

Abstract
Oxidant-induced neuronal apoptosis has been shown to involve potassium and zinc dysregulation, energetic dysfunction, activation of stress-related kinases, and caspase cleavage. The temporal ordering and interdependence of these events was investigated in primary neuronal cultures exposed to the sulfhydryl oxidizing agent 2,2′-dithiodipyridine (DTDP), a compound that induces the intracellular release of zinc. We previously observed that tetraethylammonium (TEA), high extracellular potassium, or cysteine protease inhibitors block apoptosis induced by DTDP. We now report that both p38 and extracellular signal-regulated kinase phosphorylation are evident in neuronal cultures within 2 hr of a brief exposure to 100 μm DTDP. However, only p38 inhibition is capable of blocking oxidant-induced toxicity. Cyclohexamide or actinomycin D does not attenuate DTDP-induced cell death, suggesting that posttranslational modification of existing targets, rather than transcriptional activation, is responsible for the deleterious effects of p38. Indeed, an early robust increase in TEA-sensitive potassium channel currents induced by DTDP is attenuated by p38 inhibition but not by caspase inhibition. Moreover, we found that activation of p38 is required for caspase 3 and 9 cleavage, suggesting that potassium currents enhancement is required for caspase activation. Finally, we observed that DTDP toxicity could be blocked with niacinamide or benzamide, inhibitors of poly (ADP-ribose) synthetase. Based on these findings, we conclude that oxidation of sulfhydryl groups on intracellular targets results in intracellular zinc release, p38 phosphorylation, enhancement of potassium currents, caspase cleavage, energetic dysfunction, and translationally independent apoptotic cell death.
Keywords: oxidation, apoptosis, zinc, p38, potassium, caspase
Substantive advances have been made in elucidating the cellular and molecular signaling pathways contributing to neuronal apoptosis. However, it remains largely unknown how apparently disparate events implicated in apoptosis, such as oxidative stress, ionic dysregulation, and activation of mitogen-activated protein kinases (MAPK) and caspases, signal among one another to initiate and propagate cell death. For instance, although it has been known for some time that potassium ionophores promote apoptosis, that appreciable loss of intracellular potassium occurs during apoptosis (Perregaux and Gabel, 1994; Walev et al., 1995;Bortner et al., 1997; Dallaporta et al., 1998; Bortner and Cidlowski, 1999), and that increased extracellular potassium can attenuate cell death, the precise role of the dysregulation of this ion in promoting caspase-dependent cell death has not been well defined (Ojcius et al., 1991; Beauvais et al., 1995; Hughes et al., 1997).
The loss of intracellular potassium that occurs after a variety of apoptotic stimuli can be caused by increased activation of voltage-gated potassium channels, and blocking these channels with tetraethylammonium (TEA) is neuroprotective (Yu et al., 1997). This suggests that potassium efflux may be a requisite step for propagating apoptotic signaling. Given that increased potassium conductance can be observed within 2 hr of staurosporine or tumor necrosis factor-α treatment (Maeno et al., 2000), it seems likely that potassium channel opening may be both a conserved and proximal event induced by multiple apoptotic stimuli. Although the factors that contribute to enhanced potassium channel opening and the mechanism by which the loss of intracellular potassium triggers apoptotic signal cascades have not been determined, it has been proposed that the osmotic and energetic dysfunction brought on by the loss of the cation could result in p38 activation and cell death (Yu and Choi, 2000).
p38 is a member of the MAPK family that is sensitive to a number of osmotic, oxidative, and environmental stressors. On activation, p38 catalyzes the phosphorylation and activation of specific transcription factors, including cyclic AMP-responsive element binding protein, DNA damage-inducible genes, and ATF-2 (for review, see Cobb, 1999). Although the precise mechanism by which p38 induces cell death is unclear, blockade of this kinase has been shown to be neuroprotective against a variety of apoptotic stimuli (Kawasaki et al., 1997; Clerk et al., 1998).
The purpose of this work was to define the cellular mechanisms that contribute to p38-induced cell death by determining the temporal ordering of the signaling pathways that enhance potassium channel opening, caspase activation, and cell death in neurons exposed to oxidative stress. We have demonstrated previously that exposure to the cell permeant thiol oxidant DTDP results in intracellular zinc release and apoptotic cell death in neurons that can be attenuated with the broad-spectrum cysteine protease inhibitor Boc-aspartate-fmk (BAF), the potassium channel blocker TEA, or high levels of extracellular potassium (Aizenman et al., 2000). In this study, we have used a novel and highly selective p38 inhibitor and found that oxidative stress induces early increases in p38 phosphorylation that initiate translationally independent apoptosis by increasing potassium channel activation upstream of caspase cleavage.
MATERIALS AND METHODS
Chemicals and reagents. All cysteine protease inhibitors were purchased from Enzyme Systems Inc. (Livermore, CA). Commercially available MAPK inhibitors were acquired from Calbiochem (La Jolla, CA). All media and media supplements were from Life Technologies (Grand Island, NY). Western blotting gels, transfer apparatus, and standards were from Bio-Rad (Hercules, CA), and MAPK and p38 antibodies were from Cell Signaling (Beverly, MA). Polyvinylidene difluoride (PVDF) membranes and ECL reagents for immunoblotting were from Amersham Pharmacia Biotech (Piscataway, NJ). All other chemicals were from Sigma (St. Louis, MO).
Cell culture and toxicity assays. Forebrain neuronal-enriched cultures were prepared from embryonic day 17 rat fetuses as previously described (McLaughlin et al., 1998). Dissociated cells were plated on poly-l-ornithine-treated tissue culture plates in a growth medium composed of 80% DMEM (high glucose with l-glutamine and without sodium pyruvate), 10% Ham's F12-Nutrients (Sigma), 10% bovine calf serum (heat-inactivated) with antimycotic–antibiotic mixture (with amphotericin B and streptomycin sulfate). Cultures were maintained in an incubator at 37°C, 5% CO2. Glial cell proliferation was inhibited after 48 hr in culture with 1–2 μm cytosine arabinoside. After 3 din vitro, the serum-containing medium was replaced with a serum-free solution composed of neurobasal medium (withoutl-glutamine), B27 supplement, and antimycotic–antibiotic mixture. At 2 weeks in vitro, these cultures were composed of >95% neurons as assessed by Hoescht and glial fibrillary acidic protein staining.
Toxicity assays were performed on 2-week-old cultures (14–17 din vitro) unless otherwise noted. Immediately before drug treatment, the cells were rinsed in MEM with Earle's salts (200:1) containing 0.01% bovine serum albumin, 25 mm HEPES, and N2 media supplement. For some of the experiments, cells were exposed to various compounds 1 hr before, during, and in the 18–20 hr after DTDP exposure when assessing their efficacy in altering DTDP toxicity. DTDP treatment was performed for 10 min at 37°C, 5% CO2, and terminated by serial dilution (200:1) in MEM as above. Cells were then returned to the incubator, and neuronal viability was determined 18–20 hr after exposure using a lactate dehydrogenase (LDH)-based in vitrotoxicology assay kit. Media samples (40 μl) were analyzed spectrophotometrically (490:630), according to the manufacturer's protocol, to obtain a measure of cytoplasmic LDH release from dead and dying neurons. Toxicity was assessed as the ratio of LDHDTDP/LDHvehicle and, as are all data, expressed as the mean ± SEM.
