

Background: The bacterial adhesin FimH is allosterically regulated.
Results: Mutations designed to control the allosteric state of the protein created low or high affinity variants as predicted.
Conclusion: Three regulatory regions are strongly coupled together, whereas the active site is more weakly coupled to those regions.
Significance: Allosteric regulation can be used to develop antiadhesive therapies for bacterial infections.
Keywords: Allosteric Regulation, Bacterial Adhesion, Carbohydrate-binding Protein, Protein Conformation, Protein Design, Catch Bonds
Abstract
The protein FimH is expressed by the majority of commensal and uropathogenic strains of Escherichia coli on the tips of type 1 fimbriae and mediates adhesion via a catch bond to its ligand mannose. Crystal structures of FimH show an allosteric conformational change, but it remains unclear whether all of the observed structural differences are part of the allosteric mechanism. Here we use the protein structural analysis tool RosettaDesign combined with human insight to identify and synthesize 10 mutations in four regions that we predicted would stabilize one of the conformations of that region. The function of each variant was characterized by measuring binding to the ligand mannose, whereas the allosteric state was determined using a conformation-specific monoclonal antibody. These studies demonstrated that each region investigated was indeed part of the FimH allosteric mechanism. However, the studies strongly suggested that some regions were more tightly coupled to mannose binding and others to antibody binding. In addition, we identified many FimH variants that appear locked in the low affinity state. Knowledge of regulatory sites outside the active and effector sites as well as the ability to make FimH variants locked in the low affinity state may be crucial to the future development of novel antiadhesive and antimicrobial therapies using allosteric regulation to inhibit FimH.
Introduction
The most common bacteria associated with urinary tract infections are uropathogenic strains of Escherichia coli (1). Antimicrobial drugs used to treat urinary tract infections are becoming increasingly less effective due to an increase in drug resistant E. coli (2–4). Recently, research efforts have been focused on preventing bacterial adhesion and, therefore, colonization in the urinary tract through the use of antiadhesive therapies. The protein FimH, expressed by the majority of commensal and uropathogenic strains of E. coli on the tips of type 1 fimbriae, mediates adhesion and forms receptor-ligand bonds with terminal mannosyl residues on the surface of uroepithelial cells, intestinal epithelial cells, red blood cells, neutrophils, and yeast (5). Current antiadhesive therapies targeted at FimH include ligand-like inhibitors or vaccines. For the former, the complexity of both the carbohydrate environment in vivo and mechanics of bacterial adhesion has posed concerns for developing a successful competitive inhibitor (6, 7). Several studies using FimH to immunize various animal models have shown protection against an E. coli infection, making FimH a major target in the development of vaccine against urinary tract infections. Nevertheless there still exists no Food and Drug Administration approved vaccine on the market for humans (8, 9). These observations suggest a need to understand the mechanism of FimH adhesion and how it is regulated to guide development of an effective therapy.
FimH has two domains: a lectin or mannose binding domain and a pilin domain that anchors FimH to the fimbriae. Whereas most receptor-ligand interactions dissociate under force or high flow conditions, FimH increases association under increasing tensile mechanical force. This phenomenon is known as a “catch bond.” Mechanical activation of FimH has been demonstrated to result when tensile mechanical force switches FimH from a state with low affinity for mannose to one with high (10) (see Fig. 1). This switch occurs because the pilin domain is an allosteric autoinhibitor of the lectin domain until it is pulled away by mechanical force. Tchesnokova et al. (32) points out that although antibody therapy development is an alternative to antibiotic treatment of bacterial infections, the antibodies raised against FimH stabilized the high affinity conformation of the adhesin and actually enhanced bacterial adhesion to uroepithelial cells. An alternative strategy for preventing bacterial adhesion is thus to develop allosteric inhibitors or antibodies that stabilize the low affinity state. To our knowledge, an allosteric antiadhesive that targets the low affinity state of FimH has never been reported. Characterization of the low affinity conformation may provide the means to develop a successful allosteric inhibitor or antibody.
FIGURE 1.
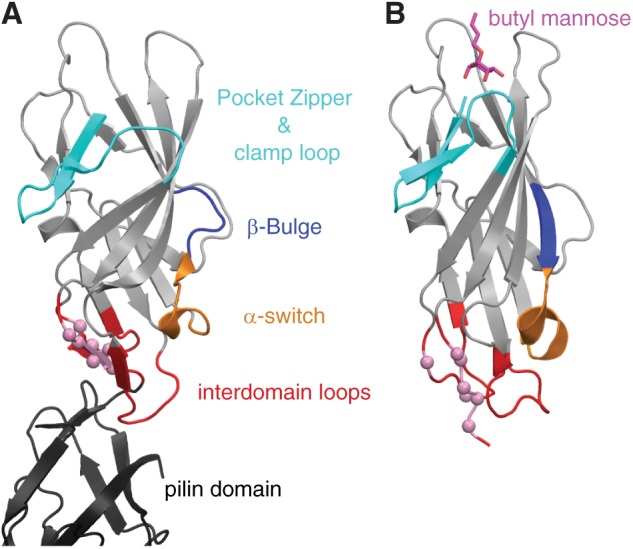
Crystal structures of the low affinity (PDB ID 3jwn) (A) and high affinity (PDB ID 1uwf) (B) lectin domains. Regions involved in the allosteric pathway are colored and labeled. Pink-colored spheres in the interdomain loops indicate the Cα atoms of the mAb21 epitope.
To design an antiadhesive therapy that targets the low affinity state of FimH, it is necessary to understand the structural basis of the allosteric switch between the low and high affinity states. FimH undergoes a “β-sheet twisting” mechanism where the β-sheet is twisted more in the low affinity than in the high affinity structure (10). FimH is still the only example in which an allosteric conformational change propagates across a β-sheet, so the mechanism of this propagation remains poorly understood. The three loops of the interdomain region, which directly contacts the effector (the pilin domain), all dramatically changed structure (Fig. 1), and at least some of these changes were shown to be due to the interaction with the pilin domain (10). On the other end of the β-sheets, the active site also changed between the two conformations. The mannose binding pocket in the low affinity structure was >3 Å wider and had lost three stabilizing backbone hydrogen bonds, making it looser than in the high affinity state. This change was caused by the presence of the pilin domain rather than by the absence of mannose in the structure (10). Thus, the existence of a thermodynamic connection between the effector and active sites has been demonstrated.
However, three additional regions also undergo conformational changes in the existing crystal structures but do not contact the effector or ligand. First, a short segment that is part of the β5-strand in the high affinity crystal structure bulges out into a loop region in the low affinity structure, which is referred to as the β-bulge. Second, a single-turn α-helix in the high affinity structure becomes a 310-helix in the low affinity structure, which is referred to as the α-switch. Third, the “pocket zipper” beneath the mannose binding pocket changes conformation with the breaking of hydrogen bonds. These regions are not part of the sites that contact either the effector pilin domain or the ligand mannose, nor do they form a complete pathway between these two sites. It remains unclear whether these structural differences are also associated with the allosteric conformational change, as they could also be caused by differences in primary structure, as residues 27, 70, and 78 differ between the two crystallized proteins and are in or nearby the β-bulge and α-switch, or to different crystallization conditions, as the low affinity structure was crystallized in 3 m salt, whereas the high affinity structures were not. Moreover, some changes in the mannose binding pocket are ligand-induced (11), whereas others are associated with the allosteric switch (10), so it cannot be assumed that all conformational changes in or near the pocket are coupled to the allosteric mechanism. To obtain a better understanding of how conformational changes can propagate across a β-sheet, we need to determine whether or not these regions are part to the allosteric mechanism.
Furthermore, although kinetic data indicate the presence of two distinct bound conformations of the FimH-mannose bond (10, 12), the structure we refer to as “low affinity” was not crystallized with mannose and may not be competent of binding mannose at all. This means that the structure of the lower affinity-bound conformation remains unknown. A better understanding of which regions are involved in the allosteric mechanism might develop testable hypotheses about the low affinity-bound structure.
In this study we used visual inspection and the protein structural analysis tool RosettaDesign (13) to identify at least one mutation in each of four regions of the lectin domain that we predicted would stabilize the low or high affinity state if the conformation of this region were connected to the allosteric switch. The mutations were introduced using site-directed mutagenesis to create 10 new variants that would together test whether each region was involved in allosteric control. The binding function of each variant was characterized by measuring binding to mannosyl and oligomannosyl surfaces under static binding conditions. An enzyme-linked immunosorbent assay (ELISA) was used to elucidate each variant's conformation. These studies elucidate which regions are involved in the allosteric mechanism of FimH. The resulting low affinity variants provide tools for future fundamental FimH biophysics studies and establish a structural map of regulatory hotspots that can serve as targets for allosteric inhibitors or as vaccines.
EXPERIMENTAL PROCEDURES
Software
Images of protein structures were generated using PyMOL (14). The RosettaDesign source code is available without charge for academic users from RosettaCommons. The MODIP server may be reached free of charge at DSDBASE. The programs CHARMM (15) and NAMD (16) were used to set up and run the MD simulations.
