

Abstract
Protein domains containing three or more ankyrin repeats (ARs) are ubiquitous in all phyla. Sequence alignments previously identified certain conserved positions, which have been shown to stabilize AR domains and promote their folding. Consensus mutations [Y254L/T257A (YLTA) and C186P/A220P (CPAP)] stabilize the naturally occuring AR domain of human IκBα to denaturation; however, only the YLTA mutations stabilize the protein to proteasomal degradation. We present results from NMR experiments designed to probe the roles of these consensus mutations in IκBα. According to residual dipolar coupling analysis, the gross structures of the AR domains of both mutants appear to be similar to the wild type (WT). Comparison of chemical shifts of mutant and WT proteins reveals that the YLTA and CPAP consensus mutations cause unexpected long-range effects throughout the AR domains. Backbone dynamics experiments reveal that the YLTA mutations in the sixth AR order the C-terminal PEST sequence on the picosecond-to-nanosecond timescale, compared to either the WT or the CPAP mutant IκBαs. This property is likely the mechanism by which the half-life of YLTA IκBα is extended in vivo.
Keywords: NMR relaxation, PEST sequence, proteasome degradation, consensus, reduced spectral density
Introduction
Ankyrin repeat (AR) motifs have been observed in proteins with a wide diversity of functions, including cell cycle, transcriptional, and developmental regulation; cytoskeletal organization; and toxins.1 AR domains, which contain as few as 3 or as many as 34 ARs, are found in the nuclei, cytoplasm, and extracellular milieu of a wide range of organisms, from mammals to bacteria and viruses.1 Currently, over 25,000 non-redundant AR-containing proteins have been identified in nature.2 The only common attribute shared by the members of this clearly important class of proteins is that AR domains are evolutionarily tuned to bind specific target proteins by the variation of non-consensus residues displayed on their surfaces.
The structure of an AR consists of a β-hairpin followed by two antiparallel α-helices separated by a loop (β-hairpin–helix–loop–helix). These modules stack upon each other to form a slightly twisted right-handed solenoid structure, with the β-hairpins protruding like the fingers of a cupped hand.3 The consensus sequence of this ~33-amino-acid motif has been well studied using statistical analyses.4–6 Although certain positions are only partially conserved, the most prominent features of the consensus sequence are shown in Fig. 1b.
Fig. 1.

(a) The crystal structure of IκBα bound to p50/p65 NFκB (Protein Data Bank ID: 1NFI). The ribbon structure of IκBα is drawn in tan, p50 is in pink, and p65 is in purple. (b) The sequence of each AR of IκBα is compared to the consensus sequence for ARs. The residues of IκBα that match the consensus sequence are in boldface. In addition, the residues mutated toward the consensus in this study, CPAP and YLTA, are underlined. (c) A ribbon model of IκBα (Protein Data Bank ID: 1NFI) with the consensus residues drawn in sticks and color coded as follows: consensus residues conserved at a particular position are drawn in blue, while consensus residues not conserved at a particular position are drawn in red. The image has been flipped 180° to show both sides of the protein.
Interpretation of the structural role that these consensus residues play in the stabilization of AR domains has primarily occurred through structural analysis, particularly of “idealized” AR domains containing only consensus residues,1,4,6 as well as through NMR studies.7 Most AR domain-containing proteins found in nature preserve a relatively low percentage of consensus residues, however. For example, Mosavi et al. found that the natural AR protein that best corresponded to their analytically determined consensus AR motif only matched with 57% sequence identity.4 One explanation for the sparse distribution of consensus residues in naturally occurring AR proteins is that too many consensus residues results in abnormally high stability,8 which would prevent the necessary turnover of these proteins in vivo. Indeed, consensus-only AR proteins are much more stable than the AR proteins found in nature.4–6 Even single and double consensus sequence substitutions in natural proteins have significant stabilizing effects.9 In fact, nearly every substitution to the consensus imparts measureable stabilization.10 In order to better understand the role that the consensus sequence plays in the stabilization of AR proteins, we chose to study the AR domain of IκBα, which consists of six ARs possessing an intermediate ratio of conserved to non-conserved consensus residues typical of natural AR domain proteins.
IκBα functions as the inhibitor of NFκB, a transcription factor that regulates apoptosis and responses to stress, infection, and inflammation.11 Crystal structures of the complex reveal that IκBα binds to NFκB through its AR domain, covering the nuclear localization sequence of NFκB and preventing the transcription factor from entering the nucleus (Fig. 1a).3,12 IκBα also has a signal response region N-terminal to the AR domain and a PEST sequence C-terminal to the AR domain. The minimal region necessary for binding and dissociation of DNA-bound NFκB is IκBα(67–287), which comprises the AR domain and includes the first seven residues of the PEST.13–16 Biophysical studies demonstrated that only AR1–AR4 fold cooperatively and show protection from amide exchange, consistent with a compact structure, whereas AR5 and AR6 exchange their amide protons within 2 min and do not fold cooperatively with AR1–AR4.9,17,18
The inherent flexibility of AR5 and AR6 compared to ARs1–4 of IκBα may result from the degree to which these ARs preserve the AR consensus sequence. Mutations of non-consensus residues toward the most prevalent signature of the AR motif, GXTPLHLA, stabilize AR domains, while mutations away from this sequence destabilize them.9,10,19–22 AR5 and AR6 of IκBα, particularly AR6, deviate from this consensus signature (Fig. 1b). Mapping of the consensus residues onto the crystal structure of NFκB-bound IκBα shows that they appear to form a cylindrical core down the center of the protein. The conserved TPLHLA sequence forms the beginning of the first α-helix closer to the concave side of the protein, while the conserved LL and GAD lie, respectively, in the second helix and the loop region preceding the β-hairpin (Fig. 1c, blue). Non-conserved consensus sequence residues (Fig. 1c, red) are found sporadically throughout the AR domain but are highly concentrated in AR6.
