

Abstract
Drug-like molecules with activity against Trypanosoma brucei are urgently required as potential therapeutics for the treatment of African sleeping sickness. Starting from known inhibitors of other glycosyltransferases, we have developed the first small molecular inhibitors of dolicholphosphate mannose synthase (DPMS), a mannosyltransferase critically involved in glycoconjugate biosynthesis in T. brucei. We show that these DPMS inhibitors prevent the biosynthesis of glycosylphosphatidylinositol (GPI) anchors, and possess trypanocidal activity against live trypanosomes.
Keywords: African sleeping sickness, Trypanosoma, Dolicholphosphate mannose synthase, Enzyme inhibitors
African sleeping sickness, also known as Human African Trypanosomiasis (HAT), is an infectious disease caused by the protozoan parasite Trypanosoma brucei (T. brucei). The parasite is transmitted by the bite of an infected tsetse fly, multiplies within the bloodstream of the mammalian host, and eventually invades the central nervous system. If untreated, HAT is invariably lethal. Despite becoming close to eradication by the late 1960s,1 the disease has made a dramatic re-emergence within the last half century, with prevalence in parts of Africa now as high as they were in the 1920s. In 2002 alone, around 48,000 deaths were reported.2 The WHO presently records around 17,500 new cases per year,3 with a cumulative rate of 50,000–70,000 cases3 and potentially over 60 million people at risk.2
The situation is further exacerbated by the growing resistance to established drug treatments for HAT, which have long been considered unsatisfactory.2 Serious side effects kill between 4% and 10% of patients who receive the arsenical melarsoprol,4 introduced in 1949, and the drug fails to cure between 10% and 30% of patients.5 The only relatively modern anti-HAT drug, the ornithine decarboxylase inhibitor eflornithine, is expensive, difficult to administer, and only effective against T. brucei gambiense.6 Thus, HAT has been described as one of the most neglected diseases of mankind,7 and novel therapeutic approaches to HAT are urgently needed.2
The T. brucei parasite is covered in a dense cell-surface coat of ~5 million variant surface glycoprotein (VSG) dimers8,9 which acts as a physical diffusion barrier for components of the innate immune system.10 The VSG coat undergoes constant antigenic variation,11,12 as the parasite switches between ~1000 immunologically distinct VSG genes,13 thus staying one step ahead of the host immune system. All VSG variants are linked to the trypanosomal plasma membrane via glycosylphosphatidylinositol (GPI) anchors (Fig. 1). Genetic14-16 and chemical17,18 studies show that GPI anchor biosynthesis is essential for viability of the bloodstream form of T. brucei, thus validating it as a drug target against HAT.19 Small molecular inhibitors of enzymes involved in GPI anchor biosynthesis therefore hold great promise as novel anti-trypanosomal agents.
Figure 1.

The central role of DPMS for the biosynthesis of GPI anchors in T. brucei. VSG: variant surface glycoprotein; GPI: glycosylphosphatidyl inositol; GDP-Man: guanosine diphosphate mannose; Dol-P-Man: dolicholphosphate mannose; DPMS: dolicholphosphate mannose synthase.
GPI anchor biosynthesis in T. brucei involves three mannosyltransferases (MTs) on the luminal face of the endoplasmic reticulum, which catalyze the assembly of the trimannoside core structure (Fig. 1).20,21 All three of these MTs use dolicholphosphate mannose (Dol-P-Man) as their donor substrate, whose biosynthesis from dolicholphosphate (Dol-P) and the sugar-nucleotide GDP-mannose (GDP-Man) is catalyzed by another mannosyltransferase, dolicholphosphate mannose synthase (DPMS). All T. brucei VSG variants also contain at least one N-glycan, which requires another four Dol-P-Man-dependant mannosyltransferases for the formation of its lipid-linked oligosaccharide precursor.22-24 Consequently, T. brucei is doubly dependant upon Dol-P-Man for the synthesis of mature, N-glycosylated and GPI-anchored VSGs, and this double dependency makes DPMS an excellent target for inhibition of VSG biosynthesis. Recently, T. brucei DPMS has also been validated genetically as a drug target.25
Despite its promise as a therapeutic target, no inhibitors for T. brucei DPMS have been reported to date. The rational design of such inhibitors is complicated by the absence of a crystal structure for T. brucei DPMS at present. In search of a suitable lead structure for the development of DPMS inhibitors, we noticed striking structural similarities among small molecular inhibitors for other glycosyltransferases26-28 and sugar-nucleotide-dependent glycoprocessing enzymes.29-31 Several such inhibitors contain a rhodanine (2-thioxothiazolidin-4-one) scaffold, and derivatives of rhodanine-3-acetic acid 1 (Scheme 1) have been reported as inhibitors of the E. coli glycosyltransferase MurG26,27 and the C. albicans protein mannosyltransferase 1 (PMT1).28 It has been suggested that the thiazolidinone ring can act as a mimic of the pyrophosphate group,26,29-31 and that this mimicry may explain the inhibitory activity of thiazolidinone derivatives towards sugar-nucleotide-dependent enzymes. As DPMS is dependent on the sugar-nucleotide donor GDP-mannose, we reasoned that the thiazolidinone scaffold may also represent a good starting point for the development of DPMS inhibitors.