RosettaDesign
Starting coordinates for the lectin domain structure were taken from the Protein Data Bank (PDB) (17) (PDB IDs 1uwf (chain A) and 3jwn (chain H) for the high and low affinity states, respectively). The crystal structures of the lectin domain have been solved with proteins from slightly different genetic backgrounds: the high and low affinity state from the K12 and KB91 background, respectively (3 amino acid changes at 27, 70, and 78). Models were created for both parent strain backgrounds. Computational mutations were made by eliminating side chain atoms beyond the Cβ atom and allowing Rosetta to rebuild the side chains according to ideal bond lengths and angles. Finally, the side chain positions of the substitutions were optimized through repacking and rotamer trials (18), and the entire structure (all atoms) was minimized by optimizing the energy of the backbone and side chains.
After repacking and minimization of the crystal structure, each of the 20 naturally occurring amino acids was substituted and scored at each sequence position in the regions of the pocket zipper (Phe-1–Ile-11), clamp loop (Gly-8–Gly-16), β-bulge (Gln-59–Ser-63), α-switch (Tyr-64–Phe-71), and interdomain loops (Ala-25–Leu-34, Pro-111–Ala-119, Asn-152–Thr-158). Side chain conformations were selected from the backbone-dependent rotamer library (19) generated from statistics of side chain conformers observed in structures found in the PDB. The backbone coordinates remained fixed, whereas the side chain identity (and position) was varied. Only one position was varied at a time, and each substitution was optimized for the local environment (all residues within 8 Å of the Cβ atom (Cα atom for glycine) of the original change were repacked). Each computational substitution was scored using the Rosetta all-atom energy function, which consists of a linear combination of the following terms: (i) Lennard-Jones attractive and repulsive, (ii) hydrogen bonding (20), (iii) solvation (21), (iv) statistical term (“pair”) that approximates electrostatics and disulfide bonds, (v) statistical term (“rot”) that estimates the backbone-dependent internal free energies of the rotamers, (vi) reference values for each amino acid that estimates the free energy of the denatured state. The energy difference from a substitution (ΔG = Gmutant − Gwild type) was computed for each state, and the difference between states was determined (ΔΔG = ΔGlow affinity − ΔGhigh affinity). Because the goal was to find mutations that stabilize the low affinity conformation relative to the high affinity state, ΔΔG was arranged so negative values reflect a bias toward the low affinity state. ΔΔG score units between −5 and +8 correlate to experimentally measured ΔΔG values with 1 score unit ≈ 1.75 kcal/mol (22). Score units are thus multiplied by 1.75 to provide predictions in kcal/mol. However, large magnitude scores usually arise from atomic clashes that are not resolved in the available computational sampling, so these are referred to as “<−5 kcal/mol” or “>+5 kcal/mol.” Mutations predicted by Rosetta to favor the low versus the high affinity structure were tested experimentally. Other mutations were chosen based on structural considerations described under “Results.”
MODIP Design
Two mutants containing double cysteines required a different technique in the design process. Briefly, MODIP evaluates a protein's geometry to identify residue pairs that could form disulfide bonds without significantly altering the crystal structure if mutated to cysteines (23, 24). Based on stereochemical parameters, each attempted residue pairing is assigned a grade from A through D with A being the best.
Mutagenesis
The recombinant parent strain KB18 was constructed and described previously (5, 25–27). Briefly, the fim null E. coli K12 derivative AAEC191A (donated by Dr. Ian Blomfield, University of Kent, UK) was transformed with the recombinant plasmid pPKL114 (donated by Dr. Per Klemm, Danish Technology University, Copenhagen, Denmark) to create KB18. Derived from the pBR322 plasmid, the pPKL114 plasmid contains the entire K12 fim gene cluster and a translational stop-linker inserted into the KpnI site of the fimH gene.
Site-directed mutagenesis followed Tchesnokova et al. (25). Briefly, primers were designed manually and ordered from Eurofins MWG Operon (Huntsville, AL). The K12 FimH-allele-containing plasmid, pGB2–24, was provided with one or two point mutations within the sequence encoding the FimH lectin domain (residues 1–160) via a QuikChange Stratagene kit. Subsequent DNA sequencing confirmed each point mutation (University of Washington DNA sequencing facility). The resulting mutant plasmid (with chloramphenicol resistance) was then transformed into KB18 (with ampicillin resistance), thereby allowing the expression of fimbriae customized with the intended point mutation.
Confirmation of Fimbrial Expression Using Fluorescence-activated Cell Sorting
We examined the expression levels of mutant fimbriae versus wild type (WT) K12 using fluorescence-activated cell sorting (FACS) and a previously published protocol with minor publications (28, 29). First, E. coli were seeded in superbroth (SB) media with proper antibiotics (ampicillin at 100 μg/ml, chloramphenicol at 30 μg/ml) and incubated overnight at 37 °C under static conditions. The next morning the bacteria were spun down, rinsed twice with 1× phosphate-buffered saline (PBS), and resuspended at A540 10. Afterward, 0.5 ml of bacteria were fixed with an equal amount of 3.7% formaldehyde for 30 min at room temperature. Fixed cells were rinsed with 1× PBS and blocked for nonspecific binding in 0.5 ml of 0.2% BSA-PBS for 30 min with light rotation at room temperature. For the identification of FimH, the bacteria were spun down and resuspended in 0.5 ml of 0.2% BSA-PBS containing 1:300 rabbit-anti-pilin-domain antibody (α-Pd PAb, Antibodies Inc., Davis, CA; immunogen: E. coli K12 FimH pilin domain). The bacteria were then incubated with light rotation for 1 h at room temperature and rinsed twice with 1× PBS. Afterward, goat-anti-rabbit immunoglobulin G (IgG) conjugated to Alexa-488 (Invitrogen) was mixed at 1:3000 in 0.2% BSA-PBS, covered, and incubated with light rotation for 1 h at room temperature. After the final incubation, the bacteria were spun down, rinsed three times with 1× PBS, resuspended in 0.5 ml of 1× PBS, and immediately analyzed. A FACScan cell sorter (3 color analyzer, BD Biosciences, University of Washington Department of Immunology's Cell Analysis Facility) quantified the expression levels based on Alexa-488 fluorescence up to 10,000 events per reading.
Assessing Variants' Function Using Radioactive-based Binding Assay
To determine the function of each variant, we performed static binding assays using radiolabeled bacteria on monomannosyl (1M)3- or trimannosyl (3M)-coated surfaces as described previously (5, 27, 30). Briefly, E. coli-expressing mutant FimH were seeded in superbroth (SB) media containing radiolabel thymidine (3H, PerkinElmer Life Sciences) and the proper antibiotics. Bacteria were incubated overnight at 37 °C under static conditions. Next, the bacteria were spun down, rinsed twice with 1× PBS, and resuspended at A540 2 with or without 1% methyl α-d-mannopyranoside (or mannose). Meanwhile, detachable 96-well plates were incubated with 100 μl of 20 μg/ml 1M yeast mannan (Sigma) and 3M bovine RNase B (Sigma) in 0.02 m NaHCO3 buffer at 37 °C for 1 h, blocked with 0.2% BSA-PBS at 37 °C for 30 min, and aspirated. Wells with 0.2% BSA-PBS were included as a control substrate. The bacteria were then added to the coated plates and incubated for 45 min at 37 °C. Next, the plates were rinsed of unbound bacteria using 1× PBS, aspirated, dried at 65 °C for 15 min, broken into separate wells, and then placed in scintillation fluid. Radioactivity was measured for each sample (using Beckman LS 3801) done in triplicate except for the BSA-PBS control, done in duplicate, and the number of cells bound was determined using calibration curves of known solution concentrations calculated with BIAevaluation software (GE Healthcare).
Assessing the Conformational Change of Variants in the Presence of mAb21
Before executing the ELISA, pili were purified from each variant according to a previously published protocol (5, 25, 31). In summary, E. coli were grown overnight at 37 °C in Terrific Broth (EMD Chemicals) media with proper antibiotics and under gentle shaking (125 rpm). The next morning the bacteria were spun down at 8000 rpm, resuspended in cold pili buffer (50 mm Tris-HCl at pH 7.0, 150 mm NaCl), and homogenized on ice 3 times for 1 min with a 1-min rest period in between (Pro 200 homogenizer, PRO Scientific Inc., Oxford, CT). Cell debris was spun down leaving the supernatant containing soluble pili. Pili were precipitated out of solution with 0.2 m MgCl2 overnight at 4 °C, spun down at 15,000 rpm, and then resuspended in cold pili buffer. Any cell debris was then spun down at 15,000. Precipitation and resuspension was repeated several times before a final resuspension in cold 1× PBS. Protein concentration was determined using a BCA™ protein assay kit (Pierce) after heating in 0.1 m HCl for 5 min at 100 °C.
A basic ELISA using mouse monoclonal antibody 21 (mAb21) was performed on the purified pili. mAb21 (from PickCell Inc., The Netherlands) is an antibody raised against E. coli K12 FimH-lectin domain (residues 1–160) and previously determined to recognize or induce the high affinity state of K12 FimH, especially in the presence of ligand (25, 32). Pili were immobilized at 0.1 mg/ml in 0.02 m NaHCO3 buffer in wells of 96-well flat-bottom plates at 37 °C for 1 h. Comparative controls consisted of FocH (high affinity with and without an excess of α-methyl-mannose) and WT K12 (high affinity in the presence of α-methyl-mannose). All samples were done in triplicate. Free protein was rinsed off with 1× PBS twice and then quenched for 30 min in 0.2% BSA-PBS. mAb21 was added at 1:1500 dilution with and without 1% α-methyl-mannose in 0.2% BSA-PBS for 137 min. Free reagent was rinsed off four times with 1× PBS. Horseradish peroxidase (HRP) conjugated to goat-anti-mouse antibodies at 1:3000 in 0.2% BSA-PBS were incubated on the plate for 1 h at 37 °C and then rinsed off 6 times with 1× PBS. Bound HRP-goat-anti-mouse was visualized with the addition of 3,3′ 5,5′ - tetramethylbenzidine peroxidase enzyme from an immunoassay substrate kit (Bio-Rad). Absorbance was read at 650 nm using a Molecular Devices Emaxx microtiter plate reader.
