

Abstract
Axonal transport is essential for the successful establishment of neuroinvasive herpesvirus infections in peripheral ganglia (retrograde transport), and the subsequent spread to exposed body surfaces following reactivation from latency (anterograde transport). We examined two components of pseudorabies virus (US3 and UL13), both of which are protein kinases, as potential regulators of axon transport. Following replication of mutant viruses lacking kinase activity, newly assembled capsids displayed an increase in retrograde motion that prevented efficient delivery of capsids to the distal axon. The aberrant increase in retrograde motion was accompanied by loss of a viral membrane marker from the transported capsids, indicating that the viral kinases allow for efficient anterograde transport by stabilizing membrane-capsid interactions during the long transit from the neuron cell body to the distal axon.
Keywords: virus, transport, neuron, axon, membrane, fusion, kinase
Neuroinvasive herpesviruses, including the human pathogens herpes simplex virus (HSV) and varicella-zoster virus (VZV) and the veterinary pathogen pseudorabies virus (PRV), establish life-long latent infections in sensory ganglia of the peripheral nervous system. These viruses have a complex infectious cycle that includes two stages of long-distance intracellular transport in axons of peripheral neurons. The first transport stage begins as extracellular virions come into contact with and enter nerve endings originating from the peripheral nervous system. Transport during this stage is dominated by retrograde microtubule-based motion that concludes when the herpesvirus capsid arrives at the nucleus of a neuronal cell resident in peripheral ganglia (i.e. trigeminal or dorsal root ganglia) (1). The second stage of transport occurs after replication of progeny viral particles in the infected peripheral neurons, usually after a prolonged period of latency. During the egress stage of infection viral particle movement is predominantly in the anterograde direction, toward the distal axon, where viral spread from the nerve endings results in infection of cells at the body surface and ultimately transmission to new hosts (2).
Transport direction in axons is dictated by factors associated with the moving viral particles and not by global changes in the infected neuron (1). Consistent with this, we recently found that targeting of PRV is coupled with membrane association. Newly infecting viruses lose the viral membrane upon entering a cell as a result of fusion of the virus envelope with the plasma membrane, and retrograde transport of the de-enveloped capsid is subsequently initiated. In contrast, PRV particles targeted to the distal axon are associated with membrane lipids and viral transmembrane proteins (3, 4). Progeny viral particles apparently need to maintain membrane association, as a subset of particles traffic back to the cell body and lack membranes. The velocities and run lengths exhibited by these misdirected particles are indistinguishable from those observed when viruses first enter neurons, suggesting that the particles have prematurely engaged the retrograde transport pathway that normally would not occur until the viral particle has spread to another cell.
How cargoes regulate their temporal trafficking in neurons is generally not well understood. PRV may use intracellular membranes to prevent capsids from engaging retrograde transport motors (i.e. dynein), while also recruiting kinesin motors to effect anterograde transport. However, the molecular events responsible for membrane recruitment as well as motor recruitment and regulation are not known. In the current study, the roles of the viral protein kinases in axon transport were examined by making mutants of PRV. Virally-encoded protein kinases are generally uncommon, but neuroinvasive herpesviruses have two, UL13 and US3, both of which are structural components of virions (5–8). Kinases have the potential to modulate intracellular virus trafficking by altering microtubule motor activity (9–11), regulating motor association to the viral particle cargo (12, 13), or by modifying viral particle composition (14).
Six mutant viruses were studied, encoding either individual or double deletions of the kinase genes or point mutations in critical catalytic residues. All viruses were made to encode an RFP-capsid fusion to allow for monitoring of virus transport dynamics in living neurons by time-lapse fluorescence microscopy. We found that viruses encoding kinase mutations exhibited an unusually high frequency of retrograde transport events during the egress phase of infection, and were inefficiently trafficked to axon terminals. Using a GFP-tagged viral membrane marker, the increase in retrograde motion was found to correlate to loss of membrane from the viral particles. These findings indicate that stable interaction of the capsid particle with a vesicular membrane is enhanced by the viral kinases and is essential for the prolonged anterograde transport ultimately necessary to spread viral infection.
Results
Isolation of viruses lacking protein kinase activity
To examine the roles of the two PRV protein kinases in axonal trafficking, viruses encoding an alanine substitution for a catalytic aspartate residue (D>A) in either or both the US3 or UL13 kinase genes were isolated (15, 16). Viruses were also made that encode deletions in the US3 and UL13 genes (Figure 1A). Each deletion allele was designed to avoid polar effects on neighboring viral gene expression. All mutations were introduced in a virus previously made to express red-fluorescent capsids to allow for imaging of individual viral particles in living neurons (referred to as “wild type” in this report) and were confirmed by restriction analysis (Supplemental Figure 1) and sequencing (data not shown) (1). UL13 and US3 are both structural components of extracellular virions, belonging to the tegument layer of proteins found between the capsid and membrane envelope (5–8, 17). Western blot analysis of purified extracellular virions confirmed that both US3 and UL13 are structurally incorporated into PRV, and documented the absence of the kinases in virions encoding deletions in either or both kinase genes (Figure 1B, and data not shown). Structural incorporation of components of the capsid (VP5), inner tegument (UL37), outer tegument (VP22) and membrane envelope (gD) were not obviously altered in the absence of kinase activity. In addition, UL13 and US3 expression and incorporation into extracellular virions were not affected by D>A substitutions (Figure 1B). Inactivation of both kinases resulted in reduced viral propagation that was comparable to that observed for the double-kinase-null virus, demonstrating the D>A substitutions were phenotypically equivalent to the loss of kinase gene expression (Figure 1C; see Supplemental Figure 2 for propagation kinetics of PRV mutated for individual kinases). The decrease in viral titers at 24 hours post-infection was comparable to that previously reported for a strain of PRV disrupted in both kinase genes (18).
Figure 1. Mutagenesis of the viral kinases.

(A) Illustration of UL13 and US3 mutations made in the PRV genome (PRV-GS847: strain Becker previously modified to express mRFP1 fused to the VP26 capsid protein). (B) Western blot analysis of extracellular purified virions. Viral kinases were detected with anti-UL13 or anti-US3 antibodies. A revertant of the double-kinase-null virus (PRV-GS1555) in which both kinase deletion alleles were repaired (rev) expressed both kinases similar to the wild type (wt). Representative proteins of each component of the virus were also examined for structural incorporation: VP5 (capsid), UL37 (inner tegument), VP22 (outer tegument), gD (envelope). (C) Kinase mutants propagate with reduced kinetics. PK15 cells were infected at MOI=5, and the cells and supernatants were harvested at the indicated times post infection. Viral titers were quantitated by plaque assay.
The viral kinases promote transport of capsids to the distal axon
Cultured dorsal root ganglia (DRG) sensory neurons were infected with either wild-type or kinase-mutant viruses. Because the US3 and UL13 kinases participate in several facets of viral infection (19–32), we sought to avoid complications from potential changes in the overall dynamics of infection that could indirectly impact bulk axon transport events by directly imaging individual capsids actively undergoing transport. Initial observations hinted at an increase number of aberrant retrograde transport events during the egress phase of infection when both kinases were absent, which was borne out by quantitative analysis (Figure 2A). These findings are noteworthy for two reasons. First, the defect was only observed when both viral kinase activities were removed. Second, the increased frequencies in retrograde motion approached the theoretical maximum of 50%, at which point an equal number of capsids moving in the anterograde and retrograde directions would imply that all progeny capsids that begin as anterograde movers leaving the cell body and moving to the distal axon would eventually reverse to aberrant retrograde motion. An alternative explanation of the mutant phenotype, that the origin of particles actively transporting to the neuronal cell body may be attributed to entry of extracellular virions late during infection is unlikely for two reasons: (i) infected cells become refractory to additional viral entry, and in DRG sensory neurons the block to infection begins at 4 hours post-infection (i.e. 8 hours before imaging was conducted) and is independent of US3 and UL13 (data not shown, and 33), and (ii) the peak amount of entering particles seen early in infection, even if sustained for 12–15 hours when imaging of egress was conducted, could not account for the number of minus-end moving particles observed during the egress phase (1).
Figure 2. Capsid transport dynamics in axons.

(A) The frequency of retrograde transport during the egress stage of infection for viruses encoding wild-type (wt and rev) or mutated kinase genes are shown. Transport of individual RFP-capsids was imaged in axons of cultured DRG sensory neurons between 12–15 hours post infection at 20 frames/s. The number of capsids undergoing retrograde events are presented as a percentage of all capsids observed actively transporting in either direction. Parental and revertant wild-type viruses are indicated (wt and rev), and reproduced findings from a previous study (2). Error bars indicate standard error (of the proportions), and asterisks designate values statistically distinct from the wild type (p < 0.0001). (n, number of capsids tracked) (B) Frequency of stalls in capsid transport during the egress stage of infection for viruses encoding wild-type (wt and rev) or mutated kinase genes. Stalls in capsid transport were scored when actively transported capsids ceased motion and failed to recover during the duration of a recording (500 frames). Infections and imaging was as described in panel A. Error bars are standard error (of the proportions). Asterisks indicate values that show deviation from the wild type (p < 0.01). (C) Retrograde and anterograde capsid transport velocities and run lengths in axons during the entry and egress phases of infection. RFP-capsid images were acquired continuously with 50 ms exposures (20 frames/s). Recordings of entry transport were acquired within the first hour post infection, while egress recordings were made between 12–14 hours post infection. Horizontal bars indicate the mean for each sample.
