

Summary
Background
Laboratory evidence has shown that cannabinoids might have a neuroprotective action. We investigated whether oral dronabinol (Δ9-tetrahydrocannabinol) might slow the course of progressive multiple sclerosis.
Methods
In this multicentre, parallel, randomised, double-blind, placebo-controlled study, we recruited patients aged 18–65 years with primary or secondary progressive multiple sclerosis from 27 UK neurology or rehabilitation departments. Patients were randomly assigned (2:1) to receive dronabinol or placebo for 36 months; randomisation was by stochastic minimisation, using a computer-generated randomisation sequence, balanced according to expanded disability status scale (EDSS) score, centre, and disease type. Maximum dose was 28 mg per day, titrated against bodyweight and adverse effects. Primary outcomes were EDSS score progression (masked assessor, time to progression of ≥1 point from a baseline score of 4·0–5·0 or ≥0·5 points from a baseline score of ≥5·5, confirmed after 6 months) and change from baseline in the physical impact subscale of the 29-item multiple sclerosis impact scale (MSIS-29-PHYS). All patients who received at least one dose of study drug were included in the intention-to-treat analyses. This trial is registered as an International Standard Randomised Controlled Trial (ISRCTN 62942668).
Findings
Of the 498 patients randomly assigned to a treatment group, 329 received at least one dose of dronabinol and 164 received at least one dose of placebo (five did not receive the allocated intervention). 145 patients in the dronabinol group had EDSS score progression (0·24 first progression events per patient-year; crude rate) compared with 73 in the placebo group (0·23 first progression events per patient-year; crude rate); HR for prespecified primary analysis was 0·92 (95% CI 0·68–1·23; p=0·57). Mean yearly change in MSIS-29-PHYS score was 0·62 points (SD 3·29) in the dronabinol group versus 1·03 points (3·74) in the placebo group. Primary analysis with a multilevel model gave an estimated between-group difference (dronabinol–placebo) of −0·9 points (95% CI −2·0 to 0·2). We noted no serious safety concerns (114 [35%] patients in the dronabinol group had at least one serious adverse event, compared with 46 [28%] in the placebo group).
Interpretation
Our results show that dronabinol has no overall effect on the progression of multiple sclerosis in the progressive phase. The findings have implications for the design of future studies of progressive multiple sclerosis, because lower than expected progression rates might have affected our ability to detect clinical change.
Funding
UK Medical Research Council, National Institute for Health Research Efficacy and Mechanism Evaluation programme, Multiple Sclerosis Society, and Multiple Sclerosis Trust.
Introduction
Multiple sclerosis is the most common cause of neurological disability in young adults. It is generally regarded as an autoimmune disease, with early episodes of inflammation associated with axonal damage, which becomes the major pathological process as the disease progresses. Initial clinical relapses are often replaced by secondary gradual progression after several years. Although therapies for the inflammatory phase are available, none has been shown to slow disease progression in the absence of relapses.
Cannabinoids are used to ameliorate multiple-sclerosis-related symptoms, particularly muscle spasticity and pain. Our previous large multicentre cannabinoids in multiple sclerosis (CAMS) trial1 focused on testing symptomatic benefits of oral cannabinoids for 15 weeks in 630 participants, 95% with progressive disease. A treatment effect on spasticity (primary outcome assessed by the Ashworth scale) was not evident, although more participants reported benefits from symptom alleviation in the active group than in the placebo group. During the study, experimental evidence emerged to suggest that cannabinoids have neuroprotective effects (with antioxidant,2 antiapoptotic,3 antiexcitatory,4 and anti-inflammatory actions5) and might also encourage remyelination.6 The CAMS follow-up study,7 in which participants, masked to treatment, could opt to continue study drug for up to 12 months, reported significant effects on spasticity in the dronabinol (Δ9-tetrahydrocannabinol) group, with some evidence for an effect on disability, measured by the expanded disability status scale (EDSS) and the Rivermead mobility index (RMI). This finding provided clinical evidence to support experimental data suggesting that cannabinoids might have a neuroprotective effect in progressive multiple sclerosis, and confirmed that this treatment continued to ameliorate patients' symptoms for up to 12 months.
In the cannabinoid use in progressive inflammatory brain disease (CUPID) trial, we aimed to test the hypothesis that oral dronabinol will slow progression of primary and secondary progressive multiple sclerosis over 3 years.
Methods
Study design and participants
We undertook a randomised, double-blind, placebo-controlled, parallel-group study. Patients were prospectively enrolled at 27 UK neurology or rehabilitation outpatient centres. Final follow-up data collection took place in January, 2012. Eligible patients were aged 18–65 years with a diagnosis of multiple sclerosis,8 either a primary progressive or a secondary progressive disease course, evidence of disease progression in the preceding year in the opinion of the local neurologist, an EDSS score of 4·0–6·5 at baseline, and willingness to abstain from other cannabis use during the trial. Exclusion criteria included use of immunomodulatory or otherwise potentially disease-modifying therapies in the previous 12 months, systemic corticosteroid use in the previous 3 months, predominant relapsing-remitting disease in the previous 12 months, multiple sclerosis relapse in the previous 6 months that was likely to have affected patients' EDSS scores, history of previous psychosis or other serious medical illness, pregnancy, serious cognitive impairment (such that the patient was unable to provide informed consent), and cannabinoid use within the previous 4 weeks (a urinary cannabinoid test was done before study entry). The study was approved by the South and West Devon Research Ethics Committee and done in accordance with Good Clinical Practice guidelines. Eligible patients provided written informed consent before participation.
Randomisation and masking
Participants were assigned via computer-generated randomisation to receive oral dronabinol in hard gelatine capsules or identically matched (in terms of appearance and smell) placebo vegetable oil capsules for 36 months in a 2:1 active-to-placebo ratio (to encourage recruitment and retention). The randomisation allocation sequence, generated by an independent statistician using a stochastic minimisation algorithm, was balanced according to EDSS score, centre, and disease type (primary or secondary progressive multiple sclerosis) with a random component. Assignment of treatment was undertaken by the central pharmacy independently of the research team to ensure allocation concealment. Participants and all other personnel directly involved in the study were masked to treatment allocation.