Scheme 1.
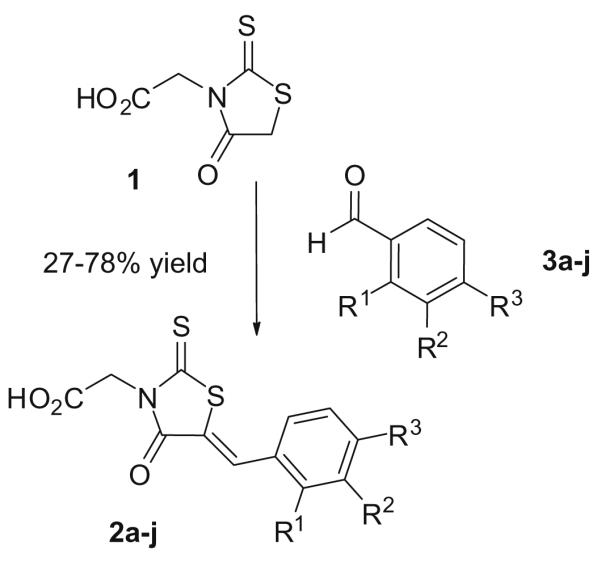
Synthesis of the target 5-benzylidene rhodanine-3-acetic acid derivatives 2a–j. Reagents and conditions: NH4OAc, DMF, 80 °C, 3 h (2a) or EtOH, piperidine, 80 °C, 3–6 h (2b–j). For substituents R1–R3 see Table 1.
Herein, we describe the successful application of this strategy. We have prepared a small library of 5-benzylidene rhodanine-3-acetic acid analogs of the general structure 2, and report herein their inhibitory activity against T. brucei DPMS and GPI anchor biosynthesis as well as their trypanocidal activity against live trypanosomes.
The target rhodanine-3-acetic acid derivatives 2a–j (Scheme 1,Table 1) were prepared by Knoevenagel condensation of rhodanine-3-acetic acid 1 and substituted benzaldehydes 3a–j. To simplify the preparation and isolation of the target compounds, we explored different solvents and catalytic bases for this reaction, including DMF/sodium acetate, toluene/piperidine and ethanol/piperidine.28,32 In our hands, the ethanol/piperidine system was the most practical one, with short reaction times and straightforward product isolation. Under these conditions, all 5-benzylidene rhodanine-3-acetic acid derivatives precipitated from the ethanolic solution upon cooling to room temperature, and could be collected by simple filtration.33 Thus, all target compounds (Table 1) were obtained as yellow or yellow-orange solids in generally good yields. Remarkably, this procedure was also applicable to benzaldehydes containing a free phenolic hydroxyl group (e.g., 2f, 2g). This was particularly important as all attempts to prepare these analogs by debenzylation of the corresponding benzyloxy derivatives (e.g., 2b, 2c) had failed.
Table 1.