Based on these findings, a detailed examination of capsid transport was conducted with the double-kinase mutant viruses. During egress, anterograde moving capsids were previously noted to sometimes stop moving for extended intervals (2). In the current study, stalling frequencies during egress were measured as actively transporting capsids that stopped moving and failed to reengage transport by the end of the recording (recordings were typically 25 s in duration). The double-kinase mutant viruses exhibited increases in the frequencies of stalling events (Figure 2B). In contrast, anterograde transport velocities and run lengths were not diminished by the absence of kinase activity. We were surprised to note small, but statistically-significant (p < 0.0001), increases in retrograde motion immediately following entry into the neurons with the mutant viruses (Figure 2C). Although increases in retrograde velocity and run length should enhance the kinetics of capsid delivery to the cell body following infection at axon terminals, the associated anterograde transport defects predict that spread of progeny viral particles to axon terminals would be inefficient in the absence of kinase activity. Examination of capsid accumulation at axon terminals late during infection provided support for this prediction (Figure 3).
Figure 3. Transport of capsids to axon terminals.
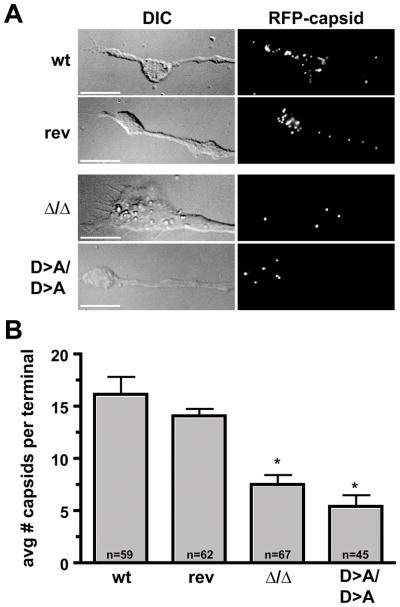
(A) DRG sensory neurons were infected with the wild-type and revertant viruses (wt and rev), the double-kinase-null virus (Δ/Δ), and the virus in which both kinases were inactivated by a single amino acid substitution (D>A/D>A). Neurons were fixed with parafomaldehyde at 14 hours post infection and axon terminal growth cones were imaged by differential interference contrast (DIC) and fluorescence microscopy. Scale bars = 10 microns. (B) Average number of capsids per growth cone (n, number of growth cones examined for each sample). Error bars are standard error (of the means) and asterisks indicate significant deviation from the wild type (p < 0.001).
The viral kinases stabilize viral particle association with membranes
In the absence of the viral kinases, the increase in retrograde motion may result from either a change in the regulation of plus- and minus-end directed microtubule motors associated with the viral cargo (which effect anterograde and retrograde transport respectively), or by a change in the composition of the viral cargo itself. PRV particles are membrane bound during anterograde transport in axons. However, sustained retrograde transport during the egress phase occurs when lipids and viral glycoproteins are absent from the viral particle (3, 4). To examine if the increase in retrograde trafficking observed in the absence of kinase activity was accompanied by loss of membrane from the transporting particles, the glycoprotein D (gD) viral transmembrane protein was fused to GFP (gD-GFP) in wild-type viruses and viruses deleted or inactivated for both kinases. The gD membrane protein is an essential component of the virion structure (34, 35). Fusion of gD to GFP did not further impair the propagation kinetics of the mutants and the gD-GFP fusion protein was structurally incorporated into extracellular virions of the mutant and wild-type viruses at equivalent levels (Supplemental Figure 3). The resulting viruses expressed RFP-capsids and the gD-GFP membrane marker, allowing for imaging of both viral components during axon transport. Following infection and replication in cultured DRG sensory neurons, progeny viral particles were predominantly detected in association with gD-GFP emissions when moving in the anterograde direction in axons (Figure 4A). We have previously estimated that the remaining subset of anterograde moving viral particles are likely also membrane bound, but with gD-GFP levels below the limit of detection (3). Consistent with the initial findings (Figure 2A), the kinase mutant viruses exhibited increased frequencies of aberrant retrograde motion. The origin of the additional retrograde moving capsids was not simple reversal events of membrane-bound viral particles, but rather was coupled to a loss of membrane from the capsids (Figure 4B). These findings support the hypothesis that membrane association is required for anterograde trafficking of viral particles.
Figure 4. Increased frequency of retrograde motion is coupled with a loss of viral membrane association.
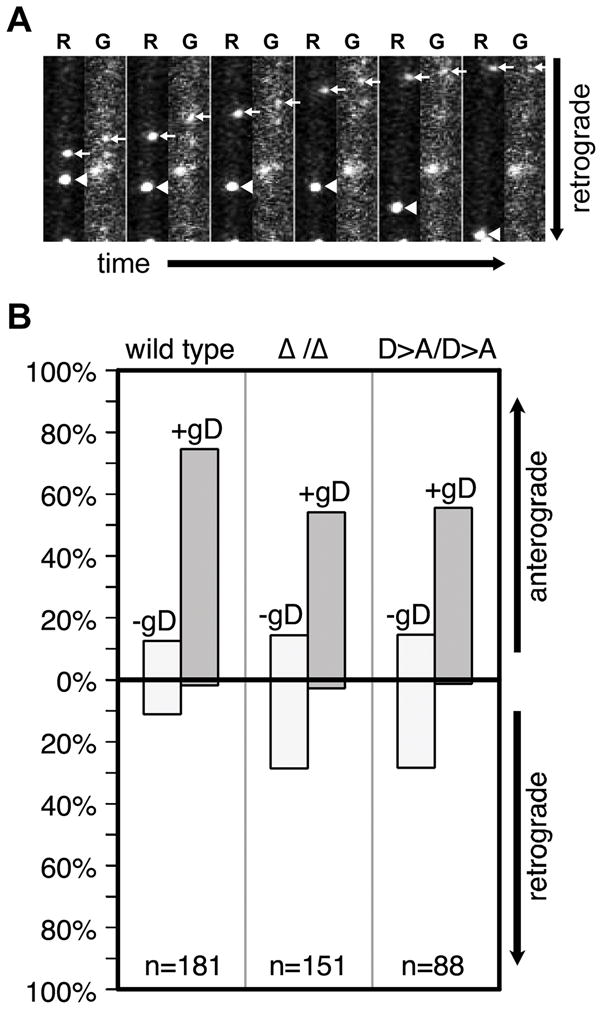
DRG sensory neurons were infected with dual-fluorescent viruses expressing mRFP1-capsids and GFP fused to the viral membrane protein, gD, and encoding either wild-type or mutated kinase genes. De novo assembled progeny virus particles were imaged and tracked in axons for red and green emissions between 12–15 hours post-infection using automated filter wheels and sequential capture of each emission spectra. This resulted in an artificial spatial shift of red and green fluorescence due to movement of the dual-fluorescent particles between red and green imaging. (A) Example of two viral particles in one axon with the double-kinase-null virus. One red capsid particle exhibits retrograde motion and lacks detectable GFP fluorescence, while the second capsid moves anterograde and is associated with GFP emissions, indicating the presence of the gD viral transmembrane protein. The frames were captured over a period of 6 seconds and are each 5.6 × 17.4 microns. (B) Quantification of mRFP1-capsid transport direction and gD association. (n, number of capsids tracked)
Retrograde transport accompanied by membrane loss is not due to the viral membrane fusion apparatus
Herpesvirus capsids mature to become membrane-bound by budding into intracellular membranes derived from the cellular secretory pathway (reviewed in 36). Because progeny viral particles undergoing anterograde transport during the egress phase of infection contain capsids, tegument proteins, lipids and transmembrane glycoproteins, these particles may be fully-assembled infectious virions (3, 4, 37). If correct, loss of membrane from the viral particle would be expected to require a membrane fusion event, for instance between the viral membrane envelope and the surrounding membrane of a cellular transport vesicle. An analogous event is required for herpesviruses to enter cells by fusing the viral envelope with the plasma membrane of a target cell (reviewed in 38). Alternatively, if capsids are bound to membranes but are not fully enveloped, loss of membrane would simply require a dissociation event (Figure 5A). To test if the viral fusion apparatus is responsible for loss of membrane from transporting viral particles during the egress stage of infection, a deletion in the gene encoding glycoprotein B (gB) was made. The gB membrane protein is required for viral membrane fusion and entry into cells (39–41). Cells made to express gB were used to propagate the gB-null virus and the resulting viral stocks were used to infected primary sensory neurons. In this way, infection of cells with a phenotypically-complemented gB-null virus can occur, but the resulting progeny viruses assembled after replication lack gB and cannot infect additional cells due to loss of membrane fusion activity (Supplemental Figure 4). When compared to neurons infected with the corresponding wild-type virus, the frequency of minus-end transport events during the egress stage of infection was not significantly changed in the absence of gB (Figure 5B). Furthermore, the increased frequency of retrograde motion observed in a virus lacking kinase activity was also gB-independent. Therefore, the loss of membrane from transporting viral particles, and the concomitant change in transport direction back to the neuronal cell body, is not dependent upon the viral fusion apparatus.