Procedures
Medical history, concomitant medication record, EDSS score, and consent were obtained at a screening visit. Dronabinol or placebo was provided at baseline 2–4 weeks later, after confirmation of eligibility and randomisation. Because of the large interindividual dose variation with oral cannabinoids, a 4 week titration period was included for both study groups, during which doses were adjusted to an appropriate level for each participant according to bodyweight and adverse events (AEs). Starting dose was one capsule (3·5 mg Δ9-tetrahydrocannabinol equivalent) twice daily. The maximum dose was 28 mg per day. Participants were instructed to increase their twice-daily dose by one capsule at weekly intervals to a maximum weight-related dose (table 1). If adverse effects developed, participants were advised not to increase the dose; if unwanted side-effects were intolerable, the dose was reduced. Dronabinol and placebo were supplied by Insys Therapeutics (Phoenix, AZ, USA) and distributed to sites by a central pharmacy.
Table 1.
Number of capsules prescribed according to weight of participants at baseline
| Week 1 | Week 2 | Week 3 | Week 4 to study end | |
|---|---|---|---|---|
| <60 kg | 1 twice a day | 2 twice a day | 2 twice a day | 2 twice a day |
| 60–80 kg | 1 twice a day | 2 twice a day | 3 twice a day | 3 twice a day |
| >80 kg | 1 twice a day | 2 twice a day | 3 twice a day | 4 twice a day |
Our study had two primary outcomes: time to EDSS score progression of at least 1 point from a baseline EDSS score of 4·0, 4·5, or 5·0 or at least 0·5 points from a baseline EDSS score of 5·5 or greater, confirmed by a physician at the next scheduled 6 monthly visit; and change from baseline to end of study in the physical impact subscale of the patient-reported 29-item multiple sclerosis impact scale (MSIS-29-PHYS). Secondary outcomes included assessment of number and nature of AEs, multiple sclerosis functional composite (MSFC) Z score change from baseline to final visit, multiple sclerosis walking scale (MSWS-12), and RMI.
Participants were reviewed after 2 and 4 weeks from when they began taking the study drug for AE monitoring and dose adjustment. Assessment visits were held at 3 and 6 months, and then every 6 months up to 36 months (visit 11) before study medication was reduced by one capsule per twice-daily dose each week (maximum period of reduction was 4 weeks). Participants who had new EDSS score progression according to the defined study endpoint at 36 months attended a further visit at 42 months (visit 12) to confirm EDSS score progression or otherwise, before discontinuing trial medication.
Each study site had a treating physician, responsible for dose adjustment and AE monitoring, and a trained assessing physician, responsible for 6-monthly EDSS assessment, but masked to AEs. The option of validated telephone EDSS assessment for participants unable or unwilling to attend clinic was introduced from June, 2010, to improve data completeness. The MSFC (consisting of a timed 25 foot walk, paced auditory serial addition test, and a nine hole peg test) was assessed yearly by a non-physician, providing training had been given. RMI was assessed every 6 months. The MSIS-29-PHYS and MSWS-12 were completed by participants every 6 months. Safety monitoring included standard clinical laboratory assessments (chemistry and haematology) at regular intervals. Random urine testing, used to detect cannabinoid use in the placebo group, was done in both groups to avoid unblinding. Brain MRI was done at baseline and yearly in participants who were chosen on the basis of quality and cost of MRI at their local centre. Images were analysed for percentage brain volume change (PBVC)9,10 and new and enlarging lesions at the Queen Square MS Centre, Nuclear Magnetic Resonance Research Unit, Institute of Neurology, University College London (London, UK).
A trial steering committee including an independent chairman, neurologist, statistician, and lay representative were responsible for trial oversight and met on a yearly basis. An independent data monitoring committee (IDMC) met yearly to review unblinded safety and efficacy data. Interim analyses of primary outcomes were produced by an independent statistician on request from the IDMC, with the predefined Haybittle-Peto boundary stopping rule. Four interim analyses were undertaken after 298 and 493 participants had been recruited and then yearly during follow-up. The IDMC recommended continuation of the trial after all interim analyses.
Statistical analysis
Previous data suggested a probable progression rate of about 70% in the control (placebo) group.11 On the basis of this progression rate and an expected 5% yearly loss to follow-up rate, recruitment of 492 patients would provide 90% power to detect a one-third reduction in hazard of progression (ie, hazard ratio [HR] of 0·67), corresponding to a relative reduction in risk of progression over 3 years of 21% (from 70% to 55% progression in the dronabinol group). For MRI effects, 261 patients allocated to dronabinol and placebo in a 2:1 ratio would provide 90% power to detect a 40% reduction in the rate of atrophy over 3 years.
Data analysis, using statistical software R (version 2.14.1),12 was by intention to treat, undertaken in accordance with a prespecified analysis plan, finalised and agreed by the trial steering committee before unmasking. We did all tests at the 5% significance level, with no adjustments for multiple testing. In models for each outcome, we considered main effects of centre, baseline EDSS score, disease type, age at registration, sex, and weight, as well as treatment.
We used Kaplan-Meier estimates to show probability of EDSS score progression in the two treatment groups and a Cox proportional hazards model to analyse time to progression. We also estimated HRs for subgroups defined by sex, baseline EDSS score, disease type, weight, and age.
The primary analysis was based on EDSS data obtained according to trial schedule; we regarded losses to follow-up before confirmed progression as missing data and treated them as censored observations at the time of last visit at which EDSS measurements were taken. Sensitivity of conclusions was assessed by repeating the analyses, treating losses to follow-up before confirmed progression as progression events.
We calculated total MSIS-29-PHYS scores and scores for each MSFC component with previously published methods.13,14 We computed MSFC component-wise Z scores15 using results from all participants at visit 2 as reference data.14 We calculated MSFC composite scores from the mean of component-wise Z scores and total scores for MSWS-1216 and RMI using an algorithm analogous to that used for the MSIS-29-PHYS for dealing with missing data.
We analysed repeated measures data for MSIS-29-PHYS with multilevel models, which included time (visit) and the other prespecified covariates. We incorporated individual differences in scores using random coefficients.17 Backward elimination was used to identify a final, reduced model, including effects significant at the 5% level, and effects of time and treatment. Repeated measures of MSFC (composite score), MSWS-12 (total score), and RMI (total score) were analysed similarly using multilevel models, with the same covariates and variable selection procedure as for MSIS-29-PHYS.