In IκBα, some of the residues that deviate from the consensus sequence involve amino acids that contact NFκB (F77, Q111, and Q255);3,12 many others, however, do not contact NFκB and can be substituted without affecting NFκB binding.9 Mutation of IκBα residues C186 in AR4 and A220 in AR5 to consensus proline residues resulted in a stability increase of ~ 1.5 kcal/mol.9 Introduction of the consensus mutations Y254L and T257A in AR6, on the other hand, increased the stability of the protein by a more modest ~0.5 kcal/mol but caused it to have a cooperative folding transition, similar to NFκB-bound IκBα.23
Intriguingly, the Y254L/T257A (YLTA) mutations significantly increase the in vivo half-life of IκBα, while the C186P/A220P (CPAP) mutations, despite increasing the overall stability, do not.23 We hypothesized that consensus mutations might variably alter the dynamics of protein, which might explain the different biophysical properties of wild type (WT), CPAP, and YLTA IκBα. Here, we present comparisons of the backbone chemical shifts and dynamics of WT, CPAP, and YLTA IκBα by NMR. Our results reveal subtle long-range chemical shift changes throughout the AR domain due to the consensus substitutions. Importantly, the lengthened in vivo half-life of the YLTA mutant is explained by a long-range effect of these mutations, which decreases the flexibility of the C-terminal PEST sequence.
Results
Backbone resonance assignments of YLTA IκBα(67–287) reflect folding of AR5–AR6
The NMR spectra of YLTA, CPAP, and WT IκBα(67–287) were assigned using a combination of assignment strategies, particularly the possibility of transferring backbone assignments from the spectra of small fragments and complexes when the fragments are structurally similar to the protein of interest.24 Assignment of the first four ARs of IκBα(67–287) was greatly facilitated by previous assignments of IκBα(67–206).25 Prior assignment of NFκB-bound IκBα(67–287)26 greatly facilitated assignment of YLTA IκBα(67–287), which then facilitated assignment of free WT IκBα(67–287). A schematic of this assignment transfer strategy is included in the Supplementary Materials (Supplementary Fig. 1). Conventional assignment strategies as well as the use of specifically labeled WT IκBα(67–287) (in which only the G, V, L, T, or A were 15N-labeled)27 were used to verify the assignments.
We were able to assign 86% of all the residues in YLTA IκBα(67–287) but could only assign 65% of residues in CPAP and WT. This inequality in the total assigned residues between the IκBα(67–287) constructs was primarily due to a more dispersed 1H,15N heteronuclear single quantum coherence (HSQC) spectrum and the greater number of total cross-peaks observed for the YLTA protein. The HSQC of YLTA IκBα(67–287) showed all 208 expected cross-peaks (Fig. 2, blue), while the HSQC of CPAP IκBα(67–287) showed 198 out of 206 expected cross-peaks, and the HSQC of WT IκBα(67–287) showed only 172 of the 208 expected cross-peaks (Fig. 2, black).
Fig. 2.
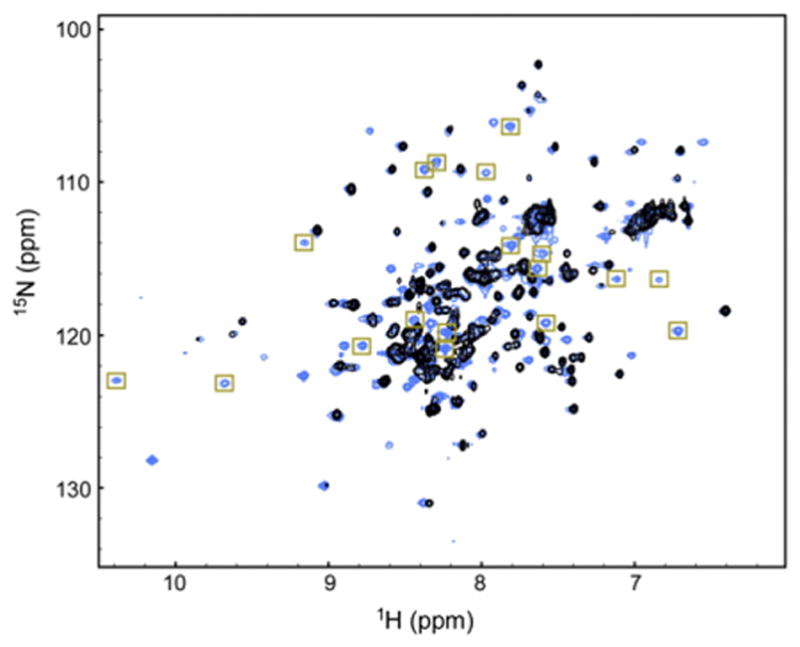
The HSQC of WT IκBα(67–287) (black) is overlaid on top of the HSQC of YLTA IκBα(67–287) (blue). The more dispersed residues of AR5–AR6 of YLTA are boxed in gold.
In almost every AR, we were able to assign more residues in the YLTA than in the WT or CPAP (Table 1). In particular, many of the additional cross-peaks observed in the HSQC of YLTA IκBα (Fig. 2, gold boxes) were assigned to residues in AR5 and AR6 (Table 1). Specifically, residues 212–231, which make up the β-finger and first α-helix of AR5, are completely missing from the WT, but most of these residues are assigned in YLTA. Residues 244–279, which constitute nearly the entire AR motif of AR6, are missing from the WT but are mostly assigned in YLTA. Only considering the residues in AR5 and AR6 and the PEST sequence, 78% of these residues were assigned in YLTA IκBα(67–287), while only 27% and 30%, respectively, were assigned in WT IκBα(67–287) and CPAP IκBα(67–287).
Table 1.
The number of assigned residues in each AR of WT, CPAP, and YLTA IκBα(67–287)
| AR | Number of assigned residues
|
||
|---|---|---|---|
| WT | CPAP | YLTA | |
| 1 | 32 | 32 | 31 |
| 2 | 28 | 29 | 31 |
| 3 | 30 | 33 | 34 |
| 4 | 29 | 25 | 34 |
| 5 | 12 | 10 | 27 |
| 6 | 1 | 7 | 27 |
RDCs reveal that WT, YLTA, and CPAP IκBα (67–287) have similar time-averaged solution structures
1H,15N residual dipolar couplings (RDCs) of WT, CPAP, and YLTA IκBα(67–287) were measured to probe for gross structural changes caused by the consensus mutations. RDCs were measured by partially aligning NMR samples with Pf1 phage and comparing the anisotropic dipolar couplings to their isotropic counterparts. Under anisotropic conditions, such as Pf1 phage, dipolar couplings report on the time-averaged direction of the amide bonds and thus RDCs are able to describe the time-averaged solution structure of a protein.28
The RDCs of all three constructs emphasize the repetitive nature of IκBα, with large negative RDCs punctuating the β-finger region and large positive RDCs in the α-helical regions, indicating that the AR motif structure of AR1–AR4 is preserved in all three proteins (Fig. 3). Due to the scarcity of cross-peaks from AR5 and AR6 of the WT and CPAP IκBα, little can be said about the conservation of structure in this region of the proteins. Notably, the RDCs of all three constructs deviate from the RDCs back-calculated from the crystal structure, with some of the largest discrepancies caused by residues from AR5 and AR6 (Supplementary Fig. 2).