Electrophysiological measurements. Recordings were conducted on a total of 78 neurons using the whole-cell configuration of the patch-clamp technique as described previously (Leszkiewicz et al., 2000). The extracellular solution contained (in mm): 115 NaCl, 2.5 KCl, 2.0 MgCl2, 10 HEPES, 0.1 1,2-bis(2-aminophenoxy)ethane-N,N,N,N,-tetraacetic acid (BAPTA), 10 d-glucose, pH was adjusted to 7.2; 0.1 μm tetrodotoxin was added to inhibit voltage-gated sodium channels. The intracellular (electrode) solution contained (in mm): 120 KCl, 1.5 MgCl2, 1 CaCl2, 2.0 Na2ATP, 1 BAPTA, 10 HEPES, pH 7.2. Measurements were obtained under voltage clamp with an Axopatch 200 amplifier (Axon Instruments, Foster City, CA) and pClamp software (Axon Instruments) using 2 MΩ electrodes. Partial compensation (≥80%) for series resistance was performed in all cases. Currents were filtered at 2 kHz and digitized at 10 kHz (Digidata; Axon Instruments). Drugs were dissolved in the extracellular solution and applied to the cells using a multibarrel fast perfusion system (Warner Instruments, Hamden, CT). Potassium currents were evoked with a series of incremental 80 msec voltage steps to 35 mV from a holding potential of −70 mV. Steady-state current amplitudes were measured relative to baseline 70 msec after the initiation of each voltage step and normalized to cell capacitance.
Caspase activity assays. Caspase 3 and 9 activities were measured using fluorometric caspase activity detection kits (R & D Systems, Minneapolis, MN). Cells were treated as above for 10 min with 100 μm DTDP, rinsed, and returned to the incubator. At various time points, cells were harvested after a wash in ice-cold PBS, and pellets were collected by centrifugation at 250 × g for 10 min at 4°C. Pellets were resuspended in standard cell lysis buffer provided by the manufacturer. Lysates were then incubated on ice for 10 min. A small aliquot of the protein suspension was removed and stored at −20°C for protein quantification using the BCA protein assay (Pierce Chemical Co., Rockford, IL). After the 10 min incubation, 50 μl of cell lysate was added to a 96 well flat-bottom plate along with an equal volume of 2× reaction buffer with freshly prepared DTT to a final concentration of 10 μm. Finally, 5 μl of caspase 9 fluorogenic substrate (LEHD-AFC) or the caspase 3 fluorogenic substrate (DEVD-AFC) was added, and the plate was incubated in the dark at 37°C for 1 hr. Fluorescence was then measured using a CytoFluor II plate reader (PerSeptive Biosystems, Framingham, MA) equipped with a 400 nm excitation filter and a 505 nm emission filter. Data are expressed as fluorescent units per microgram of protein.
Western blot assessment of MAPK activation. At various times after DTDP treatment, cells were harvested for detection of activated MAPK proteins. Cultures were placed on ice; after media aspiration, cells were washed twice in ice-cold PBS, then lysed in 50 mm Tris buffer, pH 8.0, with 1% Triton X-100, 2 mm EDTA, 150 mm NaCl, 8 mm β-glycerophosphate, and 100 μm sodium orthovanadate. Residual cells were harvested, and an aliquot of this suspension was removed for later protein determination. Then, an equal volume of laemmli buffer was added to cell lysates, and samples were sonicated for 10 sec to remove viscosity and shear DNA. Samples were heated then to 95°C for 5 min and stored at −20°C until blots were run.
Equal protein concentrations were separated using 10 or 12% SDS-PAGE minigels with prestained kaleidoscope molecular weight markers. Proteins then were transferred to PVDF membranes and blocked for 1 hr at room temperature (RT) in CHEMIblocker (Chemicon, Temecula, CA) diluted 1:1 with PBS with 0.1% Tween (PBST). Membranes were washed four times in PBST for a total of 30 min, then incubated overnight in primary antibody diluted 1:1000 in blocking solution. The following day, primary antibody was removed; blots were washed as above and then incubated for 1 hr at RT with an HRP-conjugated anti-rabbit secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:2000 in blocking buffer. Membranes were washed again as above and placed in 2 ml of ECL chemiluminescent substrate for 1 min at RT. Membranes were exposed then to Kodak X-OMAT x-ray film (VWR Scientific, Bridgeport, NJ).
RESULTS
DTDP-induced cell death in primary neuronal cultures can be attenuated with antioxidants
Brief exposure to the cell permeant oxidant DTDP induces neuronal apoptotic cell death (Aizenman et al., 2000). We observed that cell death induced by DTDP can be attenuated by the antioxidant mixture B27 (Table 1). B27 contains a number of free radical scavengers and reducing equivalents, including glutathione, α tocopherol, selenium, and ascorbate. The presence of B27 both during the 10 min exposure to DTDP and in the subsequent 18–20 hr increased survival by almost 60% in the cells exposed to 75 μmDTDP and by ∼40% in cells treated with 100 μm DTDP. This effect is most likely attributable to the antioxidant properties of B27 because the neuroprotective action of the free radical spin trap N-tert-butyl-α-phenylnitrone (PBN) was very similar to that of B27. These findings are consistent with our previous observation that DTDP results in accumulation of intracellular zinc (Aizenman et al., 2000), because alteration in zinc homeostasis and sequestration have been implicated as a causative factor in neuronal oxidative stress via production of free radicals (for review, see Weiss et al., 2000).
Table 1.
Inhibition of DTDP-induced neuronal cell death by antioxidants and PARS inhibitors
| DTDP (μm) | 10 | 25 | 50 | 75 | 100 |
|---|---|---|---|---|---|
| Control | 97 ± 1 (8) | 79 ± 2 (8) | 47 ± 3 (9) | 11 ± 5 (9) | 0 ± 3 (10) |
| + B27 | 92 ± 4 (8) | 92 ± 3* (8) | 82 ± 7* (9) | 70 ± 9* (9) | 41 ± 8* (10) |
| + PBN | nd | nd | nd | 60 ± 8* (5) | 39 ± 2* (4) |
| + Niacinamide | nd | nd | nd | 66 ± 11* (5) | 51 ± 6* (5) |
| + Benzamide | nd | nd | nd | nd | 40 ± 13* (6) |
Cultures were exposed to 100 μm DTDP for 10 min in the presence of B27 (1×), PBN (500 μm), niacinamide (3 mm), or benzamide (3 mm). Neuronal viability (mean ± SEM) expressed as a percentage of control sister cultures that were not exposed to DTDP. Numbers inparentheses represent the total number of independent observations per group, each of which was performed in at least duplicate. Asterisk denotes a significant difference at a given concentration between DTDP treatment alone and DTDP treatment with each neuroprotective agent (p < 0.05; pairedt test). nd, Not determined.
In additional experiments, we observed that 3 mmniacinamide provided significant protection against DTDP-induced apoptosis (Table 1). Niacinamide can exert neuroprotective action as an oxyradical scavenger, a precursor for NAD+synthesis, an iNOS inhibitor, as well as a competitive inhibitor of NAD+-catabolizing enzymes, including poly (ADP-ribose) synthetase (PARS) (for review, see Szabo and Dawson, 1998). Furthermore, niacinamide has recently been shown to abrogate the toxicity induced by exposing cells to high levels of extracellular zinc (Sheline et al., 2000). An additional PARS inhibitor, benzamide (3 mm), was also observed to afford a significant level of neuroprotection against 100 μm DTDP (Table 1).