Biological activity of thiazolidinones 2a-j (Scheme 1)
| Compound | R1 | R2 | R3 | Residual DPMS activity (%)a |
Trypanocidal activity (ED50)b |
|---|---|---|---|---|---|
| 2a | H | H | H | 42 (±5) | 232 (±12) |
| 2b | H | H | BnO | 10 (±2) | 338 (±31) |
| 2c | H | BnO | H | 23 (±3) | 96 (±5) |
| 2d | H | BnO | BnO | 20 (±4) | 492 (±24) |
| 2e | OH | H | H | 23 (±8) | 107 (±12) |
| 2f | H | H | OH | 70 (±4) | 427 (±19) |
| 2g | H | OH | H | 90 (±5) | 345 (±23) |
| 2h | H | H | CN | 73 (±6) | 244 (±17) |
| 2i | H | H | Cl | 86 (±5) | 398 (±19) |
| 2j | H | H | CCH | 94 (±7) | >1000 |
Residual DPMS activity in the presence of 1 mM inhibitor, relative to control experiments without inhibitor (activity = 100 ± 3%).
All ED50 values are in μM against cultured bloodstream T. brucei. All experiments were carried out at least in duplicate.
Fortuitously, our synthetic protocol provided benzylidene thiazolidinone 2b in crystalline form which allowed the unambiguous determination of its three-dimensional structure (Fig. 2).34 This is of particular interest, as in previous reports of structurally related thiazolidinones as glycosyltransferase inhibitors there had been ambiguity about the configuration of the exocyclic double bond, despite its obvious importance for structure–activity relationships.26,27 In our structure, the rhodanine acetic acid molecule 2b is essentially planar, with only the carboxy group out of plane and coordinating to one molecule of ethanol. The exocyclic double bond exists in the Z configuration, which for arylidene rhodanines has been reported as the thermodynamically stable configuration.35,36
Figure 2.

The crystal structure of the representative 5-benzylidene thiazolidinone 2b, in complex with one molecule of ethanol, shows the exocyclic double bond (C2=C20) in the Z configuration.
In an initial biological screen, all target compounds were tested for inhibition of recombinant T. brucei DPMS in E. coli membranes (Table 1)37, At 1 mM, several thiazolidinone derivatives significantly inhibited T. brucei DPMS. A large benzyloxy substituent in position R2 and/or R3 appears to be advantageous for DPMS inhibition (2b–d), while a small polar substituent is less well tolerated in these positions (2f and 2g), as are rigid R3 substituents (e.g., nitrile 2h, acetylene 2j). Intriguingly, however, a polar substituent is beneficial for inhibitory activity when placed at the 2-position (R1), and the 2-hydroxy regioisomer 2e is among the most potent DPMS inhibitors in this series.
Next, compounds 2a–j were tested for their trypanocidal activity against cultured bloodstream form T. brucei. 38 Importantly, the in vitro activity of several thiazolidinones against DPMS did translate into in vivo trypanocidal activity against live trypanosomes. In particular, the 3-benzyloxy-substituted analog 2c and the 2-hydroxy regioisomer 2e showed trypanocidal activity with ED50 values in the medium micromolar range. However, only moderate activity was observed for some of the other potent DPMS inhibitors, notably the benzyloxy-substituted analogs 2b and 2d. The limited cellular activity of these DPMS inhibitors may be due to various factors, including limited cell penetration, which are currently being investigated.
In order to assess the parasite/host selectivity of these thiazolidinones, analogs 2a–f and 2j were tested at two concentrations (0.1 and 1.0 mM final) for their cytotoxicity against HeLa cells.38 Pleasingly, none of these thiazolidinones showed any cytotoxic effect, with the exception of 2b and 2j (20% inhibition at 1.0 mM, compared to DMSO-only control), suggesting low mammalian cytotoxicity (ED50 > 1.0 mM) and a promising margin of selectivity for T. brucei.
In order to gain further insight into the mode of action of these novel DPMS inhibitors, we investigated the effect of the thiazolidinones on GPI anchor biosynthesis in a T. brucei cell-free-system (Fig. 3).39 This assay monitors the DPMS-catalyzed formation of Dol-P-Man (lane 1) as well as the downstream formation of mannosylated GPI intermediates (lane 2). As expected, the potent DPMS inhibitor 2d abolished the formation of Dol-P-Man almost completely, and significantly reduced the formation of downstream GPI intermediates. A similar effect was observed for the analog 2f (Fig. 3).
Figure 3.

