

Abstract
Alkylcarbamic acid biphenyl-3-yl esters are a class of fatty acid amide hydrolase (FAAH) inhibitors that comprises cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester (URB597), a compound with analgesic, anxiolytic-like and antidepressant-like properties in rat and mouse models. Here, we extended the structure-activity relationships (SARs) for this class of compounds by replacing the cyclohexyl ring of the parent compound cyclohexylcarbamic acid biphenyl-3-yl ester (URB524) (IC50, for FAAH = 63 nM) with a selected set of substituents of different size, shape, flexibility and lipophilicity.
Docking experiments and Linear Interaction Energy (LIE) calculations indicated that the N-terminal group of O-arylcarbamates fits within the lipophilic region of the substrate-binding site, mimicking the arachidonoyl chain of anandamide. Significant potency improvements were observed for the β-naphthylmethyl derivative 4q (IC50 = 5.3 nM) and its 3′-carbamoylbiphenyl-3-yl ester 4z (URB880, IC50 = 0.63 nM), indicating that shape complementarity and hydrogen bonds are crucial to obtain highly potent inhibitors.
Introduction
Fatty acid amide hydrolase (FAAHa)1, 2 is an intracellular serine enzyme that catalyzes the deactivating hydrolysis of the fatty-acid ethanolamide family of signaling lipids,3,4 which include endogenous ligands for cannabinoid receptors such as arachidonoylethanolamide (anandamide)5 and peroxisome proliferator-activated receptors type-α (oleoylethanolamide6,7 and palmitoylethanolamide8,9). The mechanism responsible for the inactivation of FAAH is unique among mammalian enzymes in that it involves a catalytic triad consisting of two serine residues (Ser217 and Ser241) and one lysine residue (Lys142), rather than the more common serine-histidine-aspartate triad of other serine hydrolases.10 This mechanism has been studied using both experimental11 and computational methods,12,13,14,15 and is thought to underpin the ability of FAAH to hydrolyze amides and esters at equivalent rates in vitro.
Endogenous FAAH substrates such as anandamide serve key regulatory functions in the body and have been implicated in a variety of pathological conditions, including pain, anxiety, depression and vascular hypertension.16 As a result, pharmacological inhibition of intracellular FAAH activity has been the focus of intense drug-discovery efforts.17,18 Among the different classes of FAAH inhibitors that have been recently disclosed,2 those appearing to meet basic criteria of drug-likeness are: α-ketoheterocycles,19,20,21,22,23 (thio)hydantoins,24 ureas25 and O-alkyl26 or O-aryl27,28 carbamates. The latter class includes potent and selective inhibitors based on the N-cyclohexylcarbamic acid O-aryl ester scaffold,29 such as cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester (1, URB597, Figure 1), which have been intensively investigated revealing a pharmacological profile30 characterized by a unique combination of analgesic,31,32 anxiolytic-like33 and antidepressant-like34,35 properties. Antihypertensive effects of 136 and anti-parkinsonian studies with 137 have also been described.
Figure 1.

Chemical structures of compounds 1 and 2.
A first exploration of the structure-activity relationship (SAR) for the class of O-aryl carbamates indicated that the aromatic fragment at the oxygen gave better potency when assembled to give a bent shape.27 A structural similarity between curved aromatic structures and the first portion of the arachidonoyl chain was then sought, and conformationally constrained O-aryl moieties were designed to mimic the binding conformation of the anandamide chain.38 This approach led to the identification of the lead compound cyclohexylcarbamic acid biphenyl-3-yl ester (2, URB524, Figure 1), which inhibited FAAH activity in rat brain membranes with a half-maximal inhibitory concentration (IC50) value of 63 nM. The biphenyl-3-yl motif has been recently employed in a new series of O-arylcarbamates, deriving from the structure of the known inhibitor N-arachidonoylserotonin.39
In the subsequent optimization of lead based on quantitative structure-activity relationship (QSAR) studies, it was noted that the introduction of hydrophilic residues (e.g., carbamoyl, hydroxymethyl, and hydroxyl groups) at the 3′-position of the biphenyl nucleus yielded FAAH inhibitors with potencies in the low nanomolar concentration range [e.g., 1 (IC50 = 4.6 nM)],29 a compound with a remarkable selectivity vs monoglyceride lipase (MGL),33 triacylglycerol hydrolase (TGH) and triacylglycerol lipase (TGL).40
The resolution of the crystal structure of FAAH covalently bound to methyl arachidonyl phosphonate (MAP)41 allowed molecular modeling studies of enzyme-inhibitor complexes. Docking of 1 into the active site of FAAH showed that the meta-biphenyl moiety of this compound may effectively replace the arachidonyl chain of MAP, allowing its 3′-carbamoyl group to participate in two hydrogen bonds with specific enzyme residues; we called this docking arrangement ‘orientation A’ (Figure 2). However, docking also revealed the possibility of an alternative, equally tenable binding mode29,42 in which the N-cyclohexyl group is placed into the acyl chain binding (ACB) channel and the biphenyl scaffold occupies the cytoplasmic access (CA) channel, with the carbamoyl group still being able to give hydrogen bonds with the protein (‘orientation B’, Figure 2). The synthesis of a series of cyclohexylcarbamic acid biphenyl esters with different substituents on the proximal phenyl ring did not help to discriminate between the two binding modes, as the SARs were consistent with both orientations.43 On the other hand, orientation B had been shown to be consistent with FAAH carbamoylation.15,44
Figure 2.

Representation of compound 1 docked into the FAAH binding site in two alternative orientations, named A and B, respectively. Carbons of the inhibitor are coloured in white, those of FAAH in orange. The hydrogen bonds of the carbamoyl group of compound 1 with the enzyme are evidenced in red.
In the present study, we report the synthesis and characterization of a novel series of carbamic acid biphenyl esters, designed as inhibitors of FAAH by progressive modification of the structure of compound 2. We explored the steric and lipophilic requirements of the N-substituents for FAAH inhibition and evaluated the results using QSAR and molecular modeling methods. In particular, we employed the structure-based Linear Interaction Energy method (LIE)45 implemented in the LIAISON package46 to test the hypothesis that differences in potency can be predicted from enzyme-inhibitor recognition, when the reactivity of the carbamic group can be considered essentially the same within the series. This approach had been successfully applied to several enzyme inhibitors,47,48,49 and is generally regarded as a robust computational tool for structure-based SAR rationalization50 and for the prediction of experimental binding affinity.51 Interaction energies were calculated for all compounds docked in both orientation A and B, and statistical models were built and compared.
In particular, we began our SAR investigation by gradually increasing the lipophilicity of the N-alkyl group (methyl to n-octyl). We then explored the steric requirements of the vicinal binding region by introducing allyl, isopropyl, n-butyl and cyclobutyl groups, which have comparable lipophilicity but different shapes. Particular attention was devoted to N-substitution with cyclic groups such as cycloalkyl, arylcycloalkyl and aryl groups to explore the length and size of the FAAH binding pocket. Moreover, searching for compounds able to discriminate between orientations A and B, we introduced different specific features such as a highly bulky group (1-adamantyl), a hydrophilic terminal group (6-morpholinohexyl) and a series of conformationally constrained substituents (α-naphthylmethyl, β-naphthylmethyl, para-biphenylbutyl). Finally, design of selected structures by combining the results of our present exploration with those of previous work on the biphenyl group of compound 227,29,43 led us to identify a subnanomolar FAAH inhibitor.
Chemistry
O-biphenyl-3-yl carbamates N- and 3′-substituted 4b-i,k-w,y,z were synthesized by addition of opportune 3-phenylphenol, commercially available, or 3-phenylphenol-3′-substituted to isocyanates 3b-i,k-w,y,z (Scheme 1). 3′-Hydroxybiphenyl-3-carboxylic acid amide and 1-(3′-hydroxybiphenyl-3-yl)ethanone were prepared as reported in the literature.29 Compounds 4a,j,x were obtained by reacting N,N′-carbonyldiimidazole (5) and the opportune amine and, in a subsequent step, 3-phenylphenol with the adduct previously formed (Scheme 2). An analogue procedure was followed to synthesize 2-oxooxazolidine-3-carboxylic acid biphenyl-3-yl ester (6). 2-Cyclohexylacetic acid biphenyl-3-yl ester (8) was obtained by adding 3-phenylphenol to a mixture of 2-cyclohexylacetic acid (7) and dicyclohexylcarbodiimide (Scheme 3).
Scheme 1.

a Reagents and conditions: (a) 3-phenylphenol or 3-phenylphenol-3′-substituted, toluene, Et3N, reflux, 5-27 h.
Scheme 2.

a Reagents and conditions: (a1) R-NH2, CH3CN, DMAP, reflux, 4-24 h; (a2) 3-phenylphenol, reflux, 2-30 h.
Scheme 3.

a Reagents and conditions: (a) 3-phenylphenol, CH2Cl2, DCC, DMAP, r.t., 18 h.
Isocyanates 3b-f,h,i,k-n,w were commercially available. The synthesis of isocyanates 3g,o-v,y,z (Scheme 4) was performed via Curtius rearrangement of the opportune acylazide (10), prepared by reacting the appropriate carboxylic acid (9) with diphenylphosphorylazide (DPPA) (3g,q,r,t-v,y,z) or with oxalylchloride and sodium azide (NaN3) (3o,p,s).
Scheme 4.