We used a multilevel model to analyse repeated measures data for PBVC from MRI, transformed to cumulative, relative PBVC on the log10 scale. We used logistic regression models to examine the effect of treatment and other prespecified covariates on new T1 hypointense and new or enlarging T2 hyperintense lesions during follow-up. Participants were classified as having either no or at least one new or enlarging lesion(s) during follow-up. We identified final models with a forward selection procedure and included main effects and interactions significant at the 5% level, as well as the main effect of treatment.
After prespecified subgroup analyses of time to EDSS score progression, in a post-hoc subgroup analysis we assessed the effect of treatment on time to progression in those participants with a baseline EDSS score of 5·5 or lower. We used a log-rank test to compare probability of progression between treatment groups.
Further post-hoc analyses examined the effect of treatment in the two participant groups with baseline EDSS scores of 5·5 or lower and 6·0–6·5, on both PBVC and lesions, by including separate effects of treatment in these two groups in the models.
We summarised AEs and serious AEs in terms of frequencies and relative frequencies. We used Fisher's exact test to test for associations between treatment and occurrence of serious AEs and between treatment and occurrence of each AE that affected at least 10% of all participants.
This trial is registered as an International Standard Randomised Controlled Trial (ISRCTN 62942668).
Role of the funding source
The funders had no role in study design, data collection, data analysis, data interpretation, or writing of the report. All authors had full access to all of the data in the study and the corresponding author had final responsibility for the decision to submit for publication.
Results
We recruited 498 eligible patients between May, 2006, and July, 2008. Baseline characteristics were similar in both treatment groups (table 2). Of the 493 randomly assigned participants who received treatment, 329 were assigned to dronabinol and 164 were assigned to placebo. 415 (84%) completed follow-up, of whom 119 (29%) had prematurely discontinued trial treatment (figure 1). A summary of the main results is shown in table 3.
Table 2.
Baseline characteristics of participants by treatment group and overall
| Dronabinol (n=329) | Placebo (n=164) | All(n=493) | |||
|---|---|---|---|---|---|
| Age (years) | 52·29 (7·6) | 51·97 (8·2) | 52·19 (7·8) | ||
| Weight (kg) | 74·54 (16·5) | 75·93 (16·5) | 75 (16·5) | ||
| Men | 133 (40%) | 68 (41%) | 201 (41%) | ||
| Women | 196 (60%) | 96 (59%) | 292 (59%) | ||
| Primary progressive MS | 126 (38%) | 65 (40%) | 191 (39%) | ||
| Secondary progressive MS | 203 (62%) | 99 (60%) | 302 (61%) | ||
| EDSS score* (n per score) | |||||
| 4 | 20 (6%) | 9 (5%) | 29 (6%) | ||
| 4·5 | 18 (5%) | 7 (4%) | 25 (5%) | ||
| 5 | 22 (7%) | 10 (6%) | 32 (6%) | ||
| 5·5 | 16 (5%) | 8 (5%) | 24 (5%) | ||
| 6 | 169 (51%) | 85 (52%) | 254 (52%) | ||
| 6·5 | 84 (26%) | 45 (27%) | 129 (26%) | ||
| EDSS score* | 5·8 (0·69) | 5·9 (0·67) | 5·9 (0·69) | ||
| MSFC components† | |||||
| T25-FW | |||||
| Time (s)‡ | 20·34 (30·2) | 15·25 (13·4) | 18·64 (25·9) | ||
| Speed (feet per s)§ | 2·22 (1·21) | 2·3 (1·17) | 2·25 (1·19) | ||
| Not reported | 4 (1·2%) | 1 (0·6%) | 5 (1·0%) | ||
| 9-HPT score¶ | 0·04 (0) | 0·03 (0) | 0·04 (0) | ||
| Not reported | 1 (0·3%) | 0 | 1 (0·2%) | ||
| PASAT score‖ | 41·43 (13·8) | 41·02 (13·4) | 41·29 (13·6) | ||
| Not reported | 2 (0·6%) | 3 (1·8%) | 5 (1·0%) | ||
| MSFC composite Z score** | −0·099 (0·69) | −0·108 (0·62) | −0·102 (0·67) | ||
| Not reported | 8 (2·4%) | 5 (3·0%) | 13 (2·6%) | ||
| MSIS-29-PHYS score†† | 55·0 (10·8) | 55·2 (11·0) | 55·1 (10·8) | ||
| Not reported | 3 (0·9%) | 2 (1·2%) | 5 (1·0%) | ||
Data are mean (SD), n (%), and median (IQR). MS=multiple sclerosis. EDSS=expanded disability status scale. MSFC=multiple sclerosis functional composite. T25-FW=timed 25 foot walk. 9-HPT=nine hole peg test. PASAT=paced auditory serial addition test. MSIS-29-PHYS=29-item multiple sclerosis impact scale, physical impact subscale.
A higher score indicates greater disability.
Measured during visit 2 (visit 1 was a practice visit).
A longer time indicates worse mobility and leg function.
A faster speed indicates greater mobility and leg function.
Standard score, calculated as the mean of the reciprocal of the mean of the dominant-hand score and the reciprocal of the mean of the non-dominant-hand score; a higher score indicates greater finger dexterity.
Standard score, corresponding to the number of correctly answered questions out of 60; a higher score indicates greater capacity and rate of information processing.
A higher score indicates greater physical and cognitive function.
Total score, calculated according to standard procedure for dealing with missing data, with a possible range from 20 to 80; a higher score indicates a greater physical impact of multiple sclerosis.
Figure 1.
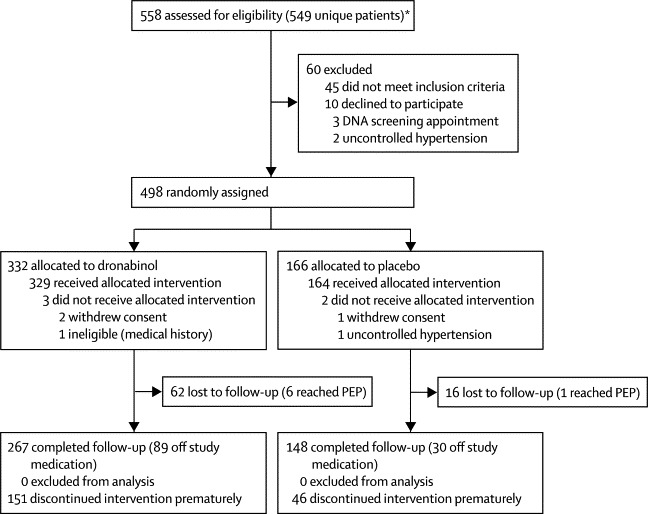
Trial profile
PEP=primary endpoint of expanded disability status scale score progression. *Nine patients initially did not fulfil entry criteria, but did so on subsequent re-screening.
