

Abstract
The progressive introduction of high-throughput molecular techniques in the clinic allows for the extensive and systematic exploration of multiple biological layers of tumors. Molecular profiles and classifiers generated from these assays represent the foundation of what the National Academy describes as the future of ‘precision medicine.’ However, the analysis of such complex data requires the implementation of sophisticated bioinformatic and statistical procedures. It is critical that oncology practitioners be aware of the advantages and limitations of the methods used to generate classifiers in order to usher them into the clinic. This article uses publicly available expression data from NSCLC patients to first illustrate the challenges of experimental design and pre-processing of data prior to clinical application and highlights the challenges of high-dimensional statistical analysis. It provides a roadmap for the translation of such classifiers to clinical practice and make key recommendations for good practice.
Introduction
As high-throughput molecular technologies become ubiquitous and as antineoplastic agents are increasingly directed against specific molecular aberrations, modeling the relationship between genomic features and prognosis or therapeutic response provides the substrate for precision medicine (1). Over the past decade, very few biomarkers have reached the required level of evidence to be implemented in the clinic (2), and a dearth of genomic signatures generated from the aforementioned technologies have been approved for clinical use (3). Ironically, as the molecular data available in repositories rapidly expand; effective, validated translation of these data to bedside diagnostics or target discovery remains a vexing challenge. Apart from the typical statistical challenges facing biomarker studies (4), there are unique issues that accompany high-dimensional genomic platforms that present obstacles to generating performant genomic signatures. Unfortunately, many of these issues are obscure to the larger oncology community.
Herein, we highlight problems associated with developing molecular signatures at each phase of development: 1) data curation and pre-processing, 2) statistical analysis, 3) and the infrastructure required for effective translation in cancer research and clinical settings. To demonstrate each of these issues we focus on gene expression data, though the discussion is applicable to many types of high dimensional data. Each section of this review includes pertinent figures of analysis performed following recommendations for best practice (Table 1). For both educational and reproducibility purposes, we provide real data (available through Synapse, the collaborative compute space developed at Sage Bionetworks, under the Synapse ID ‘syn87682’: https://www.synapse.org/#!Synapse:syn87682) and companion R scripts (available on GitHub: https://github.com/Sage-Bionetworks/Ferte-et-al-Review)
Table 1.
practical issues and recommendations for the development and the translation of molecular classifiers in oncology
| Step of development and translation | Issue | Proposal for best-practice |
|---|---|---|
| Experimental design | Selection and curation of the datasets | Cross-talk between Oncologists, Biostatisticians and Bioinformaticians is warranted to choose the most appropriate data Appropriate selection of the samples according to the clinical and biological variables Heterogeneity of the clinico-pathological variables between datasets should be evaluated and possibly adjusted Sample size assessment should be processed a priori |
| Pre-processing step | Latent unwanted structure embedded in data has the potential to dramatically impact the analysis Importance of preprocessing procedures on subsequent data analysis is neglected When translating the classifier at bedside, pre-processing a unique sample is challenging |
Raw data should be used for all subsequent analysis All the pre-processing steps should be described explicitly (including the normalization, the re-scaling and the correction for adjustment variables employed) Pre-processing code and pre-analytical plots showing the structure of the data should be provided Use of reference or housekeeper features. Use of universal or control samples as reference to be processed simultaneously to the patient sample. |
| Statistical analysis | Multiple comparisons Resubstitution bias Large p - small n Robustness of the model (multiple local optima) |
Report multiple correction adjustments for all statistics Validation data should be kept entirely and wholly separated from the training data to ensure no potential for contamination Apply methods that address the large p small n problem (e.g. ridge regression, lasso, principal component regression, partial least squares, etc.) Internal assessment of the performance of the model (cross-validation, bootstrapping) The analysis method and the code used to process it should be made publicly available |
| Performance assessment | Generalization of the model in other dataset(s) Kaplan-Meier plots and heatmaps are not adequate to assess the performance of the model Medical or biological utility |
Stability of the performance must be validated in external dataset(s) ROC AUC is relevant for binary endpoints; RMSE and R2 are relevant for continuous endpoint; time dependent AUC or concordance index are relevant for survival endpoints. The performance of the classifier must be compared with existing standard estimators |
| Clinical development | Routine measurement of molecular classifiers is limited Current clinical trial designs do not incorporate biomarkers Limits on testing of archival pathology specimens |