Fig. 3.
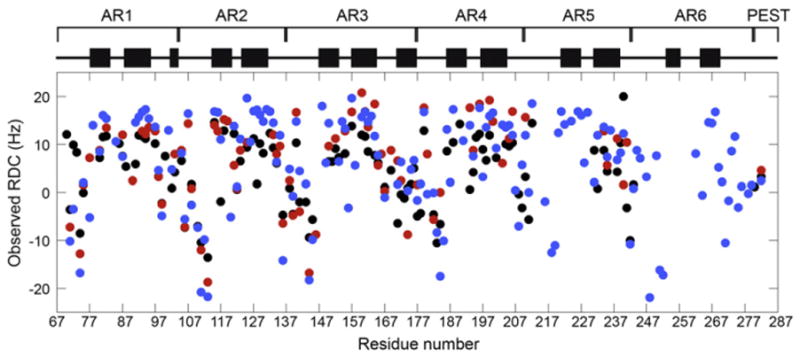
Measured RDCs of YLTA (blue), CPAP (red), and WT IκBα(67–287) (black) are plotted against residue number. Diagrammed above the RDCs are the positions of α-helical regions (black bars), as determined from the crystal structure (Protein Data Bank ID: 1NFI).
Consensus mutations cause chemical shift differences located throughout the AR domain
Because chemical shifts are a sensitive probe of the environment around a nucleus, comparison of the chemical shifts of bound IκBα to those of YLTA IκBα would indicate the degree to which these proteins are structurally similar. A comparison of the Cα and amide chemical shifts of NFκB-bound IκBα versus free YLTA IκBα revealed chemical shift differences throughout the AR domain (Fig. 4a). Cα chemical shifts that differed by more than 2 standard deviations included the following: residues 126 and 134 (AR2), 159 (AR3), 230 (AR5), and 249 and 270 (AR6). 1H–15N weighted average chemical shifts that differed by more than 1 standard deviation included the following: residues 83 (AR1), 187 and 193 (AR4), 226 (AR5), and 246, 264, 270–274, 276, 278, and 279 (AR6).
Fig. 4.

The chemical shift differences were determined for (a) bound WT-free YLTA IκBα, (b) free WT-YLTA IκBα, and (c) free WT-CPAP IκBα. The top plot of (a)–(c) gives the Cα chemical shift difference for each residue. The dotted lines in these plots denote an arbitrary cutoff for significance of 2 standard deviations from the average chemical shift difference. The bottom plot of (a)–(c) gives the weighted average of the 15N and 1H amide chemical shift differences for each residue. This value was calculated using the following equation: ([(ΔδHN)2 +(ΔδNH)2/25]/2)1/2, where ΔδHN and ΔδNH are the chemical shift differences in the amide 1H and 15N, respectively. The dotted line in these plots is an arbitrary cutoff for significance of 1 standard deviation from the average weighted chemical shift difference. Significant changes in chemical shift between WT and YLTA have bars colored blue, while those between WT and CPAP have bars colored red. On the right of each set of plots, the residues with significant chemical shift differences are drawn in sticks on a blue ribbon structure of IκBα and are color coded as follows: yellow, significant amide chemical shift difference; orange, significant Cα chemical shift difference. For residues on the ribbon structure that are colored black, chemical shifts were not available for analysis in one or both of the proteins. Residues which were mutated in CPAP or YLTA IKBa are colored magenta.
Comparison of Cα and amide chemical shift differences between the free WT protein and the YLTA mutant again showed differences throughout the AR domain (Fig. 4b). Cα chemical shift differences included the following: residues 72 (AR1), 156 (AR3), 212 (AR4), and 230 (AR5). 1H–15N weighted average chemical shift differences included the following: residues 88, 89, and 100 (AR1); 113 (AR2); 180 and 183 (AR4); and 207, 231, 234, 235, 239, and 243 (AR5).
We also compared the chemical shifts of CPAP and unbound WT in order to see if the chemical shift differences in CPAP resembled those observed in YLTA (Fig. 4c). Similar to YLTA, the CPAP mutations cause changes throughout the AR domain, but at different positions than the YLTA. Cα chemical shift differences included the following: residues 70 and 88 (AR1), 151 and 160 (AR3), and 177 (AR4). 1H–15N weighted average chemical shift differences included the following: residues 91 (AR1), 160 (AR3), 177 and 179 (AR4), and 209 and 239 (AR5).
Reduced spectral density analysis shows a less dynamic PEST in YLTA
In order to probe changes in backbone dynamics of the AR structure as a consequence of consensus mutation, we performed NMR relaxation experiments on WT, CPAP, and YLTA IκBα(67–287). R1 and R2 relaxation rates were collected at two fields (500 and 600 MHz), and 15N–{1H} nuclear Over-hauser effects (NOEs) were collected at one field (600 MHz). The data collected for YLTA are shown in Fig. 5a–c (data for WT and CPAP are provided in Supplementary Fig. 3).
Fig. 5.