Table 3.
Summary of main results
| Dronabinol (n=329) | Placebo (n=164) | Estimated treatment effect (95% CI) | p value | ||
|---|---|---|---|---|---|
| First EDSS score progression event, number of events (number of first progression events per patient-year*) | 145 (0·24) | 73 (0·23) | HR 0·92 (0·68 to 1·23)† | 0·57 | |
| MSIS-29-PHYS score, yearly change | 0·62 (3·29) | 1·03 (3·74) | −0·91 (−2·01 to 0·19)‡ | 0·11 | |
| MSFC score, yearly change | −0·17 (0·28) | −0·16 (0·30) | −0·03 (−0·19 to 0·09)‡ | 0·72 | |
| MSWS-12 score, yearly change | 0·37 (2·33) | 0·52 (2·68) | −0·19 (−0·97 to 0·60)‡ | 0·74 | |
| RMI, yearly change | −0·58 (0·96) | −0·72 (1·08) | 0·04 (−0·24 to 0·32)‡ | 0·76 | |
| MRI | n=182 | n=91 | |||
| PBVC, yearly change | −0·68 (0·95) | −0·66 (0·98) | −0·01% (−0·26 to 0·24)‡ | 0·94 | |
| New or enlarging T2 lesions | 60 (37%) | 34 (40%) | OR 0·89 (0·50 to 1·58)§ | 0·70 | |
| New or enlarging T1 lesions | 54 (34%) | 28 (33%) | OR 1·05 (0·59 to 1·88)§ | 0·87 | |
Data are n (%) or mean (SD) unless otherwise specified. EDSS=expanded disability status scale. HR=hazard ratio. MSIS-29-PHYS=29-item multiple sclerosis impact scale, physical impact subscale. MSFC=multiple sclerosis functional composite. MSWS-12=multiple sclerosis walking scale. RMI=Rivermead mobility index. PBVC=percentage brain volume change. OR=odds ratio.
Assuming progression events occur at the midpoint of the 6 months between follow-ups.
HR (dronabinol/placebo) according to Cox regression analysis (losses to follow-up regarded as censored observations); sensitivity analysis (losses to follow-up regarded as progression events) resulted in HR 1·11 (0·86–1·44), p value=0·41.
Estimated between-group difference (dronabinol–placebo) according to multilevel model.
Estimated OR according to logistic regression model (dronabinol/placebo).
Primary (ie, Cox regression) analysis showed no evidence of an effect of age (p=0·36), disease type (p=0·12), sex (p=0·56), weight (p=0·11), or treatment (p=0·57; table 3) on time to confirmed EDSS score progression. The adjusted HR for first EDSS score progression event in patients randomly assigned to dronabinol compared with those assigned to placebo was 0·92 (95% CI 0·68–1·23; table 3). At trial completion, Kaplan-Meier estimates of the probability of EDSS score progression were 0·55 (95% CI 0·46–0·63) in the dronabinol group, compared with 0·60 (0·44–0·71) in the placebo group (figure 2). Of 3812 assessments of EDSS score, 42 (1%) were by telephone (39 patients; 25 assigned to dronabinol and 14 assigned to placebo).
Figure 2.
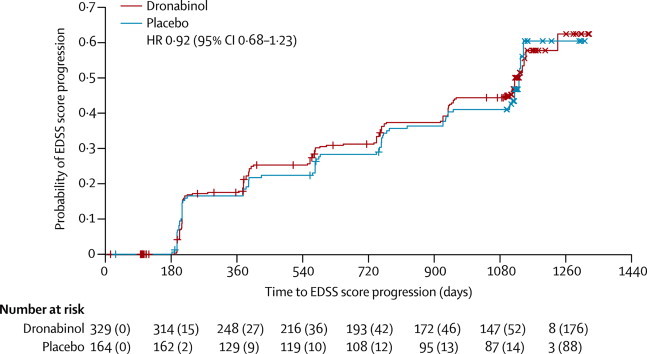
Kaplan-Meier estimates of the probability of EDSS score progression
Only EDSS score progression events that were confirmed 6 months after the first observation were included in the Kaplan-Meier analysis; plot shows timings of first events, not 6 month confirmations. Numbers accompanying the numbers at risk in parentheses are the cumulative number of censored observations. +=patients who were lost to follow-up during the trial. ×=time of last follow-up in those patients who reached the end of the trial (ie, were followed up for 3 years) without progressing. EDSS=expanded disability status scale.
We noted evidence of some centre effects (data not shown) and of an effect of baseline EDSS score on time to confirmed progression. Prespecified subgroup analyses of time to EDSS score progression suggested a differential effect of treatment between participants with lower (4·0–5·5) and higher (6·0–6·5) baseline EDSS scores (figure 3).
Figure 3.
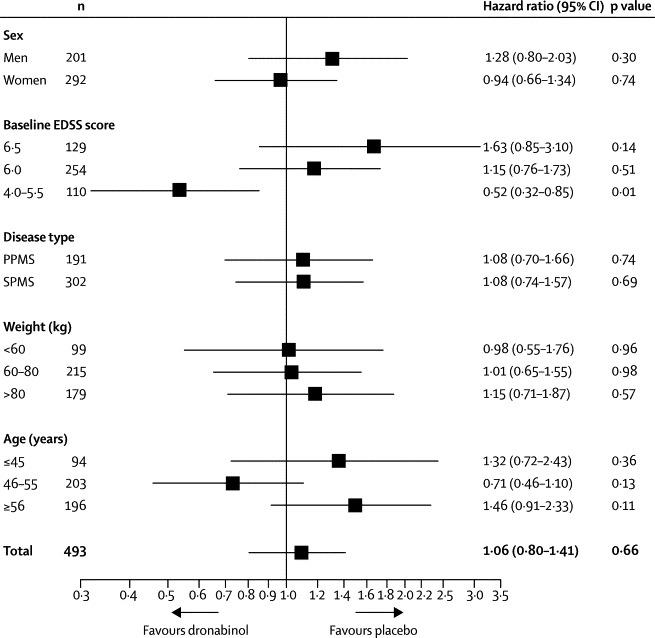
EDSS score progression by subgroup
Hazard ratio (unadjusted) of EDSS score progression in the dronabinol group compared with the placebo group. EDSS=expanded disability status scale. PPMS=primary progressive multiple sclerosis. SPMS=secondary progressive multiple sclerosis.