Expand the routine capture of pathological specimens and data in the clinic. Incorporate biomarker validation in the design of clinical trials (crosstalk with biostatistician required) Ensure the samples of the patients enrolled in ongoing and future prospective trials are preserved for subsequent and unplanned analysis provided appropriate consents are given. |
| Translation at bedside | Molecular data derived from current sampling standards are highly context specific (intra-tumoral heterogeneity, treatment effect, host effect, etc.) Uncertainty of the results of the classifiers are rarely revealed to the oncologist Poor training in modern technologies and methods is a major limit in the translation of molecular classifiers at bedside |
Increase the number of tumor samples to achieve a better representation of the disease. (Sequential biopsies, primary and metastatic sites) Confidence intervals of the results should be provided to the oncologist for the decision to be made in the patient’s context. Promote the cross-training of oncologists and cancer biologists in computational biology, systems biology and biostatistics |
PART 1: Experimental design and data pre-processing
Importance of experimental design
As in any scientific study, thoughtful experimental design increases the chance that the question being explored can be answered by the experimental data collected. A justified critique of many molecular signatures is that too little attention is paid toward typical statistical issues such as proper experimental design, sample size planning, patient selection and clinical data curation (4). As with clinical trials, appropriate selection of a patient cohort, endpoint of interest, and sample size determination must be performed a priori. Other common errors include the unbalance of clinico-pathological, treatment and survival characteristics between training and validation cohorts. Particularly, the incompatibility of follow-up between data sets results in responses that may not be comparable.
With regards to sample size calculation, several web accessible tools are available to ensure adequate statistical power (5,6). Fig. 1A presents the results of the data curation process for a gene expression classifier designed to predict three year overall survival in patients with early stage non-small cell lung cancer (NSCLC), which will be a motivating example throughout this review.
Figure 1.

Overview of the pre-processing framework. Effects on the structure of the data are represented by principle component plots for four NSCLC gene expression datasets processed separately. (A) A Table to represent the number of raw data (CEL files) included in study as a result of the data curation process. As the classifier is for early-stage patients, an explicit decision was made to only include those who are pathological stage IA to IIIA, who did not receive induction or adjuvant chemotherapy and patients for whom overall survival (OS) data are available. In addition, only patients who underwent complete tumor resection were included. Finally, gene expression outliers were identified graphically and removed from further analyses. (B) Unprocessed data analyzed by principal component analysis plot (C) Effect of five widely used unsupervised (RMA (Robust Multi-array Average) (71), gcRMA (GC Robust Multi-array Average) (72), MAS5.0 (Affymetrix Multiarray Suite 5.0) (73), dCHIP (DNA Chip Analyzer) (74) and fRMA (frozen Robust Multiarray Analysis) (75)) and one supervised (SNM) (14) normalization methods on the structure of the data. The effect of normalization on only the patients included in the Directors Challenge dataset are shown in the callouts from SNM and RMA with batch represented by different colors on principal component plot. (D) Principal component plot of SNM normalized data normalized to unit variance and 0 mean. The data and code to generate these plots are made available (see supplementary material).
Quality assessment of molecular data
Pre-analytical quality assessment of the molecular data is necessary not only when processing ‘raw’ data (data collected directly from the assay platforms prior to normalization) but continuously throughout all steps of data analysis. Methods for assessing global structure in the data, such as principal component analysis (PCA) and clustering, are used to detect outliers, or confounding artifacts in the data that must be abated before data modeling may proceed (7–10). To this end, a number of publicly available tools such as arrayQualityMetrics (9), EDASeq (10), or FastQC (Babraham institute, UK) are widely used.
Inherent biases in high dimensional data
Many high-dimensional -omic technologies estimate the abundance of targeted elements by measuring the signal of labeled probes designed to hybridize to the specific targets (features) (7,11). These signal intensities are commonly represented by a matrix of n by p elements where n is the number of samples and p is the number of molecular features. The objective of any analysis using high-dimensional molecular data is to infer the relationships between biological or clinical endpoints. Complicating these analyses is the presence of unwanted variables that arise from the specific technology platform, study design or uncontrolled biological sample heterogeneity (7,10,12–16). In most cases, technical artifacts such as dye, probe, platform, technician, and run-time batch are the most common source of latent structure in the data. Known and unknown biological variability not related to the design and endpoints of a study can also influence detecting signal above noise (14). For instance, histological grade is often associated with higher necrosis in the tumor tissue and ultimately affects signal intensities but may have little influence on the predicted clinical endpoint (17). The objectives of these pre-processing methods are (i) to remove all latent structure and technical artifacts seen in the data while (ii) preserving the influence of the biological variables of interest.
