

Summary
Background
Single nucleotide polymorphisms in the vitamin K epoxide reductase (VKOR) gene have been successfully used for warfarin dosage prediction. However, warfarin resistance studies of naturally occurring VKOR mutants do not correlate with their clinical phenotype. This discrepancy presumably arises because the in vitro VKOR activity assay is performed under artificial conditions using the non-physiological reductant dithiothreitol.
Objectives
The aim of this study is to establish an in vivo VKOR activity assay in mammalian cells(HEK293) where VKOR functions in its native milieu without interference from endogenous enzymes.
Methods
Endogenous VKOR activity in HEK293 cells was knocked out by transcription activator-like effector nucleases (TALENs)-mediated genome editing.
Results and Conclusions
Knockout of VKOR in HEK293 cells significantly decreased vitamin K-dependent carboxylation with vitamin K epoxide(KO) as substrate. However, the paralog of VKOR, VKORC1L1, also exhibits substantial ability to convert KO to vitamin K for carboxylation. Using both VKOR and VKORC1L1 knockout cells, we examined the enzymatic activity and warfarin resistance of ten naturally occurring VKOR mutants that were reported previously to have no activity in an in vitro assay. All ten mutants are fully active, five have increased warfarin resistance with the order being W59R>L128R≈W59L>N77S≈S52L. Except for the L128R mutant, this order is consistent with the clinical anticoagulant dosages. The other five VKOR mutants do not change VKOR’s warfarin sensitivity, suggesting that factors other than VKOR play important roles. In addition, we confirmed that the conserved loop cysteines in VKOR are not required for active site regeneration after each cycle of oxidation.
Keywords: Anticoagulants, Vitamin K epoxidase, VKORC1L1 protein, warfarin, Gene Knockout Techniques
Introduction
Vitamin K epoxide reductase (VKOR), the target of warfarin, is responsible for regeneration of vitamin K from its oxidized form, vitamin K epoxide (KO). Vitamin K must be further reduced to its hydroquinone form (KH2) before being utilized as a cofactor of γ-glutamyl carboxylase (GGCX) [1] (Figure 1). GGCX post-translationally modifies vitamin K-dependent proteins involved in several biological functions including blood coagulation [2]. Warfarin, the most popular oral anticoagulant (OAC), impairs the synthesis of functional clotting factors through inhibition of VKOR and is widely used for prevention and treatment of thrombosis [3]. It is the most commonly prescribed anticoagulant worldwide with annual prescriptions equaling 0.5–1.5% of the population. However, due to its narrow therapeutic index and broad individual variability in dosing requirements, warfarin is ranked among the top ten drugs with serious adverse drug events.
Figure 1. The vitamin K cycle.
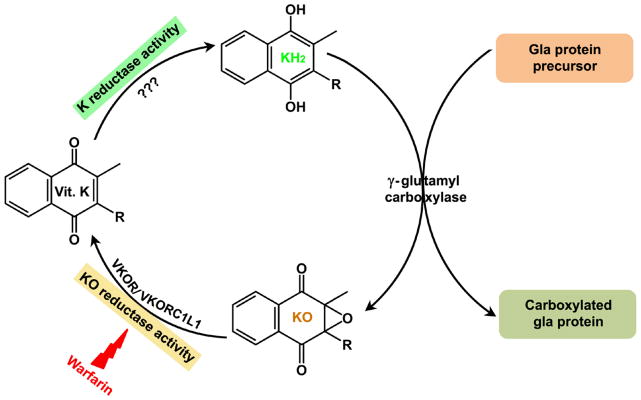
During vitamin K-dependent carboxylation, the reduced form of vitamin K (KH2) is oxidized to vitamin K epoxide (KO) by γ-glutamyl carboxylase. KO is reduced to vitamin K by VKOR or VKORC1L1 (based on this study) (KO reductase activity). This reaction is sensitive to warfarin inhibition. The reduction of vitamin K to KH2 (K reductase activity) is carried out by as yet unidentified enzyme.
Warfarin maintenance dosage can be successfully predicted by pharmacogenetic models [4]. Common genetic variation in genes of cytochrome P450 2C9 (CYP2C9) and VKOR, as well as clinical variables, explain approximately 50% of warfarin dose variability [5]. Recent studies suggest that genetic variations in VKOR have more impact than CYP2C9 on initial variability in the international normalized ratio (INR) response to warfarin [6]. Single nucleotide polymorphisms (SNPs) have been found across the entire coding as well as non-coding regions of VKOR in warfarin resistant/sensitive patients [7]. Missense mutations identified in the VKOR coding region are believed to lead to a VKOR molecule more resistant to warfarin inhibition, thus requiring higher therapeutic warfarin doses [8].