Results of sensitivity analysis showed that when we regarded losses to follow-up as progression events, rather than censored observations, the estimated HR for EDSS score progression changed to 1·11 (95% CI 0·86–1·44), but the estimated effect of treatment remained non-significant (p=0·41). This change in HR might be because the dronabinol group had a higher proportion of losses to follow-up for EDSS assessment (56 [79%] of 71) than did the placebo group (15 [21%] of 71) and represents a worst-case scenario in terms of patient deterioration and hence the potential benefit of dronabinol.
A multilevel model fitted to repeated measures of MSIS-29-PHYS score showed no evidence of an effect of treatment (estimated effect −0·9 points, 95% CI −2·0 to 0·2; p=0·11), or of disease type, sex, weight, or centre (data not shown; p>0·05 for all).
We estimated that the MSIS-29-PHYS score reduced by a mean of 1·4 points (95% CI 0·3–2·5) for every 10 year increase in age (p=0·02). In both treatment groups, mean MSIS-29-PHYS score decreased from baseline to month 3, after which it tended to increase (figure 4). With the exception of a small reduction in MSIS-29-PHYS score in patients with a baseline EDSS score of 5·0 compared with those with a score of 4·0, MSIS-29-PHYS score tended to increase with increasing baseline EDSS score (data not shown). Results remained unchanged after removal of non-significant terms from the fitted model and under an alternative analysis based on comparison of treatment groups in terms of change from baseline to last valid observation.
Figure 4.
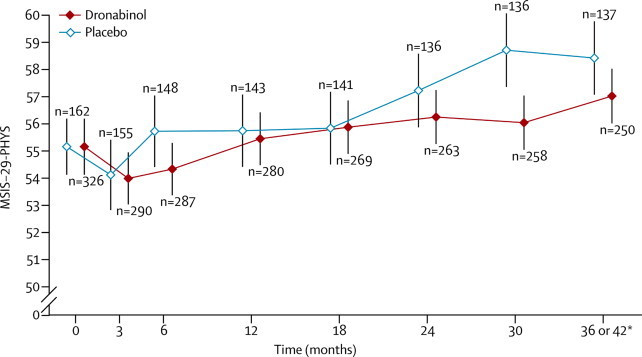
Change in MSIS-29-PHYS score over time
Datapoints show mean MSIS-29-PHYS score; vertical lines show 95% CI. MSIS-29-PHYS=29-item multiple sclerosis impact scale, physical impact subscale. n=number of patients with total scores calculated at each visit. *If deterioration was noted at 36 months, a final follow-up visit was done at 42 months to confirm progression; if a patient went on to have an assessment at 42 months, data from this visit were used; if not, data from the assessment at 36 months were used.
Results of multilevel models showed little evidence of an effect of treatment on MSFC, MSWS-12, or RMI (table 3).
Table 4 shows characteristics of the MRI substudy population. Dronabinol did not significantly affect brain atrophy compared with placebo over the course of the study (estimated effect −0·01%, 95% CI −0·26 to 0·24; p=0·94). However, atrophy did change significantly over time (p<0·0001); using a fitted model, we estimated cumulative PBVC to be a mean of −0·58% at year 1, −1·20% at year 2, and −2·02% at year 3 (figure 5).
Table 4.
Characteristics of the MRI substudy population by treatment group and overall
| Dronabinol (n=182) | Placebo(n=91) | All(n=273) | |||
|---|---|---|---|---|---|
| Baseline age (years) | 52·4 (7·3) | 52·2 (8·1) | 52·3 (7·6) | ||
| Baseline weight (kg) | 74·3 (16·1) | 75·7 (17·5) | 74·8 (16·6) | ||
| Men | 80 (44%) | 31 (34%) | 111 (41%) | ||
| Women | 102 (56%) | 60 (66%) | 162 (59%) | ||
| Primary progressive MS | 60 (33%) | 38 (42%) | 98 (36%) | ||
| Secondary progressive MS | 122 (67%) | 53 (58%) | 175 (64%) | ||
| Baseline EDSS score (n per score) | |||||
| 4 | 14 (8%) | 6 (7%) | 20 (7%) | ||
| 4·5 | 12 (7%) | 5 (5%) | 17 (6%) | ||
| 5 | 12 (7%) | 6 (7%) | 18 (7%) | ||
| 5·5 | 8 (4%) | 3 (3%) | 11 (4%) | ||
| 6 | 95 (52%) | 47 (52%) | 142 (52%) | ||
| 6·5 | 41 (23%) | 24 (26%) | 65 (24%) | ||
| Baseline EDSS score | 6 (5·6–6) | 6 (6–6·5) | 6 (6–6) | ||
| Baseline normalised brain volume | 1422 (91·0) | 1417 (85·1) | 1420 (88·9) | ||
| Not reported | 24 (13·2) | 8 (8·8) | 32 (11·7) | ||
| Cumulative atrophy (%) | |||||
| Year 1 | −0·59% (0·95) | −0·60% (0·95) | −0·59% (0·95) | ||
| Not reported or excluded | 28 (15%) | 9 (9·9%) | 37 (13·6%) | ||
| Year 2 | −1·16% (1·33) | −1·18% (1·30) | −1·17% (1·31) | ||
| Not reported or excluded | 44 (24%) | 15 (16·5%) | 59 (21·6%) | ||
| Year 3 | −1·95% (1·51) | −1·82% (1·47) | −1·90% (1·49) | ||
| Not reported or excluded | 58 (32%) | 20 (22·0%) | 78 (28·6%) | ||
| New or enlarging T1 lesions during follow-up | |||||
| 0 | 107 (59%) | 57 (63%) | 164 (60%) | ||
| 1 | 24 (13%) | 13 (14%) | 37 (14%) | ||
| 2 | 12 (7%) | 7 (8%) | 19 (7%) | ||
| 3 | 5 (3%) | 5 (5%) | 10 (4%) | ||
| ≥4 | 13 (7%) | 3 (3%) | 16 (6%) | ||
| Not reported or excluded | 21 (12%) | 6 (7%) | 27 (10%) | ||
| New or enlarging T2 lesions during follow-up | |||||
| 0 | 101 (55%) | 51 (56%) | 152 (56%) | ||
| 1 | 22 (12%) | 16 (18%) | 38 (14%) | ||
| 2 | 11 (6%) | 3 (3%) | 14 (5%) | ||
| 3 | 7 (4%) | 4 (4%) | 11 (4%) | ||
| ≥4 | 20 (11%) | 11 (12%) | 31 (11%) | ||
| Not reported or excluded | 21 (12%) | 6 (7%) | 27 (10%) | ||
Data are mean (SD), n (%), and median (IQR). MS=multiple sclerosis. EDSS=expanded disability status scale.