Figure 5. Minus-end motion is not dependent upon the virus fusion machinery.

(A) Illustration of two possible origins of unenveloped capsids that undergo aberrant retrograde transport in axons during the egress stage of infection. In the lower model, membrane fusion is required to release the enveloped capsid into the cytosol to initiate retrograde transport; the upper model does not require a fusion event to release the capsid from the associated membrane. (B) Cultured DRG sensory neurons were infected with gB-null viruses, and de novo assembled progeny viral particles were imaged and tracked in axons between 12–15 hours post infection. Frequencies of retrograde transport events are shown. The data support the upper model in panel A (see text). Error bars are standard error (of the proportions). (n, number of capsids tracked)
The viral kinases function locally
The findings up to this point indicate that the viral kinases function at the viral particle surface, presumably at the interface with cellular vesicles. To determine if the modulation of axon trafficking is specific for viral particles, DRG sensory neurons were labeled to steady-state with TMR-Dextran, which fluorescently labels retrograde and anterograde trafficking vesicles in axons. The labeled neurons were subsequently infected with PRV expressing both kinases or mock infected for 12 hours, and the anterograde vesicles were tracked and analyzed for transport velocities and run lengths. No significant changes in vesicle motility were observed (Supplemental Figure 5). We also note that the transport dynamics of anterograde moving vesicles were indistinguishable from the motion exhibited by anterograde moving viral particles (Figure 2C), as would be expected if the virus traffics anterograde by associating with endogenous cellular cargoes.
The findings also predict that the viral kinases are components of actively transporting capsids. To test this directly, recombinants of PRV were made that express RFP-capsids and GFP fused to either the US3 or UL13 kinase, with the intent of correlating the motion of the kinases with capsids in axons of living neurons (3, 37). However, the kinases are minor components of the PRV tegument (42), potentially making detection in actively transporting viral particles difficult. Nevertheless, US3-GFP emissions were sometimes detected from transporting capsids immediately following infection of neurons, as well as following replication in association with progeny capsids moving to axon terminals (Supplemental Figure 6). The results with US3 are consistent with previous reports indicating that US3 is capsid bound in mitotic cells (17, 43). Recombinants of PRV expressing UL13 fused to GFP at either the amino- or carboxy-terminus were also made. Unfortunately, neither produced sufficient GFP emissions for reliable detection in static extracellular virions, eliminating the possibility of examining UL13 localization on actively transported capsids by live cell imaging (data not shown). Although we expect that both kinases are components of the transported viral particle in axons, only the US3 kinase could be confirmed by live cell imaging.
Discussion
Intracellular transport of neuroinvasive herpesviruses, such as HSV and PRV, is dependent upon microtubules both in neurons and non-neuronal cells (44–50). The microtubule motors required for viral transport have not been identified although cytoplasmic dynein is implicated by its presence on intracellular capsids and the observation that disruption of dynactin, which is a cofactor for dynein as well as other motor complexes, interferes with capsid translocation (44, 51). Furthermore, dynein is the only motor known that can account for the retrograde velocities observed during delivery of newly infecting viral particles from the distal axon to the neuronal cell body (1). This initial stage of infection begins when the membrane envelope of a viral particle fuses with the axon plasma membrane, and the capsid is deposited into the cytosol (1, 3, 52, 53). The intracellular capsid, or a viral protein that remains capsid associated, is expected to recruit and activate the dynein motor complex (14, 37, 43, 54). Retrograde axon transport is essential for productive infection of the neuron and establishment of the life-long latent infection that is a hallmark of these viruses (47, 48).
Following reactivation from latency, progeny viral particles are assembled and transported back to the distal axon where viral spread results in infection of peripheral tissue and ultimately dissemination to new hosts. The composition of these anterograde-targeted viral particles has proved difficult to resolve. Separate studies applying electron microscopy imaging to cultured neurons infected with HSV have reported the presence of either membrane-enveloped capsids or membrane-free capsids in axons (53, 55). Electron microscopy of axons from HSV-infected animals has also produced opposing conclusions (56, 57), and a study of PRV-infected animals found capsids surrounded by compressed membranes that were not easily resolved (58). Electron microscopy studies of PRV in cultured neurons consistently reveal membrane-bound capsids in axons (4, 59), yet immunofluorescence methods provide little evidence of viral membrane glycoproteins associated with capsids in neurons infected with either HSV or PRV (2, 60, 61). A reinterpretation of these discrepancies by additional EM analysis concluded that both membrane-bound and membrane-free HSV capsids co-exist in axons, but infers that only the membrane-free capsids would have been transporting prior to fixation and imaging (62). Live imaging of capsids and membranes in sensory neurons infected with PRV supports the notion that both membrane-bound and membrane-free capsids co-exist in axons but, inconsistent with some previous interpretations, membrane-associated capsids actively transported in the anterograde direction towards axon terminals. Interestingly, membrane-free capsids moved in the retrograde direction (3, 4). Based on the PRV studies, capsid-membrane association appears to be a good predictor of virus transport direction in axons. The phenotypes of the kinase mutant viruses provided an opportunity to further examine this hypothesis.
PRV lacking kinase activity were slightly enhanced in the retrograde motion necessary to deliver viral capsids to neuronal nuclei, but suffered from a disproportional decrease in transport to axon terminals following replication. The decrease in anterograde transport resulted from an aberrant reversal to sustained retrograde motion during the egress stage of infection coupled to a concomitant loss of membrane association. These findings are consistent with the conclusion that membrane association targets capsid transport in axons (3), and further implicates the kinases as modulators of this process. We propose that dynein recruitment sites on the capsid surface (or on a capsid-associated protein) become inaccessible in a membrane bound state, and that by associating with an anterograde-trafficked vesicle the normally retrograde-trafficked capsid would gain a means to travel to the distal axon (with the kinases promoting the capsid-membrane interaction and thereby helping to exclude the capsid from cytoplasmic dynein during the voyage). Consistent with this model, we find that viral anterograde transport occurs with kinetics that are indistinguishable from that of endogenous axon vesicles.
We initially expected that the membrane-bound capsids would be fully-assembled infectious virions, and premature fusion of the virion envelope with the surrounding transport vesicle membrane would produce the membrane-free capsids that transport back to the neuronal cell body. However, the appearance of retrograde moving particles during the egress phase of infection did not require an active viral fusion apparatus. Although we cannot rule out that an unrecognized fusion mechanism could be responsible for membrane loss, an alternative hypothesis is that viral capsids become membrane bound and possibly invaginate into vesicle membranes, without becoming fully-enveloped virions. This model is supported by the finding that membrane-bound capsids purified from HSV-infected cells are frequently partially wrapped by vesicle membranes. Furthermore, these capsid-associated invaginated vesicles translocate along microtubules in vitro apparently by recruiting kinesin motors (63). Partially enveloped capsids are also detected in axons of sensory neurons infected with HSV (62). Although these particles were previously attributed to late budding events that may occur after anterograde transport has completed, the phenotypes of the kinase mutant viruses described here support that such particles are, at least in part, responsible for anterograde transport in axons.
US3 is reported to participate in several events during infection of mitotic cell lines, including: nuclear egress of capsids (22, 23, 25, 28–30, 64–66), cell-to-cell spread of virus mediated by cytoskeletal rearrangements (25–27, 32), and inhibition of apoptosis (31, 67–74). We do not expect that the axon transport defects described in this report are related to any of the known US3-dependent processes, as viruses mutated only for US3 have no defect in axon transport as reported here and in previous studies (75–78), and US3 is not required to block virus-induced apoptosis in sensory neurons and neuron-like PC12 cells (79–81). A US3-null PRV spreads with decreased kinetics in the mammalian nervous system, but this defect appears to be due to inefficient intercellular spread as intraaxonal transport occurred at wild-type velocities (75).
Of greater significance to this report, an overlap in US3 and UL13 function was previously noted based on a synergistic decrease in the propagation of PRV lacking both kinases, however, the process to which both kinases contributed was not known (18). Both kinases phosphorylate viral tegument and membrane proteins (5, 7, 19, 20, 82, 83) and in different instances promote either the association (42, 84) or disassociation (14) of viral proteins. In this regard, the kinases may effect efficient anterograde transport by modulating viral particle composition and strengthening interactions between viral transmembrane proteins and proteins in the tegument and capsid, thereby enhancing membrane association with the viral particle.
In summary, we find that the two virally-encoded protein kinases, US3 and UL13, act together to sustain long-distance transport of viral particles to the distal axon by preventing membrane disassociation from capsids and restricting capsids from engaging in retrograde transport events (as would normally occur following entry into cells). The kinases modulate what appears to be a delicate balance between retrograde and anterograde trafficking that occur at opposing stages of the herpesvirus infectious cycle, making them interesting candidates as targets to interfere with disease progression and neural dissemination. As far as we are aware, the increase in retrograde transport events associated with viruses lacking both kinase activities is the first defect reported for a herpesvirus that impairs the modulation of intracellular transport direction.