Background correction
Since the binding or hybridization events at the core of most array-based –omics technologies are stochastic, there is a degree of non-specific binding that alters the signal and must be accounted for (7,9). Many vendors provide adequate software or hardware design to explore and reduce the influence of non-specific binding. However, accepting these default corrections may also introduce additional biases.
While next generation sequencing (NGS) technologies obviate many biases present in array technologies, and therefore do not require background correction, they give rise to new unwanted biases such as base-call error and coverage biases—and others that have yet to be fully elucidated (10,11,16).
Normalization
The objective of normalization or standardization is to make the data comparable across experiments by making the distributions the same. Many studies aiming to develop oncologic molecular predictors, including the original NSCLC studies discussed in detail in this review, utilize unsupervised normalization methods (18–21). Unsupervised normalization methods (e.g. linear scaling, cross-validated splines (22), running median lines (23), loess smoothers (7) and quantile normalization (7)) remove bias across samples perceived to be due to technical variation blind to the experimental design and biological differences. The types of transformations applied in these methods vary widely and it is exceedingly important to understand that each impact the downstream model performance differently. Oncologists should note that, as with any normalization procedure, these techniques may obscure the biological signal while removing the latent structure of the data, making quality control challenging.
Several drawbacks render these methods difficult to translate into clinical practice. First, they require large datasets to perform adequately. Consequently, normalization cannot be applied on individual samples. Second, training and testing sets must be pre-processed together, necessitating simultaneous access to full training and testing datasets. As an example of the differences between five commonly used methods, we visualize the individual results on four publicly available early-stage NSCLC Affymetrix gene expression datasets (Fig. 1). PCA of the results clearly reveal that: (i) all methods transform the structure of the data (Fig. 1B), (ii) these transformations are different across normalization methods (Fig. 1B), (iii) intra-study normalization (Fig. 1C RMA and SNM call-out) does not remove artifactual segregation between studies requiring further inter-study rescaling (Fig. 1D). These differences highlight the importance of beginning with raw data when developing molecular signatures to minimize hidden biases made by previous assumptions. Unfortunately, raw data are often not available for subsequent analysis (Supplementary Table 1) (22). Searching for consistent patterns across multiple high-dimensional molecular datasets can also be done using meta-analysis techniques (24–26). Differences in individual study sample sizes and patient populations can often not be taken into account when study-level estimates are used. Pooling ‘raw’ or patient-level data and fitting appropriately stratified models across studies, while complex, is the only way to sufficiently control for these biases (25,26).
Supervised normalization
Supervised normalization incorporates information about confounding variables and variables of interest that can dramatically affect the performance of high-dimensional molecular models (12,27,28). Although experimental batch is the most frequently recognized source of latent structure, other environmental (29), genetic (30) and demographic (31) effects are inherent in each individual experiment. Several methods like SVA, SNM and ComBat (13,14,32) are designed to solve the effects of these variables on data structure, but their employment is rarely described in detail in molecular signature papers (17,33).
Specific pre-processing procedures for next generation sequencing (NGS)
NGS is a rapidly evolving field and its data are increasingly incorporated in the development of classifiers (34–36). Furthermore, these technologies have been used to elucidate many important biological differences among pediatric tumors. Recently, the International Cancer Genome Consortium (ICGC) PedBrain Tumor Project demonstrated the excellent use of NGS to elucidate the genetic complexities inherent in medulloblastoma (37). Drawbacks similar to those seen with gene expression microarrays also exist in NGS technologies and specific pre-processing methods are required (10,11). Particularly, the short length of the nucleotide sequences produced (reads are typically 50 to 150 nucleotides in length) necessitates assembly and annotation frameworks when reconstructing the genome for variant analysis purpose (such as with detecting SNPs, MNPs, InDels) (38). These technologies are also increasingly used to quantify gene expression, and just as in microarray experiments, they require close consideration of latent variables when assessing a perceived signal (39). Specific pre-processing methods to estimate gene expression have been introduced: reads per kilobase per million (RPKM), GC-content normalization, normalization to “housekeeping” genes, quantile normalization) (10,40–43).