Numerous studies have examined the in vitro activity of VKOR mutants found in warfarin-resistant patients or animals [8, 9]. Except for the hotspot mutation at position 139 (in the human VKOR sequence), other mutations do not apparently change the warfarin sensitivity of VKOR. Recently, Hodroge et al. systematically studied 25 naturally occurring VKOR mutants found in patients requiring high-dose OAC [10]. This study shows that of the 25 VKOR mutants, only six had increased resistance towards OAC. Ten had essentially no in vitro activity, making it difficult to explain how these VKOR variants function in vivo. Moreover, structure-function studies of VKOR and its homologues have led to different proposals for the mechanism of action of VKOR [11, 12]. For example, whether the conserved loop cysteines are required for active site regeneration is still controversial. These discrepancies presumably arise from the in vitro activity assay being performed in an artificial environment.
To overcome these problems, we recently developed a cell-based assay for studying vitamin K cycle enzymes in their native milieu [13]. The efficiency of carboxylation of a chimeric reporter protein, FIXgla-PC (protein C with its gla domain exchanged with that of factor IX) stably expressed in HEK293 cells was used to determine the functionality of vitamin K cycle enzymes. This approach has the advantage of assessing the function of vitamin K cycle enzymes in an environment that requires the enzymes to interact with the other endogenous components [11, 13, 14]. The disadvantage, however, is that warfarin is used to eliminate the endogenous VKOR activity; thus, all the experiments must be performed in the presence of warfarin. This makes it impossible to study warfarin resistance of naturally occurring VKOR mutations and potentially compromises functional study of the wild-type enzyme.
In the present study, we improved our previous cell-based assay by knocking out the endogenous VKOR activity using TALENs, a highly specific and efficient genome-editing tool [15]. The results indicate that both VKOR and VKORC1L1 need to be knocked out to eliminate the ability of cells using KO as substrate to support vitamin K-dependent carboxylation. Using these double gene knockout cells, we measured the enzymatic activity and warfarin sensitivity of ten VKOR mutants which were previously reported to have no activity in vitro [10]. In addition, we extended our earlier studies concerning the roles of the conserved loop cysteines in VKOR to this double gene knockout cell-based assay.
Methods
TALENs construction
TALENs recognition sites that target the start region of human VKOR or VKORC1L1 in the genomic DNA sequences were designed using TALE-NT 2.0 software (https://boglab.plp.iastate.edu/) [16]. TALENs constructs were engineered using TALE Toolbox as described previously [17]. The assembled TALENs RVD-containing repeats were cloned into the corresponding pTALEN_v2 vectors provided in the TALE Toolbox kit [17]. The integrity of each assembled TALEN was verified by sequence analysis.
Transfection of TALENs constructs into HEK293 cells
HEK293 cells stably expressing FIXgla-PC (FIXgla-PC/HEK293) [13] were used for TALENs-mediated gene knockout. To knockout the VKOR or VKORC1L1 gene, plasmid DNA containing the left and right TALENs were transiently expressed in HEK293 cells. Forty-eight hours post-transfection, cells were treated with trypsin and diluted with complete medium to obtain single cell suspensions. Diluted cells were plated into 96-well plates for functional screening of the gene knockout. Alternatively, single cell suspensions of the transfected cells were seeded into 150-mm dishes. Single colonies were picked with the aid of a microscope for functional screening.
Functional screening of TALENs-mediates VKOR knockout cell clones
To functionally screen VKOR or VKORC1L1 knockout cell colonies, we cultured the transfected cells with 5 μM KO for 48 hours. Carboxylated FIXgla-PC in the culture medium was measured by ELISA as described previously [13]. Cell colonies that produced significantly less carboxylated FIXgla-PC were then cultured with 11 μM vitamin K for 24 hours, and carboxylated FIXgla-PC was again measured by ELISA. The ratio of carboxylated FIXgla-PC produced in vitamin K and KO containing medium was used as a criterion to identify candidate cell colonies for gene knockout. To ensure that the knockout cell was clonal, further screening was performed as described above.
