum . Agric Biol Chem 54: 2761–2762. [Google Scholar]
- 27. Lynn RR, Magee PT (1970) Development of the spore wall during ascospore formation in Saccharomyces cerevisiae . J Cell Biol 44: 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szymanski KM, Binns D, Bartz R, Grishin NV, Li WP, et al. (2007) The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci U S A 104: 20890–20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Briza P, Bogengruber E, Thur A, Rutzler M, Munsterkotter M, et al. (2002) Systematic analysis of sporulation phenotypes in 624 non-lethal homozygous deletion strains of Saccharomyces cerevisiae . Yeast 19: 403–422. [DOI] [PubMed] [Google Scholar]
- 30. Deutschbauer AM, Williams RM, Chu AM, Davis RW (2002) Parallel phenotypic analysis of sporulation and postgermination growth in Saccharomyces cerevisiae . Proc Natl Acad Sci U S A 99: 15530–15535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Enyenihi AH, Saunders WS (2003) Large-scale functional genomic analysis of sporulation and meiosis in Saccharomyces cerevisiae . Genetics 163: 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marston AL, Tham WH, Shah H, Amon A (2004) A genome-wide screen identifies genes required for centromeric cohesion. Science 303: 1367–1370. [DOI] [PubMed] [Google Scholar]
- 33. Wolfe KH, Shields DC (1997) Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387: 708–713. [DOI] [PubMed] [Google Scholar]
- 34. Seoighe C, Wolfe KH (1999) Updated map of duplicated regions in the yeast genome. Gene 238: 253–261. [DOI] [PubMed] [Google Scholar]
- 35. Thomas JH (1993) Thinking about genetic redundancy. Trends Genet 9: 395–399. [DOI] [PubMed] [Google Scholar]
- 36. Krakauer DC, Nowak MA (1999) Evolutionary preservation of redundant duplicated genes. Semin Cell Dev Biol 10: 555–559. [DOI] [PubMed] [Google Scholar]
- 37. Wagner A (2000) The role of population size, pleiotropy and fitness effects of mutations in the evolution of overlapping gene functions. Genetics 154: 1389–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gregan J, Rabitsch PK, Sakem B, Csutak O, Latypov V, et al. (2005) Novel genes required for meiotic chromosome segregation are identified by a high-throughput knockout screen in fission yeast. Curr Biol 15: 1663–1669. [DOI] [PubMed] [Google Scholar]
- 39. Martin-Castellanos C, Blanco M, Rozalen AE, Perez-Hidalgo L, Garcia AI, et al. (2005) A large-scale screen in S. pombe identifies seven novel genes required for critical meiotic events. Curr Biol 15: 2056–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, et al. (2008) A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell 32: 878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Straub BK, Herpel E, Singer S, Zimbelmann R, Breuhahn K, et al. (2010) Lipid droplet-associated PAT-proteins show frequent and differential expression in neoplastic steatogenesis. Mod Pathol 23: 480–492. [DOI] [PubMed] [Google Scholar]
- 42. Straub BK, Stoeffel P, Heid H, Zimbelmann R, Schirmacher P (2008) Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology 47: 1936–1946. [DOI] [PubMed] [Google Scholar]
- 43. Melo NR, Moran GP, Warrilow AG, Dudley E, Smith SN, et al. (2008) CYP56 (Dit2p) in Candida albicans: characterization and investigation of its role in growth and antifungal drug susceptibility. Antimicrob Agents Chemother 52: 3718–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smail EH, Briza P, Panagos A, Berenfeld L (1995) Candida albicans cell walls contain the fluorescent cross-linking amino acid dityrosine. Infect Immun 63: 4078–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Baker LG, Specht CA, Lodge JK (2011) Cell wall chitosan is necessary for virulence in the opportunistic pathogen Cryptococcus neoformans . Eukaryot Cell 10: 1264–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Casadevall A, Rosas AL, Nosanchuk JD (2000) Melanin and virulence in Cryptococcus neoformans. Curr Opin Microbiol 3: 354–358. [DOI] [PubMed] [Google Scholar]
- 47.Rose MD, Fink GR (1990) Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- 48. Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, et al. (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906. [DOI] [PubMed] [Google Scholar]
- 49. Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, et al. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14: 953–961. [DOI] [PubMed] [Google Scholar]
- 50. Goldstein AL, McCusker JH (1999) Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae . Yeast 15: 1541–1553. [DOI] [PubMed] [Google Scholar]
- 51. Subach OM, Gundorov IS, Yoshimura M, Subach FV, Zhang J, et al. (2008) Conversion of red fluorescent protein into a bright blue probe. Chem Biol 15: 1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Neiman AM (1998) Prospore membrane formation defines a developmentally regulated branch of the secretory pathway in yeast. J Cell Biol 140: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Esposito RE, Dresser ME, Breitenbach M (2004) Identifying Sporulation Genes, Visualizing Synaptonemal Complexes, and Large-Scale Spore and Spore Wall Purification. In: Guthrie C, Fink GR, editors. Methods in Enzymology: Guide to yeast genetics and molecular and cell biology: Academic Press. pp. 110–130. [DOI] [PubMed]
- 55. Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, et al. (1998) The transcriptional program of sporulation in budding yeast. Science 282: 699–705. [DOI] [PubMed] [Google Scholar]
- 56. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Solid state 13C NMR spectra for chitosan, L-tyrosine, and wild-type spore walls. Spore walls were prepared as described in Methods. Chitosan and L-tyrosine are from Sigma (Chicago, Illinois).
(TIF)
Complete NMR spectra for the strains shown in Figure 7. Resonances assigned to chitosan, dityrosine and component χ are indicated above the wild type spectrum. For comparison, all the spectra have been scaled to have the same height of the Chitosan C1 peak.
(TIF)
Two examples of wild type vegetative yeast cells co-stained with BODIPY 495/503 (green) and BODIPY TR (red).
(TIF)
Analysis of double mutant strains. Three biological replicates were scored for each double mutant combination. Any replicate that displayed reduced dityrosine fluorescence is indicated by “−”. “+” indicates no loss of fluorescence observed. Double mutant combinations scored as interactions and used to generate Figure 2 are highlighted in yellow. n.d. = not determined.
(XLS)
Quantitation of fluorescence images.
(DOC)
Strains used in this study.
(DOC)
Oligonucleotides used in this study.
(DOC)