PART 2: Issues with development of classifiers in the context of high-dimensional data
Impact of high dimensional data on analysis design
In the context of molecular profiling, “high-dimensional” data are generated such that the number of features (p) is much larger than the number of samples (n) (p ≫ n). Any subsequent analysis suffers from the “curse of dimensionality,” that an association between a molecular feature and a clinical outcome of interest may occur by chance, a phenomenon known as “false discovery” (44). The most commonly utilized methods to address false discovery are based on work by Benjamini-Hochberg, Holm and Bonferroni (45–47). Additionally, there is the potential for “overfitting” of a classifier in the training dataset, which reduces the resultant classifier’s performance on new data. Until the number of samples approaches the number of features (n≈p), strategies to minimize overfitting involve reducing the dimensions of the model space (44). These methods take advantage of the high correlation between subsets of variables, virtually eliminating (principal component regression, lasso) or ‘shrinking’ (ridge regression, support vector machine, elastic net) non-essential features (48). A detailed discussion of these methods is beyond the scope of this review and has been addressed comprehensively elsewhere (49).
Little consistency across classifiers developed by different methods
Classifiers developed with different methods on the same data set often result in similar predictive performance, but exhibit little overlap in the features selected (Fig. 2). By reducing the dimensions of data, the models represent only one from multiple possible solutions (also called local optima). While we believe there is a global optimum (the best of all possible solutions), finding this solution is often computationally intractable. Sometimes, the aggregation of multiple models into a consensus classifier may result in improved predictive performance (Fig. 2). Ultimately, how the knowledgeable oncologist incorporates these different models for decision-making remains a challenging and still open question.
Figure 2.
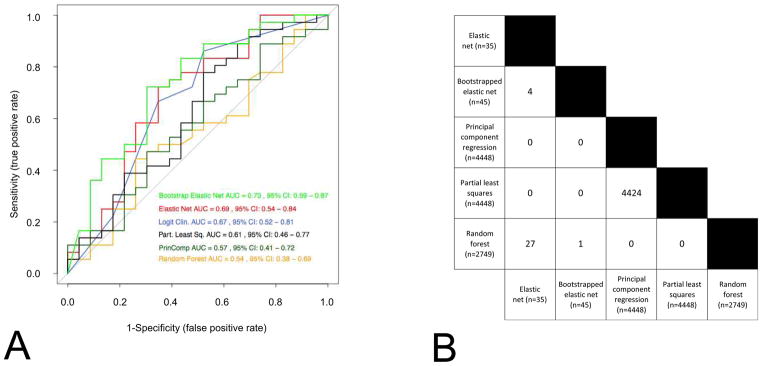
Signatures developed using different methods have similar prediction performance (Panel A) and present very little consistency with each other (Panel B). (A) Receiver-operating characteristic curves of six widely used statistical methods (logistic regression, elastic net, bootstrapped elastic net, random forest, principal component regression and partial least square regression) in predicting the probability of 3 year OS. The Director’s challenge and the Zhu et al. datasets are used as training and validation set, respectively. The ROC AUC (receiving operating characteristic curves area under the curve) and their 95% confidence interval are computed for each method. Note that all curves overlap with one another. (B) The number of features selected with each method are presented, as well as the number of genes that overlap from each method. Note the very small overlap of features across the different models, confirming that multiple and different solutions (local optima) of a same problem may lead to similar prediction results.
Internal validation performance assessment
Cross-validation and bootstrapping are the most widely used internal validation methods. These methods are performed by developing a classifier on a subset of samples and then testing the resultant classifier on held-out samples (48). Investigators must be aware that cross-validation and bootstrap methods are aimed to estimate prediction error and do not preclude testing the model in an external dataset (external validation). Internal validation methods are also used to improve the robustness of the model against noise inherent in the data.
External validation and clinical utility
Ultimately, since the clinical utility of the model is highly dependent upon its ability to correctly predict an endpoint in an external dataset, particular attention must be paid to performance metrics. A typical measure of the performance for binary classifiers (i.e. predicting binary outcome such as tumor recurrence), is based upon the receiver operating characteristic curve (ROC), which illustrates the variation of true-positive and false-positive rates along the variation of the discrimination threshold (50). In the case of predicting a continuous endpoint, root-mean squared error (RMSE) or R2 are frequently computed. When assessing time-to-event or survival endpoints the most commonly used metrics are concordance index and time-dependent AUC (4,51,52).