Oxidative stress induces rapid MAPK activation
Exposure to oxidative stress activates members of the stress-activated protein kinase family (Clerk et al., 1998). We thus measured the activation of two MAPK pathways after 100 μmDTDP treatment. Cells were harvested at various time points after DTDP exposure, and immunoblots were performed using phospho-specific antibodies to both p38 and extracellular signal-regulated kinase (ERK) p42/44. Within 30 min of exposure to DTDP, a substantial increase in p38 phosphorylation was evident (Fig. 1). After 2 hr, p38 phosphorylation decreased appreciably, and levels were indistinguishable from baseline. Similarly, phospho-ERK immunoblotting demonstrated that there was a pronounced early rise in p42/44 activation within 30 min of exposure to oxidant that decreased dramatically by 1 hr. Use of the nonphosphorylated p38 and ERK antibodies was performed as a control to ensure that equal amounts of each of the inactive MAPK proteins were present at all time points.
Fig. 1.

Activation of p38 and ERK after exposure to DTDP. Whole-cell extracts of neuronal cultures were harvested at various time points after 10 min exposure to 100 μm DTDP. Proteins were separated on 12% SDS-PAGE gels and probed with antibodies specific to the phosphorylated and nonphosphorylated forms of both p38 and ERK p42/44. Note that there was early increased p38 activation in the first 2 hr after exposure to DTDP and a similar increase in ERK activation within the first 45 min of oxidant exposure. Similar results were obtained in two additional independent experiments.
To address the contribution of MAPK activation to the observed cell death after DTDP exposure, the effects of specific inhibitors of MEK 1/2 and p38 on DTDP toxicity were assessed. Cells were preincubated for 1 hr with either 10 μm U0126 (Satoh et al., 2000) or a second-generation, highly selective, and neuroprotective inhibitor of p38, SB 239063 (20 μm)(Barone et al., 2001), and then exposed to 75 or 100 μmDTDP for 10 min in the presence of the MAPK inhibitors. Kinase inhibitors were also present during the 18–20 hr period after oxidant exposure. Neuroprotective effects of these and other compounds were assessed against both concentrations of DTDP to ensure that any protective effects of these agents were not masked by the potentially overwhelming neurotoxic action of 100 μm DTDP. Although we observed relatively large increases in p42/44 phosphorylation after DTDP exposure, this pathway did not appear to contribute to oxidant-induced cell death because U0126 did not abrogate DTDP toxicity (Fig. 2). However, treatment with the p38 inhibitor provided significant protection against both doses of DTDP tested (Fig. 2). The majority of neurons treated with DTDP in the presence of p38 inhibitor maintained a healthy phase-bright soma, yet we did observe some disruption of neuronal processes. Cells treated with SB 239063 alone did not display this phenotype (data not shown).
Fig. 2.

p38 activation contributes to DTDP-induced neurotoxicity but is not dependent on new protein synthesis. Neuronal cultures exposed to 75 or 100 μm DTDP for 10 min in the presence of inhibitors of ERK and p38 activation and protein synthesis. Both U0126 (10 μm) and SB 239063 (20 μm) were present for 60 min before, during, and 24 hr after exposure to DTDP, whereas CHX (1 μg/ml) was present only during and after the DTDP incubation. Cell viability was assessed 18–20 hr after DTDP exposure by measuring the amount of LDH released into the culture media. Data are expressed as mean percentage neuroprotection compared with the same dose of DTDP without inhibitor ± SEM. Hence, neuroprotection reflects the degree of viability relative to the extent of death induced by each concentration of DTDP. Data represent the mean ± SEM of three to six experiments performed in duplicate. *p < 0.05; paired t test.
Like other MAPKs, p38 is thought to exert its effect on cell survival through modification of transcription factors. We were surprised therefore to find that treatment with the protein synthesis inhibitor cyclohexamide (1 μg/ml) did not significantly alter the neurotoxic properties of DTDP (Fig. 2). Additionally, the transcription inhibitor actinomycin D (5 μg/ml) was completely ineffective in protecting neurons against 100 μm DTDP-induced toxicity (n = 3; data not shown). This suggests that direct or indirect modification of existing intracellular targets, rather than new protein synthesis, is the mechanism of the deleterious action of p38 after oxidant exposure.
Oxidant-induced apoptosis is accompanied by an enhancement of voltage-gated potassium currents downstream from p38 activity, but upstream from caspase activation
In a previous study (Aizenman et al., 2000), we observed that DTDP-mediated neuronal apoptosis could be abrogated by either high extracellular potassium or TEA, suggesting that activation of voltage-gated potassium channels and a concomitant putative loss of intracellular potassium were involved in the cell death process (Bortner et al., 1997; Yu et al., 1997, 2000). In the present study, we investigated whether DTDP-induced cell death was indeed accompanied by an enhancement of voltage-gated potassium currents and whether inhibitors of cell death, including SB 239063, could attenuate channel activity. Using whole-cell recordings, we detected a robust increase in the amplitudes of outward voltage-gated potassium currents ∼3–4 hr after a 10 min exposure to 100 μm DTDP, compared with vehicle-treated cells (Fig.3A). Potassium currents in both DTDP-treated cells and controls had relatively slow kinetics and were sensitive to block by 10 mm TEA, consistent with the hypothesis that apoptotic stimuli enhance delayed rectifier potassium currents in dying neurons (Yu et al., 2000).
Fig. 3.

Enhanced voltage-gated potassium currents during DTDP-induced apoptosis. A, Whole-cell potassium currents obtained in two separate cortical neurons ∼3 hr after a 10 min exposure to either vehicle (0.1% DMSO) or 100 μm DTDP. Cells were maintained in MEM at 37°C for the interval between drug exposure and the recordings. Potassium currents were evoked by a series of voltage steps to +35 mV from a holding potential of −70 mV. Note that currents are substantially larger in the DTDP-treated cell, when compared with the vehicle-treated neuron. TEA (10 mm)blocked ∼50% of the currents in both cases. B, Acute application of 100 μm DTDP or 4 mmdithiothreitol (DTT; a disulfide reducing agent) did not produce appreciable changes in the amplitude of potassium currents.
In a separate set of experiments, we confirmed that the effects of DTDP on the potassium currents were associated with the apoptotic process and were not caused by a direct interaction of the redox reagent with the channels (Aizenman et al., 1989; Gulbis et al., 2000). Acute exposure of cells to 100 μm DTDP during the recording process produced negligible changes in the amplitude of the potassium currents (Fig. 3B). No effect was seen after incubation with the reducing agent dithiothreitol (3 mm), suggesting that the aforementioned increase in potassium currents observed in dying cells is not caused by a direct redox effect of DTDP on potassium channels.
A detailed time course analysis demonstrated that the upregulation of potassium currents could be detected first ∼3 hr after DTDP exposure and continued to increase in a time-dependent fashion (Fig.4A). Recordings became particularly difficult to perform 4 hr after DTDP treatment because cell membranes became somewhat fragile and unstable. Furthermore, in most instances, the largest voltage steps in DTDP-treated cells produced currents for which amplitudes saturated our patch-clamp amplifier (>20 nA). As a result, we could not determine the absolute extent of the enhancement of these currents. Hence, all quantitative measurements were limited to a 3–4 hr postexposure time window and performed on currents evoked by voltage steps no larger than 60 mV (−70 to −10 mV), which we could reliably maintain under voltage clamp.
Fig. 4.