Verifying VKOR and VKORC1L1 gene editing in genomic DNA sequence
To verify VKOR and VKORC1L1 gene editing in the candidate cells, genomic DNA was prepared using QuickExtract DNA extraction solution as described previously [17]. The TALENs targeted region in genomic DNA was amplified by PCR and the PCR products were cloned into TOPO cloning vector for sequence analysis.
Verifying VKOR gene editing in mRNA
To verify TALENs-editing of the VKOR gene at the mRNA level, total RNA from VKOR knockout cells was extracted using Trizon. RT-PCR was performed using poly T18 and M-MLV reverse transcriptase (Invitrogen). The TALENs targeted region was amplified by PCR and the PCR product was cloned into TOPO cloning vector for sequence analysis.
Characterization of VKOR or VKORC1L1 knockout cells
To characterize VKOR or VKORC1L1 knockout cells for reporter protein carboxylation, cells were cultured with different concentrations of KO, vitamin K, or KO with different concentrations of warfarin for 48 hours. Cell culture medium was used directly for ELISA to quantitate carboxylated FIXgla-PC [13].
Functional studies of VKOR or VKORC1L1 in the double gene knockout cells
For VKOR functional studies, cDNA of wild-type or mutant VKOR or VKORC1L1 was cloned into one of the multi-cloning sites of the dual mammalian expression vector pBudCE4.1 with Metridia luciferase cDNA cloned into the other multi-cloning site [11]. Metridia luciferase was co-expressed as the internal control to normalize transfection efficiency. Plasmid DNA was transiently transfected into the double gene knockout cells. Transfected cells were cultured with complete medium containing different concentrations of vitamin K, KO, or KO with different concentrations of warfarin. The cell culture medium was directly used for measuring the concentration of carboxylated FIXgla-PC and for the luciferase activity assay to normalize the transfection efficiency [11]. Wild-type VKOR activity was normalized to 100%.
Results
TALENs-mediated knockout of VKOR in HEK293 cells
To eliminate the endogenous VKOR activity of our previous cell-based assay, we used TALENs to knockout the VKOR gene in HEK293 cells stably expressing FIXgla-PC. VKOR is thought to be the enzyme responsible for reducing KO to vitamin K (KO reductase activity) while HEK293 cells possess an “antidotal” enzyme that can reduce vitamin K to KH2 (K reductase activity) [13]. We reasoned that HEK293 cells with the VKOR gene knockout would eliminate only the KO reductase activity but leave the K reductase activity intact. Therefore, we functionally screened transfected cells by measuring the efficiency of FIXgla-PC carboxylation when the cell colonies were fed with 5 μM KO. Cell colonies with reduced carboxylation of FIXgla-PC were then fed with 11 μM vitamin K to test whether their K reductase activity was maintained. Resulting colonies with low KO but high K reductase activity indicate that VKOR function has been ablated but that the cells retain normal K reductase activity (Figure 2A).
Figure 2. TALENs-mediated VKOR knockout in HEK293 cells.

(A) Functional screening of VKOR knockout HEK293 cells. The black and gray bars represent the concentrations of carboxylated FIXgla-PC in the cell culture medium when cells were fed with KO and vitamin K, respectively. Non-transfected cells produce similar levels of carboxylated FIXgla-PC when cultured with either vitamin K or KO [13]. (B) TALENs-mediated gene editing of VKOR locus in HEK293 cells. The wild-type VKOR target sequence is shown on the top. The ATG start codon is underlined. Deletions are indicated by dashes.
To further confirm that the loss of KO reductase activity is due to TALENs-editing of the VKOR gene, we extracted genomic DNA from putative VKOR knockout cells for sequence analysis. Figure 2B shows the indels (insertions and deletions) identified in the genomic DNA sequence of the selected colonies. Most colonies have the VKOR gene edited with different length deletions including the ATG start codon. Cell colonies with both alleles of the VKOR gene edited and missing the sequence GGAACCTGGAGATAATGGGC were chosen for further study.
TALENs-mediated knockout of VKORC1L1 in HEK293 cells
Interestingly, VKOR knockout cells regained KO reductase activity after culturing for several generations (Figure 3A). In addition, this activity is sensitive to warfarin inhibition as the wild-type cells (Figure 3B). To clarify whether this regained KO reductase activity is due to the expression of VKOR, we analyzed the mRNA sequence of different knockout cell colonies and confirmed that the indels in VKOR mRNA are the same as those identified in the genomic DNA (data not shown). This result suggests that VKOR is not produced in the knockout cells. Therefore, the regained KO reductase activity in the VKOR knockout cells must result from a different enzyme.