Oncologists should be aware that Kaplan-Meier curves and log-rank comparisons estimate differences in hazard across predicted risk groups but do not assess predictive performance. The most striking example of the disconnect between discrimination and prediction is recent work that showed random signatures in breast cancer are associated with outcome (53). The discerning clinician tasked with assessing the validity of a particular signature should demand reporting of additional statistical performance metrics. An illustrative comparison of ROC-AUC, Kaplan-Meier estimates and heatmap results is in Fig. 3. Additionally, the medical utility of any molecular model must to be formally addressed with regard to clinico-pathological covariates or scores currently used in the clinic.
Figure 3.

Comparison of receiver-operating characteristic curves, Kaplan-Meier survival prediction and heatmaps for six commonly used statistical methods (bootstrapped elastic net, elastic net, logistic regression, partial least square regression, principle component regression and random forest) ordered by performance on ROC. Log-rank test is used to report p-value of the differences of good outcome and poor outcome groups (as defined by median) for Kaplan-Meier predictors. The patients included in each group in Kaplan-Meier analysis are coded in the unsupervised clustering among the validation dataset in each heatmap (magenta for good outcome, blue for poor outcome). Many clinicians and cancer biologists reading a molecular classifier paper will expect heatmaps and Kaplan-Meier curves, yet such figures are not optimal to evaluate the model performance. However, observing a significant difference in survival between the groups does not guarantee a significant performance of the model and a performant model does not guarantee true clustering of genes in the heatmaps. Notably, the ROC curves show performance differences, while the log-rank p values are less useful in this context. Furthermore, models that perform the best here (bootstrapped ElasticNet) do not exhibit marked structure in heatmaps, while poorly performing models (principle component regression) misleadingly exhibit sharper delineation of features in their heatmaps. The logistic regression analysis was performed on clinical covariates alone, and therefore no heatmaps of gene expression can be computed.
PART 3: Issues with the effective translation of the classifiers into the clinic
Translating modern classifiers to the bedside requires not only robust preprocessing and analytical methods, but also the infrastructure for incorporating high-dimensional molecular data into the clinical and translational research.
A first critical issue relates to the clinical environment of the assays used to generate the molecular data. Currently, a unique biopsy is performed on one tumor site per patient at a single time point over the course of the disease. However, a growing body of evidence demonstrates that the molecular data derived from ”single site, single time point” biopsies are highly context-specific and may provide a biased representation of the disease state (54–56). Indeed, these data can vary depending on biopsy location given intratumoral heterogeneity (54) and the discordance between the primary site and metastases (56). The time at which a sample is obtained is also a source of variation since the relevance of an oncogenic driver may change along the sequence of antineoplastic treatments (55) or during the natural history of the disease itself (56). Multiple assessments are usually not perfomed at bedside due to technical, safety and ethical constraints. However, emerging technologies, such as circulating tumor cells (57), circulating DNA (58) or next generation functional imaging could allow for dynamic sampling.
Secondly, mechanisms must be in place for appropriate clinical evaluation of classifiers to concretely translate them to the bedside. Although retrospective analysis on completed prospective trials can be performed in certain circumstances, true prospective validation remains the gold standard (59). Several prospective clinical trial designs are optimized to validate biomarkers: biomarker-stratified design, enrichment design, biomarker strategy design, multiple hypothesis design, maker based strategy design (60–65). Unfortunately, these designs were developed for the phase I–II context, and are thus not powered to capture the complexity of cancer biology and do not support inference analyses in large populations. Recently introduced adaptive signature designs address this caveat, using randomized phase III trial designs to develop and validate classifiers (66,67).
The recent scandal at Duke surrounding the use of microarrays to drive clinical trials highlights the need for peer access to both the data and methods used to generate complex biomarkers. As discussed in the recent National Academies report, “Toward Precision Medicine” (1), before a prospective clinical trial incorporating molecular classifiers is begun, the analytic process by which these classifiers are generated must be reproducible and transparent. Furthermore, all developmental assumptions about clinical endpoints and parameterization of the models should be made explicit, including explicit annotations of treatment and inclusion criteria that are distributed with the data in standard format like MIAME (68). Taking a cue from the world of software development, genomics researchers are progressively incorporating improved operating principles such as version control for scientific source code in platforms favoring open access such as GitHub, and displaying enriched content in dedicated sites such as COSMIC, cBioPortal, GenePattern.
Furthermore, making these data and methods truly accessible has the potential to dramatically increase the efficiency of research by enabling the identification of new avenues of research and avoiding the futile reproduction of strategies that may not work. To this end, dedicated compute spaces allowing for transparent and reproducible collaboration could enable the access of models in real time independent of the cycle time of peer-reviewed journals. Synapse is one such system that integrates unique data URLs, provenance, cloud computing, and markup to provide a cohesive communication of a data-intensive study (69). At a fundamental level, adequate community assessment of published models mandates that authors make data and code available (60). Publishers are also beginning to introduce new ways to share data such as with Nature: Scientific Data to improve data transparency, citation and curation.