Potassium current enhancement during DTDP-induced apoptosis is downstream from p38 activity but precedes caspase activation. A, Temporal profile of DTDP-induced enhancement of potassium currents during apoptosis. Whole-cell potassium currents were evoked by a voltage step to −10 mV from a holding voltage of −70 mV in 31 cells exposed for 10 min to 100 μm DTDP at time 0. Cells were maintained in MEM at 37°C between DTDP exposure and the establishment of the recording at various time points. Current amplitudes were normalized to cell capacitance. The line represents the average potassium current density for eight cells recorded at several random time points after exposure to vehicle alone (0.1% DMSO). Note that potassium currents begin to become enhanced, relative to baseline, ∼3 hr after DTDP exposure. B, In a separate set of recordings, neurons were exposed for 10 min to vehicle (n = 9), 100 μm DTDP alone (n = 9), or DTDP in the presence of 10 μm TPEN (n = 6), 20 μm SB 239063 (n = 14), or 10 μm BAF (n = 6). Potassium currents were evoked in these cells 3–4 hr after DTDP exposure by voltage-steps from −70 to −10 mV. Steady-state amplitudes were normalized to cell capacitance. TPEN and SB 239063, but not BAF, inhibited the enhancement of potassium currents observed in DTDP-treated cells. Values represent the mean ± SEM; asterisks denote a significant difference versus vehicle controls (p < 0.01; ANOVA followed by a Dunnett multiple comparisons test).
With this paradigm, we examined whether substances known to inhibit DTDP toxicity, namely the zinc chelatorN,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN), the p38 inhibitor SB 239063, and the caspase inhibitor BAF, could block the enhancement of the potassium currents. Coexposure of 100 μm DTDP with either 10 μm TPEN or 20 μm SB 239063 completely prevented the enhancement of the potassium currents, whereas 10 μm BAF did not (Fig.4B). These data indicate that the intracellular release of zinc (Aizenman et al., 2000) and activation of the p38 cascade precede the enhancement of channel activity. In contrast, because BAF inhibits toxicity but not potassium channel activity, this suggests that caspase activation follows potassium efflux. Importantly, these results also indicate that the enhancement of potassium currents during apoptosis is, in and of itself, not sufficient to cause neuronal cell death.
Caspases 3, 8, and 9 contribute to oxidant-induced apoptosis
The results of the potassium channel recordings strongly suggested that DTDP-induced caspase activation likely occurs subsequent to enhanced potassium efflux. Zinc neurotoxicity has been associated with mitochondrial dysfunction and, in some instances, apoptotic cell death (Skulachev et al., 1967; Treves et al., 1994; Y. H. Kim et al., 1999; Virag and Szabo, 1999; Sheline et al., 2000; Weiss et al., 2000). Thus, we sought to determine whether caspases associated with mitochondrial dysfunction were activated by DTDP, as well as the time course of their activation. We assessed the efficacy of inhibiting caspase 3, given that this protease lies downstream of caspase 9, which is activated by mitochondrial cytochrome c release, as well as a caspase 8, which has recently been localized to the mitochondria and shown to be released during cellular dysfunction (Qin et al., 2000). Cells were treated with cysteine protease inhibitors for 1 hr before, during, and 24 hr after a 10 min exposure to DTDP. As expected from previous work with relatively younger cultures (Aizenman et al., 2000), the broad-spectrum cysteine protease inhibitor BAF (10 μm) provided appreciable neuroprotection against the two doses of DTDP tested (Fig. 5). Moreover, the novel nonpeptide inhibitor of caspases 3, 7, and 9, isatin-sulfonamide-4 (IS-4; 3 μm) (Erhardt et al., 2000;Lee et al., 2000), provided 40 ± 6 and 39 ± 10% protection against 75 and 100 μm DTDP, respectively. We next treated cells with the caspase 8 inhibitor z-Leu-Glu-(OMe)Thr-Asp(OMe)-CH2F (LETD; 10 μm), which also provided appreciable neuroprotection against both doses of DTDP (p < 0.05;n = 4–5). Addition of Biotin-Phe-Ala-CH2F (zFA; 20 μm), which contains an fmk group without a caspase inhibitor peptide sequence, provided no protection on its own (data not shown).
Fig. 5.
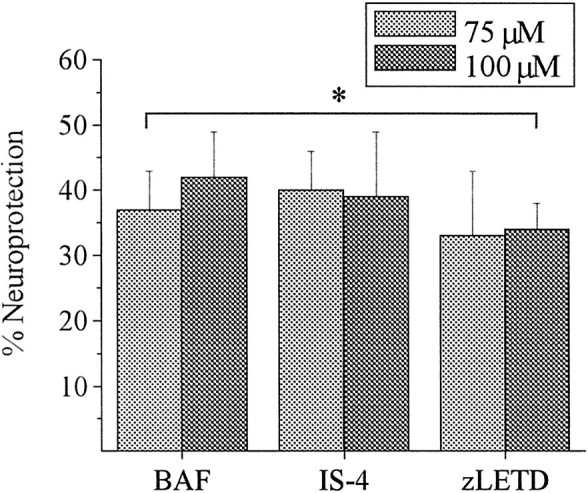
Inhibition of caspases 3, 8, and 9 blocks oxidant-induced cell death. Neuronal cultures exposed to 75 or 100 μm DTDP for 10 min in the presence of peptide and nonpeptide caspase inhibitors. The broad spectrum cysteine protease inhibitor BAF (10 μm), caspase 3, 7, and 9 selective nonpeptide inhibitor IS-4 (3 μm), and the caspase 8 selective inhibitor zLETD (10 μm) were present for 60 min before, during, and 24 hr after exposure to DTDP. Cell viability was assessed as above, and data are expressed as mean percentage neuroprotection compared with the same dose of DTDP without inhibitor ± SEM. Data represent the average of five experiments performed in duplicate. All compounds provided significant protection at both doses of DTDP tested. *p < 0.05; pairedt test.
The time course of caspase 9 and 3 activation after 10 min treatment with DTDP was then assessed using enzymatic caspase activity assays with 7-amino-4-trifluoromethyl coumarin (AFC)-labeled substrate peptides. As shown in Figure 6, there is a time-dependent increase in caspase 9 and caspase 3 activities after DTDP exposure. Notably, only very minor changes in activity were evident before the 7 hr time point, indicating that DTDP-induced caspase activation likely occurs after p38 phosphorylation and potassium efflux. This possibility was tested by examining whether antioxidants, niacinamide, and p38 inhibitors block caspase cleavage. The efficacy of each of these agents at blocking caspase activity was assessed 7 hr after treatment. The elevation in caspase 9 activity that was observed at this time point was significantly diminished by 10 μm BAF, 20 μm SB 239063, or 3 mm niacinamide (Fig.7A). Similarly, the increase in caspase 3 activity was also blocked when cells were treated with these three compounds (Fig. 7B). Additionally, the antioxidant mixture B27 and free radical spin trap PBN significantly attenuated both caspase 9 and 3 activity by 41 ± 13 and 61 ± 4%, respectively (n = 4–5;p < 0.05; one sample t test). These findings indicate that reactive oxygen species (ROS), p38, and possibly energetic stress signals are upstream effectors that elicit caspases 3 and 9 cleavage after DTDP exposure.
Fig. 6.

Caspase 9 and 3 are activated relatively late after exposure to DTDP. Neuronal cultures exposed to 100 μm DTDP for 10 min and then washed and harvested at various time points to assess the extent of caspase cleavage using fluorometric peptide substrates. Lysed cells were incubated in the presence of either the fluorogenic caspase 9 substrate (LEHD-AFC) or the caspase-3 substrate (DEVD-AFC). Activity was then calculated as fluorescent units per microgram of protein and expressed as activity in relative units compared with control. Data represent the average ± SEM of at least six experiments performed in duplicate.p < 0.05; one-way ANOVA.
Fig. 7.