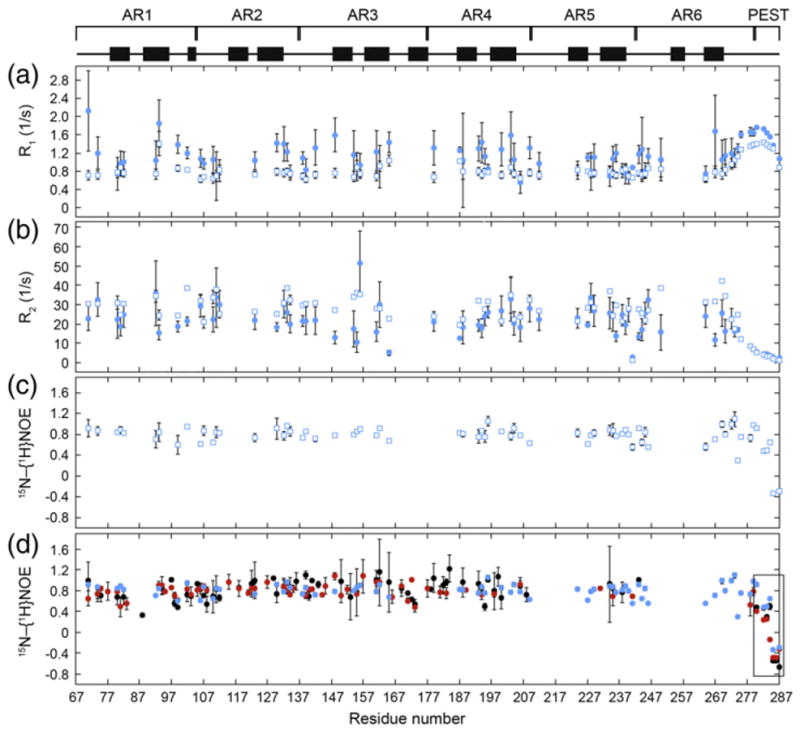
Backbone amide relaxation data collected for YLTA IκBα(67–287) at 20 °C: (a) R1, (b) R2, and (c) 15N–{1H} NOEs. R1 and R2 for all three IκBα constructs were collected at two magnetic fields: 500 MHz (filled circles) and 600 MHz (open squares). 15N–{1H}NOEs were collected at 600 MHz (open squares). (Relaxation data for WT and CPAP are provided in Supplementary Fig. 3.) (d) Overlay of 15N–{1H}NOEs for WT (black), CPAP (red), and YLTA (blue) IκBα(67–287). The PEST region is boxed for emphasis.
Backbone relaxation rates report on the picosecond-to-nanosecond timescale dynamics of amide bonds.29 Generally,15N–{1H}NOEs alone reflect fast-timescale picosecond-to-nanosecond motions, since this measure is mostly unaffected by slower timescale motions such as tumbling and microsecond-to-millisecond chemical exchange.30 In Fig. 5d, the 15N–{1H}NOE data clearly show for all three IκBα(67–287) constructs that the AR domain is relatively ordered compared to the PEST region, which is highly disordered on the picosecond-to-nanosecond timescale. Comparison of the 15N–{1H} NOEs of WT, CPAP, and YLTA IκBα(67–287) reveals that the picosecond-to-nanosecond time-scale dynamics of the AR domain do not change significantly upon consensus mutation (Fig. 5d).
Generally, R1, R2, and 15N–{1H}NOE relaxation data are analyzed by the Lipari–Szabo Model-Free formalism that assumes that the overall global motion of the protein is independent from the internal motions. Initial trials using Tensor2 modeling suggested that the Lipari–Szabo analysis was not suitable for the analysis of the IκBα(67–287) data. Reduced spectral density (RSD) analysis of the relaxation data was therefore employed to obtain information about the frequency content that makes up the motion of each amide bond.31 RSD analysis of WT, CPAP, and YLTA IκBα(67–287) relaxation data confirmed that the lower- and higher-frequency motions of AR1–AR6 of IκBα(67–287) do not change much upon consensus mutation (Fig. 6; Supplementary Fig. 4). Interestingly, the J(0.87ωH) spectral density, which probes higher-frequency motions, was significantly lower in the PEST of YLTA than in the PEST of WT (Fig. 6, blue). This indicated that the YLTA consensus mutations cause the PEST to become more rigid on this picosecond-to-nanosecond timescale. The CPAP consensus mutations, on the other hand, did not increase the rigidity of the PEST sequence (Fig. 6, red).
Fig. 6.
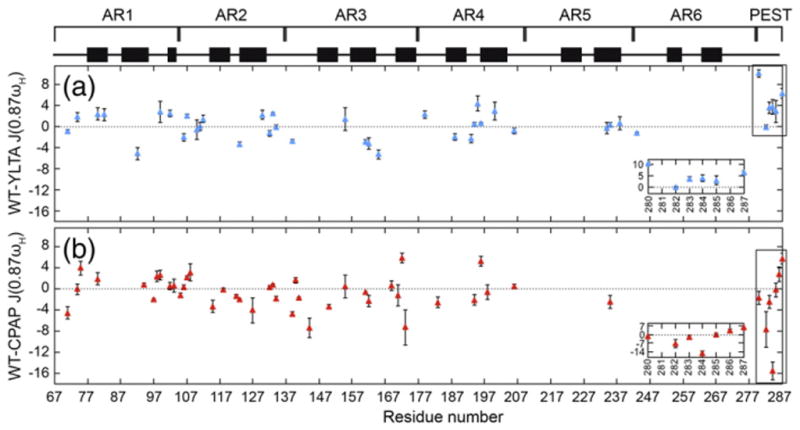
The J(0.87ωH) reduced spectral densities of WT were compared to the reduced spectral densities of (a) YLTA (blue) and (b) CPAP (red) IκBα(67–287). Differences in spectral density are plotted by residue only for those residues for which a complete set of relaxation data was available in both the WT and the respective consensus mutant protein. The PEST region is boxed and expanded in the inset of each plot.
Discussion
Consensus mutations do not significantly alter structural integrity of the AR domain
Most of the resonances seen in the HSQC spectra of the NFκB-bound WT, unbound WT, CPAP, and YLTA IκBα(67–287) had similar chemical shifts. Because of this, we were able to transfer many assignments from bound IκBα and YLTA to the unbound WT and CPAP IκBα(67–287) constructs (Supplementary Fig. 1). The congruity of cross-peak assignments between the constructs provides a strong indication that the overall AR domain structure changed very little upon consensus mutation. Little can be said, however, about AR5 and AR6 because few assignments are available for WT and CPAP. The assignments of YLTA compared well to those of bound IκBα, providing some evidence that the free structure of YLTA was not substantially different from that of the NFκB-bound WT. In addition, the RDCs of unbound WT, CPAP, and YLTA IκBα(67–287) support this conclusion, at least for AR1–AR4, by showing that these three constructs have very similar time-averaged solution structures (Fig. 3). Therefore, any long-range chemical shift perturbations found in YLTA and CPAP IκBα(67–287) are likely not due to a gross change in the structure of the AR domain.