Ideally, published models should provide simple interfaces where clinicians can quickly obtain predictions for individual patients based on their molecular and clinical features in a manner similar to Adjuvant! Online (70). Though, molecular classifiers are more that a simple combination of genes and specific issues need to be addressed when translating these tools at bedside for a unique patient setting (Fig. 4). In that regard, the modern oncologist must come to terms with the ever-changing nature of genomic science, with the ultimate risk of being left behind.
Figure 4.

Challenges in the translation at bedside of a validated molecular classifier. Described are the steps taken by the modern oncologist when obtaining a prediction of a validated classifier for a single patient. Passing through these steps, particular problems and their potential solutions are highlighted in red boxes. To embrace precision medicine, the modern oncologist needs to develop or access competencies in molecular biology and in computational biology, in addition to clinical oncology.
Conclusion
Many investigators believe that effective analysis of high dimensional molecular data is the key to controlling cancer. It is a widespread assumption that molecular classifiers represent the foundation of individualized oncologic care. To optimize the translation of these tools in to the clinic, the oncology community needs to be aware of the strengths and limitations of the specific bioinformatics and statistical methods used in the development of classifiers. Unless molecular classifiers are truly able to discern key clinical issues, the promise of precision medicine will remain elusive and the clinical impact will remain limited. The drive toward precision medicine depends on cross-disciplinary training necessary for the next generation of oncologists to lead these research endeavors. Developing competencies in cancer biology, biostatistics, computational biology, molecular biology, and computer science in addition to patient care will be crucial to training the next leaders of the field.
Supplementary Material
Acknowledgments
This work was supported by a grant U54CA149237 from the Integrative Cancer Biology Program of the National Cancer Institute. C.F. is a recipient of a fellowship from the French Cancer Research Association (ARC) (2011) and of a Young Investigator Award from the Conquer Cancer Foundation of the American Society of Clinical Oncology (2012).
Footnotes
Disclosure of conflicts of interest: The authors declare that they have no competing financial interests.
References
- 1.National Research Council of the National Academies. Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. 2011. [PubMed] [Google Scholar]
- 2.Ferté C, André F, Soria J-C. Molecular circuits of solid tumors: prognostic and predictive tools for bedside use. Nat Rev Clin Oncol. 2010;7:367–80. doi: 10.1038/nrclinonc.2010.84. [DOI] [PubMed] [Google Scholar]
- 3.Koscielny S. Why most gene expression signatures of tumors have not been useful in the clinic. Sci Transl Med. 2010;2:14ps2. doi: 10.1126/scitranslmed.3000313. [DOI] [PubMed] [Google Scholar]
- 4.Subramanian J, Simon R. Gene expression-based prognostic signatures in lung cancer: ready for clinical use? J Natl Cancer Inst. 2010;102:464–74. doi: 10.1093/jnci/djq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mukherjee S, Tamayo P, Rogers S, Rifkin R, Engle A, Campbell C, et al. Estimating dataset size requirements for classifying DNA microarray data. J Comput Biol. 2003;10:119–42. doi: 10.1089/106652703321825928. [DOI] [PubMed] [Google Scholar]
- 6.Dobbin KK, Simon RM. Sample size planning for developing classifiers using high-dimensional DNA microarray data. Biostatistics. 2007;8:101–17. doi: 10.1093/biostatistics/kxj036. [DOI] [PubMed] [Google Scholar]
- 7.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 8.Shi L, Campbell G, Jones WD, Campagne F, Wen Z, Walker SJ, et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat Biotechnol. 2010;28:827–38. doi: 10.1038/nbt.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kauffmann A, Gentleman R, Huber W. arrayQualityMetrics--a bioconductor package for quality assessment of microarray data. Bioinformatics. 2009;25:415–6. doi: 10.1093/bioinformatics/btn647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11:733–9. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:1724–35. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mecham BH, Nelson PS, Storey JD. Supervised normalization of microarrays. Bioinformatics. 2010;26:1308–15. doi: 10.1093/bioinformatics/btq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oberg AL, Bot BM, Grill DE, Poland GA, Therneau TM. Technical and biological variance structure in mRNA-Seq data: life in the real world. BMC Genomics. 