Figure 5.
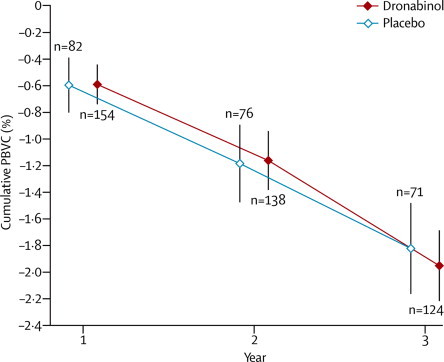
Cumulative PBVC over time
Datapoints show cumulative PBVC (%) measured at yearly MRI visits; vertical lines show 95% CI. n=number of patients with cumulative PBVC calculated at each visit. PBVC=percentage brain volume change.
We noted evidence of an effect of baseline normalised brain volume (NBV) on atrophy: for a 100 unit reduction in baseline NBV, atrophy increased by an estimated mean of 0·21% (95% CI 0·08–0·34; p=0·003).
Treatment did not significantly affect the occurrence of new or newly enlarging T2 lesions (estimated odds ratio [OR] 1·05, 95% CI 0·59–1·88; p=0·87) or new T1 lesions (0·89, 0·50–1·58; p=0·70).
Subgroup analysis of time to EDSS score progression in patients with a baseline EDSS score of 4·0–5·5 suggested a treatment effect (HR 0·52, 95% CI 0·32–0·85; figure 3) and led to a post-hoc analysis of progression in patients with this EDSS score, which provided some evidence of a potentially beneficial effect of active treatment compared with placebo (p=0·01, log-rank test; appendix). However, time to EDSS score progression in patients with a baseline EDSS score of 6·0 and those with a score of 6·5 was not significantly affected by dronabinol compared with placebo (EDSS score of 6·0, HR 1·15 [95% CI 0·76–1·73] and EDSS score of 6·5, HR 1·63 [0·85–3·10]).
Dronabinol did not significantly affect atrophy in the subgroup of patients with a baseline EDSS score of 5·5 or lower (estimated effect −0·06%, 95% CI −0·42 to 0·29; p=0·73) or those with a baseline EDSS score of 6·0–6·5 (0·01%, −0·26 to 0·28; p=0·95).
114 (35%) of 329 patients who received dronabinol had at least one serious AE compared with 46 (28%) of 164 patients who received placebo, the most common serious AE being admission to hospital for multiple-sclerosis-related events and infections. The number and nature of serious AEs reported did not significantly differ between groups (table 5).
Table 5.
Serious adverse events and most common adverse events
| Dronabinol (n=329) | Placebo (n=164) | All (n=493) | p value* | |
|---|---|---|---|---|
| Serious adverse events | ||||
| Death | 6 (1·8%) | 1 (0·6%) | 7 (1·4%) | 0·43 |
| Admission to hospital | 106 (32%) | 44 (27%) | 150 (30%) | 0·25 |
| Life-threatening or important medical event | 10 (3·0%) | 4 (2·4%) | 14 (2·8%) | 1 |
| At least one of the above | 114 (35%) | 46 (28%) | 160 (32%) | 0·15 |
| Most common adverse events | ||||
| Falls and injuries | 101 (31%) | 51 (31%) | 152 (31%) | 0·99 |
| Mobility, balance, and coordination problems | 108 (33%) | 43 (26%) | 151 (31%) | 0·16 |
| Infections (excluding urinary tract) | 95 (29%) | 47 (29%) | 142 (29%) | 0·96 |
| Fatigue and tiredness | 81 (25%) | 38 (23%) | 119 (24%) | 0·81 |
| Dizziness and lightheadedness | 105 (32%) | 12 (7%) | 117 (24%) | <0·0001 |
| Muscle disorders (spasticity, stiffness, spasms, or tremor) | 78 (24%) | 38 (23%) | 116 (24%) | 0·98 |
| Muscle disorders (weakness) | 74 (22%) | 32 (20%) | 106 (22%) | 0·52 |
| Dissociative and thinking or perception disorders | 98 (30%) | 6 (4%) | 104 (21%) | <0·0001 |
| Mood disorders (depression) | 66 (20%) | 26 (16%) | 92 (19%) | 0·31 |
| Musculoskeletal pain and aches | 49 (15%) | 41 (25%) | 90 (18%) | 0·009 |
| Constipation, diarrhoea, or faecal incontinence | 56 (17%) | 22 (13%) | 78 (16%) | 0·37 |
| Joint disorders | 47 (14%) | 28 (17%) | 75 (15%) | 0·50 |
| Urinary tract infections | 44 (13%) | 28 (17%) | 72 (15%) | 0·34 |
Data are number of patients who had at least one event of a given class (% per treatment group).
From Fisher's exact tests, with no adjustments for multiple comparisons.
Both groups had many non-serious AEs, consistent with the effects of multiple sclerosis and the known safety profile of cannabinoids. The median number of events per participant in the dronabinol group was 11 (IQR 7–17), compared with 10 (6–14) in the placebo group. Of those events judged to be either moderate or severe, the most frequent are documented in table 5.
Discussion
No treatments have as yet shown clinical efficacy in the modification of progressive multiple sclerosis in the absence of relapses (panel). Results of this study did not show an overall treatment effect of oral dronabinol on clinical disease course in progressive multiple sclerosis, nor did dronabinol affect the rate of brain atrophy.
Panel. Research in context.
