

Abstract
Cancer arises through a multistage process, but it is not fully clear how this process influences the age-specific incidence curve. Studies of colorectal and pancreatic cancer using the multistage-clonal-expansion (MSCE) model have identified two phases of the incidence curves. One phase is linear beginning about age of 60, suggesting that at least two rare rate-limiting mutations occur prior to clonal expansion of premalignant cells. A second phase is exponential, seen in earlier-onset cancers occurring before the age of 60 that are associated with premalignant clonal expansion. Here we extend the MSCE model to include clonal expansion of malignant cells, an advance that permits study of the effects of tumor growth and extinction on the incidence of colorectal, gastric, pancreatic and esophageal adenocarcinomas in the digestive tract. After adjusting the age-specific incidence for birth-cohort and calendar-year trends, we found that initiating mutations and premalignant cell kinetics can explain the primary features of the incidence curve. However, we also found that the incidence data of these cancers harbored information on the kinetics of malignant clonal expansion prior to clinical detection, including tumor growth rates and extinction probabilities on three characteristic time scales for tumor progression. Additionally, the data harbored information on the mean sojourn times for prema-lignant clones until occurrence of either the first malignant cell or the first persistent (surviving) malignant clone. Lastly, the data also harbored information on the mean sojourn time of persistent malignant clones to the time of diagnosis. In conclusion, cancer incidence curves can harbor significant information about hidden processes of tumor initiation, premalignant clonal expansion and malignant transformation, and even some limited information on tumor growth before clinical detection.
Keywords: Multistage Clonal Expansion (MSCE) model, tumor progression, colorectal cancer incidence, pancreatic cancer, esophageal cancer
Introduction
Uncontrolled cell proliferation is the sine qua non of carcinogenesis. However, long before symptoms signal cancer growth, several initiating mutations are generally required to overcome normal homeostatic regulation in a tissue allowing the gradual expansion of premalignant clones. Albeit slow and possibly stagnant, this growth enhances the probability that a premalignant cell undergoes malignant transformation generating a clone that either becomes extinct or progresses until clinical detection. Therefore, at least two distinct but overlapping clonal expansion processes are likely to occur in a tissue prior to clinical detection of cancer. In the context of the multistage-clonal-expansion (MSCE) carcinogenesis model described here, the first clonal expansion begins after normal tissue stem cells acquire two rate-limiting mutations or epigenomic changes that lead to abrogation of homeostatic tissue control, causing gradual outgrowth of occult premalignant clones over an extended time period that may range from years to decades [1]. Clonal expansion of the premalignant cell population enhances the probability that one or more of these cells suffer additional mutations or epigenomic alterations that cause malignant transformation which enables tumors to accelerate their growth and invade neighboring tissue, a process captured by the second (malignant) clonal expansion in the model.
Here we ask the basic question, how do the rate-limiting steps involved in tumor initiation, malignant transformation, and ensuing clonal expansions influence the shape of the cancer incidence curve? Conversely, what can we possibly learn from observed incidence curves about these hidden processes? In previous studies [1–4], we identified two characteristic features, or phases, in the incidence curves for colorectal and pancreatic cancers using data from the Surveillance Epidemiology and End Results (SEER) registries [5]. After adjusting for secular trends related to birth-cohort and calendar-year (period), we were able to identify an exponential phase in the incidence curve beginning in early adult life and extending to approximately the age of 60 and a linearly-increasing trend for later-onset cancers extending beyond the age of 60. In this study we ask the question whether the impact of malignant growth and fitness (defined as clone survival) on observed incidence patterns is actually discernible? To address this question, we use a MSCE model which explicitly incorporates distinct (but overlapping) clonal expansions for premalignant and malignant cells giving rise to a distribution of malignant tumors in a tissue and clinical observation of cancer via a stochastic detection event occurring in a preclinical tumor. In contrast, earlier versions of the MSCE model assumed that the first malignant cell in a tissue necessarily leads to clinical detection after a possibly random lag-time. Malignant transformations, however, are likely to occur in altered cells whose initial survival fitness may be compromised by genomic instability [6] and therefore may be prone to extinction in spite of higher cell proliferation. This is supported by comparative measurements of cell division rates and net cell proliferation (using DNA labeling and radio-graphic imaging of tumors, respectively) in a variety of carcinomas, showing large differences in the two rates which can only be explained by the frequent death of tumor cells [7].