Materials and methods
Cells
Pig kidney epithelial cells (PK15) were used for propagation of viral stocks. Single-step growth curve analysis was used to determine viral rates of propagation, and viral titers were measured by plaque assay as previously described (85). Recombinant PRV viruses were isolated following electroporation of infectious clone plasmids into PK15 cells as previously described, or using Lipofectamine 2000 (37). A Vero cell line (N7) stably expressing PRV glycoprotein B was previously described (86). N7 cells were maintained in 10% FBS and penicillin/streptomycin to which G418 (35 μg/ml) was added at every third passage.
Peripheral sensory neurons were isolated from dorsal root ganglia (DRG) of embryonic chickens (E8-E11) and maintained as previously described (2, 87). For studies of viral entry and subsequent retrograde axonal transport, intact DRG explants were seeded on coverslips treated with poly-ornithine and laminin. Neurons were maintained in culture for 3 days to allow for axon outgrowth prior to initiating infections.
Virus construction
All recombinant viruses were derived from a previously described pBecker3 derived infectious clone, pGS847, which encodes for a fusion of monomeric red-fluorescence protein (mRFP1) to the N-terminus of the capsid protein VP26 (1). Viruses deleted for either the UL13 (PRV-GS950) or US3 (PRV-GS1015), or both genes (PRV-GS1034) were previously described (88, 89). A virus encoding the catalytically-inactive US3 kinase was made using primers: 5′ CGCATCATGCATGCGGCCGTCAAGGCGGAGAAC and the reverse complement primer. The primers encode for point mutation (underlined nucleotide) that results in the substitution of an alanine in place of an aspartate in the translated protein. Primers were used in a splicing by overlap extension PCR to produce the mutant allele, which was subsequently cloned into the pGS284 allelic exchange vector and recombined with pGS847 by RecA-dependent homologous recombination (85, 90). The resulting infectious clone, pGS976, was confirmed by sequencing. Because identifying the desired mutant infectious clone by sequencing proved to be time consuming, we first replaced the UL13 open reading frame with a kanamycin-resistance cassette by RED-GAM mutagenesis using the EL250 strain of E. coli (91). Primers used were: 5′ GCGCTCGTGTTCCTCAACGGCCGCGGGCTCAGCCACCTGGACCCAGTCACGACGTTGTAAAACG and 5′ CATGTTGCCGCACGTGCGCACAAAGATGTTGCCGCCCTTGACGAAACAGCTATGACCATGATTACG. The underlined sequences provided the UL13 homology. The resulting kanamycin-resistant infectious clone plasmid was moved back to the RecA+ E. coli strain and RecA-dependent homologous recombination was used as described above to introduce an aspartate to alanine codon change. Primers used to construct the catalytically inactive UL13 allele were: 5′ GGCTCAGCCACCTGGCCGTCAAGGGCGGCAAC and the reverse complement. The resulting infectious clone, pGS1293, was initially identified as a kanamycin-sensitive isolate and confirmed by sequencing. An infectious clone encoding both mutations was made by introducing the kanamycin-resistance cassette into UL13 of pGS976, followed by replacement with the UL13 catalytically-inactive encoding allele resulting in pGS1018. A revertant of pGS1034 was constructed by sequentially repairing both deletions by RecA-dependent homologous recombination with plasmids carrying wild-type US3 and UL13 genes, resulting in pGS1555.
A derivative of pBecker3 encoding for both RFP-VP26 and gD-GFP, pGS1236, was previously described (3). The gD-GFP allele was recombined into the pGS1018 and pGS1034 infectious clones by a two-step recombination protocol resulting in pGS1892 and pGS1616 respectively (92). The ΔgB (UL27) virus was also constructed by two-step recombination using primers: 5′ ACACCACGCTGCAGCTGCGCGGGGCGCCGTCGCGCTAGCGTAGCCCCCTCCCGCGGGAAAAGGATGACGACGATAAGTAGGG and 5′ AAACAAGCGCATCTTTATTGTTTCCCGCGGGAGGGGGCTACGCTAGCGCGACGGCGCCCCGCCAACCAATTAACCAATTCTGATTAG. Each primer encodes homology to UL27 both upstream and downstream of the sequence to be deleted (underlined) and to the pEPKan-S2 template plasmid (92). The deletion leaves intact the first 130 bp of the UL27 ORF to keep intact the overlapping UL28 ORF (The UL28 stop codon is indicated in bold in the primers). The recombination was used to modify the infectious clone pGS1236 and pGS1018, resulting in pGS1901 and pGS2347 respectively.
A dual-fluorescent virus encoding mRFP1-VP26 and US3-GFP (PRV-GS1504) was isolated by recombining a PCR product encoding the kanamycin-resistance gene flanked by FLP-recombination target (FRT) sites and adjacent to the GFP coding sequence, into pGS847. Primers used for PCR amplification were: 5′ CTTCCGCTGATGAGATACTCAACTTTGGAATGTGGACCGTAGCAGACCCACGACCAGTTGCTACTATGGTGAGCAAGGGCGAGG and 5′ AAAGGTGTGTGTGTCCTACCGCTCGGAGCCGGGCCGTTTTACCGCCCGAGAAGTTCC. The underlined sequence is derived from PRV-Becker, and provided the homologies necessary to insert the GFP coding sequence in-frame with the 3′ end of the US3 open reading frame (with an eight codon linker at the fusion junction). A mRFP1-VP26/GFP-UL13 dual-fluorescent virus (PRV-GS2651) was constructed using the two-step recombination method (92). Because the 3′ end of UL14 overlaps with the 5′ end of UL13, primers were designed to preserve the 3′ end of UL14 while inserting GFP in frame with the UL13 ORF: 5′ CGACGAGGACGAGGGGGAGGAGGCGGACGAGGCCCTGCTGACCCAGTGGCTGCTGGAGGAGGCGGAGGAGGCGTGACCATGGTGAGCAAGGGCGAGGAG and 5′ GGCGCGGACGTGCCGCGGTGGATGGGCGGCCGCGCCAGCGCGGCCCGGCTCACGCCTCCTCCGCCTCCTCCAGCAGCCTTGTACAGCTCGTCCATGCC. In the first primer, the underlined sequence is homologous to UL14 and ends with the UL14 stop codon (TGA), and the highlighted base indicates a point mutation designed to destroy the endogenous UL13 start codon (ATG>GTG). In the second primer, the underlined sequence is homologous to UL13 and includes a reconstruction of the first 11 codons that were lost from the overlap with UL14. The 3′ ends of both primers are designed to amplify the GFP ORF. As a result of this recombination, the overlap between the UL13 and UL14 ORFs is removed and the GFP coding sequence is placed in-frame with the new 5′ end of the UL13 ORF. For production of the mRFP1-VP26/UL13-GFP dual-fluorescent virus (PRV-GS2639), a two-step recombination reaction was carried out with primers: 5′ CGGTGGCCCCGCTGCTCGAGCTCGTGGCCCGGTTCTGCGGCGAGGACGGCGGCGCTCGTTTTGCCGAACTCGCTGCCGTGAGCAAGGGCGAGGAGC and 5′ CCGCGCAGGAACCGCAGGAAGGTGCGCGCGGCCAGCTCCTCAGGCAGCGAGTTCGGCAAAACGAGCGCCGCCATCCTTACTTGTACAGCTCGTCCATGC. The 3′ end of UL13 overlaps with the 5′ end of UL12, so this primer pair was designed to fuse GFP to the 3′ of UL13 while reconstructing the translation start site and 5′ end of UL12 downstream of the GFP insertion. The first primer encodes homology to the 3′ end of UL13 (without the translation stop; underlined sequence) fused in-frame to the GFP sequence in the 3′ end of the primer, and also encodes a silent point mutation (indicated in bold) destroying the start codon of UL12 (ATG>ACG). In the second primer, the underlined sequence is homologous to UL12 and includes a reconstruction of the first 11 codons that were lost from the overlap with UL13, including an optimized Kozak sequence. The 3′ ends of both primers are designed to amplify the GFP ORF. As a result of this recombination, the overlap between the UL12 and UL13 ORFs is removed and the GFP coding sequence is placed in-frame with the new 3′ end of the UL13 ORF.
Fluorescence microscopy
All images were captured with an inverted widefield Eclipse TE2000-U microscope (Nikon) fitted with a 60x 1.4 numerical aperture objective, automated fluorescence filter wheels (Sutter Instruments), and a Cascade 650 camera (Roper Scientific). The microscope was housed in an environmental box maintained at 37°C (Life Imaging Services). Images were acquired and processed using the Metamorph software package (Molecular Devices).
Capsid transport in live primary sensory neurons during entry was imaged up to 1 hour post-infection and egress was imaged between 12–15 hours post-infection in chambers sealed with a 1:1:1 mixture of Vaseline, lanolin and beeswax. Time-lapse imaging of mRFP1 emissions alone was achieved by automated capture with a 50 ms exposure time, resulting in 20 frames/sec. Time-lapse imaging of mRFP1 and GFP emissions was achieved by automated sequential captures with 200 ms exposures for each channel.