a Reagents and conditions: (a) toluene, TEA, DPPA, 0.5 h (3g,q,r,t-v,y,z) or CH2Cl2, DMF, (COCl)2, 0.5 h then NaN3 in H2O, 0.5 h, 0-5 °C (3o,p,s); (b) reflux, 0.5 h.
Carboxylic acid 9t was obtained by reaction of ethyl 7-bromoheptanoate (11t) with morpholine and subsequent hydrolysis of ester 12t (Scheme 5). Carboxylic acids 9u and 9v were synthesized by a catalytic hydrogenation of the respective intermediates 15u and 15v, obtained by a Wittig reaction of the opportune aldehyde and the phosphonium salts 14u and 14v, in presence of lithium hexamethyldisylazane (LHMDS).52 Concerning the latter compounds, 14v was commercially available while 14u was prepared by reaction of 8-bromooctanoic acid (13u) and triphenylphosphine (TPP) (Scheme 6).
Scheme 5.

a Reagents and conditions: (a) toluene, morpholine, reflux, 0.5 h; (b) H2O, reflux, 72 h.
Scheme 6.

a Reagents and conditions: (a) CH3CN, TPP, 48 h; (b) THF, LHMDS 1M in THF, R-C(O)H, 24 h; (c) AcOEt, H2, Pd/C 10%, 3 h.
Results and Discussion
We measured FAAH activity in rat brain membranes, using [3H]anandamide as a substrate. The IC50 values for compounds 4a-z, 6, and 8 are reported in Tables 1a and 1b, along with that of the reference compound 2.
Table 1a.
Inhibitory Potencies (IC50) of Tested Compounds 2 and 4a-z on FAAH Activity.

| |||
|---|---|---|---|
|
| |||
| Cpds. | R- | X | IC50 (nM)± S.E.M. |
| 2 |

|
H | 63±9 |
| 4a |

|
H | 13,901±4,426 |
| 4b |

|
H | 696±38 |
| 4c |

|
H | 521±87 |
| 4d |
|
H | 113±16 |
| 4e |
|
H | 57±15 |
| 4f |
|
H | 53±8 |
| 4g |

|
H | 54±0.9 |
| 4h |

|
H | 34±7 |
| 4i |

|
H | 66±15 |
| 4j |

|
H | 244±5 |
| 4k |

|
H | 4,089±1,037 |
| 4l |

|
H | 139±34 |
| 4m |

|
H | 175±11 |
| 4n |

|
H | 481±65 |
| 4o |

|
H | 215±51 |
| 4p |

|
H | 59±7 |
| 4q |

|
H | 5.3±0.2 |
| 4r |

|
H | 9.4±2.8 |
| 4s |
|
H | 13±2 |
| 4t |
|
H | 39.9±8.5 |
| 4u |
|
H | 5.4±0.5 |
| 4v |

|
H | 7.8±1.9 |
| 4w |

|
H | 3,942±1,409 |
| 4x |

|
H | 3,091±1,880 |
| 4y |
|
C(O)CH3 | 5.2±1.8 |
| 4z |

|
C(O)NH2 | 0.63±0.04 |
| Table 1b. Inhibitory Potencies (IC50) of Tested Compounds 6 and 8 on FAAH Activity. | ||
|---|---|---|
|
| ||
| Cpds. | IC50 (nM)± S.E.M. | |
| 6 |

|
28,617±10,782 |
| 8 |

|
2,551±1,074 |
Replacing the cyclohexyl N-substituent of 2 with smaller groups such as methyl (4a), allyl (4b) and isopropyl (4c) resulted in significant decreases in inhibitory potency, which was recovered with longer alkyl chains (compounds 4d-f). Furthermore, while small cyclic N-substituents did not affect activity (4g-i), bulkier groups such as exo-2-norbornyl (4j) and adamantyl (4k) were unfavourable. The influence of a terminal alkylphenyl group strongly depended on chain length and geometry. In general, shorter alkyl chains yielded less potent inhibitors (4l, 4n, 4o), while significant increases in potency were observed when the phenyl ring was attached to an n-butyl (4r), n-hexyl (4s) and n-octyl (4u) chain. Thus, 4u (IC50 = 5.4 nM) was approximately 10 times more potent than compound 2 (IC50 = 63 nM). Substituting the phenyl ring of 4l and 4s with more hydrophilic groups had a negative effect on potency (4m, 4t). As indicated by the relatively high activity of the biphenyl n-butyl derivative 4v, and according to a QSAR model (see below), rigidification of the hydrocarbon chain was tolerated. Notably, whereas no improvement in potency was observed with the introduction of an α-napthylmethyl group (4p, IC50 = 59 nM), a β-napthylmethyl group yielded one of the most active compounds in the present series (4q, IC50 = 5.3 nM). Modification of the essential alkylcarbamic acid aryl ester scaffold led to much less active compounds (4w, 4x, 6, 8). Finally, combination of the present results with what had been previously observed about substituents 3′-acetyl (IC50 = 9.1 nM) and 3′-carbamoyl (IC50 = 4.6 nM) of the biphenyl group29 led to 4y (IC50 = 5.2 nM) and 4z (URB880, IC50 = 0.6 nM), the latter being about 100 times more potent than compound 2 and 6 times more potent than compound 1 at inhibiting FAAH activity in vitro. 4z versus rat brain MGL proved to be ineffective displaying an IC50 value of 100,000 nM.
With pIC50 values spanning almost four orders of magnitude and a large diversity in structure size, shape and lipophilicity, the twenty-two compounds listed in Table 2 constitute an ideal set for QSAR investigation. For reasons of structural consistency, compounds 4j, 4w-z, 6, and 8 were excluded from these analyses.
Table 2.
QSAR Analysis for Selected N-Substituted Carbamic Acid Biphenyl-3-yl Esters: Experimental and Calculated pIC50 Values and Residuals in Orientations A and B, Calculated by Equations 4 and 6, Respectively.
| Cpds. | Orientation A | Orientation B | |||
|---|---|---|---|---|---|
| Exp. pIC50 | Calcd. pIC50 | Residuals | Calcd. pIC50 | Residuals | |
| 2 | 7.20 | 6.49 | 0.71 | 7.25 | -0.05 |
| 4a | 4.86 | 5.37 | -0.51 | 5.52 | -0.66 |
| 4b | 6.16 | 5.54 | 0.62 | 5.93 | 0.23 |
| 4c | 6.28 | 5.94 | 0.34 | 6.45 | -0.17 |
| 4d | 6.95 | 6.09 | 0.86 | 6.74 | 0.21 |
| 4e | 7.24 | 6.64 | 0.60 | 6.59 | 0.66 |
| 4f | 7.28 | 7.29 | -0.01 | 7.00 | 0.27 |
| 4g | 7.27 | 6.13 | 1.14 | 6.92 | 0.35 |
| 4h | 7.47 | 6.35 | 1.12 | 6.76 | 0.71 |
| 4i | 7.18 | 7.08 | 0.10 | 6.88 | 0.30 |
| 4k | 5.39 | 6.94 | -1.55 | 5.95 | -0.56 |
| 4l | 6.86 | 6.57 | 0.29 | 6.50 | 0.36 |
| 4m | 6.76 | 6.11 | 0.65 | 7.01 | -0.25 |
| 4n | 6.32 | 7.01 | -0.69 | 6.88 | -0.57 |
| 4o | 6.67 | 7.41 | -0.74 | 7.24 | -0.57 |
| 4p | 7.23 | 7.48 | -0.25 | 7.21 | 0.01 |
| 4q | 8.27 | 7.38 | 0.90 | 7.34 | 0.93 |
| 4r | 8.03 | 7.44 | 0.59 | 7.53 | 0.49 |
| 4s | 7.89 | 8.31 | -0.42 | 8.39 | -0.50 |
| 4t | 7.40 | 8.35 | -0.95 | 7.63 | -0.23 |
| 4u | 8.27 | 8.90 | -0.63 | 8.66 | -0.40 |
| 4v | 8.11 | 9.04 | -0.93 | 8.54 | -0.43 |
As a first step in QSAR analysis, we searched for statistical correlation between pIC50 values and lipophilicity. A log P value was calculated for all compounds reported in Table 2 with two well known methods: the Moriguchi log P53 and the ACD/Labs log P.54 While the two calculated log P values (see Table S1 in Supporting Information) were mutually correlated (r = 0.95), poor correlations were found between pIC50 and lipophilicity, as confirmed by the regression Equations 1 and 2.
| (Eq. 1) |
| (Eq. 2) |
The use of size descriptors, such as molecular volume or molar refractivity, did not produce better models (data not shown). Therefore, while it could be qualitatively inferred that hydrophobic interactions remain crucial for obtaining highly potent inhibitors, overall lipophilicity appeared inadequate to obtain a QSAR within this series.
The FAAH crystal structure was then employed to evaluate the correlation between stereoelectronic complementarity and inhibitory potency. The structure-based linear interaction energy method, implementing a surface-generalized Born model for solvent representation (SGB-LIE),55 had been applied to several protein-ligand systems to search for relations between the magnitude of free energy changes upon binding of inhibitors to enzymes and the experimental potencies of these inhibitors.47, 48, 49 The twenty-two compounds of the current dataset were thus docked into the FAAH binding site following an iterative procedure described in the Experimental Section. Two families of complexes, corresponding to orientations A and B, were generated and SGB-LIE calculations were performed using the LIAISON program.46 The interaction energy terms, referring to van der Waals (VDW), electrostatic and cavity components, were calculated for free and bound ligands, respectively, and are reported in the supporting material (Table S2 and Table S3). The difference between these energy values (bound-free) were used to build linear interaction energy equations by multiple regression analysis (MRA).
For orientation A, no significant model was found, as indicated by the low value of r2 and the poor predictive power (q2 = 0.12) reported for Equation 3:
| (Eq. 3) |
In the data matrix used for Equation 3, the first and the third independent variables were significantly correlated (rΔUvdw,ΔUcav = 0.975); VDW energy alone gave Equation 4, with a comparable r2 and q2and better F. Equation 4, free of not significant and correlated variables, was therefore employed to calculate the expected pIC50 values reported in Table 2.
| (Eq. 4) |
A better model was obtained for orientation B using the standard SGB-LIE approach. The resulting Equation 5 explained 71% of pIC50 variation and showed a good predictive power (q2 = 0.61).
| (Eq. 5) |
Internal correlation among X variables was also present in this case (rΔUvdw,ΔUelec = -0.71; rΔUvdw,ΔUcav = 0.84; rΔUelec,ΔUcav = -0.40), probably affecting the uncertainty for the cavity term coefficient (0.375±0.513). The exclusion of ΔUcav led to Equation 6, with the same r2 and all coefficients significantly different from zero, which was employed to calculate the expected pIC50 values reported in Table 2 with the corresponding residuals.
| (Eq. 6) |
These models indicated that VDW interactions give the most significant contribution (i.e. with the largest coefficient/standard error ratio) to binding energy. VDW energy is strongly related to the closeness of ligand and enzyme surfaces. Electrostatic interactions also showed a significant effect: as chemical modulation in this set of compounds mainly regarded size and shape, this result can be a consequence of the complementarity between inhibitors and the binding site. In fact, the carbamic group of all these inhibitors may form several hydrogen bonds, and a high steric complementarity allows more efficient electrostatic interaction. However, interpretation of the ΔUelec term is complicated by the fact that it also includes interactions with the SGB solvent reaction-field energy (see Experimental Section).
Experimental and calculated pIC50 values are reported in Table 2 and graphically plotted in Figure 3. When placed in position A, the compounds with a cyclic aliphatic substituent [e.g. compounds 2, 4g, 4h and, in particular, the 1-adamantyl derivative 4k (Δobs/calc = -1.55)] generally showed a marked difference between observed and calculated pIC50. On the contrary, placing the structures in orientation B allowed better predictions (Δobs/calc = -0.56 for 4k), which can be attributed to a more accurate evaluation of the steric tolerance for the two binding pockets.
Figure 3.