For this model-driven investigation of cancer incidence we analyse SEER-9 [5] incidence data (1975-2008) for four gastrointestinal malignancies: colorectal cancer (CRC), gastric cancer (GaC), pancreatic cancer (PaC), and esophageal adenocarcinomas (EAC). We begin by adjusting for period and birth-cohort effects, using rigorous likelihood based methods to estimate model parameters for the extended MSCE model, including malignant clonal expansion rates for each cancer type. We then estimate three characteristic times: (1) the mean sojourn times for premalignant clones until occurrence of the first malignant cell regardless of its fate, (2) the analogous mean sojourn time to appearance of the first surviving (persistent) malignant clone; and (3) the mean sojourn time of persistent preclinical cancers from first malignant cell to time of cancer diagnosis.
Combined with a mathematical exploration of the MSCE model hazard function (i.e., the model-derived function which predicts the age-specific cancer incidence) our numerical findings support the hypothesis that the initiation of a benign (non-invasive) tumor, its malignant transformation, and persistence constitute major bottlenecks in the progression of a premalignant tumor to cancer. This is consistent with results from evolutionary models which find neoplastic progression to be driven mainly by mutations that confer only slight improvements in fitness [8], while the transition from a non-invasive to an invasive tumor, which expands with a significantly higher growth rate, constitutes a critical, rate-limiting event.
Materials and Methods
Model assumptions and properties
Tumor initiation
A hallmark of the MSCE model is that tumor initiation requires a number of rate-limiting mutational events before a stem cell can undergo a clonal expansion that results in a premalignant lesion (see Figure 1). For colon and pancreatic cancer we inferred previously that it takes two rare hits to transform a normal tissue stem cell into an initiated tumor cell that is no longer under homeostatic control and undergoes a (first) clonal expansion [1]. The two significant initial hits may represent biallelic inactivation of tumor suppressor genes, such as Tp53 or P16 that occur frequently in many cancers, or the Apc gene in colorectal cancer (CRC) [9]. Inactivation of TP53 is seen during early development of many digestive tract cancers, including gastric (GaC) [10], pancreatic (PaC) [11,12], and esophageal adenocarcinomas (EAC) [14–16]. Inactivation of P16 often occurs early in the development of EAC [17] and other cancers. However, the two hits may also represent activation of an oncogene such as Kras in combination with gain-of-function mutation in a tumor suppressor gene [12,13]. Additionally, EAC is associated with earlier conversion of a section of normal esophageal squamous epithelium to intestinal-type Barrett's metaplasia, called Barrett's esophagus (BE). We model the transition to BE as an additional one-time tissue alteration which occurs prior to the two initiating events leading to premalignant clonal expansions in the development of EAC [3].
Figure 1.