Consensus mutations alter long-range stacking interactions of the AR domain
Significant amide chemical shift differences between NFκB-bound IκBα and YLTA IκBα were observed near the interface where IκBα interacts with NFκB (Fig. 4a), consistent with differences in local protein environment32 due to the binding of NFκB. Amide chemical shift differences are particularly prominent in AR6, hinting that the structures of AR6 in the YLTA versus NFκB-bound forms may be slightly different. In contrast, Cα chemical shift differences, indicators of differences in secondary structure,32 are located on the face of IκBα opposite the binding interface, particularly in the less-conserved helix 2 of several ARs (Fig. 4a). These differences in secondary structure between NFκB-bound IκBα and YLTA IκBα are located along the convex side of the proteins and mainly indicate higher helical tendency in these regions of YLTA IκBα compared to NFκB-bound IκBα (Fig. 4a). Given that significant Cα chemical shift perturbations are seen around helix 2 of AR2, AR3, AR5, and AR6, one might surmise that binding may change the radius of curvature of the NFκB-bound IκBα. While the chemical shift differences of only a few residues in each of these repeats exceeded the arbitrary 2 σ cutoff we applied, lower cutoffs revealed the same trend, with more residues included.
Comparison of free WT and YLTA IκBα chemical shifts also shows differences throughout the AR domain (Fig. 4b), with only one significantly perturbed residue, V243, near the sites of mutation. Differences in amide chemical shifts are observed in AR5 (once again along the less-conserved helix 2), confirming the folding of AR5 as a consequence of the YLTA mutations, which have previously been shown to integrate AR5 and AR6 with AR1–AR4 in the cooperatively folding unit.23 The chemical shift differences observed in AR1–AR4 suggest that the YLTA consensus mutations have long-range effects on the interactions between AR subunits (Fig. 4b). Although the YLTA mutations lie in helix 1 of AR6, the chemical shift differences are observed primarily in helix 2 and the β-fingers. At lower cutoffs of significance in the chemical shift difference data, more residues with chemical shift differences confirm a pathway through the outer helices and β-fingers. Yet, the previously proposed structural roles of the consensus leucine and alanine residues do not predict these long-range effects. Rather, they focus solely on local effects such as the role of leucine in providing hydrophobic contacts within the AR subunit and the role of alanine in lengthening the first α-helix.5 It is interesting to consider how the internal stabilization of AR6 could propagate stability through the AR domain via inter- and intra-repeat interactions. These long-range effects are consistent with previous biophysical studies showing an increase in the stability of YLTA IκBα due to the consensus mutations.23 These results are also consistent with previous NMR studies showing that alteration of “capping” repeats can have long-range consequences on internal ARs.33
The chemical shift differences between CPAP and WT IκBα appear to be more localized around the sites of mutation than those observed for YLTA (Fig. 4c). In particular, amide chemical shift differences are observed in residues in the loop region between ARs near each proline. It is likely that this interaction between the conserved proline of the TPLHLA sequence and the less-conserved loop residues across the interface of neighboring ARs improves AR stacking. Similar to the YLTA mutant, chemical shift differences are also seen in AR1, far from the sites of mutation, suggesting that the CPAP consensus mutations also promote long-range interactions between AR subunits. The previously proposed structural role for this consensus proline fails to predict these long-range interactions—the primary prediction being that the proline is necessary in order to break into helix 1.4,5 Unlike YLTA, few chemical shift differences are observed in AR5 of CPAP IκBα. This result is consistent with previous biophysical studies showing that the CPAP consensus mutations of IκBα do not cause AR5 and AR6 to integrate with AR1–AR4 in the cooperatively folding unit.
Consensus mutations do not significantly alter dynamics of the AR domain
Model-Free analysis of protein dynamics necessitates the knowledge of a three-dimensional structure.34 Although crystal structures of IκBα bound to NFκB are available,3,12 amide exchange18 and single-molecule fluorescence resonance energy transfer35 analysis of free IκBα provide strong evidence that AR5 and AR6 unravel in unbound IκBα. Given the expected broad ensemble of dynamic structures, combined with its elongated and curved structure, it is not surprising that the Tensor2 Model-Free analysis program could not find an appropriate rotational diffusion tensor for unbound IκBα. In lieu of the Model-Free approach, the reduced spectral densities of WT, CPAP, and YLTA IκBα(67–287) were analyzed (Fig. 6). This analysis revealed remarkably little difference in the dynamics of the backbone NH groups of the AR domain upon introduction of consensus mutations.
Ordering of PEST in YLTA slows in vivo degradation
The misregulation of NFκB signaling is implicated in several cancers, autoimmune diseases, and inflammatory disorders, including B-cell and Hodgkin’s lymphoma,36 and rapid degradation of IκBα is critical for proper signaling through the NFκB pathway.37,38 IκBα constructs in which the PEST sequence is deleted are degraded five times more slowly in cells, suggesting that the PEST sequence of IκBα provides an essential signal for the rapid degradation of free IκBα in vivo.37 Previously, Truhlar et al. examined the half-lives of WT and YLTA IκBα in mouse embryonic fibroblast and found that the in vivo half-life of IκBα was increased threefold by the YLTA mutation.23 Here, we have shown that the PEST sequence of YLTA IκBα is more ordered on the picosecond-to-nanosecond timescale than the PEST of WT IκBα. Therefore, our results suggest that the ordering of the PEST sequence by the YLTA mutations in AR6 of IκBα makes the protein almost as resistant to degradation as deletion of the PEST sequence entirely. It is interesting to speculate whether the slowing of degradation is due to a greater fraction of the PEST sequence being folded, or whether changes in the dynamic timescales of motion of the PEST sequence actually alter access to the proteasome.
Free IκBα is degraded in vitro by the 20S proteasome,39 which is known to degrade proteins that are either intrinsically disordered throughout or which contain localized intrinsically disordered regions.40 The presence of disorder in proteins that must be degraded quickly is a common regulatory mechanism and is well documented in cell functions such as signaling, cell cycle regulation, transcription, and translation.41 It is hypothesized that the flexibility of the initiating polypeptide chain is necessary to allow physical access to the active site of the 20S proteasome, since the opening of the 20S proteasome is not very wide and because the “gate” is only open one-third of the time.40 Our studies indicate that the YLTA consensus mutations order the PEST sequence of IκBα to the extent that it is significantly less accessible to the active site of the 20S proteasome than is the PEST of WT IκBα.23
Materials and Methods
Preparation of protein NMR samples
WT human IκBα(67–287) was prepared and purified in the manner described previously for IκBα(67–317) and IκBα(67–206),17,25 with a few changes that are detailed in this section. CPAP and YLTA IκBα(67–287) mutants were prepared in a similar manner as the WT, except where indicated otherwise.