Figure 3. VKOR knockout HEK293 cells regained warfarin-sensitive KO reductase activity.

Wild-type (●) or VKOR knockout (○) cells were cultured with increasing concentrations of KO (A) or 5 μM KO and increasing concentrations of warfarin (B) for 48 hours. The concentrations of carboxylated FIXgla-PC in the cell culture medium was determined (A) and presented as percentage activities (B).
The best candidate for an enzyme that might function as VKOR is VKORC1L1 which shares 50% sequence identity with VKOR [9] (Supplementary Figure S1). To examine the contribution of VKORC1L1 to the regained KO reductase activity, we designed TALENs pairs targeting VKORC1L1 to eliminate the expression of VKORC1L1 in the VKOR knockout cells. Significantly decreased KO reductase activities in the functional screening results of the cell colonies suggest that VKORC1L1 is responsible for the regained KO reductase activity (Supplementary Figure S2). Genomic DNA sequence analysis of the candidate knockout cell colonies show that the indels found in the VKORC1L1 targeting region result in frame-shift knockouts of VKORC1L1. Interestingly, we found an insertion of a 343-base pair sequence from chromosome 5 in one of the alleles (Supplementary Figure S3).
VKORC1L1 is a warfarin-sensitive KO reductase
Cells with both VKOR and VKORC1L1 knocked out (the double gene knockout) no longer support FIXgla-PC carboxylation with KO as substrate (Figure 4A). These cells also produce significantly less carboxylated FIXgla-PC with vitamin K as the substrate (Figure 4B). These results agree with our previous results when warfarin was used to inactivate endogenous VKOR [13]. We then tested whether we could restore KO reductase activity by expressing VKOR for its function study. We transiently expressed VKOR in the double gene knockout cells and fed the cells with 5 μM KO in the presence and absence of 0.25 μM warfarin. As shown in Figure 5, no carboxylated FIXgla-PC was detected from cells that transfected with only the expression vector (control). However, expression of VKOR significantly increases the production of carboxylated FIXgla-PC. In addition, this activity is sensitive to warfarin inhibition, as expected.
Figure 4. Effect of the double gene knockout on FIXgla-PC carboxylation using vitamin K or KO as substrate.

Wild-type (●) or double gene knockout (○) cells were cultured with increasing concentrations of KO (A) or vitamin K (B) for 48 hours.
Figure 5. VKOR and VKORC1L1 in vivo activity assay in the double gene knockout cells.

VKOR or VKORC1L1 each were transiently expressed in the double gene knockout cells. Transfected cells were cultured with 5 μM KO in the absence (black bars) or presence (gray bars) of 0.25 μM warfarin. The control is transfection of the expression vector alone. Data are presented as mean ± S.D. (n=3).
To further confirm that VKORC1L1 can complement the function of VKOR, we transiently expressed VKORC1L1 in the double gene knockout cells. Figure 5 shows that VKORC1L1 can fully support FIXgla-PC carboxylation as efficiently as VKOR. This activity is abolished in the present of warfarin. These results suggest that VKORC1L1 is a warfarin sensitive KO reductase, further supporting the conclusion that VKORC1L1 is responsible for the regained KO reductase activity in the VKOR knockout cells.
Conserved loop cysteines in VKOR are not required for its function
We next sought to clarify the disputed role of the conserved loop cysteines (C43 and C51) in the function of VKOR using these double gene knockout cells. These two cysteines have been proposed to regenerate VKOR’s active site by an intra-molecular electron transfer pathway according to the in vitro studies [12, 18], but not in our previous in vivo study [14]. One possible concern with our in vivo results is that warfarin was used to inactivate the endogenous VKOR, presumably by blocking the active site but not the loop cysteines. Therefore, it is possible that the loop cysteines of the endogenous VKOR could interact with the active site of exogenously expressed loop cysteine mutated VKOR for electron transfer. Our double gene knockout cells, however, provide a clean background for clarifying this issue. Figure 6 shows that the loop cysteine mutants of VKOR behave similarly in both cell lines, confirming our previous conclusion that the loop cysteines are not required for the regeneration of VKOR’s active site.
Figure 6. In vivo activity assay of VKOR loop cysteine mutants.