Differential effects of 5-benzylidene thiazolidinones 2a–j on DPMS and GPI anchor biosynthesis (M1: Manα1-4GlcNH2-PI; M2: Manα1-6Manα1-4GlcNH2-PI; M3: Manα1-2Manα1-6Manα1-4GlcNH2-PI; aM3: Manα1-2Manα1-6Manα1-4GlcNH2-(acyl)PI; A′: ethanolamine-P-Manα1-2Manα1-6Manα1-4GlcNH2-PI).
Interestingly, thiazolidinones 2b and 2e did not affect Dol-P-Man production in the cell-free system, but did potently inhibit the formation of mannosylated GPI intermediates. Thus, the strong inhibitory effect of 2b and 2e on GPI anchor biosynthesis in the cell-free system may be due to inhibition of one or more GPI biosynthetic enzymes early in the pathway, for example, the GlcNAc transferase, de-N-acetylase or the first mannosyltransferase (MT1). Importantly, all of these enzymes represent therapeutic targets in their own right, and their inhibition may contribute to the trypanocidal activity of 2b and 2e.
In summary, we have identified the first small molecular inhibitors of trypanosomal DPMS. We demonstrate that DPMS inhibitors compromise trypanosomal GPI anchor biosynthesis in a cell-free system, and that inhibition of DPMS is a promising strategy for the development of anti-trypanosomal agents. Work on the optimization of these initial lead compounds with regard to their DPMS inhibitory potency as well as cellular activity is ongoing. Thiazolidinones 2b, 2d and 2e in particular are promising candidates for further development because of their respective activities against trypanosomal DPMS and GPI anchor biosynthesis.
Acknowledgments
We thank the EPSRC National Mass Spectrometry Service, Swansea, and Mr. Thomas Pesnot (UEA) for the recording of spectra, and the 2008 MPharm mini-project students in the Wagner group for technical assistance. T.K.S. is supported by a Wellcome Trust Senior Research Fellowship (067441).
References and notes
- 1.Delespaux V, de Koning HP. Drug Resist. Update. 2007;10:30. doi: 10.1016/j.drup.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Pink R, Hudson A, Annick-Mouries M, Bendig M. Nat. Rev. Drug Discov. 2005;4:727. doi: 10.1038/nrd1824. [DOI] [PubMed] [Google Scholar]
- 3.The World Health Organisation . African trypanosomiasis. WHO publications; Geneva: Fact sheet No. 259 (revised August 2006) [Google Scholar]
- 4.Pepin J, Guern C, Ethier L, Milord F, Mpia B, Mansinsa D. Lancet. 1989;1:1246. doi: 10.1016/s0140-6736(89)92340-4. [DOI] [PubMed] [Google Scholar]
- 5.Burri C, Keiser J. Trop. Med. Int. Health. 2001;6:412. doi: 10.1046/j.1365-3156.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- 6.Burri C, Brun R. Parasitol. Res. 2003;90:S49. doi: 10.1007/s00436-002-0766-5. [DOI] [PubMed] [Google Scholar]
- 7.Stich A, Barrett MP, Krishna S. Trends Parasitol. 2003;19:195. doi: 10.1016/s1471-4922(03)00055-2. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson MAJ, Cross GA. J. Biol. Chem. 1984;259:3011. [PubMed] [Google Scholar]
- 9.Ferguson MAJ, Low MG, Cross GA. J. Biol. Chem. 1985;260:14547. [PubMed] [Google Scholar]
- 10.Ferguson MAJ. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10673. [Google Scholar]
- 11.Cross GAM. BioEssays. 1996;18:283. doi: 10.1002/bies.950180406. [DOI] [PubMed] [Google Scholar]
- 12.Aitcheson N, Talbot S, Shapiro J, Hughes K, Adkin C, Butt T, Sheader K, Rudenko G. Mol. Microbiol. 2005;57:1608. doi: 10.1111/j.1365-2958.2005.04795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blum ML, Down JA, Gurnett AM, Carrington M, Turner MJ, Wiley DC. Nature. 1993;362:603. doi: 10.1038/362603a0. [DOI] [PubMed] [Google Scholar]
- 14.Chang T, Milne KG, Guther ML, Smith TK, Ferguson MAJ. J. Biol. Chem. 2002;277:50176. doi: 10.1074/jbc.M208374200. [DOI] [PubMed] [Google Scholar]