Molecular Dynamics (MD) Simulations; Initial Conformations
The simulations with the WT were started from the crystallographic structure of the isolated FimH lectin domain (PDB ID 1uwf (33)), which is in the high affinity state (10) and contains α-d-mannose in the binding pocket. The mutant A10P was built from the x-ray structure of the WT using the program CHARMM (15), whereby the coordinates of the Pro-10 side chain atoms were constructed from internal coordinates (except for the Cβ atom, whose coordinates were taken from Ala-10). Positional restraints were then applied to all atoms of the protein except amino acids 9–11, and 100 steps of steepest descent minimization were performed with the program CHARMM (15).
MD Simulations; Setup
The MD simulations were performed with the program NAMD (16) using the CHARMM all-hydrogen force field (PARAM22) (34) and the TIP3P model of water. The protein was solvated in a cubic water box with a side length of 86 Å. Chloride and sodium ions were added to neutralize the system and approximate a salt concentration of 150 mm. To avoid finite size effects, periodic boundary conditions were applied. After solvation, the system underwent 500 steps of minimization, whereas the coordinates of the heavy atoms of the protein were held fixed with a subsequent 500 steps with no restraints. Electrostatic interactions were calculated within a cutoff of 10 Å, whereas long range electrostatic effects were taken into account by the Particle Mesh Ewald summation method (35). Van der Waals interactions were treated with the use of a switch function starting at 8 Å and turning off at 10 Å. The dynamics were integrated with a time step of 2 fs using the SHAKE algorithm to rigidly constrain hydrogen atoms. Snapshots were saved every 10 ps for trajectory analysis. Before production runs, harmonic constraints were applied to the positions of all heavy atoms of the protein to equilibrate the system at 300 K during a time length of 0.2 ns. In the case of the mutant, harmonic constraints were kept on all heavy atoms except those of the mutated residue and of the neighboring amino acids, and equilibration was continued for another 2 ns. After this equilibration phase, the harmonic constraints were released. Two simulations were performed with the WT and two with the A10P mutant. Each run lasted 50 ns, but the first 10 ns were considered part of the equilibration process and not used for analysis. During all runs, the temperature was kept constant at 300 K by using the Langevin thermostat (36) with a damping coefficient of 1 ps−1, whereas the pressure was held constant at 1 atm by applying a pressure piston (37).
MD Simulations; Analysis of Hydrogen Bonds
The simulation trajectories were used to analyze the formation of hydrogen bonds within the backbone of the clamp loop and between mannose and the lectin domain. To define a hydrogen bond, a H·O distance cutoff of 2.7 Å and a D-H·O angle cutoff of 120 degrees was used, where a donor D could either be an oxygen or a nitrogen. Within the clamp loop, only state-dependent backbone hydrogen bonds were analyzed. State-dependent hydrogen bonds are those that are formed in either only the crystallographic structure of the high affinity but not of the low affinity state and vice versa (10). On the other hand, to study the interaction between mannose and the lectin domain, only those hydrogen bonds were considered that were observed to be formed in at least 66% of the simulation frames of at least one of the two simulations with the WT. In the case of hydrogen bonds involving the side chain of Asp-54, no distinction was made as to which of the two oxygens was the acceptor. Similarly, no distinction was made as to which hydrogen was donated by the N-terminal amide group of Phe1. This resulted in a total of six persistent hydrogen bonds between mannose and the lectin domain: Phe-1 NH, 1M O2; Phe-1 NH, 1M O6; Asp-47 NH, 1M O6; Asp-54 Oδ, 1M O4H; Asp-54 Oδ, 1M O6H; Asn-133 NϵH, 1 m O4 (1M stands for monomannose). To better visualize the strength of the interaction between mannose and the lectin domain, the distances between the hydrogen atom and the acceptor of all six hydrogen bonds in every frame of the last 40 ns of both simulations with the WT and both simulations with the mutant, respectively, were averaged, and the S.D. was calculated. If multiple hydrogen bonds were formed between the Asp-54 side chain and the same donor in mannose or between the amide nitrogen of Phe1 and the same acceptor in mannose, then only the hydrogen bond with the shortest distance between the donated hydrogen and the acceptor was considered.
RESULTS
Summary of Studies
In this study we used the crystal structures of FimH in two different states to predict point mutations that should stabilize one conformational state or the other. The amino acid substitutions were selected within four regions that we call the pocket zipper, β-Bulge, α-switch, and interdomain loops (Fig. 2). To determine which of these regions are part of the allosteric pathway, we predicted point mutations that should stabilize one conformational state or the other. We made a special effort to predict mutations that would stabilize the low affinity state as almost all prior mutations have increased affinity, and both types of variants provide useful tools for future studies. In most cases we analyzed the structures visually to identify amino acids that were solvent-exposed in one conformation but buried in the other. We then used RosettaDesign computational analysis to predict specific substitutions at these locations that would cause the largest ΔΔG or difference in energy between the two conformations.
FIGURE 2.

Close-up of regions and residues selected to explore the allosteric pathway between the low and high affinity structures of the lectin domain of FimH. A, two states of the pocket zipper show the backbone hydrogen bonding (dashed lines) and residue Ala-10, which was selected for mutagenesis. B, shown are both conformations for the sequential stretch of 13 amino acids encompassing the β-bulge (purple) and α-switch (gold). Highlighted amino acids selected for mutagenesis to bias FimH toward one conformational state are shown as sticks. The two side chain conformations for Arg-60 shown in the low affinity structure reflect the 50/50 occupancy status observed in the crystal structure. C, shown is spatial orientation of selected residues from the interdomain loops. All residues shown interact with different neighbors when they shift between the low and high affinity structures, with many residues switching from being buried to solvent-exposed.
We quantified the expression of FimH for each variant by detecting the amount of pilin domain in fluorescence-activated cell sorting of whole-cell E. coli. All variants demonstrated expression averaging 74–82% of wild type K12 (data not shown) and thus were concluded to not alter pilus biogenesis.
To determine function, we measured binding to the 1M glycan yeast mannan, as binding of FimH to 1M glycans mimics that to glycoproteins on host cells (38, 39). However, although 1M affinity can be measured easily for FimH in which a chaperone protein or truncation prevents pilin domain autoinhibition (33, 40), native wild type FimH has a barely measurable affinity for 1M (31). Because of this, affinity could not be measured for the low affinity variants that are of most interest in this paper. Instead, we took advantage of avidity by measuring binding of whole bacteria to 1M-coated surfaces. An accepted assay is to calculate the ratio of bacteria binding to 1M- versus 3M-coated surfaces (Fig. 3A). The 3M structure has additional interactions with FimH (41) that mediate strong binding of both high and low affinity variants (5, 30) so that binding to 3M occurs at essentially the rate bacteria are transported to the surface (e.g. by gravitational settling). Thus, when assays are stopped before surface saturation, the 1M/3M ratio approximates the fraction of bacteria that binds after encountering the surface. A low ratio relative to WT reflects low affinity, whereas a high ratio reflects high affinity (5, 30) (Fig. 3B). This assay thus provides a quantitative measurement of avidity. The assay is unlikely to reflect mutation-induced changes in specificity of FimH for 1M versus 3M, because none of the mutations were in the binding pocket for either 1M (42) or 3M (41).
FIGURE 3.