Systematic review
We did a systematic review by searching Medline (1950 to May 14, 2013), Embase (1980 to May 14, 2013), and all Cochrane databases, with the search terms “cannabinoid”, “tetrahydrocannabinol”, “THC”, “multiple sclerosis”, “clinical trial”, “progression”, “primary progressive”, “secondary progressive”, and “disease course”. We included all human clinical trials, focusing on changes of disease course rather than symptomatic benefit. These searches confirmed that no clinical trial of the effects of cannabinoids on disease course in progressive multiple sclerosis had been published. Although several other treatments have been tested in phase 2 and 3 clinical trials in progressive disease, including β interferons, rituximab, intravenous pooled immunoglobulins, myelin basic protein peptide, and glatiramer acetate, none has shown convincing clinical benefit.
Interpretation
Results of previous studies have provided some evidence for a symptomatic benefit from this group of drugs,1,7,19,20 but none has previously explored a potential role in disease modification. We did not set out to assess symptom benefit in CUPID, and our current results do not support a role for cannabinoids in slowing progression of multiple sclerosis. However, future studies should consider focusing on patients with the greatest likelihood of continued deterioration over the course of the study, ideally recruiting people earlier in their progressive course.
Several factors might have reduced the chance of finding any potential treatment effect, particularly treatment discontinuation, loss to follow-up, and less overall progression than expected. Long-term studies in progressive neurological diseases are notoriously difficult to undertake: loss to follow-up might hinder interpretation of results, and low event rates and discontinuation of study medication also decrease the power to detect a treatment effect. Overall, loss to follow-up in CUPID was around the level expected, but more attrition occurred in the dronabinol group. This coincided with a higher number of AEs than in the placebo group, which also largely accounted for premature treatment discontinuation, although no major safety concerns were reported. As in previous studies of cannabinoids, most AEs occurred during the dose titration period, but the high lipid solubility of cannabinoids means that long-term build-up can occur some months after treatment initiation. Any future long-term studies to investigate disease-modifying effects of cannabinoids should use lower doses to reduce the risk of AEs, which should increase compliance and reduce potential error in any intention-to-treat analysis.
Low progression rates make the identification of a treatment effect less likely, and further work is necessary to establish optimum inclusion criteria for trials of progressive multiple sclerosis. Our study population was skewed towards the higher end of the EDSS score range (52% had a score of 6·0; 26% had a score of 6·5). Mean EDSS score at baseline was 5·9 (SD 0·69), higher than all other recent studies of treatments in progressive disease. Some studies have taken account of the slower progression rates in patients with higher EDSS scores by adjusting recruitment to ensure a lower overall mean EDSS score. The PROMiSe study18 was adjusted to 40% recruitment of patients with an EDSS score of 3·0–5·0, producing a mean EDSS score of 4·9 and yearly progression rate of 16% in patients with primary progressive multiple sclerosis, compared with 24% estimated in CUPID. Other outcome measures also did not show expected rates of progression. Although recent studies have tried to be more specific about monitoring pretrial progression to fulfil inclusion criteria, CUPID was designed to be a pragmatic study for people with progressive multiple sclerosis, testing some of the findings from the CAMS extension phase, which suggested that dronabinol might have an effect on multiple sclerosis progression. Future studies should ensure that the population recruited has a high chance of progression.
Prespecified and exploratory subgroup analyses suggested that dronabinol might have a slight potentially beneficial effect in terms of EDSS score progression and other outcome measures in less disabled patients (baseline EDSS score <6·0). Conversely, dronabinol might have a slight potentially negative effect in patients with higher EDSS scores. One possible explanation for these findings is that cannabinoids have been shown to reduce muscle stiffness;1,19,20 although an antispasticity effect might improve function at lower levels of disability, if walking is compromised by weakness, dronabinol might reduce muscle tone to the point where muscle power becomes affected and walking is more difficult. This side-effect is well known with agents such as baclofen. Distinguishing a symptomatic effect from a disease-modifying effect can be a difficult task, although the fact that no effect on brain atrophy was seen argues against a substantial neuroprotective effect of cannabinoids in progressive multiple sclerosis.1,7,19,20
This independent study provides the largest dataset on cannabinoid exposure over time currently available. Most outcomes, both primary and secondary, did not provide any evidence for a treatment effect with dronabinol. The suggestion of a potential effect in lower disability groups might have relevance for inclusion criteria in future clinical trials in progressive multiple sclerosis. CUPID data, when used to calculate sample sizes for further studies recruiting patients with an EDSS score of 4·0–5·5, suggest that a sample of around 375 participants would have 90% power to detect a difference of around 18% between dronabinol and placebo. In view of the shortage of treatments for multiple sclerosis progression, further trials in progressive multiple sclerosis focusing on patients whose disease seems most likely to progress are indicated.
Acknowledgments
Acknowledgments
We thank the patients who took part in this study and their families, and we acknowledge the contribution of all principal investigators and their study site teams. Particular thanks go to the coordinating team in Plymouth (especially Jacqui Mathews, Chris Hayward, Margie Berrow, Brian Wainman, Mark Warner, Linda Sutcliffe, Chris Rollinson, Lorraine Underwood, Caroline Snelgrove, Corinna Phillips, and Liz Ford) and also to Mike Marner, Suzi Reilly, Nick Pilkington, Adrian Pace, Richard Hosking, and Claire Fox.
Contributors
JZ contributed to the study concept and design and participated in study conduct, patient enrolment, and interpretation of data. DW contributed to the study design, planned and supervised the statistical analysis, and participated in the interpretation of data. SB did the statistical analysis and contributed to the interpretation of data. AN contributed to the study design and participated in study conduct and revision of the report. JH contributed to the study design, conduct, patient enrolment, data analysis and interpretation. JV coordinated the study. MGC participated in the statistical analysis. DMi designed the MRI substudy and supervised the analysis of imaging outcomes. SM undertook the MRI analysis. DMc supervised MRI data collection and quality assurance. All authors contributed to the writing of the paper and have seen and approved the final version.