- 15.Nagamune K, Nozaki T, Maeda Y, Ohishi K, Fukuma T, Hara T, Schwarz RT, Sutterlin C, Brun R, Riezman H, Kinoshita T. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10336. doi: 10.1073/pnas.180230697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lillico S, Field MC, Blundell P, Coombs GH, Mottram JC. Mol. Biol. Cell. 2003;14:1182. doi: 10.1091/mbc.E02-03-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TK, Crossman A, Brimacombe JS, Ferguson MAJ. EMBO J. 2004;23:4701. doi: 10.1038/sj.emboj.7600456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crossman A, Smith TK, Ferguson MAJ, Brimacombe JS. Tetrahedron Lett. 2005;46:7419. [Google Scholar]
- 19.Ferguson MAJ, Brimacombe JS, Brown JR, Crossman A, Dix A, Field RA, Guther MLS, Milne KG, Sharma DK, Smith TK. Biochim. Biophys. Acta. 1999;1455:327. doi: 10.1016/s0925-4439(99)00058-7. [DOI] [PubMed] [Google Scholar]
- 20.Menon AK, Mayor S, Schwarz RT. EMBO J. 1990;9:4249. doi: 10.1002/j.1460-2075.1990.tb07873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarz RT, Mayor S, Menon AK, Cross GAM. Biochem. Soc. Trans. 1988;17:746. [Google Scholar]
- 22.Helenius A, Aebi M. Annu. Rev. Biochem. 2004;73:1019. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 23.Orlean P. Biochem. Cell Biol. 1992;70:438. doi: 10.1139/o92-067. [DOI] [PubMed] [Google Scholar]
- 24.Schutzbach JS. Glycoconjugate J. 1997;14:175. doi: 10.1023/a:1018533620002. [DOI] [PubMed] [Google Scholar]
- 25.Denton, H.; Fyffe, S. A.; Smith, T. K., submitted for publication.
- 26.Helm JS, Hu Y, Chen L, Gross B, Walker SJ. Am. Chem. Soc. 2003;125:11168. doi: 10.1021/ja036494s. [DOI] [PubMed] [Google Scholar]
- 27.Hu Y, Helm JS, Chen L, Ginsberg C, Gross B, Kraybill B, Tiyanont K, Fang X, Wu T, Walker S. Chem. Biol. 2004;11:703. doi: 10.1016/j.chembiol.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 28.Orchard MG, Neuss JC, Galley CMS, Carr A, Porter DW, Smith P, Scopes DIC, Haydon D, Vousden K, Stubberfield CR, Young K, Page M. Bioorg. Med. Chem. Lett. 2004;14:3975. doi: 10.1016/j.bmcl.2004.05.050. [DOI] [PubMed] [Google Scholar]
- 29.Soltero-Higgin M, Carlson EE, Phillips JH, Kiessling LL. J. Am. Chem. Soc. 2004;126:10532. doi: 10.1021/ja048017v. [DOI] [PubMed] [Google Scholar]
- 30.Carlson EE, May JF, Kiessling LL. Chem. Biol. 2006;13:825. doi: 10.1016/j.chembiol.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 31.Andres CJ, Bronson JJ, D’Andrea SV, Deshpande MS, Falk PJ, Grant-Young KA, Harte W, Ho H-T, Misco PF, Robertson JG, Stock D, Sun Y, Walsh AW. Bioorg. Med. Chem. Lett. 2000;10:715. doi: 10.1016/s0960-894x(00)00073-1. [DOI] [PubMed] [Google Scholar]
- 32.Irvine MW, Patrick GL, Kewney J, Hastings SF, MacKenzie SJ. Bioorg. Med. Chem. Lett. 2008;18:2032. doi: 10.1016/j.bmcl.2008.01.117. [DOI] [PubMed] [Google Scholar]
- 33.Representative procedure for 2b (all thiazolidinones 2a–j were characterized by NMR and HRMS): A mixture of rhodanine-3-acetic acid 1 (0.51 g, 2.61 mmol), 4-benzyloxybenzaldehyde 3b (0.60 g, 2.83 mmol) and a drop of piperidine in absolute ethanol (25 ml) was heated to reflux for 2 h. Upon cooling of the reaction to room temperature, a precipitate formed which was collected by filtration, washed with cold ethanol, and dried under vacuum to provide 0.78 g (76%) of 2b as yellow crystals. Melting point: 202–205°. δH (400 MHz, d6-DMSO) 4.72 ppm (s, 3H), 5.21 (s, 3H), 7.20–7.22 (m, 2H), 7.34–7.50 (m, 5H), 7.64–7.66 (m, 2H), 7.86 (s, 1H). δC (75.5 MHz, d6-DMSO) 45.0, 69.7, 116.1, 118.8, 125.7, 128.0, 128.3, 128.7, 133.3, 134.3, 136.6, 161.1, 166.7, 167.6, 193.4. m/z (ESI) 384.0370 [M–H]−, C10H14O4NS2 requires 384.0365.