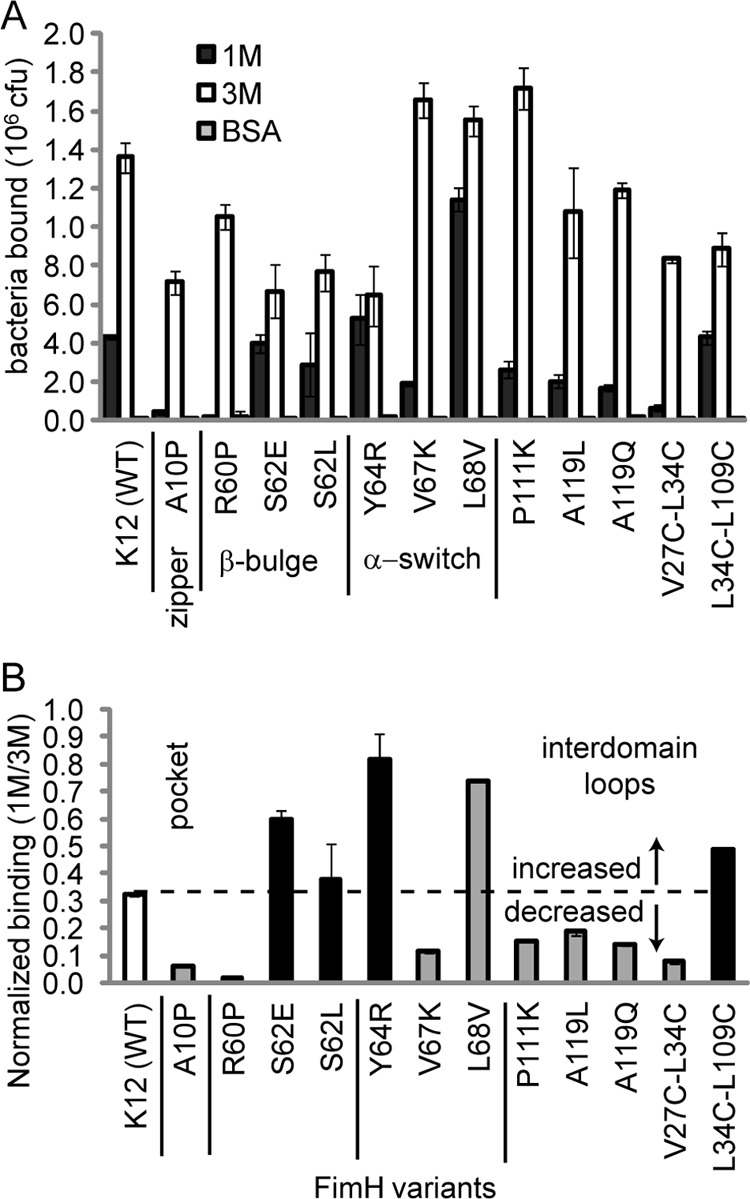
A, shown is binding of E. coli to 1M and 3M substrates. All E. coli were mannose-inhibitable (data not shown), and control bovine serum albumin substrate indicated no nonspecific binding. Error bars represent S.D. B, shown is the ratio of 1M/3M binding. Colors indicate predicted function. White is WT, gray is low affinity, and black is high affinity). The dotted line shows the WT 1M/3M ratio (0.32) so that position relative to this line shows actual function. Error bars represent propagation of the errors from panel A.
To determine the conformation of each variant, we performed an ELISA using conformation-specific mAb21 in the presence and absence of the ligand mannose (Fig. 4). The antibody mAb21 recognizes the high affinity state of FimH, which can be induced either by ligand binding or by activating mutations (25). In fact, a particular high affinity variant of FimH, which is referred to as FocH (27), is strongly recognized by mAb21 even without ligand (25). On the other hand, WT FimH requires ligand for strong recognition by mAb21, demonstrating that it switches to the high affinity state when it binds ligand (25). These studies allowed us to determine which regions exert allosteric control over FimH, as described in detail below.
FIGURE 4.
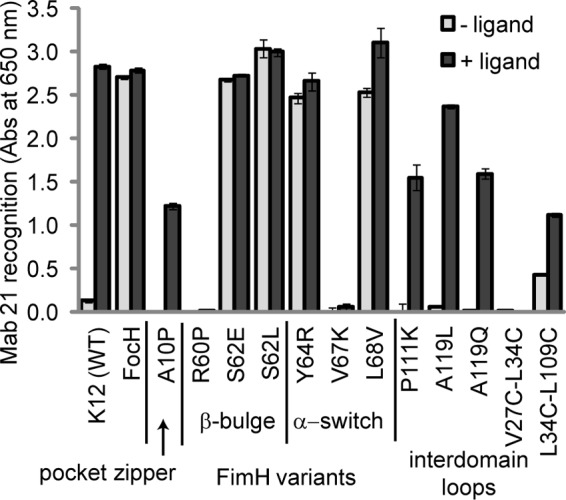
mAb21 recognition of FimH in pili with and without ligand mannose in ELISA assay. mAb21 recognition in the absence of mannose reflects whether each variant is high affinity in its native state. mAb21 recognition in the presence of mannose measures the ability of each variant to change into the high affinity state upon binding ligand. Error bars represent propagation of error for S.D. of sample minus that of blank.
Pocket Zipper (Phe-1–Ile-11) and Clamp Loop (Gly-8–Gly-16)
A major difference in the mannose-binding site among the various crystallographic structures is that the pocket zipper, a two-strand β-hairpin, is “zipped” (tight) in the high affinity conformation and is “unzipped” (loose) in the low affinity conformation (Fig. 2A). Furthermore, the unzipped state in low affinity induces the segment comprising residues 8–16 to move away from mannose. We previously referred to the 8–16 segment as the “clamp loop” because in the high affinity state it “tightens” the binding pocket around mannose (10). Analyses of the crystal structure suggests that the backbone hydrogen bond between the NH atom of residue C3 and the O of residue I11 is critical for closing the angle (C3:N, N7:N, I11:N) in the tight conformation. We hypothesized that substituting proline for alanine at position 10 (A10P) would bias the structure toward the low affinity conformation because the φ/ψ angles for this residue are outside of the allowed region of the Ramachandran map for proline in the high affinity conformation. We calculated using RosettaDesign that the predicted ΔΔG for this mutation is <−5 kcal/mol, where a negative value indicates that the mutation should stabilize the low affinity conformation. The 1M/3M ratio experiment showed that the A10P variant is a significantly weaker binder than WT and, therefore, demonstrates a low affinity function. In the absence of ligand, mAb21 binds more poorly to the A10P variant than to WT, suggesting that the A10P mutation stabilizes the low affinity conformation rather than simply damaging the pocket. Ligand enhanced recognition by mAb21 for the A10P variant but not to the levels observed for WT. The function and conformation assays together suggest that A10P stabilizes the low affinity conformation in a way that propagates from the mutation in the hairpin to the mAb21 epitope situated in the interdomain region.
β-Bulge (Gln-59–Ser-63)
The β-bulge is a short segment that is part of a β-strand in the high affinity conformation but bulges out into a small loop in the low affinity conformation (Fig. 2B). This bulge is hypothesized to act as a release valve for the strain caused by the more twisted conformation of the main β-sheet in the low affinity state. Mutations were selected to stabilize the bulged configuration to test the importance of the bulge to the allosteric pathway by determining whether the overall structure should shift toward the low affinity state. We hypothesized that a proline in position 60 would destabilize the high affinity conformation as prolines destabilize β-sheets. The RosettaDesign calculation was a ΔΔG of <−5 kcal/mol, favoring low affinity as hypothesized. In experiments, the R60P variant demonstrated much weaker binding than WT or even A10P and, therefore, reveals a low affinity function. mAb21 failed to recognize R60P even with ligand, demonstrating that the R60P variant remained locked in the low affinity state under these conditions.
The bulged conformation also requires an extra amino acid, which is provided by amino acids 62 through 63 shifting upward and rotating 180°. For example, the side chain of residue 62 is buried in the low affinity conformation and solvent-exposed in the high affinity conformation. To test whether this shift was coupled to the allosteric pathway, we hypothesized that the solvent-facing configuration would tolerate a change in amino acid more readily. RosettaDesign predicted that S62L and S62E would each result in ΔΔG >+5 kcal/mol, favoring the high affinity conformation. Both variants bound to mannose strongly relative to WT and were recognized strongly by mAb21, suggesting that these mutations indeed favored the high affinity conformation. In summary, the mutations R60P, S62L, and S62E alter the overall function and conformation of FimH, thereby demonstrating the importance of the β-Bulge to the FimH allosteric pathway.
α-Switch (Tyr-64–Phe-71)
The α-switch is a single-turn 310-helix in the low affinity conformation that switches to an α-helix in the high affinity conformation (Fig. 2B). Structurally, this helical rearrangement is directly connected to the bulge segment because the 310-helix conformation uses one less amino acid, allowing residue A-63 to shift toward the β-bulge. To test whether this region is connected to the allosteric pathway, we selected mutations that were favorable for the 310-helix while being unfavorable for the α-helix. Position 67 switches from solvent exposed in the low affinity conformation to buried in the high affinity conformation. RosettaDesign predicted that the V67K mutation would have the largest energy difference between conformations and favored the low affinity conformation (ΔΔG <−5 kcal/mol). In experiments, the V67K variant binds significantly less than WT and was not recognized by mAb21 in the absence or presence of ligand. The side chain at position 68 is buried in the low affinity conformation and solvent-exposed in the high affinity. If packing is already optimized, any change should favor the high affinity conformation. However, RosettaDesign predicted that ΔΔG was <−5 kcal/mol for the L68V mutation, favoring the low affinity conformation. L68V demonstrated a high affinity function as indicated by the 1M/3M ratio as well as by mAb21 recognition in the absence and presence of ligand. These inconsistencies might be due to insufficient sampling by the Rosetta protocol used here and emphasizes the importance of using human insight as well as computational prediction when designing mutations to test structures. We also selected a mutation predicted to be favorable to the α-helix. The side chain of Tyr-64 is buried in the 310-helix conformation and exposed to solvent in the α-helix. RosettaDesign predicted that Y64R favored the high affinity conformation with ΔΔG >+5 kcal/mol. The Y64R variant did indeed show a high affinity function and was recognized by mAb21 in the absence and presence of ligand. Even with the one contradiction to Rosetta calculations, the fact that all three mutations in the α-switch significantly alter mannose binding in correlation with conformational state indicates that this region is also part of the allosteric pathway.