NAD+ depletion and p38 activation occur before caspase cleavage. Neuronal cultures were exposed to 100 μm DTDP for 10 min in the presence of niacinamide (Nia; 3 mm), BAF (10 μm), or SB 239063 (20 μm) as above and harvested 7 hr later. Data were analyzed and expressed as in Figure 6and represent the mean ± SEM of four to six independent experiments performed in duplicate. There was a statistically significant decrease in caspase cleavage by blocking either NAD+-catabolization or p38 on caspase cleavage, suggesting that activation of these pathways occurs before caspase involvement in DTDP toxicity. Values represent the mean ± SEM;asterisks denote a significant difference versus matched DTDP treatment (*p < 0.05; paired ttest). All DTDP treatment groups were pooled for presentation purposes.
Oxidant and zinc dysregulation are the earliest apoptotic signals induced by DTDP, and their dysfunction leads to p38 activation
The sum of the biochemical and electrophysiological data suggested that p38 activation is both a proximal and necessary event for both potassium channel opening and caspase cleavage. However, we had not yet determined the temporal ordering and interdependence of oxidative stress and zinc dysregulation in relation to MAPK phosphorylation. Therefore, we performed immunoblots for p38 activation on cells that had been treated with neuroprotective agents. Because 2 hr after DTDP treatment was the latest time point at which we observed maximal p38 phosphorylation, we chose this time to assess the efficacy of the neuroprotective agents at blocking p38 activation. Cells exposed for 10 min to 25 μm TPEN in the presence of DTDP were provided the greatest inhibition (>90%) of p38 induction (Fig.8). The free radical spin trap PBN (500 μm) also substantially decreased p38 phosphorylation (60%). This finding indicates that oxidative stress and zinc release from intracellular stores are required for DTDP induction of p38. However, niacinamide (3 mm) decreased p38 phosphorylation to a much lesser extent (25%), as did the potassium channel blocker TEA (25 mm; <20%), suggesting that although these drugs can block cell death, their effects mostly lay downstream of p38 phosphorylation.
Fig. 8.

p38 activation occurs upstream of K+ channel opening and is induced by zinc release and oxidative stress. Neuronal cultures were exposed to 100 μm DTDP for 10 min in the presence of niacinamide (3 mm), TEA (25 mm), TPEN (25 μm), or PBN (500 μm) as above, and whole- cell extracts were harvested 2 hr later. Proteins were separated by SDS-PAGE and transferred to PVDF membranes that were then probed with an antibody selective for the phosphorylated form of p38. Extracts from C-6 glioma cells after anisomycin (Anis) treatment were used as a positive control. Note that p38 activation induced by DTDP was substantially attenuated by both ion chelation (TPEN) and free radical scavenging (PBN), but only marginally by TEA blockade of K+ efflux. Similarly, inhibition of NAD+ catabolism by niacinamide provided only marginal attenuation of p38 activation. Quantification of levels with NIH Image software revealed the following mean band densities (± SD): control, 109.9 ± 36.9; DTDP, 226.1 ± 13.9; DTDP + niacinamide, 196.6 ± 21.9; DTDP + TEA, 202.3 ± 18.2; DTDP + TPEN, 114.4 ± 18.3; DTDP + PBN, 175.5 ± 26.3. Comparable results were obtained in two other independent experiments.
DISCUSSION
We show here that oxidative stress induced by DTDP induces apoptotic cell death via activation of the stress-related protein kinase p38. We present evidence that p38 phosphorylation is a proximal event leading to the enhancement of voltage-gated potassium currents, a phenomenon that has previously been associated with apoptosis (Yu et al., 1997). This process occurs in the absence of new protein synthesis, suggesting that p38 causes direct or indirect phosphorylation of the potassium channels themselves, or an associated structure. We also confirm a previous report that the enhancement of the potassium currents precedes the activation of caspases (Maeno et al., 2000). Furthermore, the observation that caspase inhibitors block cell death but not potassium current enhancement suggests that the putative loss of intracellular potassium after DTDP treatment is not, in and of itself, a lethal event.
We reported previously that DTDP induces intracellular zinc release (Aizenman et al., 2000). This process contributes to the observed p38 activation because it is inhibited by TPEN, a high-affinity zinc chelator. Zinc release may also be critically important in other forms of oxidation-induced apoptosis in which p38 has been implicated, such as that occurring after NO exposure (Ghatan et al., 2000). Indeed, NO has been shown previously to trigger zinc release from metallothionein (Pearce et al., 2000), a metalloprotein that also releases zinc after DTDP oxidation (Jiang et al., 1998; Maret and Vallee, 1998). We also find evidence that the intracellular signaling cascades activated by DTDP appear to be similar to those observed by other researchers after exposure to extracellular zinc. Samet et al. (1998) reported p38 and ERK activation in human bronchial cells after zinc exposure. Although we observed increases in both ERK and p38 after DTDP treatment, only p38 activation contributes significantly to the observed neurotoxicity.
Exposure to zinc may induce energetic stress via inhibition of glycolytic enzymes (Ikeda et al., 1980; Krotkiewska and Banas, 1992;Kukimoto et al., 1996), because the resultant cell death can be attenuated by addition of 3 mm niacinamide (Sheline et al., 2000). Similarly, we found that niacinamide provides significant protection against DTDP toxicity. The most widely described neuroprotective properties of niacinamide have been attributed to the fact that it is a precursor for NAD+synthesis and an inhibitor of the NAD+-catabolizing enzyme PARS. PARS is activated by DNA damage after oxidative stress (for review, see Pieper et al., 1999), and depletion of NAD+ by PARS places an energetic stress on cells (Satoh and Lindahl, 1992). Niacinamide inhibits PARS with an IC50 of 100 μmin vitro (Rankin et al., 1989). However, at the doses used in our work and the work by Sheline et al. (2000), niacinamide can also act as an oxyradical scavenger, an iNOS inhibitor, and an inhibitor of other NAD+-catabolizing enzymes (Ziegler et al., 1996; for review, see Szabo and Dawson, 1998). We believe that this and our previous work suggest that the neuroprotective action of niacinamide can be attributed most likely to its ability to increase cellular energetic status. That is, niacinamide is far less effective than the free radical spin trap PBN in blocking p38 activation, suggesting that the oxyradical scavenging activity of niacinamide is not overly dramatic. Furthermore, we have previously observed appreciable DNA damage within 3 hr of exposure to DTDP (Aizenman et al., 2000), suggesting that PARS activation and subsequent energetic dysfunction may also be triggered by the production of free radicals after DTDP exposure. Finally, another PARS inhibitor, benzamide, was also effective in inhibiting DTDP-induced cell death.
Our findings that the earliest manifestations of DTDP-induced cell stress are associated with oxidative injury are supported by the neuroprotective action of both PBN and B27, as well as by the finding that PBN partially blocks p38 activation. This result supports previous studies in which PBN attenuates oxidative stress-induced p38 activation in other in vitro systems (Floyd, 1999). There is evidence suggesting that rapid rise in intracellular zinc, such as that which occurs after DTDP exposure (Aizenman et al., 2000), may itself lead to ROS generation and lipid peroxidation (Sensi et al., 1999). The ability of zinc to directly elicit oxidative stress remains somewhat controversial however, given that the ability of antioxidants to attenuate zinc toxicity has been inconsistent (E. Y. Kim et al., 1999; Y. H. Kim et al., 1999; but also see Sensi et al., 1999;Sheline et al., 2000). It is also possible that critical cellular redox agents, including thioredoxin, glutathione, cysteine, andN-acetyl cysteine, are directly altered by DTDP. These cellular antioxidants are important not only as free radical scavengers but also as metal chelators and substrates for redox reactions (for review, see Deneke, 2000).