For protein samples used in backbone triple-resonance experiments, expression of 2H,13C,15N-labeled IκBα(67–287) was carried out in M9 minimal medium containing 99% D2O, supplemented with 15NH4Cl (2 g/L), and [13C] glucose (8 g/L). Cells were first acclimated in 30 mL of M9ZB media for 5 h at 37 °C. These cells were then pelleted and resuspended in 10 mL of M9 minimal media and grown for 1 h at 37 °C. A portion (1 mL) of this culture was then taken and diluted with 1 mL of M9 minimal media prepared in D2O; this 2-mL culture was grown at 37 °C for 2 h. M9 minimal media prepared in D2O (18 mL) were added to this culture and grown for another 8 h. The entire 20-mL culture was then used to inoculate a 1-L culture of M9 minimal media prepared in D2O, which had been supplemented for labeling as described above. Because the YLTA mutant in particular does not express well, 3 L of culture was grown for each NMR sample rather than 1 L.
For protein samples with specific 15N amino acid labeling, expression was carried out in M9 minimal medium supplemented with 14N-labeled amino acids, which were added to the medium in proportion to their abundance in the amino acid sequence to a final total amount of 10 g/L culture. In order to prevent scrambling of the label, we added only 1/10th of the above amount of the 15N-labeled amino acid.42
Purification proceeded as previously described, except that a 1-h HiLoadQ gradient ranging from 225 mM to 700 mM NaCl was used instead. The final protein samples were exchanged into buffer composed of 25 mM 2H-Tris, pH 7.5, 50 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 5 mM 3-[(3-cholamidopropyl)dimethylammonio]pro-panesulfonic acid, 2 mM NaN3, 2 mM DTT, 10% D2O, and 1% protease inhibitor cocktail. Protein for NMR experiments was concentrated down to 0.1 mM, as described previously.25
Backbone resonance experiments
Resonance assignments for IκBα free and in complex with NFκB were performed using 2H,13C,15N-labeled IκBα. HNCA,43,44 HN(CO)CA,43,44 HN(CA)CB,45 HN(COCA) CB,44 and HNCO43 spectra, or their transverse relaxation optimized spectroscopy (TROSY) equivalents for larger constructs,46 were acquired.
NMR spectra were acquired at 20 °C on a Bruker DRX600 spectrometer equipped with a cryoprobe [HNCA, HNCO, HN(CO)CA] and an Avance 800 spectrometer for TROSY spectra (HNCA, HNCB). The following parameters were used in three-dimensional experiments: for HNCA, data size=2048 (t3)×32 (t2)×88 (t1) complex points, number of scans=64; for HN(CO)CA, data size=2048 (t3)×32 (t2)×88 (t1) complex points, number of scans=24; for HNCB, data size=2048 (t3)×32 (t2)×90 (t1) complex points, number of scans=128; for HNCO, data size=1024 (t3)×128 (t2)×256 (t1) complex points, number of scans= 72. The delay time between each scan was 1.2 s. Data were processed using NMRpipe47 and analyzed using NMRView.48
RDC experiments
Spin-state selected TROSY two-dimensional [1H–15N] correlation experiments were collected for isotropic and Pf1 phage-aligned 15N,2H-labeled IκBα samples at 25 °C on a Varian VS 800 MHz equipped with a cryoprobe.49 Experiments were collected in an interleaved manner with 72 scans and 2048 (t2)×128 (t1) complex data points. RDCs were extracted by calculating the differences between the TROSY and 1H anti-TROSY peaks in isotropic and anisotropic data sets. In order to optimize alignment conditions, we titrated WT, CPAP, and YLTA IκBα(67–287) samples with Pf1 phage (Asla Biotech) from 5 to 11 mg/mL in 2-mg/mL increments. Pf1 phage concentrations between 9 and 11 mg/mL provided significant alignment for all three constructs with RDCs ranging from −21.91 to 20.79 Hz. Observed RDCs were compared to RDCs back-calculated from the crystal structure (Protein Data Bank ID: 1NFI) using the program PALES (Supplementary Fig. 2).50 The correlations between observed and calculated RDCs were, as expected, weaker than those found previously for IκBα(67–206).25 Many of the largest discrepancies were from residues in AR5–AR6, but some were from charged residues displayed on the surface of IκBα, possibly due to interaction with the Pf1 molecules.
Backbone relaxation dynamics experiments
Measurements of the T1, T2, and 15N–{1H}NOEs for uniformly 15N,2H-labeled WT, CPAP, and YLTA IκBα(67–287) were made at 20 °C. T1 and T2 values were collected at both 500 MHz and 600 MHz on a Bruker Avance 501 and Bruker DRX600 using standard Bruker programs. 15N–{1H}NOEs were collected at 600 MHz also using a standard Bruker program. For 15N–{1H}NOEs, independent saturated and unsaturated spectra were recorded in an interleaved manner for each sample. T1 delays were 12 (duplicated), 45, 89, 177, 353, 705 (duplicated), 1057, 1409, 1761, and 2201 (duplicated) ms. T2 delays were 9 (duplicated), 13, 21, 29, 37 (duplicated), 45, 53, 69, 85, and 101 (duplicated) ms. The delay time between each scan was 3 s. Data were processed using NMRPipe and analyzed using Sparky and Sparky2rate‡.47,51
RSD analysis
Backbone amide relaxation data were analyzed using RSD mapping, explained in further detail elsewhere.30,52,53 In brief, the spectral density function, J(ω), describes the contributions of different frequencies to the motion of an amide bond vector. In a spectral density analysis, the magnitude of J(ω) at five frequencies is calculated. However, in the RSD approach, it is assumed that dJ(ω)/dω2 is relatively constant over the linear combinations of (ωH + ωN) and (ωH − ωN).30,52–54 Therefore, the relaxation measures can be expressed as linear combinations of only three spectral densities, J(0), J(ωN), and J(0.87ωH), which can then be rewritten in terms of the relaxation measures:
where σ=[(NOE-1)R1γN/γH] is the cross-relaxation rate of the spin pair, d = [μohγHγN/8π2]〈rNH−3〉, , μo is the permeability of free space, h is Planck’s constant, γH and γN are the gyromagnetic ratios of 1H and 15N nuclei, ωH and ωN are the Larmor frequencies of the 1H and 15N nuclei, rNH is the nitrogen–hydrogen bond length (1.04 Å),55 and Δσ is the chemical shift anisotropy of 15N spins (−160 ppm).56
Biological Magnetic Resonance Bank accession number
The 1H, 15N, and Cα chemical shift assignments of WT, CPAP, and YLTA IκBα(67–287) have been deposited in the Biological Magnetic Resonance Bank (accession numbers: 18756, 18759, and 18760, respectively).