Plot of the observed pIC50 values vs those calculated by SGB-LIE models in orientations A (Equation 4, left panel) and B (Equation 6, right panel).
Visual inspection of the FAAH active site reveals that the ACB pocket is more lipophilic than the CA channel, in that it is mainly lined by aliphatic and aromatic residues.41 The introduction of a polar substituent at the nitrogen atom, as in the case of compound 4t, represented a further attempt to discriminate between the two binding modes. When the morpholinohexyl derivative was placed in orientation B, its potency was accurately calculated (Δobs/calc = -0.23). Furthermore, Equation 6 accounted for the different potencies of compounds 4s and 4t. Conversely, compound 4t in orientation A was an outlier, with a calculated pIC50 higher by one unit than the experimental one and with no variation of calculated potency with respect to 4s. Thus, the SGB-LIE approach, considering orientation B, can appropriately evaluate the effect of “lipophilicity” for these compounds.
However, even considering the better orientation, the SGB-LIE model did not reproduce potency variations with complete accuracy (r2 = 0.71). In fact, it could not account for the 10-fold higher potency of the β-naphthylmethyl derivative 4q over the α-naphthylmethyl one 4p. Compound 4q had been designed as an improved derivative of the benzyl derivative 4l and visual inspection and manual docking of it, according to orientation B, indicated that 4q would fit the ACB cavity better than 4p. Somewhat unexpectedly, while experimental potency confirmed the effect, LIAISON underestimated this difference, calculating similar potencies for the two compounds. However, also in this case, orientation B worked slightly better than orientation A.
Despite model weaknesses, probably due to the fact that recognition is only partially responsible for experimental IC50 and to the approximations mentioned in the Experimental Section, additional observations supported the consistency of the SGB-LIE model with orientation B. Compound 4j, which contains a racemic norbornyl group, was excluded from the training set; the calculated pIC50 for both enantiomers was 7.3 ± 0.1, reasonably close to the experimental value of 6.6. Compound 4v was designed as a less flexible derivative of 4u, representing the conformation it assumed in orientation B; as correctly predicted by Equation 6, the two compounds actually showed similar potencies.
The consistency of the model described above with the SAR previously reported for the biphenyl moiety29 was tested by selecting one of the most potent compounds of the series, 4q, and placing at the 3′-position of its biphenyl nucleus a polar hydrogen bonding group. Even if the two moieties did not show a purely additive effect on pIC50, the subnanomolar potency showed by 4z confirmed that concurrent positioning of the lipophilic N-alkyl group within the ACB pocket and of the biphenyl moiety within the more polar CA cavity (represented in Figure 4) favors inhibitory potency.
Figure 4.
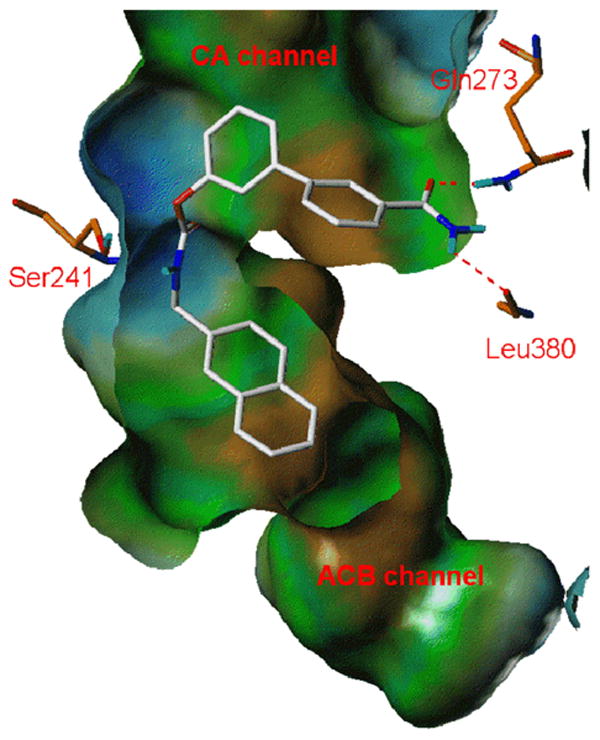
Compound 4z (white carbons) docked into FAAH (orange carbons) binding site in orientation B. The hydrogen bonds between the carbamoyl group of 4z and the CA channel of FAAH are evidenced with dashed red lines.
Conclusions
The potency of carbamic FAAH inhibitors, such as those described in the present study, is expected to depend on two factors: first, the ability of the inhibitor to bind to the enzyme recognition site; and, second, the propensity of the carbamate group to react with the Ser241 nucleophile. However, in the case of the carbamic acid biphenyl esters investigated here, recognition events appear to predominate. In the present series of compounds, alteration of the N-alkyl structure (as in compounds 4w, 4x, 6, 8) led to a decrease of inhibitory potency, probably reflecting negative effects upon reactivity. On the other hand, good correlations between calculated and experimental potency could be achieved by limiting SAR analysis to compounds that share the alkylcarbamic acid biphenyl ester scaffold, provided that the binding mode is rightly chosen. Our findings rule out the hypothesis that the biphenyl nucleus mimics the fatty acyl chain of FAAH substrates, and describe a protocol that, at affordable computational costs, provides a robust and sufficiently accurate QSAR model.
The QSAR model based on SGB-LIE estimation of binding propensity supported the rational design of new FAAH inhibitors, comprising the subnanomolar inhibitor 4z. Such a model, being based on complex stereoelectronic complementarity, has sufficient accuracy to be considered as a tool to estimate FAAH inhibitor potency for different N-alkyl carbamates in a process of combined pharmacokinetic, toxicologic and pharmacodynamic optimization of this promising class of inhibitors. Notably, generic descriptors of lipophilicity or steric bulk performed less well in predicting the ability of the N-alkyl portion to bind a lipophilic cavity.
The present results confirm the hypothesis that carbamic acid biphenyl esters inhibit FAAH by assuming an orientation in which the N-substituent is positioned within the ACB channel of the enzyme (‘orientation B’). While previous studies44 based this hypothesis on the evidence of enzyme carbamoylation, and exploited the role of lipophilicity for the N-alkyl group, the present work describes the SAR requirements for potent inhibitors and underlines steric similarities between the two binding pockets, as shown by the generally better prediction of pIC50 values in the orientation B.
In brief, rational exploration of the chemical space, convergent consideration of SARs and structural information allowed us to find appropriate stereoelectronic features and to obtain a subnanomolar inhibitor of FAAH activity, named 4z which is currently undergoing in vivo tests. The definition of these requirements may also be useful to the development of new classes of FAAH inhibitors, aimed at selectivity and pharmacokinetic optimization.
Experimental Section
(a) Chemicals, Materials and Methods
All reagents were purchased from Sigma-Aldrich or Lancaster in the highest quality commercially available. Solvents were RP grade unless otherwise indicated. Chromatographic separations were performed on silica gel (Kieselgel 60, 0.040-0.063 mm, Merck). TLC analyses were performed on precoated silica gel on aluminium sheets (Kieselgel 60 F254, Merck). Melting points were determined on a Büchi SMP-510 capillary melting point apparatus. The structure of the unknown compounds was unambiguously assessed by MS, 1H NMR, IR, and elemental analysis. EI-MS spectra were recorded with a Fisons Trio 1000 (70 eV) spectrometer; only molecular ions (M+) and base peaks are given. 1H NMR spectra were recorded on a Bruker AC 200 spectrometer and analyzed using the WIN-NMR software package; chemical shifts were measured by using the central peak of the solvent. IR spectra were obtained on a Nicolet Atavar 360 FT spectometer. Elemental analyses were performed on a Carlo Erba analyzer. All products had satisfactory (within ± 0.4 of theoretical values) C, H, N analyses results.
General Procedure for the Synthesis of N-Substituted Carbamic Acid Biphenyl-3-yl Ester 4b-i,k-w,y,z