Cysteine mutants of VKOR were transiently expressed in the double gene knockout cells (black bars), and transfected cells were cultured with 5 μM KO. The gray bars are data from our previous study using non-knockout cells and all VKOR variants bearing an extra warfarin resistant mutation (Y139F) [11]. Transfected cells were cultured with 5 μM KO and 4 μM warfarin.
Warfarin resistance evaluation of the naturally occurring VKOR mutants
Recently, Hodroge et al have systematically studied 25 naturally occurring VKOR mutants in intact microsomes prepared from Pichia pastoris expressing these VKOR variants [10]. Ten of these mutants had essentially no in vitro activity; therefore their warfarin resistance could not be tested. To test whether these ten VKOR mutants are active in vivo, we transiently expressed these mutants in the double gene knockout cells. Figure 7A shows that all these VKOR mutants are fully active. Mutations W59L and L128R are approximately twice as active as the wild-type enzyme.
Figure 7. Warfarin resistance studies of VKOR mutants using the double gene knockout cells.

VKOR variants were transiently expressed in the double gene knockout cells. Transfected cells were cultured with 5 μM KO (A) or 5 μM KO and increasing concentrations of warfarin (B).
To test warfarin resistance, we cultured cells transiently expressing these ten VKOR variants with 5 μM KO and increasing concentrations of warfarin. Five VKOR mutants showed increased warfarin resistance compared with wild-type VKOR (Figure 7B). The W59R mutant shows significantly increased warfarin resistance, but its activity is similar to wild-type VKOR. Furthermore, the W59R mutant has lower activity but higher warfarin resistance than the W59L mutant. These results indicate that mutations in VKOR have different effects on KO reduction and warfarin inhibition. This is consistent with a recent clinical study showing that patients with warfarin resistant VKOR mutations have normal vitamin K-dependent clotting factors in the absence of warfarin [19]. The other five VKOR mutants have similar warfarin sensitivity as the wild-type enzyme (data not shown). This result suggests that not all the naturally occurring VKOR mutants are resistant to warfarin inhibition; therefore, other factors also play important roles in warfarin resistance.
Discussion
Warfarin resistance studies of naturally occurring VKOR mutants have been hampered by the lack of an appropriate enzymatic activity assay. As an endoplasmic reticulum membrane protein, VKOR has more than half of its residues either buried inside the membrane or located in a hydrophobic region [20]. Results from characterization of the purified protein suggest that VKOR requires an intact membrane structure for its function [21]. However, most warfarin resistance studies are done in vitro in the presence of detergent with dithiothreitol to provide the reducing equivalents normally provided by unknown endogenous enzymes. These unknowns make it difficult to evaluate the effects of mutations on the activity of VKOR. Our recently described cell-based assay is a useful tool for in vivo functional studies of VKOR [11, 13, 14]; however, endogenous VKOR activity in HEK293 cells prevents its application for warfarin resistance studies, especially for those VKOR mutations with marginal resistance. Our objective in this study is to improve the previous cell-based assay for functional studies of VKOR without interference from endogenous enzymes.
For this purpose, we knocked out VKOR in HEK293 cells stably expressing the chimeric reporter protein, FIXgla-PC [13]. Unexpectedly, HEK293 cells with their VKOR knocked out can still reduce KO to support vitamin K-dependent carboxylation (Figure 3A). The retention of the KO reducatase activity was shown to be the result of VKORC1L1. Recently, Westhofen et al reported that VKORC1L1 reduces KO 18,054-fold slower than does VKOR [22]. Based on their results, they hypothesized that VKORC1L1’s primary function is to reduce vitamin K to KH2. As KH2 has been recognized as a potential biological antioxidant [23], they further proposed that VKORC1L1 functions as a housekeeping protein that mediates vitamin K-dependent intracellular anti-oxidative damage [22].