Supplementary Material
Acknowledgments
We thank Brian Fuglestad, Gerard Kroon, and Brendan Borin for their expertise and help with the use of NMR spectrometers. This work was supported by National Institutes of Health grant P01 GM71862. C.F.C. is grateful for support from an individual National Research Service Award pre-doctoral fellowship (F31 GM081897). L.D.H. is grateful for support from a Molecular Biophysics Training Grant (T32 GM008326).
Abbreviations used
- AR
ankyrin repeat
- CPAP
C186P/A220P
- YLTA
Y254L/T257A
- WT
wild type
- HSQC
heteronuclear single quantum coherence
- RDC
residual dipolar coupling
- NOE
nuclear Overhauser effect
- RSD
reduced spectral density
- TROSY
transverse relaxation optimized spectroscopy
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2012.12.012
References
- 1.Sedgwick SG, Smerdon SJ. The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem Sci. 1999;24:311–316. doi: 10.1016/s0968-0004(99)01426-7. [DOI] [PubMed] [Google Scholar]
- 2.Letunic I, Doerks T, Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40:302–305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95:749–758. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 4.Mosavi LK, Minor DL, Peng ZY. Consensus-derived structural determinants of the ankyrin repeat motif. Proc Natl Acad Sci USA. 2002;99:16029–16034. doi: 10.1073/pnas.252537899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Binz HK, Stumpp MT, Forrer P, Amstutz P, Plückthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol. 2003;332:489–503. doi: 10.1016/s0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- 6.Kohl A, Binz HK, Forrer P, Stumpp MT, Plückthun A, Grütter MG. Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc Natl Acad Sci USA. 2003;100:1700–1705. doi: 10.1073/pnas.0337680100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan C, Li J, Mahajan A, Poi MJ, Byeon IJ, Tsai MD. Solution structure of the human oncogenic protein gankyrin containing seven ankyrin repeats and analysis of its structure–function relationship. Biochemistry. 2004;43:12152–12161. doi: 10.1021/bi049116o. [DOI] [PubMed] [Google Scholar]
- 8.Wetzel SK, Settanni G, Kenig M, Binz HK, Plückthun A. Folding and unfolding mechanism of highly stable full-consensus ankyrin repeat proteins. J Mol Biol. 2008;376:241–257. doi: 10.1016/j.jmb.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 9.Ferreiro DU, Cervantes CF, Truhlar SM, Cho SS, Wolynes PG, Komives EA. Stabilizing IkappaBalpha by “consensus” design. J Mol Biol. 2007;365:1201–1216. doi: 10.1016/j.jmb.2006.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeVries I, Ferreiro DU, Sánchez IE, Komives EA. Folding kinetics of the cooperatively folded subdomain of the IκBα ankyrin repeat domain. J Mol Biol. 2011;408:163–176. doi: 10.1016/j.jmb.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 12.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell. 1998;95:759–770. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 13.Malek S, Huxford T, Ghosh G. Ikappa Balpha functions through direct contacts with the nuclear localization signals and the DNA binding sequences of NF-kappaB. J Biol Chem. 1998;273:25427–25435. doi: 10.1074/jbc.273.39.25427. [DOI] [PubMed] [Google Scholar]
- 14.Phelps CB, Sengchanthalangsy LL, Malek S, Ghosh G. Mechanism of kappa B DNA binding by Rel/NF-kappa B dimers. J Biol Chem. 2000;275:24392–24399. doi: 10.1074/jbc.M003784200. [DOI] [PubMed] [Google Scholar]
- 15.Bergqvist S, Croy CH, Kjaergaard M, Huxford T, Ghosh G, Komives EA. Thermodynamics reveal that helix four in the NLS of NF-kappaB p65 anchors IkappaBalpha, forming a very stable complex. J Mol Biol. 2006;360:421–434. doi: 10.1016/j.jmb.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergqvist S, Ghosh G, Komives EA. The IkappaBalpha/NF-kappaB complex has two hot spots, one at either end of the interface. Protein Sci. 2008;17:2051–2058. doi: 10.1110/ps.037481.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Croy CH, Bergqvist S, Huxford T, Ghosh G, Komives EA. Biophysical characterization of the free IkappaBalpha ankyrin repeat domain in solution. Protein Sci. 2004;13:1767–1777. doi: 10.1110/ps.04731004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Truhlar SM, Torpey JW, Komives EA. Regions of IkappaBalpha that are critical for its inhibition of NF-kappaB. DNA interaction fold upon binding to NF-kappaB. Proc Natl Acad Sci USA. 2006;103:18951–18956. doi: 10.1073/pnas.0605794103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zweifel ME, Leahy DJ, Hughson FM, Barrick D. Structure and stability of the ankyrin domain of the Drosophila Notch receptor. Protein Sci. 2003;12:2622–2632. doi: 10.1110/ps.03279003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowe AR, Itzhaki LS. Rational redesign of the folding pathway of a modular protein. Proc Natl Acad Sci USA. 2007;104:2679–2684. doi: 10.1073/pnas.0604653104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang KS, Guralnick BJ, Wang WK, Fersht AR, Itzhaki LS. Stability and folding of the tumour suppressor protein p16. J Mol Biol. 1999;285:1869–1886. doi: 10.1006/jmbi.1998.2420. [DOI] [PubMed] [Google Scholar]
- 22.Tripp KW, Barrick D. Enhancing the stability and folding rate of a repeat protein through the addition of consensus repeats. J Mol Biol. 2007;365:1187–1200. doi: 10.1016/j.jmb.2006.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Truhlar SM, Mathes E, Cervantes CF, Ghosh G, Komives EA. Pre-folding IkappaBalpha alters control of NF-kappaB signaling. J Mol Biol. 2008;380:67–82. doi: 10.1016/j.jmb.2008.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiaux J, Bertelsen EB, Horwich AL, Wuthrich K. NMR analysis of a 900K GroEL–GroES complex. Nature. 2002;418:207–211. doi: 10.1038/nature00860. [DOI] [PubMed] [Google Scholar]