Interdomain Loops (Ala-25–Leu-34, Pro-111–Ala-119, 152-158)
The interdomain region consists of three separate sequences in the mannose binding lectin domain that contact the anchoring pilin domain (Fig. 2C). The interdomain region changes configuration in the low affinity state to enable interactions with the pilin domain, as previously described (10). The pilin domain is known to control affinity (31) and allosteric conformation (25), strongly suggesting that the interdomain loops are part of the allosteric pathway. In addition, two variants in this region affected both mannose binding and mAb21 recognition (10). Residue 34 shifts dramatically between the two configurations, so pilin binding (docked) configuration was locked by a disulfide bond introduced with the double V27C/L34C mutation, whereas the undocked configuration was locked by a disulfide bond introduced with the double L34C/L109C mutation. As controls, we included the two variants from Le Trong et al. (10) for comparison in our experiments. Consistent with prediction, the V27C/L34C variant displayed a low affinity in both function and conformation, and the L34C/L109C variant displayed a high affinity function. Moreover, mAb21 recognized L34C/L109C better than WT in the absence of ligand. Together, this suggests that the two variants are in the predicted allosteric conformations. Surprisingly, mAb21 recognized L34C/L109C less than WT in presence of ligand. In the discussion, we explain our conclusion that this is because L109C subtly distorts the mAb21 epitope.
In addition, we predicted several new mutations in this region. We noted that Ala-119 is buried in the high affinity conformation and exposed in the low affinity conformation. Accordingly, RosettaDesign predicted that long side chains at this position with point mutations A119L and A119Q would favor the low affinity conformation (ΔΔG <−5 kcal/mol for each). Indeed, both the A119L and A119Q variants demonstrated a low affinity function as well as a low affinity conformation in the absence of ligand. However, in the presence of ligand, mAb21 recognized both variants, demonstrating that this variant can switch to high affinity in presence of ligand. Similar to residue 119, Pro-111 is buried in the high affinity conformation and exposed in the low affinity conformation. A charged side chain that is large (e.g. P111K) was predicted to strongly favor the low affinity conformation (ΔΔG <−5 kcal/mol). Similar to A119L and A119Q, P111K indicates a low affinity function and low affinity conformation in the absence of ligand, but mAb21 recognizes P111K in the presence of ligand. In summary, A119L, A119Q, and P111K contribute to the allosteric pathway.
Comparison of Function Versus Conformation
For the most part, the function (1M/3M ratio) correlates with the conformation (mAb21 binding), as indicated by the trendlines in Fig. 5. The WT variant is at a key transition point in the correlation graph. Because WT already has maximum mAb21 binding with mannose, variants with higher 1M/3M ratios, which are expected to bind mAb21 better, can best be distinguished by mAb21 binding without mannose (Fig. 5A). Conversely, because WT already has little mAb21 binding without mannose, variants with lower 1M/3M ratios (which are expected to bind even more poorly) are best distinguished by mAb21 binding with mannose (Fig. 5B). Thus, although all the low affinity variants (A10P, R60P, V27C/L34C, V67K, A119(L/Q), P111K) are not recognized by mAb21 in the absence of ligand, only R60P and V27C/L34C show an insignificant shift in mAb21 recognition in the presence of ligand (p > 0.05, V67K had p < 0.05), indicating that they are locked in the low affinity state even in the activating conditions of this assay. Overall, the strong correlation of function and conformation indicates that the primary determinant of binding strength is the allosteric state of the variants.
FIGURE 5.
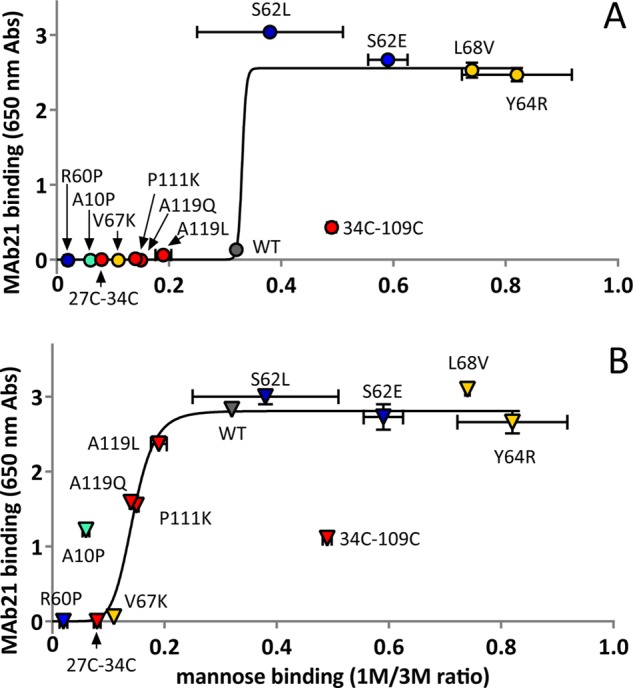
Correlation of mannose and mAb21 binding. A, shown is the allosteric coupling between the mannose-binding site (1M/3M) and the interdomain region (mAb21), with the latter in the absence (panel A) or presence (panel B) of ligand mannose. The data and error bars are taken directly from Figs. 3B and 4. The lines show an empirical trend rather than a mechanistic model. Abs, absorbance units.
We suggest that the position of a variant along the trendline is determined by the overall strength of its mutation, meaning the degree to which that mutation stabilizes the low or high affinity conformation of the region containing the mutation. However, deviations from the trendline are likely to indicate whether that region is more tightly coupled to the mannose versus the mAb21-binding site. As noted above, the low recognition of L34C/L109C by mAb21 is likely due to epitope distortion, so we do not try to draw conclusions from it, but the other clear outlier is worth considering. The A10P mutation lies above the trendline in Fig. 5B. That is, relative to the other low affinity variants, A10P binds more strongly to mAb21 than to mannose. This suggests that the mutation is effective in stabilizing the low affinity conformation of the nearby mannose-binding site but that this conformational change does not propagate as strongly to the interdomain region. That is, the mannose-binding site may be weakly coupled to the interdomain loops, α-switch, and β-bulge.
MD Simulations
To further test the idea that the pocket zipper is weakly coupled to the remaining allosteric regions, we performed MD simulations with WT and A10P variants. We sought to address two structural issues with these simulations. First, we have assumed that the A10P mutation destabilizes the high affinity conformation of the mannose-binding site, but an alternative possibility is that A10P interferes directly with nearby mannose binding. Second, we wanted to test for weak allosteric coupling by observing whether the conformation of the mannose-binding site could indeed change conformation without instantly changing the conformation of the interdomain region. In simulations of WT in the high affinity state with mannose bound (PDB ID 1uwf), the structure of the pocket zipper and clamp loop remained stable throughout the simulations by two measures. First, the three hydrogen bonds (C3 NH[bond]Ile-11 O, Gly-16 NH[bond]Gln-143 O, and Pro-12 O[bond]Gly15 NH) that are unique to the high affinity conformation of the pocket (10) remained stable at close to their native length (Fig. 6A). Second, the root mean square deviation of this region remained within 1 Å of the high affinity crystal structure (Fig. 6B). In contrast, in simulations where the A10P mutation was computationally introduced, each of the three hydrogen bonds were destabilized, and the root mean square deviation of this region also fluctuated extensively and increased by 72% on average (Fig. 6). Thus, the A10P mutation destabilized the high affinity conformation of the pocket zipper and clamp loop, consistent with our initial design prediction and with the experimental results. Despite these changes, all six hydrogen bonds (see “Experimental Procedures”) that persistently bind FimH to mannose (43) were strong and stable in A10P as well as WT (Fig. 6A). This demonstrates that the A10P mutation does not directly disrupt mannose binding. Finally, the conformational change in the clamp loop in A10P did not propagate to the interdomain loops (Fig. 6B), as both stayed within 1 Å root mean square deviation of the high affinity structure most of the time. This indicates that allosteric propagation between these two regions occurs on a longer time scale than probed by the standard MD simulations presented here. This in turn supports the concept that the pocket zipper and clamp loop are relatively weakly coupled to the interdomain region, which includes the mAb21 epitope.
FIGURE 6.
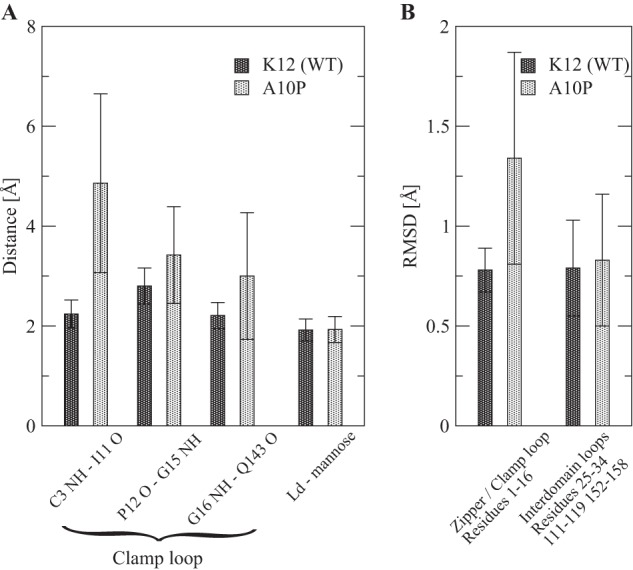
MD simulations with WT and A10P variants. A, shown is the average distance between atoms involved in hydrogen bonds during the last 40 ns. A length less than 2.7 Å is considered a strong bond, between 2.7 and 3 Å is considered a weak bond, and over 3 Å is considered broken. B, shown is average root mean square deviation (RMSD) from the initial structure averaged over the last 40 ns for residues contained in the pocket zipper and clamp loop and for residues contained in the interdomain loops. All values are averages of the measurement taken during the last 40 ns of two 50-ns simulations. The error bars indicate S.D. over time in both trajectories in order to illustrate the fluctuations.