CUPID trial investigators
J Zajicek (Derriford Hospital, Plymouth)*, R J Coleman (Aberdeen Royal Infirmary, Aberdeen)*, C Ko-Ko (West Midlands Rehabilitation Centre, Birmingham), N J Scolding (Frenchay Hospital, Bristol)*, A Coles (Addenbrooke's Hospital, Cambridge), N Robertson (University Hospital of Wales, Cardiff)*, A Shehu (University Hospitals Coventry and Warwickshire, Coventry), B Weller (Western General Hospital, Edinburgh)*, R J Martin (Gloucestershire Royal Hospital, Gloucester), D Kidd (Hertford County Hospital, Hertfordshire), P G Mattison (Ayrshire Central Hospital, Irvine), B Kendall (Leicester General Hospital, Leicester)*, O Malik and R Nicholas (Charing Cross Hospital, London)*, P Talbot (Hope Hospital, Manchester), D Bates and M Duddy (Royal Victoria Infirmary, Newcastle)*, M Lee (Norfolk and Norwich University Hospital, Norwich)*, C Constantinescu (University Hospital, Nottingham), J Palace (John Radcliffe Hospital, Oxford), C Hillier (Poole General Hospital, Dorset), P Tidswell (Royal Preston Hospital, Preston), S J L Howell (Royal Hallamshire Hospital, Sheffield)*, C P Hawkins (University Hospital of North Staff, Stoke on Trent), E Fathers (Musgrove Park Hospital, Taunton)*, B McLean (Royal Cornwall Hospital, Truro)*, G Giovannoni (Barts and the Royal London, London)*, G Mazibrada (Queen Elizabeth Hospital, Birmingham), A Weir (Royal Berkshire Hospital, Reading). *Also participated in the MRI component.
Trial steering and data monitoring committees
Trial steering committee members were Nigel Leigh, Carl Counsell, David Jones, Andrew Nunn, Nicola Russell, and Alan Thompson. Independent data monitoring committee members were Richard Gray, Ian Bone, and Michael Hutchinson.
Conflicts of interest
We declare that we have no conflicts of interest.
Supplementary Material
References
- 1.Zajicek J, Fox P, Sanders H, on behalf of the UK MS Research Group Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): multicentre randomised placebo-controlled trial. Lancet. 2003;362:1517–1526. doi: 10.1016/S0140-6736(03)14738-1. [DOI] [PubMed] [Google Scholar]
- 2.Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA. 1998;95:8268–8273. doi: 10.1073/pnas.95.14.8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molina-Holgado E, Vela JM, Arevalo-Martin A. Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J Neurosci. 2002;22:9742–9753. doi: 10.1523/JNEUROSCI.22-22-09742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skaper SD, Buriani A, Dal Toso R. The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc Natl Acad Sci USA. 1996;93:3984–3989. doi: 10.1073/pnas.93.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang YH, Lee ST, Lin WW. Effects of cannabinoids on LPS stimulated inflammatory mediator release from macrophages: involvement of eicosanoids. J Cell Biochem. 2001;81:715–723. doi: 10.1002/jcb.1103. [DOI] [PubMed] [Google Scholar]
- 6.Arevalo-Martin A, Vela JM, Molina-Holgado E. Therapeutic action of cannabinoids on a murine model of multiple sclerosis. J Neurosci. 2003;23:2511–2516. doi: 10.1523/JNEUROSCI.23-07-02511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zajicek J, Fox P, Teare L. Cannabinoids in multiple sclerosis (CAMS) study, safety and efficacy data from up to 12-months follow-up. J Neurol Neurosurg Psychiatry. 2005;76:1664–1669. doi: 10.1136/jnnp.2005.070136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonald WI, Compston A, Edan G. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50:121–127. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- 9.Smith SM, De Stefano N, Jenkinson M, Matthews PM. Normalised accurate measurement of longitudinal brain change. J Comput Assist Tomogr. 2001;25:466–475. doi: 10.1097/00004728-200105000-00022. [DOI] [PubMed] [Google Scholar]
- 10.Smith SM, Zhang Y, Jenkinson M. Accurate, robust and automated longitudinal and cross sectional brain change analysis. Neuroimage. 2002;17:479–489. doi: 10.1006/nimg.2002.1040. [DOI] [PubMed] [Google Scholar]
- 11.SPECTRIMS Study Group Randomised controlled trial of interferon-beta-1a in secondary progressive MS: clinical results. Neurology. 2001;56:1496–1504. doi: 10.1212/wnl.56.11.1496. [DOI] [PubMed] [Google Scholar]
- 12.R development Core Team . R: a language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2011. [Google Scholar]
- 13.Hobart J, Cano S. Improving the evaluation of therapeutic interventions in multiple sclerosis: the role of new psychometric methods. Health Technol Assess. 2009;13:1–177. doi: 10.3310/hta13120. [DOI] [PubMed] [Google Scholar]
- 14.Fischer JS, Jak AJ, Kniker JE, Rudick RA, Cutter GR. Multiple sclerosis functional composite (MSFC) administration and scoring manual. National Multiple Sclerosis Society; New York, NY: 2001. [Google Scholar]
- 15.Cohen JA, Cutter GR, Fischer JS. Use of the multiple sclerosis functional composite as an outcome measure in a phase 3 clinical trial. Arch Neurol. 2001;58:961–967. doi: 10.1001/archneur.58.6.961. [DOI] [PubMed] [Google Scholar]
- 16.Molt RW, McAuley E, Mullen S. Longitudinal measurement invariance of the multiple sclerosis walking scale-12. J Neurol Sci. 2011;305:75–79. doi: 10.1016/j.jns.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 17.Pinheiro JC, Bates DM. Mixed-effects models in S and S-PLUS. Springer Verlag; New York: 2000. [Google Scholar]
- 18.Wolinsky JS, Narayana PA, O'Connor P. Glatiramer acetate in primary progressive multiple sclerosis: results from a multinational multicenter double blind placebo controlled trial. Ann Neurol. 2007;61:14–24. doi: 10.1002/ana.21079. [DOI] [PubMed] [Google Scholar]
- 19.Novotna A, Mares J, Ratcliffe S. A randomized double blind placebo-controlled, parallel group, enriched design study of nabiximols (Sativex) as add-on therapy in subjects with refractory spasticity caused by multiple sclerosis. Eur J Neurol. 2011;18:1122–1131. doi: 10.1111/j.1468-1331.2010.03328.x. [DOI] [PubMed] [Google Scholar]
- 20.Zajicek J, Hobart J, Slade A. Multiple sclerosis and extract of cannabis: results from the MUSEC trial. J Neurol Neurosurg Psychiatry. 2012;83:1125–1132. doi: 10.1136/jnnp-2012-302468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.