To a stirred solution of the opportune 3-phenylphenol or 3-phenylphenol-3′-substituted (1 mmol) in toluene (5 mL) were added the suitable isocyanate (1.1 mmol) and Et3N (0.05 mmol, 0.005 g, 0.007 mL). After the reactants were refluxed for 5 h a further amount of the opportune isocyanate was added [4w, 0.27 mmol, 0.033 g; 4b,d,e,h,l,n, 0.55 mmol: 4b, 0.046 g; 4d, 0.055 g; 4e, 0.070 g; 4h, 0.061 g; 4n, 0.081 g; 4f, 0.825 mmol, 0.128 g; 4c, 0.90 mmol, 0.076 g; 4i, 1.1 mmol, 0.153 g; 4l: 0.081 g (no such further addition was necessary in the cases of 4g,k,m,o-v,y,z)] and the mixture was again reacted (4y, 2 h; 4c,w, 3 h; 4b, 4 h; 4i, 6 h; 4l, 12 h; 4d,e,h, 14 h; 4z, 15 h; 4n, 17 h; 4t, 19 h; 4f, 22 h) then cooled and concentrated. Purification of the residue by column cromatography (cyclohexane/EtOAc 85:15 for 4c,e, 8:2 for 4h,i,k,w, 4:6 for 4z; CH2Cl2/cyclohexane 9:1 for 4y, 8:2 for 4n-s,u,v, 9:1 for 4g,m, 95:5 for 4f, 99.5:0.5 for 4b; CH2Cl2 for 4d,l; CH2Cl2/MeOH 95:5 for 4t) and recrystallization gave 4b-i,k-w,y,z.
Allylcarbamic Acid Biphenyl-3-yl Ester (4b)
White crystals. Yield: 91% (0.467 g). Mp 64-65 °C (Et2O/petroleum ether). MS (EI): m/z 253 (M+), 170 (100). 1H NMR (CDCl3): δ 3.91-3.97 (t, 2H), 5.13 (br d, 1H), 5.19-5.35 (m, 2H), 5.84-6.02 (m, 1H), 7.10-7.16 (m, 1H), 7.32-7.35 (m, 6H), 7.61-7.62 (m, 2H) ppm. IR (Nujol): 3370, 1712 cm-1. Anal. (C16H15NO2) C, H, N.
Isopropylcarbamic Acid Biphenyl-3-yl Ester (4c)56
White crystals. Yield: 58% (0.148 g). Mp 86-89 °C (EtOH). MS (EI): m/z 255 (M+), 170 (100). 1H NMR (CDCl3): δ 1.26 (d, 6H), 3.84-4.01 (m, 1H), 4.90 (br d, 1H), 7.08-7.23 (m, 1H), 7.31-7.49 (m, 5H), 7.56-7.62 (m, 3H) ppm. IR (Nujol): 3400, 1697 cm-1. Anal. (C16H17NO2) C, H, N.
n-Butylcarbamic Acid Biphenyl-3-yl Ester (4d)
White crystals. Yield: 79% (0.212 g). Mp 68-70 °C (EtOH). MS (EI): m/z 269 (M+), 170 (100). 1H NMR (CDCl3): δ 0.97 (t, 3H), 1.44-1.56 (m, 4H), 3.30 (q, 2H), 5.04 (br s, 1H), 7.08-7.16 (m, 1H), 7.31-7.48 (m, 6H), 7.56-7.62 (m, 2H) ppm. IR (Nujol): 3323, 1704 cm-1. Anal. (C17H19NO2) C, H, N.
n-Hexylcarbamic Acid Biphenyl-3-yl Ester (4e)
White crystals. Yield: 90% (0.267 g). Mp 71-73 °C (petroleum ether). MS (EI): m/z 297 (M+), 170 (100). 1H NMR (CDCl3): δ 0.88-1.64 (m, 11H), 3.29 (m, 2H), 5.06 (br s, 1H), 7.08-7.18 (m, 1H), 7.31-7.48 (m, 6H), 7.56-7.62 (m, 2H) ppm. IR (Nujol): 3326, 1702 cm-1. Anal. (C19H23NO2) C, H, N.
n-Octylcarbamic Acid Biphenyl-3-yl Ester (4f)
White crystals. Yield: 91% (0.590 g). Mp 80 °C (EtOH). MS (EI): m/z 326 (M+), 170 (100). 1H NMR (CDCl3): δ 0.87-0.93 (m, 3H), 1.29-1.33 (m, 10H), 1.59 (q, 2H), 3.29 (q, 2H), 5.04 (br s, 1H), 7.08-7.19 (m, 1H), 7.31-7.48 (m, 6H), 7.56-7.62 (m, 2H) ppm. IR (Nujol): 3326, 1701 cm-1. Anal. (C21H27NO2) C, H, N.
Cyclobutylcarbamic Acid Biphenyl-3-yl Ester (4g)
White crystals. Yield: 54% (0.382 g). Mp 114-118 °C (EtOH). MS (EI): m/z 267 (M+), 169 (100). 1H NMR (CDCl3): δ 1.63-2.08 (m, 4H), 2.33-2.48 (m, 2H), 4.16-4.36 (m, 1H), 5.21-5.22 (br d, 1H), 7.09-7.61 (m, 9H) ppm. IR (Nujol): 3444, 3310, 1704 cm-1. Anal. (C17H17NO2) C, H, N.
Cyclopentylcarbamic Acid Biphenyl-3-yl Ester (4h)
White scales. Yield: 57% (0.160 g). Mp 142-145 °C (EtOH). MS (EI): m/z 281 (M+), 170 (100). 1H NMR (CDCl3): δ 1.50-1.74 (m, 6H), 2.01-2.10 (m, 2H), 4.04-4.14 (m, 1H), 5.01 (br d, 1H), 7.10-7.17 (m, 1H), 7.31-7.48 (m, 6H), 7.56-7.62 (m, 2H) ppm. IR (Nujol): 3311, 1706 cm-1. Anal. (C18H19NO2) C, H, N.
Cyclohexylmethylcarbamic Acid Biphenyl-3-yl Ester (4i)
White crystals. Yield: 81% (0.500 g). Mp 118 °C (EtOH). MS (EI): m/z 310 (M+), 170 (100). 1H NMR (CDCl3): δ 0.95-1.82 (m, 11H), 3.15 (t, 2H), 5.09 (br s, 1H), 7.10-7.69 (m, 9H) ppm. IR (Nujol): 3426, 3311, 1701 cm-1. Anal. (C20H23NO2) C, H, N.
Adamant-1-ylcarbamic Acid Biphenyl-3-yl Ester (4k)
White crystals. Yield: 24% (0.083 g). Mp 128-131 °C (EtOH). MS (EI): m/z 347 (M+), 170 (100). 1H NMR (CDCl3): δ 1.70 (m, 6H), 2.03-2.13 (m, 9H), 4.93 (br s, 1H), 7.07-7.18 (m, 1H), 7.35-7.48 (m, 6H), 7.57-7.62 (m, 2H) ppm. IR (Nujol): 3309, 1704 cm-1. Anal. (C23H25NO2) C, H, N.
Benzylcarbamic Acid Biphenyl-3-yl Ester (4l)
White solid. Yield: 98% (0.297 g). Mp 97-98 °C (EtOH). MS (EI): m/z 303 (M+), 170 (100). 1H NMR (CDCl3): δ 4.49 (d, 2H), 5.38 (br s, 1H), 7.12-7.18 (m, 1H), 7.32-7.49 (m, 11H), 7.57-7.62 (m, 2H) ppm. IR (Nujol): 3325, 1707 cm-1. Anal. (C19H15NO2)C, H, N.
Furan-2-ylmethylcarbamic Acid Biphenyl-3-yl Ester (4m)
White crystals. Yield: 62% (0.365 g). Mp 86-87 °C (Et2O/petroleum ether). MS (EI): m/z 293 (M+), 170 (100). 1H NMR (CDCl3): δ 4.48 (d, 2H), 5.40 (br s, 1H), 6.31-6.38 (m, 2H), 7.09-7.24 (m, 1H), 7.31-7.49 (m, 7 H), 7.56-7.62 (m, 2H) ppm. IR (Nujol): 3328, 1701 cm-1. Anal. (C18H15NO3) C, H, N.
Phenethylcarbamic Acid Biphenyl-3-yl Ester (4n)
White crystals. Yield: 65% (0.206 g) Mp 137-138 °C (EtOH). MS (EI): m/z 317 (M+), 170 (100). 1H NMR (CDCl3): δ 2.89-2.96 (t, 2H), 3.53-3.63 (q, 2H), 5.08 (br s, 1H), 7.08-7.14 (m, 1H), 7.22-7.67 (m, 13H) ppm. IR (Nujol): 3418, 3359, 1732, 1712 cm-1. Anal. (C21H19NO2) C, H, N.
Indan-2-ylcarbamic Acid Biphenyl-3-yl Ester (4o)
White crystals. Yield: 71% (0.930 g). Mp 140-141 °C (EtOH). MS (EI): m/z 329 (M+), 170 (100). 1H NMR (CDCl3): δ 2.89-3.00 (dd, 2H), 3.32-3.44 (m, 2H), 4.55-4.70 (m, 1H), 5.30 (br d, 1H), 7.10-7.62 (m, 13H) ppm. IR (Nujol): 3302, 1699 cm-1. Anal. (C22H19NO2) C, H, N.
Naphthalen-1-ylmethylcarbamic Acid Biphenyl-3-yl Ester (4p)
White solid. Yield: 74% (0.260 g). Mp 101-102 °C (EtOH). MS (EI): m/z 353 (M+), 170 (100). 1H NMR (CDCl3): δ 4.96 (d, 2H), 5.36 (br s, 1H), 7.12-7.21 (m, 1H), 7.32-8.14 (m, 15H) ppm. IR (Nujol): 3316, 1699 cm-1. Anal. (C24H19NO2) C, H, N.
Naphthalen-2-ylmethylcarbamic Acid Biphenyl-3-yl Ester (4q)
White crystals. Yield: 11% (0.153 g). Mp 160 °C (EtOH). MS (EI): m/z 353 (M+), 266 (100), 170 (100). 1H NMR (CDCl3): δ 4.66 (d, 2H), 5.47 (br s, 1H), 7.14-7.62 (m, 13H), 7.79-7.90 (m, 3H) ppm. IR (Nujol): 3266, 1704 cm-1. Anal. (C24H19NO2) C, H, N.
4-Phenylbutylcarbamic Acid Biphenyl-3-yl Ester (4r)
White crystals. Yield: 13% (0.180 g). Mp 112 °C (EtOH). MS (EI): m/z 345 (M+), 170 (100). 1H NMR (CDCl3): δ 1.61-1.74 (m, 4H), 2.65-2.72 (t, 2H), 3.27-3.37 (t, 2H), 5.03 (br s, 1H), 7.09-7.49 (m, 12 H), 7.55-7.61 (m, 2H) ppm. IR (Nujol): 3311, 1716 cm-1. Anal. (C23H23NO2) C, H, N.
6-Phenylhexylcarbamic Acid Biphenyl-3-yl Ester (4s)
White crystals. Yield: 68% (0.254 g). Mp 75-76 °C (EtOH). MS (EI): m/z 374 (M+), 170 (100). 1H NMR (CDCl3): δ 1.37-1.50 (m, 4H), 1.53-1.73 (m, 4H), 2.63 (t, 2H), 3.24-3.34 (m, 2H), 5.04 (br s, 1H), 7.10-7.49 (m, 12H), 7.56-7.62 (m, 2H) ppm. IR (Nujol): 3327, 1704 cm-1. Anal. (C25H27NO2) C, H, N.
6-Morpholinohexylcarbamic Acid Biphenyl-3-yl Ester (4t)
White crystals. Yield: 13% (0.382 g). Mp 93-95 °C (Et2O). MS (EI): m/z 382 (M+), 170 (100). 1H NMR (CDCl3): δ 1.40-1.60 (m, 8H), 2.30-2.47 (m, 6H), 3.24-3.34 (q, 2H), 3.71-3.76 (m, 4H), 5.15 (br d, 1H), 7.09-7.15 (m, 1H), 7.35-7.47 (m, 6H), 7.57-7.61 (m, 2H) ppm. IR (Nujol): 3300, 1712 cm-1. Anal. (C23H20N2O3) C, H, N.
8-Phenyloctylcarbamic Acid Biphenyl-3-yl Ester (4u)
White crystals. Yield: 16% (0.141 g). Mp 77-79 °C (EtOH). MS (EI): m/z 402 (M+), 170 (100). 1H NMR (CDCl3): δ 1.27-1.36 (m, 8H), 1.59-1.63 (m, 4H), 2.58-2.66 (t, 2H), 3.24-3.34 (m, 2H), 5.05 (br s, 1H), 7.10-7.49 (m, 12H), 7.56-7.62 (m, 2H) ppm. IR (Nujol): 3325, 1704 cm-1. Anal. (C27H31NO2) C, H, N.
4′-Phenyl-4-phenylbutylcarbamic Acid Biphenyl-3-yl Ester (4v)
White crystals. Yield: 76% (0.320 g). Mp 127-130 °C (EtOH). MS (EI): m/z 421 (M+), 167 (100). 1H NMR (CDCl3): δ 1.61-1.73 (m, 4H), 2.69-2.76 (m, 2H), 3.30-3.39 (m, 2H), 4.91-5.12 (m, 1H), 7.27-7.61 (m, 18 H) ppm. IR (Nujol): 3292, 1712 cm-1. Anal. (C29H27NO2) C, H, N.
Phenylcarbamic Acid Biphenyl-3-yl Ester (4w)57
White scales. Yield: 95% (0.274 g). Mp 133-136 °C (EtOH). MS (EI): m/z 289 (M+), 170 (100). 1H NMR (CDCl3): δ 7.02 (br s, 1H), 7.09-7.22 (m, 2H), 7.32-7.63 (m, 12H) ppm. IR (Nujol): 3352, 1720 cm-1. Anal. (C19H15NO2) C, H, N.
Naphthalen-2-ylmethylcarbamic Acid 3′-Acetylbiphenyl-3-yl Ester (4y)
White crystals. Yield: 20% (0.081 g). Mp 151-152 °C (EtOH). MS (EI): m/z 395 (M+), 212 (100). 1H NMR (CDCl3): δ 2.67 (s, 3H), 4.64-4.68 (d, 2H), 5.47-5.50 (br s, 1H), 7.18-7.23 (m, 1H), 7.44-7.58 (m, 7H), 7.78-7.97 (m, 6H), 8.18 (s, 1H) ppm. IR (Nujol): 3282, 1708 cm-1. Anal. (C26H21NO3) C, H, N.