Endogenous carrier vesicles were imaged using similar parameters. Three days post seeding DRG sensory neurons onto poly-ornithine treated coverslips, the cultures were incubated with 0.2 mg/ml of TMR-Dextran for 12 hours. For studies of infected cells, neurons were infected with GFP-capsid virus (PRV-GS443) (2) at the time of TMR-Dextran addition. The neurons were washed three times with prewarmed culture media, and imaging as described above using 50 ms exposure times. Anterograde and retrograde carrier vesicles were labeled by this method, but only the anterograde trafficked vesicles were analyzed for transport dynamics.
Transport dynamics of run length, velocity, and stalls were measured using the kymograph function provided as part of the Metamorph imaging package. Continuous runs of transport were identified as uninterrupted vertical lines in the resulting plots of distance versus time. Each run was measured for distance traveled and average velocity (slope).
For quantifying capsid accumulation at axon terminals, dissociated DRG from and E9 chicken embryos were infected with wild type (PRV-GS847), revertant (PRV-GS1555), Δ/Δ (PRV-GS1034), or D>A/D>A (PRV-GS1018) viruses. Neurons were washed with phospho-buffered saline (PBS) and fixed at 14 hours post-infection with 3.2% formalin, and the number of capsids (identified as red fluorescence punctae) were counted for each axon terminal.
Extracellular virus purification and western blot analysis
For each mutant virus examined, one 100 cm dish of confluent PK15 cells was infected at a multiplicity of 10. The supernatant was harvested at 17 hours post infection and the cell debris was removed by centrifugation at 5000 rpm for 30 min at 4°C. The cleared supernatant was transferred to a Beckman SW28 centrifugation tube, and 6 ml of 30% sucrose was layered underneath. The sample was spun at 13000 × g for 30 min at 4°C. The pellet was resuspended in 100 μl of TNE (50 mM Tris-HCl, 150 mM NaCl, 10 mM EDTA) supplemented with protease inhibitor cocktail (Sigma). Infected cell lysates were made by infecting one 10 cm dish of confluent PK15s at a multiplicity of infection of 10. The cell media was removed at 17 hours post infection and the cells were harvested and washed three times with PBS at 4°C. Cells were resuspended in 1 ml of 2x final sample buffer (6.25 mM Tris [pH 6.8], 10% glycerol, 0.01% bromophenol blue, 2% sodium dodecyl sulfate, 10% β-mercaptoethanol) and boiled for 3 min.
An aliquot of purified virus was combined with an equal volume of 2x final sample buffer and boiled for 3 min. Ten microliters of the sample was separated on a 8.0% sodium dodecyl sulfate-polyacrylamidegel (Lifegels) and then transferred to a Hybond ECL membrane (Amersham Pharmacia). The VP5 capsid protein was detected with mouse monoclonal antibody 3C10 (a gift from Lynn Enquist) at a dilution of 1:1000. The UL37 tegument protein was detected with rabbit polyclonal antibody D1789 at a dilution of 1:2500 (93). The VP22 tegument protein was detected with rabbit polyclonal antibody PAS236 (a gift from Lynn Enquist) at a dilution of 1:5000. The gD envelope protein was detected with mouse monoclonal antibody SC1 (a gift from Gary Cohen, Roz Eisenberg and Sarah Connolly) at a dilution of 1:50,000. The tegument protein kinase, UL13, was detected with a rabbit polyclonal anitsera (a gift from Lynn Enquist) at a dilution of 1:1000. The tegument protein kinase, US3, was detected with a rabbit antisera at a dilution of 1:8000 (32). For detection of gD-GFP expression in infectious viral particles and infected cell lysates, a mouse monoclonal GFP antibody (Santa Cruz) was used at a dilution of 1:1000. Secondary goat anti-mouse or goat anti-rabbit antibodies conjugated to horseradish peroxidase (Jackson ImmunoResearch) were used at a 1:10,000 dilution, followed by incubation with a luminol-coumericacid-H2O2 chemiluminescence solution. Exposed film was digitized with an EDAS 290 documentation system (Kodak).
Supplementary Material
Figure S1. Viral genotype confirmation by restriction enzyme analysis. Mutations of the US3 and UL13 genes were introduced into a derivative of the infectious viral clone, pBecker3 (a clone of PRV strain Becker), that was previously modified to express mRFP1 fused to the VP26 capsid protein (encoded by the UL35 gene). Shown are restriction enzyme digests of the wild type, double point mutation (D>A/D>A), and double deletion (Δ/Δ) infectious clones. D>A point mutations do not result in predicted changes in the restriction patterns, but the analysis nevertheless demonstrates that no unexpected alterations in the restriction fragments have occurred. For the kinase deletions, restriction fragments are predicted and confirmed to be altered as follows. For US3: BamHI 3984 bp decreases to 2880 bp; NcoI 6774 bp decreases to 5670 bp; PstI 6139 bp and 1328 bp are overlapped by the deletion and fuse to produce a hybrid fragment of 6363bp; SalI 2430 bp and 2587 bp are overlapped by the deletion and fuse to produce a hybrid fragment of 3913 bp. For UL13:BamHI 16303 bp decreases to 15505 bp; NcoI 3461 bp decreases to 2663 bp; PstI 3139 bp and 437 bp are overlapped by the deletion and fuse to produce a hybrid fragment of 2778 bp; SalI 5778 bp decreases to 4980 bp. Numbers at left indicate lengths of DNA standards in kbp.
Figure S2. Single-step growth curve analysis of single kinase mutants. PK15 cells were infected at MOI=5, and the cells and media were harvested together at the indicated times post infection. Viral titers were quantitated by plaque assay.
Figure S3. Propagation and structural incorporation of gD-GFP expressing viruses. (A) Fusion of GFP to the carboxy terminus of gD does not impact the already reduced propagation kinetics displayed by the kinase mutants (compare with Figure 1). PK15 cells were infected at an MOI=5, and the cells and media were harvested together at the indicated times post infection. Viral titers were quantitated by plaque assay. (B) Western blot analysis demonstrates that gD-GFP fusion protein expression (infected cell lysates) and structural incorporation (extracellular viral particles) are comparable in viruses possessing and lacking kinase activity. Detection of the PRV major capsid protein (VP5) is included as a loading control. (C) Fluorescence analysis of gD-GFP incorporation into individual extracellular capsid-containing particles is consistent with results shown in panel B. Extracellular viral particles released from infected Vero cells infected at an MOI=0.01 were imaged on cover glass with a 60x/1.4NA objective 2 days post infection. Individual viral particles were identified by mRFP1-capsid emissions, and the presence of corresponding GFP emissions were scored for each viral particle. Error bars represent standard error (of the proportions).
Figure S4. Confirmation of the gB-null phenotype. An RFP-capsid virus deleted for the gB gene (PRV-GS1901) required cells stably expressing gB to propagate and spread (by trans-complementation). A derivative of the RFP-capsid/gB-null virus encoding inactive UL13 and US3 kinases behaved similarly (not shown). The gB protein is an essential component of the viral membrane fusion apparatus.
Cultures of DRG sensory neurons were labeled to steady state with 0.2 mg/ml TMR-Dextran as a tracer of the endocytic and recycling pathways. Velocities and run lengths of labeled vesicles moving toward the distal axon (anterograde transport) were determined by kymograph analysis and plotted both for uninfected neurons and neurons infected with wild-type PRV (PRV-GS443). Horizontal bars indicate the mean for each sample.
A recombinant of PRV expressing mRFP1-capsids and US3-GFP (PRV-GS1504) was imaged following infection of PK15 cells or DRG sensory neurons. (A) PK15 cells were infected at a MOI = 0.1 and a representative plaque was imaged for fluorescence emissions 2 days post infection with a 10x/0.3 NA objective. (B) DRG explants were imaged prior to 1 hour post infection to track incoming viral capsids moving to the nucleus (top) or 12–14 hours post infection to track progeny capsids moving to the distal axon (bottom). Frames were captured with a 60x/1.4NA objective and an electron multiplying charged coupled device (EMCCD) set to high gain. Entry frames are 2.02 × 9.66 microns, and egress frames are 2.54 × 11.83 microns. The montages shown represent the brightest US3-GFP emissions that were observed. Because of the overall low emission levels of US3-GFP from single particles, a quantitative analysis of co-transport frequencies was not performed.
Acknowledgments
We are grateful to George Shubeita, Steven Gross, Lynn Enquist and Sarah Antinone for critical discussions of the data, Klaus Osterrieder for his gift of “En Passant” plasmids, Lynn Enquist for VP5, VP22 and UL13 antibodies, Gary Cohen, Roz Eisenberg and Sarah Connolly for gD antibody, Bruce Banfield for US3 antibody, and Patricia Spear for providing the N7 cell line. This work was supported by National Institutes of Health grants 1RO1AI056346 (to G.A.S.). K.E.C. received support from the training program in Immunology and Molecular Pathogenesis NIHT32AI07476.