The observation that DTDP induces apoptotic cell death, which can be attenuated with caspase inhibitors, high levels of extracellular potassium, or TEA, is consistent with the observation that loss of intracellular potassium and cell shrinkage are early requisite features of apoptosis (Barbiero et al., 1995; Benson et al., 1996; Bortner et al., 1997; Hughes et al., 1997). However, the signaling pathways that contribute to enhanced potassium efflux are ill defined. Kinase signaling pathways have been implicated in apoptotic ionic dysregulation as Gomez-Angelats et al. (2000) recently demonstrated that suppression of PKC occurs upstream of potassium channel opening in Fas-induced apoptosis. Furthermore, Fas-induced cell shrinkage could be blocked by PKC stimulation or enhanced by PKC inhibition. Preliminary findings in our laboratory suggest moderate protection against 100 μm DTDP can be induced by the PKC activator PMA (40 nm) (B. A. McLaughlin and E. Aizenman, unpublished observation). The role of PKC and other kinases on p38 signaling remains to be elucidated because the temporal ordering and reliance of these phosphorylation events is unclear in other systems (Jun et al., 1999a,b; Shimizu et al., 1999; Yu et al., 2000). For instance, PKC activation has been linked to increased reactive oxygen species generation and has been suggested to be deleterious to neuronal cultures exposed to zinc (Noh et al., 1999). This discrepancy may be an important point of divergence between intracellular and extracellular zinc-induced toxicity (Cuajungco and Lees, 1997).
Our observation that p38 activation is an early and requisite event for potassium channel opening strongly suggests that SAPK phosphorylation leads to a volume change in neurons. Maeno and coworkers (2000)recently reported that apoptotic volume decrease occurs before DNA fragmentation, cytochrome c release, and caspase activation, apoptotic events that are reminiscent of those reported here and previously (Aizenman et al., 2000). Nonetheless, because p38 is activated by osmotic stress in other systems (for review, see Ono and Han, 2000), one could easily envision a scenario in which the putative loss of intracellular potassium activates p38 and other proapoptotic signaling molecules, thus placing enhanced potassium channel activation upstream of p38 phosphorylation. Indeed, Benson et al. (1996) have shown that cell shrinkage correlates with activation of ERK and p38 in kidney cells and that use of p38 blockers largely inhibits the resulting protective cellular volume recovery, whereas ERK inhibitors do not. However, our observation that p38 inhibitors completely block the enhancement of TEA-sensitive potassium channels after DTDP exposure and that TEA only marginally attenuates the observed increase in p38 activation supports a very different temporal ordering of MAPK phosphorylation and potassium channel activation, at least in neurons. It is likely that the combination of oxidative stress and zinc dysregulation elicited by DTDP, and possibly other oxidants, is sufficient to activate p38, which then leads to enhanced activation of TEA-sensitive potassium channels and a subsequent, requisite volumetric dysregulation observed in other forms of apoptosis.
We were intrigued by the observation that although p38 activation plays a prominent role in DTDP toxicity, it does not require new protein synthesis. The finding that p38 enhances voltage-gated potassium currents associated with apoptosis suggests that these channel, or closely associated proteins, may be direct or indirect targets of phosphorylation by this kinase. It has been reported that p38 can alter activity of intracellular proteins in a transcriptionally independent manner. For example, Kusuhara and coworkers (1998) have shown that the activity of the membrane-bound Na+/H+exchanger can be modified in a transcriptionally independent manner by both ERK and p38. Given that the delayed rectifying potassium channels have multiple phosphorylation sites that alter activity (Levitan, 1999), it is thus possible that they may also be modified by MAPKs. Indeed, Adams et al. (2000) have shown that the Kv4.2 potassium channel can be directly phosphorylated by ERK, suggesting that the activity of this and the potassium channels associated with apoptotic cell death may also be modified by p38.
In conclusion, our work suggests that oxidant-induced zinc dysregulation and ROS generation are the most proximal events in an apoptotic cascade in which p38 activation leads to potassium efflux, and subsequently, energetic dysfunction and caspase activation. This novel temporal ordering of apoptotic signal transduction is important not only in furthering our understanding of the complex interplay between posttranslational protein modification and ionic dysfunction in apoptosis, but also as a model (Fig. 9) in which to address the importance of these events in neurodegenerative conditions involving zinc release and oxidative stress, such as cerebral ischemia.
Fig. 9.

Model of early and late events in oxidant-induced neuronal injury. Based on the findings in this and our previous work, the cellular and molecular signaling pathways that contribute to oxidant-induced neurotoxicity have been summarized. The “early” events (left panel) in DTDP toxicity include the rapid loss of zinc homeostasis within 10 min of exposure to DTDP (Aizenman et al., 2000), a primary oxidative cell injury resulting in a rapid increase in p38 phosphorylation within 30 min of oxidant exposure, and then subsequent DNA damage within 3 hr of DTDP treatment (Aizenman et al., 2000). p38 activation can be blocked by the zinc chelator TPEN and, to some degree, by the free radical spin trap, PBN. Also by 3 hr, enhanced activation of TEA-sensitive potassium channels associated with apoptosis can be observed. p38 is directly responsible for the loss of intracellular potassium because the p38 inhibitor SB 239063 can attenuate channel activity, whereas potassium channel blockers have a very small effect on p38 activity. The “late” events (right panel) in this cascade likely include energetic dysfunction brought on by zinc dysregulation (Skulachev et al., 1967; for review, see Weiss et al., 2000), NADH depletion, and, ultimately, caspase activation within 7 hr. Blockade of any of these events with compounds such as niacinamide, benzamide, and peptide and nonpeptide caspase inhibitors provides substantial neuroprotection.
Footnotes
This work was supported by a grant from the Alzheimer's Disease Research Centre, University of Pittsburgh (B.A.M), National Institutes of Health Grant NS29365 (E.A.), and an American Heart Association Grant-in-Aid (E.A.). We thank Drs. Randall Pittman, Ian Reynolds, Donald DeFranco, and Gregg Stanwood for helpful comments and suggestions, and Daniel Leszkiewicz, Shen Du, and Annalhees Rodriguez for assistance.
Correspondence should be addressed to Dr. Elias Aizenman, Department of Neurobiology, University of Pittsburgh School of Medicine, E1456 BST, Pittsburgh, PA 15261. E-mail: redox@pitt.edu.