Our results, however, show that VKORC1L1 reduces KO as efficiently as VKOR to support vitamin K-dependent carboxylation (Figure 3A and Figure 5). It should be noted that the conclusion of Westhofen et al is based on the comparison of in vitro data from whole-cell lysates [22] to that of purified VKOR reconstituted into intact membranes [21]. Nevertheless, it appears that VKORC1L1 is not a key enzyme for KO reduction under physiological conditions. If it were, a significant decrease of KO reductase activity in the VKOR knockout cells would not have been observed initially (Figure 2A), and the murine knockout of VKOR would be viable [24]. The delay in onset of VKORC1L1 activity in VKOR knockout cells is difficult to explain. It may reflect the fact that overexpressing vitamin K-dependent reporter protein and/or the growth of the cells in the presence of vitamin K or KO results in the induction of VKORC1L1. However, we were unable to detect differences in mRNA level for VKORC1L1 before and after the cells regained KO reductase activity. Nevertheless, for the purpose of this study, the delay in onset of VKORC1L1 activity is immaterial since our goal was to produce a cell line without KO reductase activity.
With both VKOR and VKORC1L1 knocked out in HEK293 cells, we are able to study warfarin resistance of VKOR mutants without interference from endogenous enzymes. During the preparation of this manuscript, Fregin et al. published a warfarin resistance study [25] that used a cell-based assay similar to what we previously described [13]. Instead of using a conformation-specific antibody to assess carboxylation of FIX’s gla domain of the chimeric reporter protein, they used FIX activity to evaluate the carboxylation of full length FIX. In general, Fregin et al reached a conclusion similar to that of this study. However, they reported that 50% inhibition (IC50) of wild-type VKOR and the L128R mutant occurred at 24.7 nM and 1226.4 nM warfarin, respectively. These values are ~10-fold higher than our results from the double gene knockout cells (3.48 nM and 107 nM, respectively) (Figure 7B). The higher IC50 in their cell-based assay is probably, at least partially, caused by endogenous VKOR. An additional complication may arise from their use of vitamin K instead of KO as the substrate because HEK293 cells possess significant warfarin resistant vitamin K reductase activity (antidotal enzyme) [13].
We examined ten naturally occurring VKOR mutants that had essentially no in vitro activity [10]. All, however, are fully active in our assay. Five showed increased warfarin resistance with the order of resistance being W59R>L128R≈W59L>N77S≈S52L (Figure 7B). Except for the L128R mutant, this order is consistent with the corresponding clinical OAC dosage. As for mutations in the same position, patients with the W59R mutation required a daily dose of 18–21 mg phenprocoumon to achieve the desired anticoagulation (INR 2.5–3.5) [26], while patients with the mutation W59L required 15 mg phenprocoumon day−1 (INR 1.2) [7]. In addition, patients with S52L [27] and N77S [7] mutations, which have similar warfarin resistance in our assay, were treated with 9 mg phenprocoumon day−1 and both have an INR of <2. As the therapy was aborted for patients with mutations W59L and N77S and each patient reached a different INR level, it is hard to compare the clinical OAC dosage and the warfarin resistance in this study. However, our studies on warfarin resistance agree with the current clinical OAC dosage applied to patients.
On the other hand, a daily dose of 30 mg phenprocoumon was insufficient to achieve stable anticoagulation in a patient carrying the L128R mutation [28]. This contrasts with our cell-based assay where L128R is not the most warfarin resistant mutation (Figure 7B). In addition, there are 5 mutations that exhibit no warfarin resistance in our assay but that are reported to require high dose OAC in patients. These results suggest that factors other than VKOR play important roles in warfarin resistance.
Since the identification of the VKOR gene, SNPs in VKOR have been extensively studied for their application in managing warfarin dosage as well as understanding the mechanism of warfarin inhibition [4, 29]. The distribution of the naturally occurring VKOR mutations around the conserved loop cysteine (supplementary Figure S4) has also been used to rationalize the proposed four-transmembrane domain topology model for VKOR and the intra-molecular electron transfer pathway for active site regeneration [7, 10, 12]. However, our previous studies show that the loop cysteines are not required for VKOR activity [11, 14]. This has been further confirmed by the result from the in vivo assay with the double gene knockout cells (Figure 6). Importantly, not all the naturally occurring VKOR mutants are resistant to warfarin inhibition (Figure 7). Therefore, it is not appropriate to deduce VKOR structure-function relationship from these naturally occurring mutations without knowing the in vivo activity and warfarin resistance.
In summary, we have established a stable cell line with both VKOR and VKORC1L1 knocked out for the functional study of enzymes involved in KO reduction. We demonstrated that this double gene knockout cell line is a useful tool for characterization and evaluation of the naturally occurring VKOR mutants found in individuals requiring high-dose of anticoagulant.
Supplementary Material
Acknowledgments
We thank Dr. David Straight for helpful discussions and Dr. Laura Gabiger for editing the manuscript. This work was supported by National Institutes of Health Grants HL048318 (to D.W.S.).