Identification of Low Affinity FimH Mutants
In the course of this work, we identified six new low affinity variants that provide tools to complement the large number of previously identified high affinity variants (5, 31, 38, 44–46) for future studies about FimH allostery. Of these, A119L, A119Q, and P111K have only a slightly decreased 1M/3M ratio and switch at least partially to the high affinity conformation in the presence of mannose in the mAb21 assays. A10P, R60P, V67K, and V27C/L34C variants have the lowest affinity for mannose, and each represents a different region of the FimH lectin domain. However, the A10P variant can switch to the high affinity conformation with mannose, whereas the other three remain locked in the low affinity conformation in all conditions tested here. Thus, R60P, V67K, and V27C/L34C are the most promising candidates for future studies requiring low affinity variants.
DISCUSSION
In this study we applied visual inspection and RosettaDesign to crystal structures of FimH in two allosteric conformations to predict 10 mutations that would stabilize one or the other conformation. We genetically engineered variants with these mutations using site-directed mutagenesis. We evaluated them for function by testing the ability to bind to mannose-coated surfaces and for conformation by testing mAb21 binding, which specifically recognizes the high affinity state. Every variant that was designed using both human and computational analysis demonstrated the predicted function. In one case, human insight and RosettaDesign, as used here, contradicted each other, and this variant demonstrated the function predicted by human insight. In every case, binding of the conformation-sensitive antibody correlated with function, demonstrating that the changes in ligand binding strength were caused by a change in the allosteric conformation. This suggests that the combination of human and computational analysis is needed for successful prediction of mutations that affect the allosteric state of a protein. The fact that all predicted variants affected function demonstrates that all four regions are part of the allosteric pathway. Previously, only the role of the interdomain region was tested with point mutations based on the crystal structures (10).
To draw the conclusions above, we assumed that binding to mAb21 directly reflected the conformational state of FimH, so it is worth evaluating this assumption in depth. Two other mechanisms could cause loss of mAb21 binding. First, a mutation may directly interfere with the mAb21 epitope. However, none of our mutations is part of the mAb21 epitope, which is restricted to residues 26, 29, and 153–157 (32). Moreover, mAb21 recognized all variants given the right conditions. A10P, R60P, and V67K were recognized normally by mAb21 when expressed with the FocH pilin domain and incubated with mannose, which lacks the ability of the wild type pilin domain to stabilize the low affinity state (32). V27C/L34C was recognized normally by mAb21 in the presence of mannose and DTT, which breaks the disulfide bond that maintains the low affinity conformation for this variant (10). It might be noted that the L34C/L109C only reaches 40–80% of mAb21 binding compared with WT even with mannose (Fig. 4 and Ref. 10) and even when expressed with the FocH pilin domain (not shown), suggesting that the disulfide bond in the hydrophobic core subtly distorts the nearby mAb21 epitope. However, the slightly reduced mAb21 binding does not interfere with our ability to observe the increased recognition of mAb21 in the absence of mannose, which demonstrates the allosteric activation by this double mutation. A second alternative explanation could be that some variants are misfolded or fail to express so that both mannose binding and mAb21 binding would be lost but not because FimH is in the low affinity state. However, the FACS data demonstrated that expression levels are comparable, and the ability to bind RNase B (Fig. 3) demonstrates that the proteins are folded properly. Thus, the level of mAb21 binding reflects the population of FimH in the high versus the low affinity state.
The interdomain region was previously shown to exert allosteric control over the mannose-binding site of FimH (10, 45, 46), and a novel β-sheet-twisting allosteric mechanism was proposed in which a semi-rigid β-sheet takes on a narrow and straight or wider and twisted conformation that forms a mechanical connection between the effector and active sites (10). However, no prior data addressed the role of three other regions, the pocket zipper, the β-bulge, and the α-switch, in the allosteric mechanism. All three regions lie between the active and effective sites but do not form a complete pathway of localized conformational changes. Instead, the pocket zipper and clamp loop are spatially separated from the β-bulge, α-switch, and interdomain loops, as illustrated in Fig. 7. Nevertheless, the effect of mutations in these three regions on mannose binding and mAb21 binding demonstrate that these regions control the conformation of both active and effector sites as illustrated in the central panel of Fig. 7. It is possible that the regions, like the effector and active sites themselves, simply stabilize either the twisted or straight conformation of the β-sheet. The involvement of the pocket zipper, β-bulge, and α-switch in the allosteric mechanism expands the targets for allosteric antiadhesives. Indeed, it may be advantageous to target an allosteric antiadhesive to a region other than the interdomain region so that it does not compete with the pilin domain natural allosteric inhibition (10, 31), allowing the two to work in synchrony to stabilize FimH in the low affinity state.
FIGURE 7.
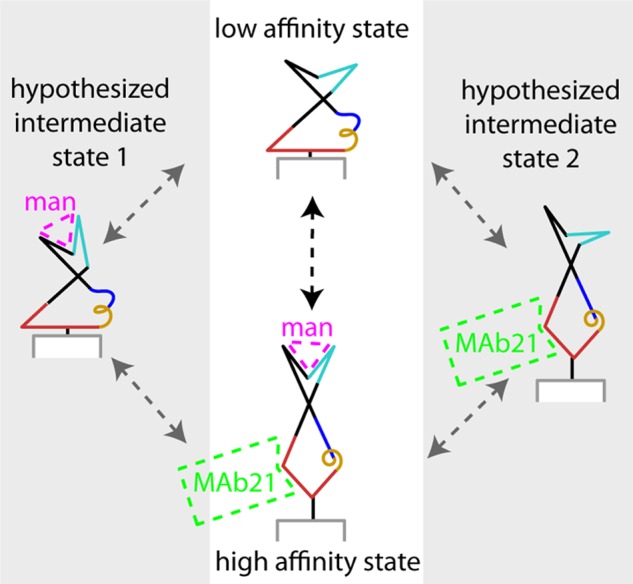
Schematic of allosteric coupling. The stable low and high affinity conformations are shown in the central white panel. The different conformations of the clamp loop and pocket zipper (cyan), interdomain loops (red) α-helix (gold) and β-bulge (blue) illustrate the conclusions that all are involved in the allosteric mechanism. The gray side panels illustrate the hypothesis that weak coupling of the clamp loop and pocket zipper to the interdomain loops, α-helix and β-bulge, could result in intermediate states in which one of these regions is in the high affinity conformation and the other low. If so, the allosteric transition would occur through one of these intermediates (gray arrows) rather than in a single concerted reaction (black arrow). When the clamp loop and pocket zipper (cyan) are in the high affinity conformation, mannose is shown binding in magenta. When the interdomain loops, α-helix and β-bulge (red, gold, and blue, respectively) are in the high affinity conformation, mAb21 is shown binding in green. Part of the pilin domain is shown in gray and white.
Our data can also be used to suggest how tightly each of these regions is coupled to the conformation of the mannose binding versus pilin binding regions. The comparative effect of mutations on binding mannose versus mAb21 suggests that the β-bulge, α-helix, and interdomain loops are more tightly coupled to the pilin binding effector region, whereas the pocket zipper is more tightly coupled to mannose binding active site. Interestingly, this biochemical coupling reflects the pathway of localized conformational changes in the crystal structures; although such a pathway exists between the β-bulge, α-helix, and pilin binding interdomain loops as well as between the pocket zipper and mannose-binding site, no such pathway exists between these two groups. This information should be useful in choosing binding sites for computational design of allosteric inhibitors or of structures to test as vaccines to prevent an E. coli infection.
The weak coupling of the mannose-binding site to the rest of the allosteric pathway suggests the existence of one or more intermediate states, as illustrated in Fig. 7, in which either the active site or the effector site has switched conformations but not both. These hypothesized states provide new structural insight to physiologically relevant FimH properties. Previous experiments have demonstrated that FimH-mannose bonds undergo a force-dependent transition from a short-lived and a long-lived state (10, 12, 45). The long-lived state is critical for strong adhesion (45), and its structure is almost certainly the high affinity structure that has been repeatedly co-crystallized with mannose (33, 42). The short-lived state is necessary for rolling adhesion (45, 47), which in turn allows rapid colonization of surfaces (48). However, the structure of this state is unknown, as the low affinity crystal structure lacks mannose (10). Here we hypothesize that these short-lived bonds may form when the clamp loop and pocket zipper close around mannose in the high affinity conformation, but the β-bulge, α-helix, and interdomain loops remain in the low affinity conformation, as in intermediate state 1 of Fig. 7. This is significant because it predicts that a mutation, antibody, or molecule that stabilizes the low affinity state of the pocket would eliminate rolling as well as firm adhesion if the short-lived bonds involve intermediate state 1 but would allow rolling adhesion if the short-lived bonds involve the low affinity state. Intermediate state 2 of Fig. 7 may also play a significant role by providing a pathway for dissociation of mannose. If unbinding of high affinity bonds occurs through intermediate state 2, then a modulator (such as a mutation, antibody, or molecule) that stabilizes the high affinity state of the mannose-binding pocket would increase bond lifetime. However, in conditions where unbinding from the high affinity state occurs by a previously described pathway in which mannose is pulled out of a still-tight pocket (43), these modulators would not affect bond lifetime. Thus, the existence and significance of both hypothesized intermediate states could be tested in future studies.