Naphthalen-2-ylmethylcarbamic Acid 3′-Carbamoylbiphenyl-3-yl Ester (4z)
White crystals. Yield: 22% (0.087 g). Mp 168-170 °C (EtOH). MS (EI): m/z 396 (M+), 183 (100). 1H NMR (CDCl3): δ 4.64-4.67 (d, 2H), 5.41-5.55 (br s, 1H), 5.61-5.76 (m, 1H), 6.08-6.22 (m, 1H), 7.17-7.23 (m, 1H), 7.44-7.56 (m, 7H), 7.74-7.90 (m, 6H), 8.02-8.04 (m, 1H) ppm. IR (Nujol): 3373, 2361, 1718 cm-1. Anal. (C25H20N2O3) C, H, N.
General Procedure for the Synthesis of N-Substituted Carbamic Acid Biphenyl-3-yl Ester 4a,j,x,6
To a stirred solution of the opportune amine (1 mmol) and diimidazol-1-ylmethanone (5) (4a, 1 mmol; 4j,x,6, 2 mmol) in dry CH3CN (4a, 5.5 mL; 4j,x,6, 11 mL), DMAP (0.044 mmol, 0.006 g) was added and the mixture was refluxed under N2 atmosphere (4j,6, 4 h; 4x, 20 h; 4a, 24 h). Then 3-phenylphenol (1 mmol, 0.170 g) was added and the mixture was refluxed again (4x, 2 h; 6, 18 h; 4j, 20 h; 4a, 30 h), cooled and concentrated. Purification of the residue by column cromatography (cyclohexane/EtOAc 8:2 for 4a,x, 4:6 for 6; CH2Cl2/cyclohexane 8:2 for 4j) and recrystallization gave 4a,j,x,6.
Methylcarbamic Acid Biphenyl-3-yl Ester (4a)58
White crystals. Yield: 9% (0.020 g). Mp 111-115 °C (Et2O/petroleum ether). MS (EI): m/z 227 (M+), 170 (100). 1H NMR (CDCl3): δ 2.93 (d, 3H), 5.02 (br s, 1H), 7.09-7.18 (m, 1H), 7.33-7.62 (m, 8H) ppm. IR (Nujol): 3338, 1709 cm-1. Anal. (C14H13NO2) C, H, N.
(±) Bicyclo[2.2.1]hept-2-ylcarbamic Acid Biphenyl-3-yl Ester (4j)
White crystals. Yield: 25% (0.157 g). Mp 151-152 °C (EtOH). MS (EI): m/z 307 (M+), 170 (100). 1H NMR (CDCl3): δ 1.09-1.55 (m, 7H), 1.81-1.93 (m, 1H), 2.33 (m, 2H), 3.58-3.67 (m, 1H), 4.92 (br d, 1H), 7.09-7.15 (m, 1H), 7.35-7.48 (m, 6H), 7.57-7.62 (m, 2H) ppm. IR (Nujol): 3331, 1707 cm-1. Anal. (C20H21NO2) C, H, N.
Biphenyl-3-ylcarbamic Acid Biphenyl-3-yl Ester (4x)59
Pearly crystals. Yield: 18% (0.066 g). Mp, MS (EI), 1H NMR and IR are according to the literature.59 Anal. (C25H19NO2) C, H, N.
2-Oxooxazolidine-3-carboxylic Acid Biphenyl-3-yl Ester (6)
White crystals. Yield: 32% (0.091 g). Mp 123-125 °C (EtOH). MS (EI): m/z 283 (M+), 170 (100). 1H NMR [CD3C(O) CD3]: δ 4.22-4.31 (m, 2H), 4.48-4.56 (m, 2H), 7.22-7.70 (m, 9H) ppm. IR (Nujol): 3327, 1823, 1719 cm-1. Anal. (C16H13NO4) C, H, N.
Synthesis of 2-Cyclohexylacetic Acid Biphenyl-3-yl Ester (8)
To a stirred solution of 2-cyclohexylacetic acid (7) (1 mmol, 0.142 g), dicyclohexylcarbodiimide (1.1 mmol, 0.227 g) and DMAP (0.2 mmol, 0.024 g) in CH2Cl2 (2 mL) was added a mixture of 3-phenylphenol (1.2 mmol, 0.204 g) in CH2Cl2 (1 mL). The mixture was stirred at room temperature for 18 h, and concentrated. The residue was diluted with EtOAc and filtered on Celite. The purification of the residue by column chromatography (cyclohexane/EtOAc 9:1) gave 8 as an oil that solidifies in freezer to a white-yellow solid. Yield 81% (0.239 g). Mp <35 °C. MS (EI): m/z 294 (M+), 170 (100). 1H NMR (CDCl3): δ 1.02-2.01 (m,11H), 2.47 (d, 2H), 7.05-7.62 (m, 9H) ppm. IR (nujol): 1762 cm-1. Anal. (C20H22O2) C, H, N.
General Procedure for the Synthesis of Aryl- or Arylalkylisocyanates 3g,q,r,t-v,y,z
To a stirred suspension of the suitable acid (1.1 mmol) in toluene (5.5 mL), DPPA (1.276 mmol, 0.351 g, 0.275 mL) and, dropwise, Et3N (1.32 mmol, 0.134 g, 0.184 mL) were added. After 10 min the mixture was first stirred at room temperature for 30 min, then refluxed (3v,y,z, 2 h; 3u, 3 h; 3q,r, 5 h; 3g, 22 h; 3t, 24 h) and finally cooled and concentrated. The products obtained were not characterized because of their high instability and were used as such for the next reaction. (Yields: 96-98%).
General Procedure for the Synthesis of Aryl- or Arylalkylisocyanates 3o,p,s
To a stirred solution of the suitable acid (1.1 mmol) in CH2Cl2 (10 mL), DMF (0.1 mL) and (COCl)2 (1.6 mmol, 0.202 g, 0.14 mL) were added. After 10 min the mixture was first stirred at room temperature for 15 min, then concentrated by keeping the temperature below 30 °C and finally diluted with dry CH3C(O)CH3. This solution was added dropwise to a stirred and ice-cold solution of NaN3 (2.53 mmol, 0.164 g) in H2O (1.65 mL). The resulting mixture was stirred for 30 min, then diluted with CHCl3 and finally washed with brine. The combined organic layers were dried (Na2SO4), concentrated by keeping the temperature below 30 °C, refluxed for 30 min (vigorous gas development) and then concentrated again to give 3o,p,s as colourless oils. The products obtained were not characterized because of their high instability and were used as such for the next reaction. (Yields: 96-98%).
Synthesis of 7-morpholinoheptanoic acid (9t)
To a stirred solution of ethyl 7-bromo heptanoate (11t) (5 mmol, 1.18 g) in toluene (3 mL), a solution of morpholine (9.5 mmol, 0.828 g, 0.832 mL) in toluene (3 mL) was added dropwise. The mixture was refluxed for 30 min and filtered on Celite. The filtrate was washed with brine and Et2O. The combined organic layers were dried and concentrated. Purification of the residue by column chromatography (EtOAc/MeOH 96:4) gave 12t as an yellow-orange oil. 12t (3.9 mmol, 0.949 g) was refluxed for 72 h in H2O (245 mL); the solvent was then evaporated and the residue diluted with EtOAc and washed with H2O. The combined aqueous layers were again washed with EtOAc (5x30 mL) and concentrated. 9t was obtained as orange oil. Yield: 23% (0.555 g). MS (EI): m/z 215 (M+), 100 (100). 1H NMR (CDCl3): δ 1.26-1.54 (m, 8H), 2.16-2.23 (m, 2H), 2.43-2.67 (m, 6H), 3.71-3.76 (m, 4H), 13.04 (s, 1H) ppm.
Synthesis of 9-phenylnonanoic acid (9u) and 4′-phenyl-5-phenylpentanoic acid (9v)
To a stirred solution of 8-bromooctanoic acid (13u) (2 mmol, 0.483 g) in dry CH3CN (6.7 mL), TPP (2.12 mmol, 0.57 g) was added and the mixture was refluxed under N2 atmosphere for 48 h, then cooled and concentrated. The residue was washed with dry Et2O (4x7 mL) then concentrated. The white gummy solid obtained, dried under vacuum, constitutes the phosphonium salt 14u. (Yield: 98%). To a stirred suspension of the opportune phosphonium salt (14u,v, 1.96 mmol) in dry THF, a 1 M solution of lithium hexamethyldisilazane (LHMDS, 4.3 mmol, 0.725 g) in dry THF (4.3 mL) was slowly added under N2 atmosphere. The resulting mixture was stirred for 15 min and then, after the addiction of the opportune aldehyde (1.58 mmol), stirred again for 24 h. Then HCl 2 N was added until acid pH and the aqueous phase was extracted with EtOAc. The organic layer was dried over Na2SO4, filtrated and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 7:3 for 15u, 1:1 for 15v) gave 15u,v. (Yield: 55% and 63%, respectively). Compounds 15u and 15v were hydrogenated by Pd on charcoal (10%) to give 9u,v. (Yield: 98%).
9-Phenylnonanoic Acid (9u)60
White crystals. Yield: 53% (0.125 g). MS (EI) and 1H NMR are according to the literature.52
4′-Phenyl-5-phenylpentanoic Acid (9v)61
White crystals. Yield: 62% (0.155 g). MS (EI) and 1H NMR are according to the literature.61
(b) Pharmacology
Membrane fractions were prepared from brain homogenates, and FAAH activity was assayed using anandamide[ethanolamine-3H] [American Radiolabeled Chemicals, ARC (St. Louis, MO), 60 Ci/mmol] as a substrate.43 Inhibition of MGL was assessed on purified rat recombinant MGL, using 2-oleoylglycerol as substrate, as previously described.59
(c) Linear Interaction Energy Methods
The Linear Interaction Energy (LIE) method is based on the Linear Response Approximation, which estimates ΔG of the binding of a small molecule to a protein as a function of polar and non-polar energy components, that are considered linearly related to electrostatic and VDW interactions between the ligand and its environment.45 The free energy of binding for the protein-ligand complex is calculated considering two states: the “free” ligand, in a solvent environment, and the ligand bound to the solvated protein.