2012;13:304. doi: 10.1186/1471-2164-13-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taub MA, Corrada Bravo H, Irizarry RA. Overcoming bias and systematic errors in next generation sequencing data. Genome Med. 2010;2:87. doi: 10.1186/gm208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen C, Grennan K, Badner J, Zhang D, Gershon E, Jin L, et al. Removing batch effects in analysis of expression microarray data: an evaluation of six batch adjustment methods. PloS One. 2011;6:e17238. doi: 10.1371/journal.pone.0017238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shedden K, Taylor JMG, Enkemann SA, Tsao M-S, Yeatman TJ, et al. Director’s Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14:822–7. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu C-Q, Ding K, Strumpf D, Weir Ba, Meyerson M, Pennell N, et al. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J Clin Oncol. 2010;28:4417–24. doi: 10.1200/JCO.2009.26.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou J, Aerts J, Den Hamer B, Van Ijcken W, Den Bakker M, Riegman P, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PloS One. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schadt EE, Li C, Ellis B, Wong WH. Feature extraction and normalization algorithms for high-density oligonucleotide gene expression array data. J Cell Biochem Suppl. 2001;(Suppl 37):120–5. doi: 10.1002/jcb.10073. [DOI] [PubMed] [Google Scholar]
- 23.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:RESEARCH0032. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A. 2004;101:9309–14. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marot G, Foulley J, Mayer C, Jaffrézic F. Moderated effect size and P-value combinations for microarray meta-analyses. Bioinformatics. 2009;25:2692–9. doi: 10.1093/bioinformatics/btp444. [DOI] [PubMed] [Google Scholar]
- 26.Campain A, Yang YH. Comparison study of microarray meta-analysis methods. BMC Bioinformatics. 2010;11:408. doi: 10.1186/1471-2105-11-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spielman RS, Bastone LA, Burdick JT, Morley M, Ewens WJ, Cheung VG. Common genetic variants account for differences in gene expression among ethnic groups. Nat Genet. 2007;39:226–31. doi: 10.1038/ng1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572–7. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- 29.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schadt EE, Lamb J, Yang X, Zhu J, Edwards S, Guhathakurta D, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nature Genet. 2005;37:710–7. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 32.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–27. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 33.Scherer A, editor. Batch Effects and Noise in Microarray Experiments: Sources and Solutions. Chichester: John Wiley & Sons; 2009. [Google Scholar]
- 34.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–8. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones DTW, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–5. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pop M, Salzberg SL. Bioinformatics challenges of new sequencing technology. Trends Genet. 2008;24:142–9. doi: 10.1016/j.tig.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansen KD, Wu Z, Irizarry RA, Leek JT. Sequencing technology does not eliminate biological variability. Nat Biotech. 2011;29:572–3. doi: 10.1038/nbt.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinformatics. 2011;12:480. doi: 10.1186/1471-2105-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 42.Hansen KD, Irizarry RA, Wu Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics. 2012;13:204–16. doi: 10.1093/biostatistics/kxr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83. doi: 10.1186/gb-2012-13-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clarke R, Ressom HW, Wang A, Xuan J, Liu MC, Gehan EA, et al. The properties of high-dimensional data spaces: implications for exploring gene and protein expression data. Nat Rev Cancer. 2008;8:37–49. doi: 10.1038/nrc2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9:811–8. doi: 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- 46.Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- 47.Dunn O. Multiple Comparisons Among Means. J Am Statist Assoc. 1961;56:52–64. [Google Scholar]
- 48.Hastie T, Tibshirani R, Friedman JH. The elements of statistical learning: data mining, inference, and prediction: with 200 full-color illustrations. New York: Springer; 2001. [Google Scholar]
- 49.Ein-Dor L, Kela I, Getz G, Givol D, Domany E. Outcome signature genes in breast cancer: is there a unique set? Bioinformatics. 2005;21:171–8. doi: 10.1093/bioinformatics/bth469. [DOI] [PubMed] [Google Scholar]