Here we also characterized six new low affinity variants. A previous study showed that antibodies raised against high affinity FimH structures increased rather than decreased bacterial adhesion to uroepithelial cells because they stabilized the high affinity state of FimH (32). This suggests that antibodies raised against the low affinity variants may inhibit bacterial adhesion because they stabilize the low affinity state of FimH. If so, these low affinity variants may be used as vaccines. The variants A10P, R60P, V67K, and V27C/L34C represent all four regions investigated herein and provided the strongest stabilization of a low affinity function and conformation. However, weak coupling between the effector and active sites could mean that these variants may raise fundamentally different antibodies. Because the A10P mutation had a greater effect on mannose binding than on Mab-21 binding, it may preferentially raise antibodies that recognize the low affinity conformation of the binding pocket, whereas R60P, V67K, and V27C/L34C may preferentially raise antibodies to the low affinity conformation of the effector region. Although both types of antibodies would be expected to inhibit FimH, the kinetics of inhibition could be quite different. These low affinity variants may also be useful to screen for allosteric inhibitors or antibodies. It may be important to use more than one low affinity variant for both immunogen and screening to address the issue that some antibodies may recognize an epitope that includes one of the mutations and thus be ineffective against WT FimH.
The low affinity variants identified here should also be useful for understanding FimH-mediated bacterial adhesion. Many pathogenic clinical isolates express variants that have a high affinity for mannose (30, 38, 44), and more high affinity variants have been engineered (27, 31, 46). These were critical for showing that FimH forms strong slip bonds if it is already allosterically activated. These variants also mediated strong stationary adhesion that is not shear-enhanced, demonstrating the role of catch bonds in shear-enhanced adhesion. These studies were essential for understanding the biological importance of the allosteric inhibition of FimH. However, the corresponding questions have not been asked to understand the importance of the allosteric activation of FimH. It is possible that the low affinity variants can not be activated by force or that they will simply require more force for activation, like a loss-of-function variant of the catch bond-forming protein von Willebrand Factor (49). It has been suggested that FimH that is locked in the low affinity state could still mediate shear-enhanced adhesion due to mechanical properties of the type 1 pili that anchor FimH (47, 50). The variants developed here are likely to have unique kinetic properties. The native versus high affinity FimH structures differ even more in bond lifetime than in affinity (10), but the kinetics of a stabilized low affinity variant are not known. Similarly, mutations that decrease dissociation rates lead to more stationary adhesion (45, 47), but the effect of mutations that increase bond kinetics is not known. The low affinity variants developed here can be used to address these and other fundamental questions about bacterial adhesion.
Most bacterial adhesins have never been tested in controlled flow conditions, so the prevalence of shear-enhanced adhesion is still being determined. Nevertheless, this phenomenon has already been reported for the collagen receptor of Staphylococcus aureus (51), P-pili (52), and CfaI (53) of E. coli, Hsa and GspB of Streptococcus gordonii (54), and type 1 fimbriae of Salmonella enterica (55). These adhesins contribute to virulence and so are also targets of antiadhesive therapies. Although none of these other adhesins have been shown to have allosteric properties, E. coli FimH was studied for >100 years and crystallized in 6 unique crystals with nearly identical conformations both with and without ligand (33, 41, 42, 56) before the alternative allosteric conformation was finally crystallized (10). This suggests that allostery may also be overlooked in other bacterial adhesins, and our success in using the crystal structure for allosteric design demonstrates the value of obtaining alternative structures.
This work was supported, in whole or in part, by National Institutes of Health Grant 1RO1 AI50940 and Molecular Biophysics Training Grant 1RO1 AI50940 (University of Washington; to B. A. K. and V. B. R.). This work was also supported by National Science Foundation (NSF) Grant CBET-1132860, Integrative Graduate Education and Research Traineeship Fellowship Award NSF #DGE-0504573 (to B. A. K.), and the University of Washington Initiatives Fund (to B. A. K.).
- 1M
- monomannose
- 3M
- trimannose
- MD
- molecular dynamics.
REFERENCES
- 1. Foxman B. (2010) The epidemiology of urinary tract infection. Nat. Rev. Urol. 7, 653–660 [DOI] [PubMed] [Google Scholar]
- 2. Karlowsky J. A., Kelly L. J., Thornsberry C., Jones M. E., Sahm D. F. (2002) Trends in antimicrobial resistance among urinary tract infection isolates of Escherichia coli from female outpatients in the United States. Antimicrob. Agents Chemother. 46, 2540–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schito G. C., Naber K. G., Botto H., Palou J., Mazzei T., Gualco L., Marchese A. (2009) The ARESC study. An international survey on the antimicrobial resistance of pathogens involved in uncomplicated urinary tract infections. Int. J. Antimicrob. Agents 34, 407–413 [DOI] [PubMed] [Google Scholar]
- 4. Zhanel G. G., Hisanaga T. L., Laing N. M., DeCorby M. R., Nichol K. A., Palatnik L. P., Johnson J., Noreddin A., Harding G. K., Nicolle L. E., Hoban D. J., and NAUTICA Group (2005) Antibiotic resistance in outpatient urinary isolates. Final results from the North American Urinary Tract Infection Collaborative Alliance (NAUTICA). Int. J. Antimicrob. Agents 26, 380–388 [DOI] [PubMed] [Google Scholar]
- 5. Sokurenko E. V., Chesnokova V., Doyle R. J., Hasty D. L. (1997) Diversity of the Escherichia coli type 1 fimbrial lectin. Differential binding to mannosides and uroepithelial cells. J. Biol. Chem. 272, 17880–17886 [DOI] [PubMed] [Google Scholar]
- 6. Hartmann M., Lindhorst T. K. (2011) The bacterial lectin FimH, a target for drug discovery: carbohydrate inhibitors of type 1 fimbriae-mediated bacterial adhesion. Eur. J. Org. Chem. 3583–3609 [Google Scholar]
- 7. Thomas W. (2008) Catch bonds in adhesion. Annu Rev. Biomed Eng. 10, 39–57 [DOI] [PubMed] [Google Scholar]
- 8. Langermann S., Möllby R., Burlein J. E., Palaszynski S. R., Auguste C. G., DeFusco A., Strouse R., Schenerman M. A., Hultgren S. J., Pinkner J. S., Winberg J., Guldevall L., Söderhäll M., Ishikawa K., Normark S., Koenig S. (2000) Vaccination with FimH adhesin protects cynomolgus monkeys from colonization and infection by uropathogenic Escherichia coli. J. Infect. Dis. 181, 774–778 [DOI] [PubMed] [Google Scholar]
- 9. Langermann S., Palaszynski S., Barnhart M., Auguste G., Pinkner J. S., Burlein J., Barren P., Koenig S., Leath S., Jones C. H., Hultgren S. J. (1997) Prevention of mucosal Escherichia coli infection by FimH-adhesin-based systemic vaccination. Science 276, 607–611 [DOI] [PubMed] [Google Scholar]
- 10. Le Trong I., Aprikian P., Kidd B. A., Forero-Shelton M., Tchesnokova V., Rajagopal P., Rodriguez V., Interlandi G., Klevit R., Vogel V., Stenkamp R. E., Sokurenko E. V., Thomas W. E. (2010) Structural basis for mechanical force regulation of the adhesin FimH via finger trap-like β-sheet twisting. Cell 141, 645–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nilsson L. M., Yakovenko O., Tchesnokova V., Thomas W. E., Schembri M. A., Vogel V., Klemm P., Sokurenko E. V. (2007) The cysteine bond in the Escherichia coli FimH adhesin is critical for adhesion under flow conditions. Mol. Microbiol. 65, 1158–1169 [DOI] [PubMed] [Google Scholar]
- 12. Thomas W., Forero M., Yakovenko O., Nilsson L., Vicini P., Sokurenko E., Vogel V. (2006) Catch bond model derived from allostery explains force-activated bacterial adhesion. Biophys. J. 90, 753–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rohl C. A., Strauss C. E., Misura K. M., Baker D. (2004) Protein structure prediction using Rosetta. Methods Enzymol. 383, 66–93 [DOI] [PubMed] [Google Scholar]
- 14. DeLano W. L. (2002) The PyMOL User's Manual, DeLano Scientific, Palo Alto, CA [Google Scholar]
- 15. Brooks B. R., Brooks C. L., 3rd, Mackerell A. D., Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M. (2009) CHARMM. The Biomolecular Simulation Program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuhlman B., Baker D. (2000) Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. U.S.A. 97, 10383–10388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dunbrack R. L., Jr., Cohen F. E. (1997) Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 6, 1661–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morozov A. V., Kortemme T., Tsemekhman K., Baker D. (2004) Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations. Proc. Natl. Acad. Sci. U.S.A. 101, 6946–6951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lazaridis T., Karplus M. (1999) Effective energy function for proteins in solution. Proteins 35, 133–152 [DOI] [PubMed] [Google Scholar]