The present application of the LIE method implements the formulation proposed by Carlson and Jorgensen,62 where the Surface Generalized Born (SGB) continuum model is used for solvent representation.63 In the resulting SGB-LIE approach the free energy of binding is calculated as:
| (Eq. 7) |
In this equation, (<Ubvdw> - <Ufvdw>) estimates, by means of a Lennard-Jones potential, the variation of steric energy associated with ligand binding; (<Ubelec> - <Ufelec>) describes the change of electrostatic energy due to ligand desolvation and its accommodation into the protein binding site; the last term (<Ubcav> - <Ufcav>) accounts for the energy penalty due to the formation of a cavity within the solvent.64 The <> bracket notation indicates that an ensemble average of the energy terms should be taken into account for binding energy calculations. Molecular dynamics simulation or Monte Carlo sampling should be applied to generate a significant number of configurations for LIE calculations. However, local sampling with energy minimization had been shown to provide reasonable results in several cases, with limited or no reduction in the accuracy of ΔG estimation,47,48,49 and this approach was applied to the extended set of data of this study.
In the SGB-LIE equation (Eq. 7), all the terms are evaluated for the interaction between ligand, both in the free and bound state, and its environment. α, β and γ are free coefficients which were calculated by fitting the experimental free energies of binding for a training set of ligands with known protein affinity values. The transferability and dependence of such parameters on the choice of force field and protein-ligand system are still subject to debate. Nevertheless, this empirical fitting can be useful to compensate part of the limits of the method, due to neglection of conformational changes, intramolecular strain and entropic effects.48 In the case of covalent binding of inhibitors to FAAH, we lack an accurate measure of the free energy of binding; another approximation inherent to our approach is that, for a series of compounds with similar reactivity towards the enzyme catalytic center, the pIC50 values should be linearly related to the free energy of the recognition process. For this reason only compounds belonging to the unsubstituted O-biphenyl carbamates were included in the training set.
It should be pointed out that in the SGB approach the electrostatic energy (Uelec) is obtained by combining the pure Coulombic (Ucoul) interaction energy with the SGB solvent reaction-field energy (Urxn)55
| (Eq. 8) |
(d) Protein Preparation and Inhibitor Docking
Molecular models of the inhibitors were built by Sybyl version 7.2.65 Energy minimizations were performed employing the Merck molecular force field (MMFF94s)66 implemented in Sybyl, with the dielectric constant set to 1 to an energy gradient of 0.01 kcal/(mol·Å). FAAH structure coordinates were taken from those of the covalent adduct with methyl arachidonyl phosphonate (MAP), deposited in the Protein Data Bank (PDB code: 1MT5).41 The first structural refinement (addition of missing side chains and hydrogens) was done by the Biopolymer module of Sybyl, and it was followed by a visual inspection of histidines, choosing the tautomeric states that maximize the number of hydrogen bonds. The geometry of added atoms was then relaxed by energy minimization to a gradient of 0.1 kcal/(mol·Å), the MAP atoms were removed, and the hydrogen atoms of the catalytic site residues were reassigned, forming a hydrogen bond between Ser241 and Ser217, and one between Ser217 and the neutral NH2 group of Lys142.
Two families of complexes were then generated for the inhibitors, corresponding to the two possible orientations already reported for cyclohexylcarbamic acid biphenyl-3-yl esters within the catalytic site.29,43 The inhibitors were interactively docked into the enzyme channel, optimizing their position and conformation by the Dock_minimize procedure. Docking was followed by energy minimization of the complexes, allowing movements of the side chain of the residues at maximum 8 Å from the inhibitor, using MMFF94s force field to a gradient of 0.1 kcal/(mol·Å). In the first orientation (orientation A), all the docked ligands place their biphenyl fragment in the acyl binding pocket (also occupied by the acyl chain of covalently bound MAP in the 1MT5 pdb structure) and the substituent at the nitrogen atom in the cytosolic access channel. In the second one (orientation B), the meta-biphenyl fragment lies in the cytosolic access channel, whereas the carbamate nitrogen substituent occupies the acyl binding pocket. In both orientations, the carbonyl oxygen closely interacts with the NH groups of the oxyanion hole, whereas the NH of the carbamate forms a hydrogen bond with the backbone oxygen of Met191. At the end of this preliminary optimization cycle, the energy-minimized complexes were transported to the LIAISON package for subsequent SGB-LIE calculations.
(e) LIE Calculation
To compute the interaction terms required in the LIE model, a second energy minimization was performed with LIAISON, applying OPLS2001 force field,67 in combination with the Surface Generalized Born continuum model. The Truncated Newton (TN) technique was applied to an energy gradient of 0.01 kcal/(mol·Å) using a residue based cut-off distance of 10 Å for the free ligands, and to 0.05 kcal/(mol·Å) for FAAH-inhibitor complexes, with a residue based cut-off distance of 15 Å. During energy minimization of the complexes, only the side chains of the protein residues within 20 Å from the catalytic Ser241 were allowed to move. The protein backbone was maintained with fixed geometry to preserve the structure of the binding site. The energy terms reported in Equations 7 and 8 were collected by LIAISON after energy minimization and used for further MRA analysis. Their values are reported in the supporting information (Tables S2 and S3).
(f) QSAR
Multiple regression analysis (MRA) calculations were performed with an Excel (Microsoft Co., version 97) spreadsheet, employing the built-in statistical functions and automated macro procedures to determine the empirical value for regression coefficients and statistical parameters. Moriguchi Log P53 were calculated employing an external script implemented in Sybyl 7.2, whereas ACD Log P54 where calculated by using ACD/Labs suite software.68
Standard deviation of the errors in prediction (SDEP), and the relative predictivity parameter, q2, were calculated by cross-validation, omitting one compound at a time from the set, according to the leave-one-out technique.69
Supplementary Material
Acknowledgments
This work was supported by Italian MiUR (Ministero dell'Università e della Ricerca) (Grant N° 2005032713_002), Universities of Parma and Urbino “Carlo Bo”, and the National Institute of Drug Abuse (to D.P.). The S.I.T.I. (Settore Innovazione Tecnologie Informatiche) and C.I.M. (Centro Interdipartimentale Misure) of the University of Parma are gratefully acknowledged for supplying the Sybyl software license.
Footnotes
Abbreviations: FAAH: fatty acid amide hydrolase; SAR: structure-activity relationship; QSAR: quantitative structure-activity relationship; MGL: monoglyceride lipase; TGH: triacylglycerol hydrolase; TGL: triacylglycerol lipase; MAP: methyl arachidonylphosphonate; ACB: acyl chain binding; CA: cytoplasmic access; LIE: linear interaction energy; DPPA: diphenylphosphorylazide; LHMDS: lithium hexamethyldisylazane; TPP: triphenylphosphine; SGB: surface-generalized Born; MRA: multiple regression analysis; VDW: van der Waals; SDP: standard deviation of error in prediction.