REFERENCES
- 1.Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, Sweatt JD. The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem. 2000;75:2277–2287. doi: 10.1046/j.1471-4159.2000.0752277.x. [DOI] [PubMed] [Google Scholar]
- 2.Aizenman E, Lipton SA, Loring RH. Selective modulation of NMDA responses by reduction and oxidation. Neuron. 1989;2:1257–1263. doi: 10.1016/0896-6273(89)90310-3. [DOI] [PubMed] [Google Scholar]
- 3.Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin BA, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- 4.Barbiero G, Duranti F, Bonelli G, Amenta JS, Baccino FM. Intracellular ionic variations in the apoptotic death of L cells by inhibitors of cell cycle progression. Exp Cell Res. 1995;217:410–418. doi: 10.1006/excr.1995.1104. [DOI] [PubMed] [Google Scholar]
- 5.Barone FC, Irving EA, Lee JC, Kassis S, Kumar S, White RF, Nelson AH, Ohtstein EH, Hunter AJ, Ward K, Smith BR, Adams JL, Parsons AA. SB 239069 a second generation p38 mitogen-activated protein kinase inhibitor reduces brain injury and neurological deficits in rat focal stroke. J Pharmacol Exp Ther. 2001;296:312–321. [PubMed] [Google Scholar]
- 6.Beauvais F, Michel L, Dubertret L. Human eosinophils in culture undergo a striking and rapid shrinkage during apoptosis. Role of K+ channels. J Leukoc Biol. 1995;57:851–855. doi: 10.1002/jlb.57.6.851. [DOI] [PubMed] [Google Scholar]
- 7.Benson RSP, Heer S, Dive C, Watson AJM. Characterization of cell volume loss in CEM-C7A cells during dexamethasone-induced apoptosis. Am J Physiol. 1996;270:C1190–C1203. doi: 10.1152/ajpcell.1996.270.4.C1190. [DOI] [PubMed] [Google Scholar]
- 8.Bortner CD, Cidlowski JA. Caspase independent/dependent regulation of K(+), cell shrinkage, and mitochondrial membrane potential during lymphocyte apoptosis. J Biol Chem. 1999;274:21953–21962. doi: 10.1074/jbc.274.31.21953. [DOI] [PubMed] [Google Scholar]
- 9.Bortner CD, Hughes FM, Jr, Cidlowski JA. A primary role for K+ and Na+ efflux in the activation of apoptosis. J Biol Chem. 1997;272:32436–32442. doi: 10.1074/jbc.272.51.32436. [DOI] [PubMed] [Google Scholar]
- 10.Clerk A, Fuller SJ, Michael A, Sugden PH. Stimulation of “stress-regulated” mitogen-activated protein kinases (stress-activated protein kinases/c-Jun N-terminal kinases and p38-mitogen-activated protein kinases) in perfused rat hearts by oxidative and other stresses. J Biol Chem. 1998;273:7228–7234. doi: 10.1074/jbc.273.13.7228. [DOI] [PubMed] [Google Scholar]
- 11.Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71:479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 12.Cuajungco MP, Lees GJ. Zinc metabolism in the brain: relevance to human neurodegenerative disorders. Neurobiol Dis. 1997;4:137–169. doi: 10.1006/nbdi.1997.0163. [DOI] [PubMed] [Google Scholar]
- 13.Dallaporta B, Hirsch T, Susin SA, Zamzami N, Larochette N, Brenner C, Marzo I, Kroemer G. Potassium leakage during the apoptotic degradation phase. J Immunol. 1998;160:5605–5615. [PubMed] [Google Scholar]
- 14.Deneke SM. Thiol-based antioxidants. Curr Top Cell Regul. 2000;36:151–180. doi: 10.1016/s0070-2137(01)80007-8. [DOI] [PubMed] [Google Scholar]
- 15.Erhardt JA, Wong V, Legos JJ, Lee D, Nutell M, Parsons AA, Barone FC. Novel caspase inhibitor IS4 elucidates critical role for caspase-3/-7 in PC-12 cell death following tropic factor removal. Soc Neurosci Abstr. 2000;26:1013. [Google Scholar]
- 16.Floyd RA. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med. 1999;222:236–245. doi: 10.1046/j.1525-1373.1999.d01-140.x. [DOI] [PubMed] [Google Scholar]
- 17.Ghatan S, Larner S, Kinoshita Y, Hetman M, Patel L, Xia Z, Youle RJ, Morrison RS. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol. 2000;150:335–347. doi: 10.1083/jcb.150.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomez-Angelats M, Bortner CD, Cidlowski JA. Protein kinase C (PKC) inhibits fas receptor-induced apoptosis through modulation of the loss of K+ and cell shrinkage. A role for PKC upstream of caspases. J Biol Chem. 2000;275:19609–19619. doi: 10.1074/jbc.M909563199. [DOI] [PubMed] [Google Scholar]
- 19.Gulbis JM, Zhou M, Mann S, MacKinnon R. Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science. 2000;289:123–127. doi: 10.1126/science.289.5476.123. [DOI] [PubMed] [Google Scholar]
- 20.Hughes FM, Jr, Bortner CD, Purdy GD, Cidlowski JA. Intracellular K+ suppresses the activation of apoptosis in lymphocytes. J Biol Chem. 1997;272:30567–30576. doi: 10.1074/jbc.272.48.30567. [DOI] [PubMed] [Google Scholar]
- 21.Ikeda T, Kimura K, Morioka S, Tamaki N. Inhibitory effects of Zn2+ on muscle glycolysis and their reversal by histidine. J Nutr Sci Vitaminol (Tokyo) 1980;26:357–366. doi: 10.3177/jnsv.26.357. [DOI] [PubMed] [Google Scholar]
- 22.Jiang LJ, Maret W, Vallee BL. The glutathione redox couple modulates zinc transfer from metallothionein to zinc-depleted sorbitol dehydrogenase. Proc Natl Acad Sci USA. 1998;95:3483–3488. doi: 10.1073/pnas.95.7.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jun CD, Pae HO, Kwak HJ, Yoo JC, Choi BM, Oh CD, Chun JS, Paik SG, Park YH, Chung HT. Modulation of nitric oxide-induced apoptotic death of HL-60 cells by protein kinase C and protein kinase A through mitogen-activated protein kinases and CPP32-like protease pathways. Cell Immunol. 1999a;194:36–46. doi: 10.1006/cimm.1999.1480. [DOI] [PubMed] [Google Scholar]
- 24.Jun CD, Oh CD, Kwak HJ, Pae HO, Yoo JC, Choi BM, Chun JS, Park RK, Chung HT. Overexpression of protein kinase C isoforms protects RAW 264.7 macrophages from nitric oxide-induced apoptosis: involvement of c-Jun N-terminal kinase/stress-activated protein kinase, p38 kinase, and CPP-32 protease pathways. J Immunol. 1999b;162:3395–3401. [PubMed] [Google Scholar]
- 25.Kawasaki H, Morooka T, Shimohama S, Kimura J, Hirano T, Gotoh Y, Nishida E. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem. 1997;272:18518–18521. doi: 10.1074/jbc.272.30.18518. [DOI] [PubMed] [Google Scholar]
- 26.Kim EY, Koh JY, Kim YH, Sohn S, Joe E, Gwag BJ. Zn2+ entry produces oxidative neuronal necrosis in cortical cell cultures. Eur J Neurosci. 1999;11:327–334. doi: 10.1046/j.1460-9568.1999.00437.x. [DOI] [PubMed] [Google Scholar]
- 27.Kim YH, Kim EY, Gwag BJ, Sohn S, Koh JY. Zinc-induced cortical neuronal death with features of apoptosis and necrosis: mediation by free radicals. Neuroscience. 1999;89:175–182. doi: 10.1016/s0306-4522(98)00313-3. [DOI] [PubMed] [Google Scholar]
- 28.Krotkiewska B, Banas T. Interaction of Zn2+ and Cu2+ ions with glyceraldehyde-3-phosphate dehydrogenase from bovine heart and rabbit muscle. Int J Biochem. 1992;24:1501–1505. doi: 10.1016/0020-711x(92)90078-f. [DOI] [PubMed] [Google Scholar]