- 25.Cervantes CF, Markwick PR, Sue SC, McCammon JA, Dyson HJ, Komives EA. Functional dynamics of the folded ankyrin repeats of I kappa B alpha revealed by nuclear magnetic resonance. Biochemistry. 2009;48:8023–8031. doi: 10.1021/bi900712r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sue SC, Cervantes C, Komives EA, Dyson HJ. Transfer of flexibility between ankyrin repeats in IkappaB* upon formation of the NF-kappaB complex. J Mol Biol. 2008;380:917–931. doi: 10.1016/j.jmb.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cervantes CF. PhD Thesis. UC San Diego; 2010. NMR Studies of the transcriptional Inhibitor I Kappa B alpha and its interaction with the transcription factor NF kappa B. [Google Scholar]
- 28.Prestegard JH, Al-Hashimi HM, Tolman JR. NMR structures of biomolecules using field oriented media and residual dipolar couplings. Q Rev Biophys. 2000;33:371–424. doi: 10.1017/s0033583500003656. [DOI] [PubMed] [Google Scholar]
- 29.Palmer AG., III NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 30.Ishima R, Nagayama K. Protein backbone dynamics revealed by quasi spectral density function analysis of amide N-15 nuclei. Biochemistry. 1995;34:3162–3171. doi: 10.1021/bi00010a005. [DOI] [PubMed] [Google Scholar]
- 31.Peng JW, Wagner G. Mapping of the spectral densities of N–H bond motions in eglin c using heteronuclear relaxation experiments. Biochemistry. 1992;31:8571–8586. doi: 10.1021/bi00151a027. [DOI] [PubMed] [Google Scholar]
- 32.Wishart DS, Case DA. Use of chemical shifts in macromolecular structure determination. Methods Enzymol. 2001;338:3–34. doi: 10.1016/s0076-6879(02)38214-4. [DOI] [PubMed] [Google Scholar]
- 33.Wetzel SK, Ewald C, Settanni G, Jurt S, Plückthun A, Zerbe O. Residue-resolved stability of full-consensus ankyrin repeat proteins probed by NMR. J Mol Biol. 2010;402:241–258. doi: 10.1016/j.jmb.2010.07.031. [DOI] [PubMed] [Google Scholar]
- 34.Palmer AG., III NMR probes of molecular dynamics: overview and comparison with other techniques. Annu Rev Biophys Biomol Struct. 2001;30:129–155. doi: 10.1146/annurev.biophys.30.1.129. [DOI] [PubMed] [Google Scholar]
- 35.Lamboy JA, Kim H, Lee KS, Ha T, Komives EA. Visualization of the nanospring dynamics of the IkappaBalpha ankyrin repeat domain in real time. Proc Natl Acad Sci USA. 2011;108:10178–10183. doi: 10.1073/pnas.1102226108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Courtois G, Gilmore TD. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 37.Mathes E, O’Dea EL, Hoffmann A, Ghosh G. NF-kappaB dictates the degradation pathway of IkappaBalpha. EMBO J. 2008;27:1357–1367. doi: 10.1038/emboj.2008.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Dea EL, Barken D, Peralta RQ, Tran KT, Werner SL, Kearns JD, et al. A homeostatic model of IkappaB metabolism to control constitutive NF-kappaB activity. Mol Syst Biol. 2007;3:111. doi: 10.1038/msb4100148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alvarez-Castelao B, Castaño JG. Mechanism of direct degradation of IkappaBalpha by 20S proteasome. FEBS Lett. 2005;579:4797–4802. doi: 10.1016/j.febslet.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 40.Suskiewicz MJ, Sussman JL, Silman I, Shaul Y. Context-dependent resistance to proteolysis of intrinsically disordered proteins. Protein Sci. 2011;20:1285–1297. doi: 10.1002/pro.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 42.Truhlar SM, Cervantes CF, Torpey JW, Kjaergaard M, Komives EA. Rapid mass spectrometric analysis of 15N-Leu incorporation fidelity during preparation of specifically labeled NMR samples. Protein Sci. 2008;17:1636–1639. doi: 10.1110/ps.036418.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grzesiek S, Bax A. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J Magn Reson. 1992;96:432–440. [Google Scholar]
- 44.Yamazaki T, Lee W, Arrowsmith CH, Muhandiram DR, Kay LE. A suite of triple-resonance NMR experiments for the backbone assignment of 15N, 13C, 2H labeled proteins with high sensitivity. J Am Chem Soc. 1994;116:11655–11666. [Google Scholar]
- 45.Wittekind M, Mueller L. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha- and beta-carbon resonances in proteins. J Magn Reson. 1993;101:201–205. [Google Scholar]
- 46.Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K. TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci USA. 1998;95:13585–13590. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 48.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 49.Ding K, Gronenborn AM. Sensitivity-enhanced 2D IPAP, TROSY-anti-TROSY, and E.COSY experiments: alternatives for measuring dipolar 15N–1HN couplings. J Magn Reson. 2003;163:208–214. doi: 10.1016/s1090-7807(03)00081-8. [DOI] [PubMed] [Google Scholar]
- 50.Zweckstetter M. NMR: prediction of molecular alignment from structure using the PALES software. Nat Protoc. 2008;3:679–690. doi: 10.1038/nprot.2008.36. [DOI] [PubMed] [Google Scholar]
- 51.Goddard TD, Kneller DG. Sparky 3. University of California; San Francisco: 2008. [Google Scholar]
- 52.Peng JW, Wagner G. Frequency spectrum of NH bonds in eglin c from spectral density mapping at multiple field. Biochemistry. 1995;34:16733–16752. doi: 10.1021/bi00051a023. [DOI] [PubMed] [Google Scholar]
- 53.Farrow NA, Zhang O, Forman-Kay JD, Kay LE. Comparison of the backbone dynamics of a folded and an unfolded SH3 domain existing in equilibrium in aqueous buffer. Biochemistry. 1995;34:868–878. doi: 10.1021/bi00003a021. [DOI] [PubMed] [Google Scholar]
- 54.Krishnan VV, Sukumar M, Gierasch LM, Cosman M. Dynamics of cellular retinoic acid binding protein I on multiple time scales with implications for ligand binding. Biochemistry. 2000;39:9119–9129. doi: 10.1021/bi000296l. [DOI] [PubMed] [Google Scholar]
- 55.Ottiger M, Bax A. Determination of Relative N–HN, N–C′, Calpha–C′, and Calpha–Halpha effective bond lengths in a protein by NMR in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120:12334–12341. [Google Scholar]
- 56.Prieto JH, Sampoli Benitez BA, Melacini G, Johnson DA, Wood MJ, Komives EA. Dynamics of the fragment of thrombomodulin containing the fourth and fifth epidermal growth factor-like domains correlate with function. Biochemistry. 2005;44:1225–1233. doi: 10.1021/bi0478852. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.