Supporting Information Available: Tables of calculated Log P values, SGB-LIE components, elemental analytical data. This material is available free of charge via the Internet address http://pubs.acs.org.
References
- 1.Piomelli D. The Molecular Logic of Endocannabinoid Ligands. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 2.Labar G, Michaux C. Fatty Acid Amide Hydrolase: From Characterization to Therapeutics. Chem Biodiv. 2007;4:1882–1902. doi: 10.1002/cbdv.200790157. [DOI] [PubMed] [Google Scholar]
- 3.Fowler CJ, Jonsson KO, Tiger G. Fatty acid amide hydrolase: biochemistry, pharmacology, and therapeutic possibilities for an enzyme hydrolyzing anandamide, 2-arachidonoylglycerol, palmitoylethanolamide, and oleamide. Biochem Pharmacol. 2001;62:517–526. doi: 10.1016/s0006-2952(01)00712-2. [DOI] [PubMed] [Google Scholar]
- 4.Patricelli MP, Cravatt BF. Proteins Regulating the Biosynthesis and Inactivation of Neuromodulatory Fatty Acid Amides. Vitam Horm. 2001;62:663–674. doi: 10.1016/s0083-6729(01)62002-8. [DOI] [PubMed] [Google Scholar]
- 5.Devane WA, Hanuš L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and Structure of a Brain Constituent That Binds to the Cannabinoid Receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 6.Rodríguez de Fonseca F, Navarro M, Gómez R, Escuredo L, Nava F, Fu J, Murillo-Rodríguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. An anorexic lipid mediator regulated by feeding. Nature. 2001;414:209–212. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- 7.Fu J, Gaetani S, Oveisi F, LoVerme J, Serrano A, Rodríguez de Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 8.Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- 9.Lambert DM, Vandevoorde S, Jonsson KO, Fowler CJ. The Palmitoylethanolamide Family: A New class of Anti-Inflammatory Agents? Curr Med Chem. 2002;9:663–674. doi: 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]
- 10.McKinney MK, Cravatt BF. Evidence for Distinct Roles in Catalysis for Residues of the Serine-Serine-Lysine Catalytic Triad of Fatty Acid Amide Hydrolase. J Biol Chem. 2003;278:37393–37399. doi: 10.1074/jbc.M303922200. [DOI] [PubMed] [Google Scholar]
- 11.McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 12.Lodola A, Mor M, Hermann JC, Tarzia G, Piomelli D, Mulholland AJ. QM/MM modelling of oleamide hydrolysis in fatty acid amide hydrolase (FAAH) reveals a new mechanism of nucleophile activation. Chem Commun. 2005;35:4399–4401. doi: 10.1039/b503887a. [DOI] [PubMed] [Google Scholar]
- 13.Tubert-Brohman I, Acevedo O, Jorgensen WL. Elucidation of Hydrolysis Mechanisms for Fatty Acid Amide Hydrolase and its Lys142Ala Variant via QM/MM Simulations. J Am Chem Soc. 2006;128:16904–16913. doi: 10.1021/ja065863s. [DOI] [PubMed] [Google Scholar]
- 14.Lodola A, Mor M, Zurek J, Tarzia G, Piomelli D, Harvey JN, Mulholland AJ. Conformational Effects in Enzyme Catalysis: Reaction via a High Energy Conformation in Fatty Acid Amide Hydrolase. Biophys J. 2007;92:L20–L22. doi: 10.1529/biophysj.106.098434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lodola A, Mor M, Rivara S, Christov C, Tarzia G, Piomelli D, Mulholland AJ. Identification of productive inhibitor binding orientation in fatty acid amide hydrolase (FAAH) by QM/MM mechanistic modelling. Chem Commun. 2008;38:214–216. doi: 10.1039/b714136j. [DOI] [PubMed] [Google Scholar]
- 16.Piomelli D. The Endocannabinoid System: From Cell Biology to Therapy. In: Madras BK, editor. Cell Biology of Addiction. Cold Spring Harbor Laboratory Press; Woodbury, NY: 2006. pp. 223–237. [Google Scholar]
- 17.Cravatt BJ, Lichtman AH. Fatty acid amide hydrolase: an emerging therapeutic target in the endocannabinoid system. Curr Opin Chem Biol. 2003;7:469–475. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]
- 18.Lambert DM, Fowler CJ. The Endocannabinoid System: Drug Targets, Lead Compounds, and Potential Therapeutic Applications. J Med Chem. 2005;48:5059–5087. doi: 10.1021/jm058183t. [DOI] [PubMed] [Google Scholar]
- 19.Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Acevedo O, Guimaraes CRW, Jorgensen WL, Cravatt BF. Discovery of a Potent, Selective, and Efficacious Class of Reversible α-ketoheterocycle Inhibitors of Fatty Acid Amide Hydrolase Effective as Analgesics. J Med Chem. 2005;48:1849–1856. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romero FA, Hwang I, Boger DL. Delineation of a Fundamental α-Ketoheterocycle Substituent Effect for Use in the Design of Enzyme Inhibitors. J Am Chem Soc. 2006;128:14004–14005. doi: 10.1021/ja064522b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romero FA, Du W, Hwang I, Rayl TJ, Kimball FS, Leung D, Hoover HS, Apodaca RL, Breitenbucher JG, Cravatt BF, Boger DL. Potent and Selective α-Ketoheterocycle-Based Inhibitors of the Anandamide and Oleamide Catabolizing Enzyme, Fatty Acid Amide Hydrolase. J Med Chem. 2007;50:1058–1068. doi: 10.1021/jm0611509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardouin C, Kelso MJ, Romero FA, Rayl TJ, Leung D, Hwang I, Cravatt BF, Boger DL. Structure-Activity Relationships of α-Ketooxazole Inhibitors of Fatty Acid Amide Hydrolase. J Med Chem. 2007;50:3359–3368. doi: 10.1021/jm061414r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myllymaeki MJ, Saario SM, Kataja AO, Castillo-Melendez JA, Nevalainen T, Juvonen RO, Jaervinen T, Koskinen AMP. Design, Synthesis, and In Vitro Evaluation of Carbamate Derivatives of 2-Benzoxazolyl- and 2-Benzothiazolyl-(3-hydroxyphenyl)-methanones as Novel Fatty Acid Amide Hydrolase Inhibitors. J Med Chem. 2007;50:4236–4242. doi: 10.1021/jm070501w. [DOI] [PubMed] [Google Scholar]
- 24.Muccioli GG, Fazio N, Scriba EKG, Poppitz W, Cannata F, Poupaert HJ, Wouters J, Lambert MD. Substituted 2-Thioxoimidazolidin-4-ones and Imidazolidine-2,4-diones as Fatty Acid Amide Hydrolase Inhibitors Templates. J Med Chem. 2006;49:417–425. doi: 10.1021/jm050977k. [DOI] [PubMed] [Google Scholar]
- 25.Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Novel Mechanistic Class of Fatty Acid Amide Hydrolase Inhibitors with Remarkable Selectivity. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 26.Hertzog DL. Recent advances in the cannabinoids. Expert Opin Ther Patents. 2004;14:1435–1452. [Google Scholar]
- 27.Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. Design, Synthesis, and Structure-Activity Relationships of Alkylcarbamic Acid Aryl Esters, a New Class of Fatty Acid Amide Hydrolase Inhibitors. J Med Chem. 2003;46:2352–2360. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]