- 50.Berrar D, Flach P. Caveats and pitfalls of ROC analysis in clinical microarray research (and how to avoid them) Brief Bioinform. 2012;13:83–97. doi: 10.1093/bib/bbr008. [DOI] [PubMed] [Google Scholar]
- 51.Heagerty PJ, Zheng Y. Survival model predictive accuracy and ROC curves. Biometrics. 2005;61:92–105. doi: 10.1111/j.0006-341X.2005.030814.x. [DOI] [PubMed] [Google Scholar]
- 52.Kattan MW. Evaluating a new marker’s predictive contribution. Clin Cancer Res. 2004;10:822–4. doi: 10.1158/1078-0432.ccr-03-0061. [DOI] [PubMed] [Google Scholar]
- 53.Venet D, Dumont JE, Detours V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput Biol. 2011;7:e1002240. doi: 10.1371/journal.pcbi.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bai H, Wang Z, Chen K, Zhao J, Lee JJ, Wang S, et al. Influence of chemotherapy on EGFR mutation status among patients with non-small-cell lung cancer. J Clin Oncol. 2012;30:3077–83. doi: 10.1200/JCO.2011.39.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eifert C, Powers RS. From cancer genomes to oncogenic drivers, tumour dependencies and therapeutic targets. Nat Rev Cancer. 2012;12:572–8. doi: 10.1038/nrc3299. [DOI] [PubMed] [Google Scholar]
- 57.Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359:366–77. doi: 10.1056/NEJMoa0800668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwarzenbach H, Hoon DSB, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–37. doi: 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 59.Buyse M, Sargent DJ, Grothey A, Matheson A, De Gramont A. Biomarkers and surrogate end points--the challenge of statistical validation. Nat Rev Cancer. 2010;7:309–17. doi: 10.1038/nrclinonc.2010.43. [DOI] [PubMed] [Google Scholar]
- 60.Alsheikh-Ali AS, Qureshi W, Al-Mallah MH, Ioannidis JP. Public availability of published research data in high-impact journals. PloS One. 2011;6:e24357. doi: 10.1371/journal.pone.0024357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sargent DJ, Conley BA, Allegra C, Collette L. Clinical trial designs for predictive marker validation in cancer treatment trials. J Clin Oncol. 2005;23:2020–7. doi: 10.1200/JCO.2005.01.112. [DOI] [PubMed] [Google Scholar]
- 62.Simon R. The use of genomics in clinical trial design. Clin Cancer Res. 2008;14:5984–93. doi: 10.1158/1078-0432.CCR-07-4531. [DOI] [PubMed] [Google Scholar]
- 63.Karuri SW, Simon R. A two-stage Bayesian design for co-development of new drugs and companion diagnostics. Stat Med. 2012;31:901–14. doi: 10.1002/sim.4462. [DOI] [PubMed] [Google Scholar]
- 64.Redman MW, Crowley JJ, Herbst RS, Hirsch FR, Gandara DR. Design of a phase III clinical trial with prospective biomarker validation: SWOG S0819. Clin Cancer Res. 2012;18:4004–12. doi: 10.1158/1078-0432.CCR-12-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simon R, Maitournam A. Evaluating the efficiency of targeted designs for randomized clinical trials. Clin Cancer Res. 2004;10:6759–63. doi: 10.1158/1078-0432.CCR-04-0496. [DOI] [PubMed] [Google Scholar]
- 66.Matsui S, Simon R, Qu P, Shaughnessy JD, Barlogie B, Crowley J. Developing and Validating Continuous Genomic Signatures in Randomized Clinical Trials for Predictive Medicine. Clin Cancer Res. 2012:6065–73. doi: 10.1158/1078-0432.CCR-12-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Freidlin B, Simon R. Adaptive signature design: an adaptive clinical trial design for generating and prospectively testing a gene expression signature for sensitive patients. Clin Cancer Res. 2005;11:7872–8. doi: 10.1158/1078-0432.CCR-05-0605. [DOI] [PubMed] [Google Scholar]
- 68.Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat Genet. 2001;29:365–71. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- 69.Derry JMJ, Mangravite LM, Suver C, Furia MD, Henderson D, Schildwachter X, et al. Developing predictive molecular maps of human disease through community-based modeling. Nat Genet. 2012;44:127–30. doi: 10.1038/ng.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ravdin PM, Siminoff LA, Davis GJ, Mercer MB, Hewlett J, Gerson N, et al. Computer program to assist in making decisions about adjuvant therapy for women with early breast cancer. J Clin Oncol. 2001;19:980–91. doi: 10.1200/JCO.2001.19.4.980. [DOI] [PubMed] [Google Scholar]
- 71.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J Am Statist Assoc. 2004;99:909–17. [Google Scholar]
- 73.Statistical Algorithms Description Document. 2002. Affymetrix. [Google Scholar]
- 74.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–6. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA) Biostatistics. 2010;11:242–53. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.