- 34.The X-ray coordinates for 2b have been deposited at the Cambridge Crystallographic Data Centre, deposition number CCDC 715650.
- 35.Ishida T, In Y, Inoue M, Ueno Y, Tanaka C. Tetrahedron Lett. 1989;30:959. [Google Scholar]
- 36.Ohishi Y, Mukai T, Nagahara M, Yajima M, Kajikawa N, Miyahara K, Takano T. Chem. Pharm. Bull. 1990;38:1911. doi: 10.1248/cpb.38.1911. [DOI] [PubMed] [Google Scholar]
- 37.Full experimental details will be published elsewhere. Briefly, E. coli membranes containing recombinantly expressed, full-length (membrane-bound) T. brucei DPMS (70 ng/ml total protein) were added to a reaction mix (60 ml) of Na–Hepes (50 mM, pH 7.4), KCl (25 mM), MgCl2 (5 mM), MnCl2 (5 mM), dehydrofarnesol-phosphate (1 mg), GDP-[3H]Man (0.25 mCi/1 mM), and inhibitor (1 mM), all in the presence of 0.1% n-octyl-glucopyranoside. The reaction was incubated at 30 °C for 10 min, quenched, and lipids extracted by the addition of CHCl3:MeOH (1:1). The resulting supernatant was dried and partitioned between butan-1-ol and water, a portion butan-1-ol was counted as a measure of dehydrofarnesol-phosphate-[3H]Man product.
- 38.The trypanocidal activity of all inhibitors against cultured bloodstream T. brucei (strain 427) and their cytotoxicity against HeLa cells was determined using the Alamar Blue™ viability test as described in Mikus J, Steverding D. Parasitol. Int. 2000;48:265. doi: 10.1016/s1383-5769(99)00020-3.
- 39.Cell-free system assay of GPI biosynthesis in brief: Membranes of bloodstream T. brucei (variant 117) were prepared as described previously (Smith TK, et al. EMBO J. 1999;18:5922. doi: 10.1093/emboj/18.21.5922. snap frozen in liquid nitrogen and stored at −80 °C until needed. Per assay, 1 × 107 cell equivalents were used. Membranes were washed twice in 10 ml of wash buffer (50 mM Na–Hepes, pH 7.4, 25 mM KCl, 5 mM MgCl2, 100 mM tosyl-l-lysine chloromethyl ketone (TLCK), and 1 μg/ml leupeptin), resuspended by sonication in 20 ml of 2× incorporation buffer (2 times concentrated wash buffer containing 10 mM MnCl2, 1 mg/ml tunicamycin and 1 mM dithiothreitol) and added to a reaction tube containing an equal volume of GDP-[3H]Man (0.3 mCi per 107 cell equivalents), 1 mM UDP-GlcNAc and thiazolidinone inhibitor candidate (in DMSO, final concentration 1 mM). The cell-free system was then sonicated briefly, incubated at 30 °C for 1 h, and the reaction stopped by the addition of 267 ml of CHCl3:MeOH (1:1). Glycolipid products were recovered by extraction into CHCl3:MeOH:H2O (10:10:3), evaporated to dryness, partitioned between butan-1-ol and water, and analyzed by HPTLC (CHCl3:MeOH:NH4OAc (1 M):conc. NH3:H2O 180:140:9:9:23). Dried HPTLC plates were sprayed with En3Hance™, and radiolabeled components were visualized by fluorography at −80 °C using Kodak Biomax MS films with an intensifying screen.