- 28.Sit SY, Conway C, Bertekap R, Xie K, Bourin C, Burris K, Deng H. Novel inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett. 2007;17:3287–3291. doi: 10.1016/j.bmcl.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. Cyclohexylcarbamic Acid 3′- or 4′-Substituted Biphenyl-3-yl Esters as Fatty Acid Amide Hydrolase Inhibitors: Synthesis, Quantitative Structure-Activity Relationships, and Molecular Modelling Studies. J Med Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 30.Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, Dasse O, Monaghan EP, Parrott JA, Putman D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597) CNS Drug Rev. 2006;12:21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan WC. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russo R, LoVerme J, La Rana G, Compton T, Parrott J, Duranti A, Tontini A, Mor M, Tarzia G, Calignano A, Piomelli D. The fatty-acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neurophatic pain after oral administration. J Pharmacol Exp Ther. 2007;322:236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- 33.Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 34.Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Goldberg SR, Cuomo V, Piomelli D. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. 2006, 103, 2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bortolato M, Mangieri RA, Fu J, Kim JH, Arguello O, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry. 2007;62:1103–1110. doi: 10.1016/j.biopsych.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 36.Bátkai S, Pacher P, Osei-Hyiaman D, Radaeva S, Liu J, Harvey-White J, Offertàler L, Mackie K, Rudd MA, Bukoski RD, Kunos G. Endocannabinoids Acting at Cannabinoid-1 Receptors Regulate Cardiovascular Function in Hypertension. Circulation. 2004;110:1996–2002. doi: 10.1161/01.CIR.0000143230.23252.D2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- 38.Mor M, Rivara S, Lodola A, Lorenzi S, Bordi F, Plazzi PV, Spadoni G, Bedini A, Duranti A, Tontini A, Tarzia G. Application of 3D-QSAR in the Rational Design of Receptor Ligands and Enzyme Inhibitors. Chem Biodiv. 2005;2:1438–1451. doi: 10.1002/cbdv.200590117. [DOI] [PubMed] [Google Scholar]
- 39.Ortar G, Cascio MG, De Petrocellis L, Morera E, Rossi F, Schiano-Moriello A, Nalli M, de Novellis V, Woodward DF, Maione S, Di Marzo V. New N-Arachidonoylserotonin Analogues with Potential “Dual” Mechanism of Action against Pain. J Med Chem. 2007;50:6554–6569. doi: 10.1021/jm070678q. [DOI] [PubMed] [Google Scholar]
- 40.Clapper JR, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. The fatty-acid amide hydrolase inhibitor URB597 does not affect triacylglycerol hydrolysis in rat tissues. Pharmacol Res. 2006;54:341–344. doi: 10.1016/j.phrs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 41.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural Adaptation in a Membrane Enzyme that Terminates Endocannabinoid Signaling. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 42.Basso E, Duranti A, Mor M, Piomelli D, Tontini A, Tarzia G, Traldi P. Tandem mass spectrometric data-FAAH inhibitory activity relationships of some carbamic acid O-aryl esters. J Mass Spectrom. 2004;39:1450–1455. doi: 10.1002/jms.729. [DOI] [PubMed] [Google Scholar]
- 43.Tarzia G, Duranti A, Gatti G, Piersanti G, Tontini A, Rivara S, Lodola A, Plazzi PV, Mor M, Kathuria S, Piomelli D. Synthesis and Structure-Activity Relationships of FAAH Inhibitors: Cyclohexylcarbamic Acid Biphenyl Esters with Chemical Modulation at the Proximal Phenyl Ring. ChemMedChem. 2006;1:130–139. doi: 10.1002/cmdc.200500017. [DOI] [PubMed] [Google Scholar]
- 44.Alexander JP, Cravatt BF. Mechanism of Carbamate Inactivation of FAAH: Implications for the Design of Covalent Inhibitors and In Vivo Functional Probes for Enzymes. Chem Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Åqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7:385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 46.Liaison, version 3.5. Schrödinger. LLC; New York, NY: 2005. [Google Scholar]
- 47.Singh P, Mhaka AM, Christensen SB, Gray JJ, Denmeade SR, Isaacs JT. Applying Linear Interaction Energy Method for Rational Design of Noncompetitive Allosteric Inhibitors of the Sarco- and Endoplasmic Reticulum Calcium-ATPase. J Med Chem. 2005;48:3005–3014. doi: 10.1021/jm049319a. [DOI] [PubMed] [Google Scholar]
- 48.Bortolato A, Moro S. In Silico Binding Free Energy Predictability by Using the Linear Interaction Energy (LIE) Method: Bromobenzimidazole CK2 Inhibitors as a Case Study. J Chem Inf Model. 2007;47:572–582. doi: 10.1021/ci600369n. [DOI] [PubMed] [Google Scholar]
- 49.Tounge BA, Reynolds CH. Calculation of the Binding Affinity of β-Secretase Inhibitors Using the Linear Interaction Energy Method. J Med Chem. 2003;46:2074–2082. doi: 10.1021/jm020513b. [DOI] [PubMed] [Google Scholar]
- 50.Stjernschantz E, Marelius J, Medina C, Jacobsson M, Vermeulen NPE, Oostenbrink C. Are Automated Molecular Dynamics Simulations and Binding Free Energy Calculations Realistic Tools in Lead Optimization? An Evaluation of the Linear Interaction Energy (LIE) Method. J Chem Inf Model. 2006;46:1972–1983. doi: 10.1021/ci0601214. [DOI] [PubMed] [Google Scholar]
- 51.Åqvist J, Marelius J. The Linear Interaction Energy Method for Predicting Ligand Binding Free Energies. Combin Chem High Throughput Screen. 2001;4:613–626. doi: 10.2174/1386207013330661. [DOI] [PubMed] [Google Scholar]
- 52.Bambal RB, Hanzlik RP. Effects of Steric Bulk and Conformational Rigidity on Fatty Acid Omega Hydroxylation by a Cytochrome P450 4A1 Fusion Protein. Arch Biochem Biophys. 1996;334:59–66. doi: 10.1006/abbi.1996.0429. [DOI] [PubMed] [Google Scholar]
- 53.Moriguchi I, Hirono S, Liu Q, Nakagome I, Matsushita Y. Simple Method of Calculating Octanol/Water Partition Coefficient. Chem Pharm Bull. 1992;40:127–130. [Google Scholar]
- 54.Petrauskas AA, Kolovanov EA. ACD/Log P method description. Perspect Drug Disc Des. 2000;19:99–116. [Google Scholar]
- 55.Zhou R, Friesner RA, Ghosh A, Rizzo RC, Jorgensen WL, Levy RM. New Linear Interaction Method for Binding Affinity Calculations Using a Continuum Solvent Model. J Phys Chem B. 2001;105:10388–10397. [Google Scholar]
- 56.Kauch M, Snieckus V, Hoppe D. Substitution of Hydroxybiaryls via Directed ortho-Lithiation of N-Silylated O-Aryl N-Isopropylcarbamates. J Org Chem. 2005;70:7149–7158. doi: 10.1021/jo0506938. [DOI] [PubMed] [Google Scholar]
- 57.Woods GF, Tucker IW. Reaction of β-cyclohexanedione (dihydroresorcinol) and its ethyl enol ether with phenylmagnesium bromide. J Am Chem Soc. 1948;70:2174–2177. doi: 10.1021/ja01186a054. [DOI] [PubMed] [Google Scholar]
- 58.Kohn GK, Ospenson JN, Moore JE. Some Structural Relationships of a Group of Simple Alkyl Phenyl N-Methylcarbamates to Anticholinesterase Activity. J Agr Food Chem. 1965;13:232–235. [Google Scholar]
- 59.King AR, Rosengarth A, Tontini A, Duranti A, Mor M, Rivara S, Clapper JR, Luecke H, Tarzia G, Piomelli D. URB602 Inhibits Monoacylglycerol Lipase and Selectively Blocks 2-Arachidonoylglycerol Degradation in Intact Brain Slices. Chem Biol. 2007;14:1357–1365. doi: 10.1016/j.chembiol.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huisgen R, Rapp W, Ugi I, Walz H, Glogger I. Medium Sized Rings. IV. Preparation and Properties of 1,2,3,4-Benzocycla-1,3-dien-5-ones. Ann. 1954;586:52–69. [Google Scholar]
- 61.Grovenstein E, Jr, Akabori S. Carbanions. XVI. Reactions of 4-Chloro-1-p-biphenylylbutane with Alkali Metals. Formation of a Spiro Anion. J Am Chem Soc. 1975;97:4620–4626. [Google Scholar]
- 62.Carlson HA, Jorgensen WL. An Extended Linear Response Method for Determining Free Energies of Hydration. J Phys Chem. 1995;99:10667–10673. [Google Scholar]
- 63.Ghosh A, Sendrovic Rapp C, Friesner RA. Generalized Born Model Based on a Surface Integral Formulation. J Phys Chem B. 1998;102:10983–10990. [Google Scholar]
- 64.Jones-Hertzog DK, Jorgensen WL. Binding Affinities for Sulfonamide Inhibitors with Human Thrombin Using Monte Carlo Simulations with Linear Response Method. J Med Chem. 1997;40:1539–1549. doi: 10.1021/jm960684e. [DOI] [PubMed] [Google Scholar]
- 65.Sybyl version 7.2. Tripos Inc; 1699 South Hanley Rd., St. Louis, MO, 63144: [Google Scholar]
- 66.Halgren TA. Merck Molecular Force Field. I. Basis, Form, Scope, Parametrization, and Performance of MMFF94*. J Comput Chem. 1996;17:490–519. [Google Scholar]
- 67.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 68.ACD/Log P, version 8.00. Advanced Chemistry Development, Inc; Toronto ON, Canada: 2004. www.acdlabs.com. [Google Scholar]
- 69.Wold S, Eriksson L. Statistical Validation of QSAR Results. In: van de Waterbeemd H, editor. Chemometric Methods in Molecular Design. VCH Publishers Inc; New York: 1995. pp. 309–318. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.