- 29.Kukimoto I, Hoshino S, Kontani K, Inageda K, Nishina H, Takahashi K, Katada T. Stimulation of ADP-ribosyl cyclase activity of the cell surface antigen CD38 by zinc ions resulting from inhibition of its NAD+ glycohydrolase activity. Eur J Biochem. 1996;239:177–182. doi: 10.1111/j.1432-1033.1996.0177u.x. [DOI] [PubMed] [Google Scholar]
- 30.Kusuhara M, Takahashi E, Peterson TE, Abe J, Ishida M, Han J, Ulevitch R, Berk BC. p38 Kinase is a negative regulator of angiotensin II signal transduction in vascular smooth muscle cells: effects on Na+/H+ exchange and ERK1/2. Circ Res. 1998;83:824–831. doi: 10.1161/01.res.83.8.824. [DOI] [PubMed] [Google Scholar]
- 31.Lee D, Long SA, Adams JL, Chan G, Vaidya KS, Francis TA, Kikly K, Winkler JD, Sung CM, Debouck C, Richardson S, Levy MA, DeWolf WE, Jr, Keller PM, Tomaszek T, Head MS, Ryan MD, Haltiwanger RC, Liang PH, Janson CA, McDevitt PJ, Johanson K, Concha NO, Chan W, Abdel-Meguid SS, Badger AM, Lark MW, Nadeau DP, Suva LJ, Gowen M, Nuttall ME. Potent and selective nonpeptide inhibitors of caspases 3 and 7 inhibit apoptosis and maintain cell functionality. J Biol Chem. 2000;275:16007–16014. doi: 10.1074/jbc.275.21.16007. [DOI] [PubMed] [Google Scholar]
- 32.Leszkiewicz DN, Kandler K, Aizenman E. Enhancement of NMDA receptor-mediated currents by light in rat neurones in vitro. J Physiol (Lond) 2000;524:365–374. doi: 10.1111/j.1469-7793.2000.t01-1-00365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levitan IB. Modulation of ion channels by protein phosphorylation. How the brain works. Adv Second Messenger Phosphoprotein Res. 1999;33:3–22. doi: 10.1016/s1040-7952(99)80003-2. [DOI] [PubMed] [Google Scholar]
- 34.Maeno E, Ishizaki Y, Kanaseki T, Hazama A, Okada Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc Natl Acad Sci USA. 2000;97:9487–9492. doi: 10.1073/pnas.140216197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maret W, Vallee BL. Thiolate ligands in metallothionein confer redox activity on zinc clusters. Proc Natl Acad Sci USA. 1998;95:3478–3482. doi: 10.1073/pnas.95.7.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLaughlin BA, Nelson D, Silver IA, Erecinska M, Chesselet MF. Methylmalonate toxicity in primary neuronal cultures. Neuroscience. 1998;86:279–290. doi: 10.1016/s0306-4522(97)00594-0. [DOI] [PubMed] [Google Scholar]
- 37.Noh K-M, Kim YH, Koh J-Y. Mediation by membrane protein kinase C of zinc-induced oxidative neuronal injury in mouse cortical cultures. J Neurochem. 1999;72:1609–1616. doi: 10.1046/j.1471-4159.1999.721609.x. [DOI] [PubMed] [Google Scholar]
- 38.Ojcius DM, Zychlinsky A, Zheng LM, Young JD. Ionophore-induced apoptosis: role of DNA fragmentation and calcium fluxes. Exp Cell Res. 1991;197:43–49. doi: 10.1016/0014-4827(91)90477-c. [DOI] [PubMed] [Google Scholar]
- 39.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 40.Pearce LL, Gandley RE, Han W, Wasserloos K, Stitt M, Kanai AJ, McLaughlin MK, Pitt BR, Levitan ES. Role of metallothionein in nitric oxide signaling as revealed by a green fluorescent fusion protein. Proc Natl Acad Sci USA. 2000;97:477–482. doi: 10.1073/pnas.97.1.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem. 1994;269:15195–15203. [PubMed] [Google Scholar]
- 42.Pieper AA, Verma A, Zhang J, Snyder SH. Poly (ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci. 1999;20:171–181. doi: 10.1016/s0165-6147(99)01292-4. [DOI] [PubMed] [Google Scholar]
- 43.Qin Z-H, Wang Y, Kikly KK, Sapp E, Kegel KB, Aronin N, DiFiglia M. Pro-caspase-8 is predominately localized in mitochondria and released into cytoplasm upon apoptotic stimulation. J Biol Chem. 2001;276:8079–8086. doi: 10.1074/jbc.M007028200. [DOI] [PubMed] [Google Scholar]
- 44.Rankin PW, Jacobson EL, Benjamin RC, Moss J, Jacobson MK. Quantitative studies of inhibitors of ADP-ribosylation in vitro and in vivo. J Biol Chem. 1989;264:4312–4317. [PubMed] [Google Scholar]
- 45.Samet JM, Graves LM, Quay J, Dailey LA, Devlin RB, Ghio AJ, Wu W, Bromberg PA, Reed W. Activation of MAPKs in human bronchial epithelial cells exposed to metals. Am J Physiol. 1998;275:L551–L558. doi: 10.1152/ajplung.1998.275.3.L551. [DOI] [PubMed] [Google Scholar]
- 46.Satoh MS, Lindahl T. Role of poly (ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 47.Satoh T, Nakatsuka D, Watanabe Y, Nagata I, Kikuchi H, Namura S. Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci Lett. 2000;288:163–166. doi: 10.1016/s0304-3940(00)01229-5. [DOI] [PubMed] [Google Scholar]
- 48.Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci USA. 1999;96:2414–2419. doi: 10.1073/pnas.96.5.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheline CT, Behrens MM, Choi DW. Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J Neurosci. 2000;20:3139–3146. doi: 10.1523/JNEUROSCI.20-09-03139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimizu T, Kato T, Jr, Tachibana A, Sasaki MS. Coordinated regulation of radioadaptive response by protein kinase C and p38 mitogen-activated protein kinase. Exp Cell Res. 1999;251:424–432. doi: 10.1006/excr.1999.4582. [DOI] [PubMed] [Google Scholar]
- 51.Skulachev VP, Chistyakov VV, Jasaitis AA, Smirnova EG. Inhibition of the respiratory chain by zinc ions. Biochem Biophys Res Commun. 1967;26:1–6. doi: 10.1016/0006-291x(67)90242-2. [DOI] [PubMed] [Google Scholar]
- 52.Szabo C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- 53.Treves S, Trentini PL, Ascanelli M, Bucci G, Di Virgilio F. Apoptosis is dependent on intracellular zinc and independent of intracellular calcium in lymphocytes. Exp Cell Res. 1994;211:339–343. doi: 10.1006/excr.1994.1096. [DOI] [PubMed] [Google Scholar]
- 54.Virag L, Szabo C. Inhibition of poly(ADP-ribose) synthetase (PARS) and protection against peroxynitrite-induced cytotoxicity by zinc chelation. Br J Pharmacol. 1999;126:769–777. doi: 10.1038/sj.bjp.0702332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J. 1995;14:1607–1614. doi: 10.1002/j.1460-2075.1995.tb07149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weiss JH, Sensi SL, Koh JY. Zn2+: a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci. 2000;21:395–401. doi: 10.1016/s0165-6147(00)01541-8. [DOI] [PubMed] [Google Scholar]
- 57.Yu SP, Choi DW. Ions, cell volume, and apoptosis. Proc Natl Acad Sci USA. 2000;97:9360–9362. doi: 10.1073/pnas.97.17.9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- 59.Ziegler M, Jorcke D, Zhang J, Schneider R, Klocker H, Auer B, Schweiger M. Characterization of detergent-solubilized beef liver mitochondrial NAD+ glycohydrolase and its truncated hydrosoluble form. Biochemistry. 1996;35:5207–5212. doi: 10.1021/bi9527698. [DOI] [PubMed] [Google Scholar]