References
- 1.Smith GA, Pomeranz L, Gross SP, Enquist LW. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc Natl Acad Sci U S A. 2004;101(45):16034–16039. doi: 10.1073/pnas.0404686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith GA, Gross SP, Enquist LW. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci USA. 2001;98(6):3466–3470. doi: 10.1073/pnas.061029798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antinone SE, Smith GA. Two modes of herpesvirus trafficking in neurons: membrane acquisition directs motion. J Virol. 2006;80(22):11235–11240. doi: 10.1128/JVI.01441-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feierbach B, Bisher M, Goodhouse J, Enquist LW. In vitro analysis of transneuronal spread of an alphaherpesvirus infection in peripheral nervous system neurons. J Virol. 2007;81(13):6846–6857. doi: 10.1128/JVI.00069-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coulter LJ, Moss HW, Lang J, McGeoch DJ. A mutant of herpes simplex virus type 1 in which the UL13 protein kinase gene is disrupted. J Gen Virol. 1993;74(Pt 3):387–395. doi: 10.1099/0022-1317-74-3-387. [DOI] [PubMed] [Google Scholar]
- 6.Overton HA, McMillan DJ, Klavinskis LS, Hope L, Ritchie AJ, Wong-kai-in P. Herpes simplex virus type 1 gene UL13 encodes a phosphoprotein that is a component of the virion. Virology. 1992;190(1):184–192. doi: 10.1016/0042-6822(92)91204-8. [DOI] [PubMed] [Google Scholar]
- 7.Cunningham C, Davison AJ, Dolan A, Frame MC, McGeoch DJ, Meredith DM, Moss HW, Orr AC. The UL13 virion protein of herpes simplex virus type 1 is phosphorylated by a novel virus-induced protein kinase. J Gen Virol. 1992;73(Pt 2):303–311. doi: 10.1099/0022-1317-73-2-303. [DOI] [PubMed] [Google Scholar]
- 8.Zhang G, Stevens R, Leader DP. The protein kinase encoded in the short unique region of pseudorabies virus: description of the gene and identification of its product in virions and in infected cells. J Gen Virol. 1990;71(Pt 8):1757–1765. doi: 10.1099/0022-1317-71-8-1757. [DOI] [PubMed] [Google Scholar]
- 9.Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. Embo J. 2002;21(3):281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaughan PS, Leszyk JD, Vaughan KT. Cytoplasmic dynein intermediate chain phosphorylation regulates binding to dynactin. J Biol Chem. 2001;276(28):26171–26179. doi: 10.1074/jbc.M102649200. [DOI] [PubMed] [Google Scholar]
- 11.Dillman JF, 3rd, Pfister KK. Differential phosphorylation in vivo of cytoplasmic dynein associated with anterogradely moving organelles. J Cell Biol. 1994;127(6 Pt 1):1671–1681. doi: 10.1083/jcb.127.6.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guillaud L, Wong R, Hirokawa N. Disruption of KIF17-Mint1 interaction by CaMKII-dependent phosphorylation: a molecular model of kinesin-cargo release. Nat Cell Biol. 2008;10(1):19–29. doi: 10.1038/ncb1665. [DOI] [PubMed] [Google Scholar]
- 13.Karcher RL, Roland JT, Zappacosta F, Huddleston MJ, Annan RS, Carr SA, Gelfand VI. Cell cycle regulation of myosin-V by calcium/calmodulin-dependent protein kinase II. Science. 2001;293(5533):1317–1320. doi: 10.1126/science.1061086. [DOI] [PubMed] [Google Scholar]
- 14.Morrison EE, Wang YF, Meredith DM. Phosphorylation of structural components promotes dissociation of the herpes simplex virus type 1 tegument. J Virol. 1998;72(9):7108–7114. doi: 10.1128/jvi.72.9.7108-7114.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241(4861):42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 16.Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. Faseb J. 1995;9(8):576–596. [PubMed] [Google Scholar]
- 17.Granzow H, Klupp BG, Mettenleiter TC. The pseudorabies virus US3 protein is a component of primary and of mature virions. J Virol. 2004;78(3):1314–1323. doi: 10.1128/JVI.78.3.1314-1323.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Wind N, Domen J, Berns A. Herpesviruses encode an unusual protein-serine/threonine kinase which is nonessential for growth in cultured cells. J Virol. 1992;66(9):5200–5209. doi: 10.1128/jvi.66.9.5200-5209.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng TI, Ogle WO, Roizman B. UL13 protein kinase of herpes simplex virus 1 complexes with glycoprotein E and mediates the phosphorylation of the viral Fc receptor: glycoproteins E and I. Virology. 1998;241(1):37–48. doi: 10.1006/viro.1997.8963. [DOI] [PubMed] [Google Scholar]
- 20.Kato A, Yamamoto M, Ohno T, Tanaka M, Sata T, Nishiyama Y, Kawaguchi Y. Herpes simplex virus 1-encoded protein kinase UL13 phosphorylates viral Us3 protein kinase and regulates nuclear localization of viral envelopment factors UL34 and UL31. J Virol. 2006;80(3):1476–1486. doi: 10.1128/JVI.80.3.1476-1486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Overton H, McMillan D, Hope L, Wong-Kai-In P. Production of host shutoff-defective mutants of herpes simplex virus type 1 by inactivation of the UL13 gene. Virology. 1994;202(1):97–106. doi: 10.1006/viro.1994.1326. [DOI] [PubMed] [Google Scholar]
- 22.Klupp BG, Granzow H, Mettenleiter TC. Effect of the pseudorabies virus US3 protein on nuclear membrane localization of the UL34 protein and virus egress from the nucleus. J Gen Virol. 2001;82(Pt 10):2363–2371. doi: 10.1099/0022-1317-82-10-2363. [DOI] [PubMed] [Google Scholar]
- 23.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol. 2002;76(17):8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calton CM, Randall JA, Adkins MW, Banfield BW. The pseudorabies virus serine/threonine kinase Us3 contains mitochondrial, nuclear and membrane localization signals. Virus Genes. 2004;29(1):131–145. doi: 10.1023/B:VIRU.0000032796.27878.7f. [DOI] [PubMed] [Google Scholar]
- 25.Schumacher D, Tischer BK, Trapp S, Osterrieder N. The protein encoded by the US3 orthologue of Marek’s disease virus is required for efficient de-envelopment of perinuclear virions and involved in actin stress fiber breakdown. J Virol. 2005;79(7):3987–3997. doi: 10.1128/JVI.79.7.3987-3997.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Minnebruggen G, Favoreel HW, Jacobs L, Nauwynck HJ. Pseudorabies virus US3 protein kinase mediates actin stress fiber breakdown. J Virol. 2003;77(16):9074–9080. doi: 10.1128/JVI.77.16.9074-9080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Favoreel HW, Minnebruggen G, Adriaensen D, Nauwynck HJ. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc Natl Acad Sci U S A. 2005;102(25):8990–8995. doi: 10.1073/pnas.0409099102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagenaar F, Pol JM, Peeters B, Gielkens AL, de Wind N, Kimman TG. The US3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J Gen Virol. 1995;76 ( Pt 7):1851–1859. doi: 10.1099/0022-1317-76-7-1851. [DOI] [PubMed] [Google Scholar]
- 29.Ryckman BJ, Roller RJ. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J Virol. 2004;78(1):399–412. doi: 10.1128/JVI.78.1.399-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bjerke SL, Roller RJ. Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology. 2006;347(2):261–276. doi: 10.1016/j.virol.2005.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leopardi R, Van Sant C, Roizman B. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc Natl Acad Sci U S A. 1997;94(15):7891–7896. doi: 10.1073/pnas.94.15.7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demmin GL, Clase AC, Randall JA, Enquist LW, Banfield BW. Insertions in the gG gene of pseudorabies virus reduce expression of the upstream Us3 protein and inhibit cell-to-cell spread of virus infection. J Virol. 2001;75(22):10856–10869. doi: 10.1128/JVI.75.22.10856-10869.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banfield BW, Kaufman JD, Randall JA, Pickard GE. Development of pseudorabies virus strains expressing red fluorescent proteins: new tools for multisynaptic labeling applications. J Virol. 2003;77(18):10106–10112. doi: 10.1128/JVI.77.18.10106-10112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ligas MW, Johnson DC. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J Virol. 1988;62(5):1486–1494. doi: 10.1128/jvi.62.5.1486-1494.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rauh I, Mettenleiter TC. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J Virol. 1991;65(10):5348–5356. doi: 10.1128/jvi.65.10.5348-5356.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mettenleiter TC. Budding events in herpesvirus morphogenesis. Virus Res. 2004;106(2):167–180. doi: 10.1016/j.virusres.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 37.Luxton GW, Haverlock S, Coller KE, Antinone SE, Pincetic A, Smith GA. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc Natl Acad Sci U S A. 2005;102(16):5832–5837. doi: 10.1073/pnas.0500803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spear PG, Longnecker R. Herpesvirus entry: an update. J Virol. 2003;77(19):10179–10185. doi: 10.1128/JVI.77.19.10179-10185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai WH, Gu B, Person S. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J Virol. 1988;62(8):2596–2604. doi: 10.1128/jvi.62.8.2596-2604.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turner A, Bruun B, Minson T, Browne H. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J Virol. 1998;72(1):873–875. doi: 10.1128/jvi.72.1.873-875.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klupp BG, Nixdorf R, Mettenleiter TC. Pseudorabies virus glycoprotein M inhibits membrane fusion. J Virol. 2000;74(15):6760–6768. doi: 10.1128/jvi.74.15.6760-6768.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michael K, Klupp BG, Mettenleiter TC, Karger A. Composition of pseudorabies virus particles lacking tegument protein US3, UL47, or UL49 or envelope glycoprotein E. J Virol. 2006;80(3):1332–1339. doi: 10.1128/JVI.80.3.1332-1339.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Granzow H, Klupp BG, Mettenleiter TC. Entry of pseudorabies virus: an immunogold-labeling study. J Virol. 2005;79(5):3200–3205. doi: 10.1128/JVI.79.5.3200-3205.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sodeik B, Ebersold MW, Helenius A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol. 1997;136:1007–1021. doi: 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mabit H, Nakano MY, Prank U, Saam B, Dohner K, Sodeik B, Greber UF. Intact microtubules support adenovirus and herpes simplex virus infections. J Virol. 2002;76(19):9962–9971. doi: 10.1128/JVI.76.19.9962-9971.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. The Pseudorabies Virus VP1/2 Tegument Protein Is Required for Intracellular Capsid Transport. J Virol. 2006;80(1):201–209. doi: 10.1128/JVI.80.1.201-209.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kristensson K, Lycke E, Röyttä M, Svennerholm B, Vahlne A. Neuritic transport of herpes simplex virus in rat sensory neurons in vitro. Effects of substances interacting with microtubular function and axonal flow [nocodazole, taxol and erythro-9-3-(2-hydroxynonyl)adenine] J Gen Virol. 1986;67:2023–2028. doi: 10.1099/0022-1317-67-9-2023. [DOI] [PubMed] [Google Scholar]
- 48.Topp KS, Meade LB, LaVail JH. Microtubule polarity in the peripheral processes of trigeminal ganglion cells: relevance for the retrograde transport of herpes simplex virus. J Neurosci. 1994;14(1):318–325. doi: 10.1523/JNEUROSCI.14-01-00318.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Topp KS, Bisla K, Saks ND, Lavail JH. Centripetal transport of herpes simplex virus in human retinal pigment epithelial cells in vitro. Neurosci. 1996;71(4):1133–1144. doi: 10.1016/0306-4522(95)00497-1. [DOI] [PubMed] [Google Scholar]
- 50.Miranda-Saksena M, Armati P, Boadle RA, Holland DJ, Cunningham AL. Anterograde transport of herpes simplex virus type 1 in cultured, dissociated human and rat dorsal root ganglion neurons. J Virol. 2000;74(4):1827–1839. doi: 10.1128/jvi.74.4.1827-1839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dohner K, Wolfstein A, Prank U, Echeverri C, Dujardin D, Vallee R, Sodeik B. Function of Dynein and dynactin in herpes simplex virus capsid transport. Mol Biol Cell. 2002;13(8):2795–2809. doi: 10.1091/mbc.01-07-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lycke E, Hamark B, Johansson M, Krotochwil A, Lycke J, Svennerholm B. Herpes simplex virus infection of the human sensory neuron. An electron microscopy study. Arch Virol. 1988;101(1–2):87–104. doi: 10.1007/BF01314654. [DOI] [PubMed] [Google Scholar]
- 53.Lycke E, Kristensson K, Svennerholm B, Vahlne A, Ziegler R. Uptake and transport of herpes simplex virus in neurites of rat dorsal root ganglia cells in culture. J Gen Virol. 1984;65(Pt 1):55–64. doi: 10.1099/0022-1317-65-1-55. [DOI] [PubMed] [Google Scholar]
- 54.Wolfstein A, Nagel CH, Radtke K, Dohner K, Allan VJ, Sodeik B. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic. 2006;7(2):227–237. doi: 10.1111/j.1600-0854.2005.00379.x. [DOI] [PubMed] [Google Scholar]
- 55.Penfold ME, Armati PJ, Mikloska Z, Cunningham AL. The interaction of human fetal neurons and epidermal cells in vitro. In Vitro Cell Dev Biol Anim. 1996;32(7):420–426. doi: 10.1007/BF02723004. [DOI] [PubMed] [Google Scholar]
- 56.LaVail JH, Tauscher AN, Hicks JW, Harrabi O, Melroe GT, Knipe DM. Genetic and molecular in vivo analysis of herpes simplex virus assembly in murine visual system neurons. J Virol. 2005;79(17):11142–11150. doi: 10.1128/JVI.79.17.11142-11150.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.LaVail JH, Topp KS, Giblin PA, Garner JA. Factors that contribute to the transneuronal spread of herpes simplex virus. J Neurosci Res. 1997;49(4):485–496. [PubMed] [Google Scholar]
- 58.Card JP, Rinaman L, Lynn RB, Lee BH, Meade RP, Miselis RR, Enquist LW. Pseudorabies virus infection of the rat central nervous system: ultrastructural characterization of viral replication, transport, and pathogenesis. J Neurosci. 1993;13(6):2515–2539. doi: 10.1523/JNEUROSCI.13-06-02515.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ch’ng TH, Enquist LW. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J Virol. 2005;79(17):10875–10889. doi: 10.1128/JVI.79.17.10875-10889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Snyder A, Wisner TW, Johnson DC. Herpes simplex virus capsids are transported in neuronal axons without an envelope containing the viral glycoproteins. J Virol. 2006;80(22):11165–11177. doi: 10.1128/JVI.01107-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Snyder A, Bruun B, Browne HM, Johnson DC. A herpes simplex virus gD-YFP fusion glycoprotein is transported separately from viral capsids in neuronal axons. J Virol. 2007;81(15):8337–8340. doi: 10.1128/JVI.00520-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saksena MM, Wakisaka H, Tijono B, Boadle RA, Rixon F, Takahashi H, Cunningham AL. Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J Virol. 2006;80(7):3592–3606. doi: 10.1128/JVI.80.7.3592-3606.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee GE, Murray JW, Wolkoff AW, Wilson DW. Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J Virol. 2006;80(9):4264–4275. doi: 10.1128/JVI.80.9.4264-4275.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Purves FC, Spector D, Roizman B. The herpes simplex virus 1 protein kinase encoded by the US3 gene mediates posttranslational modification of the phosphoprotein encoded by the UL34 gene. J Virol. 1991;65(11):5757–5764. doi: 10.1128/jvi.65.11.5757-5764.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol. 2001;75(18):8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leach N, Bjerke SL, Christensen DK, Bouchard JM, Mou F, Park R, Baines J, Haraguchi T, Roller RJ. Emerin is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both UL34 and US3. J Virol. 2007;81(19):10792–10803. doi: 10.1128/JVI.00196-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galvan V, Roizman B. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type-dependent manner. Proc Natl Acad Sci U S A. 1998;95(7):3931–3936. doi: 10.1073/pnas.95.7.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Asano S, Honda T, Goshima F, Watanabe D, Miyake Y, Sugiura Y, Nishiyama Y. US3 protein kinase of herpes simplex virus type 2 plays a role in protecting corneal epithelial cells from apoptosis in infected mice. J Gen Virol. 1999;80 ( Pt 1):51–56. doi: 10.1099/0022-1317-80-1-51. [DOI] [PubMed] [Google Scholar]
- 69.Jerome KR, Fox R, Chen Z, Sears AE, Lee H, Corey L. Herpes simplex virus inhibits apoptosis through the action of two genes, Us5 and Us3. J Virol. 1999;73(11):8950–8957. doi: 10.1128/jvi.73.11.8950-8957.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Munger J, Roizman B. The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc Natl Acad Sci U S A. 2001;98(18):10410–10415. doi: 10.1073/pnas.181344498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cartier A, Komai T, Masucci MG. The Us3 protein kinase of herpes simplex virus 1 blocks apoptosis and induces phosporylation of the Bcl-2 family member Bad. Exp Cell Res. 2003;291(1):242–250. doi: 10.1016/s0014-4827(03)00375-6. [DOI] [PubMed] [Google Scholar]
- 72.Ogg PD, McDonell PJ, Ryckman BJ, Knudson CM, Roller RJ. The HSV-1 Us3 protein kinase is sufficient to block apoptosis induced by overexpression of a variety of Bcl-2 family members. Virology. 2004;319(2):212–224. doi: 10.1016/j.virol.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 73.Geenen K, Favoreel HW, Olsen L, Enquist LW, Nauwynck HJ. The pseudorabies virus US3 protein kinase possesses anti-apoptotic activity that protects cells from apoptosis during infection and after treatment with sorbitol or staurosporine. Virology. 2005;331(1):144–150. doi: 10.1016/j.virol.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 74.Hata S, Koyama AH, Shiota H, Adachi A, Goshima F, Nishiyama Y. Antiapoptotic activity of herpes simplex virus type 2: the role of US3 protein kinase gene. Microbes and infection/Institut Pasteur. 1999;1(8):601–607. doi: 10.1016/s1286-4579(99)80059-8. [DOI] [PubMed] [Google Scholar]