- 22. Kellogg E. H., Leaver-Fay A., Baker D. (2011) Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 79, 830–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sowdhamini R., Srinivasan N., Shoichet B., Santi D. V., Ramakrishnan C., Balaram P. (1989) Stereochemical modeling of disulfide bridges. Criteria for introduction into proteins by site-directed mutagenesis. Protein Eng. 3, 95–103 [DOI] [PubMed] [Google Scholar]
- 24. Dani V. S., Ramakrishnan C., Varadarajan R. (2003) MODIP revisited. Re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein Eng. 16, 187–193 [DOI] [PubMed] [Google Scholar]
- 25. Tchesnokova V., Aprikian P., Yakovenko O., Larock C., Kidd B., Vogel V., Thomas W., Sokurenko E. (2008) Integrin-like allosteric properties of the catch bond-forming FimH adhesin of Escherichia coli. J. Biol. Chem. 283, 7823–7833 [DOI] [PubMed] [Google Scholar]
- 26. Blomfield I. C., McClain M. S., Eisenstein B. I. (1991) Type 1 fimbriae mutants of Escherichia coli K12. Characterization of recognized afimbriate strains and construction of new fim deletion mutants. Mol. Microbiol. 5, 1439–1445 [DOI] [PubMed] [Google Scholar]
- 27. Sokurenko E. V., Schembri M. A., Trintchina E., Kjaergaard K., Hasty D. L., Klemm P. (2001) Valency conversion in the type 1 fimbrial adhesin of Escherichia coli. Mol. Microbiol. 41, 675–686 [DOI] [PubMed] [Google Scholar]
- 28. Kisiela D., Laskowska A., Sapeta A., Kuczkowski M., Wieliczko A., Ugorski M. (2006) Functional characterization of the FimH adhesin from Salmonella enterica serovar Enteritidis. Microbiology 152, 1337–1346 [DOI] [PubMed] [Google Scholar]
- 29. Humphries A. D., Raffatellu M., Winter S., Weening E. H., Kingsley R. A., Droleskey R., Zhang S., Figueiredo J., Khare S., Nunes J., Adams L. G., Tsolis R. M., Bäumler A. J. (2003) The use of flow cytometry to detect expression of subunits encoded by 11 Salmonella enterica serotype typhimurium fimbrial operons. Molecular Microbiology 48, 1357–1376 [DOI] [PubMed] [Google Scholar]
- 30. Sokurenko E. V., Chesnokova V., Dykhuizen D. E., Ofek I., Wu X. R., Krogfelt K. A., Struve C., Schembri M. A., Hasty D. L. (1998) Pathogenic adaptation of Escherichia coli by natural variation of the FimH adhesin. Proc. Natl. Acad. Sci. U.S.A. 95, 8922–8926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aprikian P., Tchesnokova V., Kidd B., Yakovenko O., Yarov-Yarovoy V., Trinchina E., Vogel V., Thomas W., Sokurenko E. (2007) Interdomain interaction in the FimH adhesin of Escherichia coli regulates the affinity to mannose. J. Biol. Chem. 282, 23437–23446 [DOI] [PubMed] [Google Scholar]
- 32. Tchesnokova V., Aprikian P., Kisiela D., Gowey S., Korotkova N., Thomas W., Sokurenko E. (2011) Type 1 fimbrial adhesin FimH elicits an immune response that enhances cell adhesion of Escherichia coli. Infect Immun. 79, 3895–3904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bouckaert J., Berglund J., Schembri M., De Genst E., Cools L., Wuhrer M., Hung C. S., Pinkner J., Slättegård R., Zavialov A., Choudhury D., Langermann S., Hultgren S. J., Wyns L., Klemm P., Oscarson S., Knight S. D., De Greve H. (2005) Receptor binding studies disclose a novel class of high affinity inhibitors of the Escherichia coli FimH adhesin. Mol. Microbiol. 55, 441–455 [DOI] [PubMed] [Google Scholar]
- 34. MacKerell A. D., Bashford D., Bellot M., Dunbrack R. L., Jr., Evansec J., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kucknir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher I. W. E., Roux B., Schlenkrick M., Smith J., Sote R., Straub J., Watanabe M., Wiorkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empircal potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 35. Darden T., York D., Pedersen L. (1993) Particle mesh ewald. An n.log(n) method for ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 36. Schneider T., Stoll E. (1978) Molecular-dynamics study of a three-dimensional one-component model for distortive phase-transitions. Phys. Rev. B 17, 1302–1322 [Google Scholar]
- 37. Feller S. E., Zhang Y. H., Pastor R. W., Brooks B. R. (1995) Constant pressure molecular dynamics simulation. The langevin piston method. J. Chem. Phys. 103, 4613–4621 [Google Scholar]
- 38. Weissman S. J., Chattopadhyay S., Aprikian P., Obata-Yasuoka M., Yarova-Yarovaya Y., Stapleton A., Ba-Thein W., Dykhuizen D., Johnson J. R., Sokurenko E. V. (2006) Clonal analysis reveals high rate of structural mutations in fimbrial adhesins of extraintestinal pathogenic Escherichia coli. Mol. Microbiol. 59, 975–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aprikian P., Interlandi G., Kidd B. A., Le Trong I., Tchesnokova V., Yakovenko O., Whitfield M. J., Bullitt E., Stenkamp R. E., Thomas W. E., Sokurenko E. V. (2011) The bacterial fimbrial tip acts as a mechanical force sensor. PLoS Biol. 9, e1000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bouckaert J., Mackenzie J., de Paz J. L., Chipwaza B., Choudhury D., Zavialov A., Mannerstedt K., Anderson J., Piérard D., Wyns L., Seeberger P. H., Oscarson S., De Greve H., Knight S. D. (2006) The affinity of the FimH fimbrial adhesin is receptor-driven and quasi-independent of Escherichia coli pathotypes. Mol. Microbiol. 61, 1556–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wellens A., Garofalo C., Nguyen H., Van Gerven N., Slättegård R., Hernalsteens J. P., Wyns L., Oscarson S., De Greve H., Hultgren S., Bouckaert J. (2008) Intervening with urinary tract infections using anti-adhesives based on the crystal structure of the FimH-oligomannose-3 complex. PLoS ONE 3, e2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hung C. S., Bouckaert J., Hung D., Pinkner J., Widberg C., DeFusco A., Auguste C. G., Strouse R., Langermann S., Waksman G., Hultgren S. J. (2002) Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection. Mol. Microbiol. 44, 903–915 [DOI] [PubMed] [Google Scholar]
- 43. Nilsson L. M., Thomas W. E., Sokurenko E. V., Vogel V. (2008) Beyond induced-fit receptor-ligand Interactions. Structural changes that can significantly extend bond lifetimes. Structure 16, 1047–1058 [DOI] [PubMed] [Google Scholar]
- 44. Sokurenko E. V., Courtney H. S., Maslow J., Siitonen A., Hasty D. L. (1995) Quantitative differences in adhesiveness of type 1 fimbriated Escherichia coli due to structural differences in fimH genes. J. Bacteriol. 177, 3680–3686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thomas W. E., Nilsson L. M., Forero M., Sokurenko E. V., Vogel V. (2004) Shear-dependent “stick-and-roll” adhesion of type 1 fimbriated Escherichia coli. Molecular Microbiology 53, 1545–1557 [DOI] [PubMed] [Google Scholar]
- 46. Thomas W. E., Trintchina E., Forero M., Vogel V., Sokurenko E. V. (2002) Bacterial adhesion to target cells enhanced by shear force. Cell 109, 913–923 [DOI] [PubMed] [Google Scholar]
- 47. Whitfield M., Ghose T., Thomas W. (2010) Shear-stabilized rolling behavior of E. coli examined with simulations. Biophys. J. 99, 2470–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Anderson B. N., Ding A. M., Nilsson L. M., Kusuma K., Tchesnokova V., Vogel V., Sokurenko E. V., Thomas W. E. (2007) Weak rolling adhesion enhances bacterial surface colonization. J. Bacteriol. 189, 1794–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Auton M., Sedlák E., Marek J., Wu T., Zhu C., Cruz M. A. (2009) Changes in thermodynamic stability of von Willebrand factor differentially affect the force-dependent binding to platelet GPIbα. Biophys. J. 97, 618–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Björnham O., Axner O. (2010) Catch-bond behavior of bacteria binding by slip bonds. Biophys. J. 99, 1331–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li Z. J., Mohamed N., Ross J. M. (2000) Shear stress affects the kinetics of Staphylococcus aureus adhesion to collagen. Biotechnol. Prog 16, 1086–1090 [DOI] [PubMed] [Google Scholar]
- 52. Nilsson L. M., Thomas W. E., Trintchina E., Vogel V., Sokurenko E. V. (2006) Catch bond-mediated adhesion without a shear threshold. Trimannose versus monomannose interactions with the FimH adhesin of Escherichia coli. J. Biol. Chem. 281, 16656–16663 [DOI] [PubMed] [Google Scholar]
- 53. Tchesnokova V., McVeigh A. L., Kidd B., Yakovenko O., Thomas W. E., Sokurenko E. V., Savarino S. J. (2010) Shear-enhanced binding of intestinal colonization factor antigen I of enterotoxigenic Escherichia coli. Mol. Microbiol. 76, 489–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ding A. M., Palmer R. J., Jr., Cisar J. O., Kolenbrander P. E. (2010) Shear-enhanced oral microbial adhesion. Appl. Environ Microbiol. 76, 1294–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kisiela D. I., Kramer J. J., Tchesnokova V., Aprikian P., Yarov-Yarovoy V., Clegg S., Sokurenko E. V. (2011) Allosteric catch bond properties of the FimH adhesin from Salmonella enterica serovar typhimurium. J. Biol. Chem. 286, 38136–38147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Choudhury D., Thompson A., Stojanoff V., Langermann S., Pinkner J., Hultgren S. J., Knight S. D. (1999) X-ray structure of the FimC-FimH chaperone-adhesin complex from uropathogenic Escherichia coli. Science 285, 1061–1066 [DOI] [PubMed] [Google Scholar]