Footnotes
Addendum
J.K. Tie and D. Stafford designed the experiments, analyzed the data and wrote the paper. D.Y. Jin performed the experiments and analyzed data. K. Tie (current undergraduate student of Duke University) performed the experiments.
Disclosure of conflict of interests
The authors declare no competing financial interests.
References
- 1.Stafford DW. The vitamin K cycle. J Thromb Haemost. 2005;3:1873–8. doi: 10.1111/j.1538-7836.2005.01419.x. [DOI] [PubMed] [Google Scholar]
- 2.Chatrou ML, Reutelingsperger CP, Schurgers LJ. Role of vitamin K-dependent proteins in the arterial vessel wall. Hamostaseologie. 2011;31:251–7. doi: 10.5482/ha-1157. [DOI] [PubMed] [Google Scholar]
- 3.Moualla H, Garcia D. Vitamin K antagonists--current concepts and challenges. Thromb Res. 2011;128:210–5. doi: 10.1016/j.thromres.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Eriksson N, Wadelius M. Prediction of warfarin dose: why, when and how? Pharmacogenomics. 2012;13:429–40. doi: 10.2217/pgs.11.184. [DOI] [PubMed] [Google Scholar]
- 5.Gage BF, Eby C, Johnson JA, Deych E, Rieder MJ, Ridker PM, Milligan PE, Grice G, Lenzini P, Rettie AE, Aquilante CL, Grosso L, Marsh S, Langaee T, Farnett LE, Voora D, Veenstra DL, Glynn RJ, Barrett A, McLeod HL. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther. 2008;84:326–31. doi: 10.1038/clpt.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lund K, Gaffney D, Spooner R, Etherington AM, Tansey P, Tait RC. Polymorphisms in VKORC1 have more impact than CYP2C9 polymorphisms on early warfarin International Normalized Ratio control and bleeding rates. Br J Haematol. 2012;158:256–61. doi: 10.1111/j.1365-2141.2012.09150.x. [DOI] [PubMed] [Google Scholar]
- 7.Watzka M, Geisen C, Bevans CG, Sittinger K, Spohn G, Rost S, Seifried E, Muller CR, Oldenburg J. Thirteen novel VKORC1 mutations associated with oral anticoagulant resistance: insights into improved patient diagnosis and treatment. J Thromb Haemost. 2011;9:109–18. doi: 10.1111/j.1538-7836.2010.04095.x. [DOI] [PubMed] [Google Scholar]
- 8.Pelz HJ, Rost S, Hunerberg M, Fregin A, Heiberg AC, Baert K, MacNicoll AD, Prescott CV, Walker AS, Oldenburg J, Muller CR. The genetic basis of resistance to anticoagulants in rodents. Genetics. 2005;170:1839–47. doi: 10.1534/genetics.104.040360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hortnagel K, Pelz HJ, Lappegard K, Seifried E, Scharrer I, Tuddenham EG, Muller CR, Strom TM, Oldenburg J. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427:537–41. doi: 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- 10.Hodroge A, Matagrin B, Moreau C, Foure I, Hammed A, Benoit E, Lattard V. Vkorc1 mutations detected in patients resistant to vitamin k antagonists are not all associated with a resistant vkor activity. J Thromb Haemost. 2012;10:2535–43. doi: 10.1111/jth.12019. [DOI] [PubMed] [Google Scholar]
- 11.Tie JK, Jin DY, Stafford DW. Human vitamin k epoxide reductase and its bacterial homologue have different membrane topologies and reaction mechanisms. J Biol Chem. 2012;287:33945–55. doi: 10.1074/jbc.M112.402941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature. 2010:507–12. doi: 10.1038/nature08720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tie JK, Jin DY, Straight DL, Stafford DW. Functional study of the vitamin K cycle in mammalian cells. Blood. 2011;117:2967–74. doi: 10.1182/blood-2010-08-304303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tie JK, Jin DY, Stafford DW. Mycobacterium tuberculosis vitamin K epoxide reductase homologue supports vitamin K-dependent carboxylation in mammalian cells. Antioxid Redox Signal. 2012;16:329–38. doi: 10.1089/ars.2011.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 16.Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, Bogdanove AJ. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 2012;40:W117–22. doi: 10.1093/nar/gks608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc. 2012;7:171–92. doi: 10.1038/nprot.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schulman S, Wang B, Li W, Rapoport TA. Vitamin K epoxide reductase prefers ER membrane-anchored thioredoxin-like redox partners. Proc Natl Acad Sci U S A. 2010;107:15027–32. doi: 10.1073/pnas.1009972107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrington DJ, Siddiq S, Allford SL, Shearer MJ, Mumford AD. More on: endoplasmic reticulum loop VKORC1 substitutions cause warfarin resistance but do not diminish gamma-carboxylation of the vitamin K-dependent coagulation factors. J Thromb Haemost. 2011;9:1093–5. doi: 10.1111/j.1538-7836.2011.04249.x. [DOI] [PubMed] [Google Scholar]
- 20.Tie JK, Nicchitta C, von Heijne G, Stafford DW. Membrane topology mapping of vitamin K epoxide reductase by in vitro translation/cotranslocation. J Biol Chem. 2005;280:16410–6. doi: 10.1074/jbc.M500765200. [DOI] [PubMed] [Google Scholar]
- 21.Chu PH, Huang TY, Williams J, Stafford DW. Purified vitamin K epoxide reductase alone is sufficient for conversion of vitamin K epoxide to vitamin K and vitamin K to vitamin KH2. Proc Natl Acad Sci U S A. 2006;103:19308–13. doi: 10.1073/pnas.0609401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westhofen P, Watzka M, Marinova M, Hass M, Kirfel G, Muller J, Bevans CG, Muller CR, Oldenburg J. Human vitamin K 2,3-epoxide reductase complex subunit 1-like 1 (VKORC1L1) mediates vitamin K-dependent intracellular antioxidant function. J Biol Chem. 2011;286:15085–94. doi: 10.1074/jbc.M110.210971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Lin JC, Wang H, Peterson JW, Furie BC, Furie B, Booth SL, Volpe JJ, Rosenberg PA. Novel role of vitamin k in preventing oxidative injury to developing oligodendrocytes and neurons. J Neurosci. 2003;23:5816–26. doi: 10.1523/JNEUROSCI.23-13-05816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spohn G, Kleinridders A, Wunderlich FT, Watzka M, Zaucke F, Blumbach K, Geisen C, Seifried E, Muller C, Paulsson M, Bruning JC, Oldenburg J. VKORC1 deficiency in mice causes early postnatal lethality due to severe bleeding. Thromb Haemost. 2009;101:1044–50. [PubMed] [Google Scholar]
- 25.Fregin A, Czogalla KJ, Gansler J, Rost S, Taverna M, Watzka M, Bevans CG, Muller CR, Oldenburg J. A new cell culture-based assay quantifies VKORC1 function and reveals warfarin resistance phenotypes not shown by the DTT-driven VKOR assay. J Thromb Haemost. 2013 doi: 10.1111/jth.12185. [DOI] [PubMed] [Google Scholar]
- 26.Wilms EB, Touw DJ, Conemans JM, Veldkamp R, Hermans M. A new VKORC1 allelic variant (p.Trp59Arg) in a patient with partial resistance to acenocoumarol and phenprocoumon. J Thromb Haemost. 2008;6:1224–6. doi: 10.1111/j.1538-7836.2008.02975.x. [DOI] [PubMed] [Google Scholar]
- 27.Schmeits PC, Hermans MH, van Geest-Daalderop JH, Poodt J, de Sauvage Nolting PR, Conemans JM. VKORC1 mutations in patients with partial resistance to phenprocoumon. Br J Haematol. 2010;148:955–7. doi: 10.1111/j.1365-2141.2009.08017.x. [DOI] [PubMed] [Google Scholar]
- 28.Bodin L, Perdu J, Diry M, Horellou MH, Loriot MA. Multiple genetic alterations in vitamin K epoxide reductase complex subunit 1 gene (VKORC1) can explain the high dose requirement during oral anticoagulation in humans. J Thromb Haemost. 2008;6:1436–9. doi: 10.1111/j.1538-7836.2008.03049.x. [DOI] [PubMed] [Google Scholar]
- 29.Jorgensen AL, Fitzgerald RJ, Oyee J, Pirmohamed M, Williamson PR. Influence of CYP2C9 and VKORC1 on Patient Response to Warfarin: A Systematic Review and Meta-Analysis. PLoS One. 2012;7:e44064. doi: 10.1371/journal.pone.0044064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.