- 75.Olsen LM, Ch’ng TH, Card JP, Enquist LW. Role of pseudorabies virus Us3 protein kinase during neuronal infection. J Virol. 2006;80(13):6387–6398. doi: 10.1128/JVI.00352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klopfleisch R, Klupp BG, Fuchs W, Kopp M, Teifke JP, Mettenleiter TC. Influence of pseudorabies virus proteins on neuroinvasion and neurovirulence in mice. J Virol. 2006;80(11):5571–5576. doi: 10.1128/JVI.02589-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kimman TG, de Wind N, Oei-Lie N, Pol JM, Berns AJ, Gielkens AL. Contribution of single genes within the unique short region of Aujeszky’s disease virus (suid herpesvirus type 1) to virulence, pathogenesis and immunogenicity. J Gen Virol. 1992;73(Pt 2):243–251. doi: 10.1099/0022-1317-73-2-243. [DOI] [PubMed] [Google Scholar]
- 78.Meignier B, Longnecker R, Mavromara-Nazos P, Sears AE, Roizman B. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus 1. Virology. 1988;162(1):251–254. doi: 10.1016/0042-6822(88)90417-5. [DOI] [PubMed] [Google Scholar]
- 79.Geenen K, Favoreel HW, Nauwynck HJ. Cell type-specific resistance of trigeminal ganglion neurons towards apoptotic stimuli. Vet Microbiol. 2006;113(3–4):223–229. doi: 10.1016/j.vetmic.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 80.Geenen K, Favoreel HW, Nauwynck HJ. Higher resistance of porcine trigeminal ganglion neurons towards pseudorabies virus-induced cell death compared with other porcine cell types in vitro. J Gen Virol. 2005;86(Pt 5):1251–1260. doi: 10.1099/vir.0.80760-0. [DOI] [PubMed] [Google Scholar]
- 81.Aiamkitsumrit B, Zhang X, Block TM, Norton P, Fraser NW, Su YH. Herpes simplex virus type 1 ICP4 deletion mutant virus d120 infection failed to induce apoptosis in nerve growth factor-differentiated PC12 cells. J Neurovirol. 2007;13(4):305–314. doi: 10.1080/13550280701361490. [DOI] [PubMed] [Google Scholar]
- 82.Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J Virol. 2005;79(14):9325–9331. doi: 10.1128/JVI.79.14.9325-9331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Asai R, Ohno T, Kato A, Kawaguchi Y. Identification of proteins directly phosphorylated by UL13 protein kinase from herpes simplex virus 1. Microbes and infection/Institut Pasteur. 2007;9(12–13):1434–1438. doi: 10.1016/j.micinf.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 84.Matsuzaki A, Yamauchi Y, Kato A, Goshima F, Kawaguchi Y, Yoshikawa T, Nishiyama Y. US3 protein kinase of herpes simplex virus type 2 is required for the stability of the UL46-encoded tegument protein and its association with virus particles. J Gen Virol. 2005;86(Pt 7):1979–1985. doi: 10.1099/vir.0.80949-0. [DOI] [PubMed] [Google Scholar]
- 85.Smith GA, Enquist LW. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J Virol. 1999;73(8):6405–6414. doi: 10.1128/jvi.73.8.6405-6414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mettenleiter TC, Spear PG. Glycoprotein gB (gII) of pseudorabies virus can functionally substitute for glycoprotein gB in herpes simplex virus type 1. J Virol. 1994;68(1):500–504. doi: 10.1128/jvi.68.1.500-504.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smith CL. Cultures from chick peripheral ganglia. In: Banker G, Goslin K, editors. Culturing Nerve Cells. Cambridge: MIT Press; 1998. pp. 261–287. [Google Scholar]
- 88.Antinone SE, Shubeita GT, Coller KE, Lee JI, Haverlock-Moyns S, Gross SP, Smith GA. The Herpesvirus capsid surface protein, VP26, and the majority of the tegument proteins are dispensable for capsid transport toward the nucleus. J Virol. 2006;80(11):5494–5498. doi: 10.1128/JVI.00026-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Coller KE, Lee JI, Ueda A, Smith GA. The capsid and tegument of the alpha herpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J Virol. 2007;81(21):11790–11797. doi: 10.1128/JVI.01113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Horton RM, Cai Z, Ho SN, Pease LR. Gene splicing by overlap extension: Tailor-made genes using the polymerase chain reaction. Biotechniques. 1990;8:528–535. [PubMed] [Google Scholar]
- 91.Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG. A Highly Efficient Escherichia coli-Based Chromosome Engineering System Adapted for Recombinogenic Targeting and Subcloning of BAC DNA. Genomics. 2001;73(1):56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- 92.Tischer BK, von Einem J, Kaufer B, Osterrieder N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques. 2006;40(2):191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 93.Lee JI, Luxton GW, Smith GA. Identification of an essential domain in the herpesvirus VP1/2 tegument protein: the carboxy terminus directs incorporation into capsid assemblons. J Virol. 2006;80(24):12086–12094. doi: 10.1128/JVI.01184-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Viral genotype confirmation by restriction enzyme analysis. Mutations of the US3 and UL13 genes were introduced into a derivative of the infectious viral clone, pBecker3 (a clone of PRV strain Becker), that was previously modified to express mRFP1 fused to the VP26 capsid protein (encoded by the UL35 gene). Shown are restriction enzyme digests of the wild type, double point mutation (D>A/D>A), and double deletion (Δ/Δ) infectious clones. D>A point mutations do not result in predicted changes in the restriction patterns, but the analysis nevertheless demonstrates that no unexpected alterations in the restriction fragments have occurred. For the kinase deletions, restriction fragments are predicted and confirmed to be altered as follows. For US3: BamHI 3984 bp decreases to 2880 bp; NcoI 6774 bp decreases to 5670 bp; PstI 6139 bp and 1328 bp are overlapped by the deletion and fuse to produce a hybrid fragment of 6363bp; SalI 2430 bp and 2587 bp are overlapped by the deletion and fuse to produce a hybrid fragment of 3913 bp. For UL13:BamHI 16303 bp decreases to 15505 bp; NcoI 3461 bp decreases to 2663 bp; PstI 3139 bp and 437 bp are overlapped by the deletion and fuse to produce a hybrid fragment of 2778 bp; SalI 5778 bp decreases to 4980 bp. Numbers at left indicate lengths of DNA standards in kbp.
Figure S2. Single-step growth curve analysis of single kinase mutants. PK15 cells were infected at MOI=5, and the cells and media were harvested together at the indicated times post infection. Viral titers were quantitated by plaque assay.
Figure S3. Propagation and structural incorporation of gD-GFP expressing viruses. (A) Fusion of GFP to the carboxy terminus of gD does not impact the already reduced propagation kinetics displayed by the kinase mutants (compare with Figure 1). PK15 cells were infected at an MOI=5, and the cells and media were harvested together at the indicated times post infection. Viral titers were quantitated by plaque assay. (B) Western blot analysis demonstrates that gD-GFP fusion protein expression (infected cell lysates) and structural incorporation (extracellular viral particles) are comparable in viruses possessing and lacking kinase activity. Detection of the PRV major capsid protein (VP5) is included as a loading control. (C) Fluorescence analysis of gD-GFP incorporation into individual extracellular capsid-containing particles is consistent with results shown in panel B. Extracellular viral particles released from infected Vero cells infected at an MOI=0.01 were imaged on cover glass with a 60x/1.4NA objective 2 days post infection. Individual viral particles were identified by mRFP1-capsid emissions, and the presence of corresponding GFP emissions were scored for each viral particle. Error bars represent standard error (of the proportions).
Figure S4. Confirmation of the gB-null phenotype. An RFP-capsid virus deleted for the gB gene (PRV-GS1901) required cells stably expressing gB to propagate and spread (by trans-complementation). A derivative of the RFP-capsid/gB-null virus encoding inactive UL13 and US3 kinases behaved similarly (not shown). The gB protein is an essential component of the viral membrane fusion apparatus.
Cultures of DRG sensory neurons were labeled to steady state with 0.2 mg/ml TMR-Dextran as a tracer of the endocytic and recycling pathways. Velocities and run lengths of labeled vesicles moving toward the distal axon (anterograde transport) were determined by kymograph analysis and plotted both for uninfected neurons and neurons infected with wild-type PRV (PRV-GS443). Horizontal bars indicate the mean for each sample.
A recombinant of PRV expressing mRFP1-capsids and US3-GFP (PRV-GS1504) was imaged following infection of PK15 cells or DRG sensory neurons. (A) PK15 cells were infected at a MOI = 0.1 and a representative plaque was imaged for fluorescence emissions 2 days post infection with a 10x/0.3 NA objective. (B) DRG explants were imaged prior to 1 hour post infection to track incoming viral capsids moving to the nucleus (top) or 12–14 hours post infection to track progeny capsids moving to the distal axon (bottom). Frames were captured with a 60x/1.4NA objective and an electron multiplying charged coupled device (EMCCD) set to high gain. Entry frames are 2.02 × 9.66 microns, and egress frames are 2.54 × 11.83 microns. The montages shown represent the brightest US3-GFP emissions that were observed. Because of the overall low emission levels of US3-GFP from single particles, a quantitative analysis of co-transport frequencies was not performed.