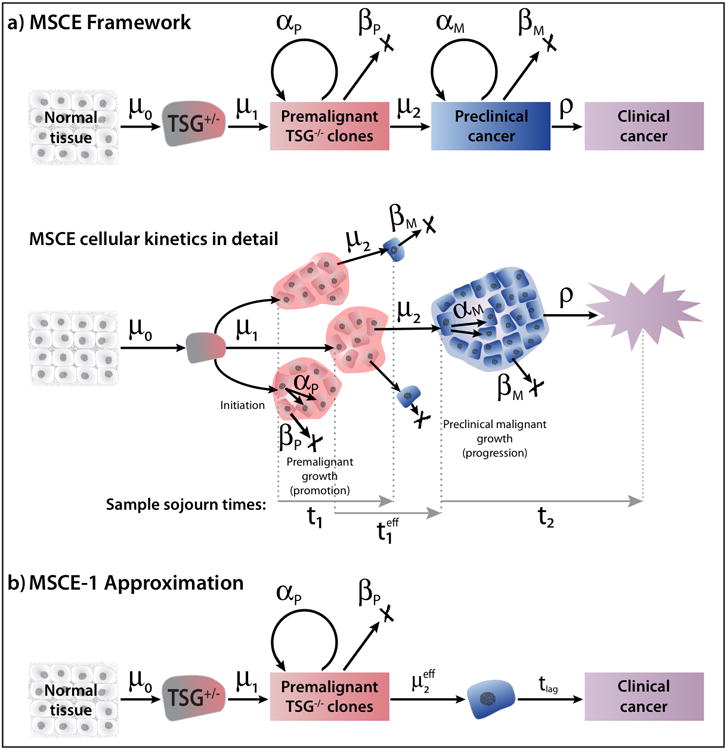
(a) The multistage-clonal expansion (MSCE) model for cancer with two stochastic birth-death-migration processes representing clonal expansions of premalignant and malignant cells. The model assumes a ‘two-hit’ tumor initiation process with Poisson initiation rates μ0, μ1 which leads to the stochastic appearance of premalignant progenitor cells in the tissue. In the lower sample MSCE realization of the cellular process, premalignant cells undergo a first clonal expansion described by a birth-death-migration process with cell division rate αP, cell death-or-differentiation rate βP, and malignant transformation rate μ2. Malignant cells, in turn, undergo a second clonal expansion with cell division and death rates αM, and βM, respectively, allowing for stochastic growth and possibly extinction of the malignant tumor. Clinical detection occurs through a size-based detection process with parameter ρ. The sample sojourn time t1 represents the time from the initiation of a premalignant clone until first malignant transformation. The sample sojourn time represents the time from the initiation of a premalignant clone to the first malignant cell in that clone which results in a persistent tumor which escapes extinction. Lastly, the sample sojourn time t2 represents the time for a persistent tumor to develop from a single malignant cell to detected, clinical cancer. The MSCE model is well approximated by (b) MSCE-1 Approximation which includes an effective malignant transformation rate (see text) and a constant lag-time for tumor progression.
A mathematical consequence of the two-hit hypothesis for (premalignant) tumor initiation is that the hazard function of the model (which represents the age-specific incidence) has a linearly increasing trend for older ages [1]. The presence of such a linear phase in the incidence curves for colorectal and pancreatic cancer could indeed be demonstrated by likelihood-based comparisons of models with two (or more hits) for initiation. Models with single-hit tumor initiations do not give rise to a linear phase in the hazard function [1, 2].
Tumor promotion
Prior to the transition into the initiation-associated linear phase, the MSCE hazard function increases exponentially with a rate which is approximately given by the net cell proliferation rate of premalignant P cells [1]. The transition from the exponential phase to the linear phase occurs around the age of 60 for colorectal and pancreatic cancer. Clonal expansion of P cells is represented by a stochastic birth-death-mutation (bdm) process with cell division rate αP, death-or-differentiation rate βP, and mutation rate μ2. The net cell proliferation rate of P cells is given by αP – βP – μ2 and the asymptotic probability of extinction of P cells by the ratio βP/αP [18], which is the probability that a premalignant cell, together with its progeny, will ultimately become extinct. Premalignant P cells may suffer further mutations with rate μ2 which transform them into malignant (M) cancer stem cells. Although the premalignant cell population is likely to undergo a complex evolutionary process involving multiple mutations in critical regulatory pathways before acquiring a malignant phenotype [6], only two initial rate-limiting mutations prior to clonal expansion appear necessary to adequately describe the main shape of the incidence curve for the four digestive tract cancers studied here.
Tumor progression and cancer detection
Development of a preclinical tumor in the MSCE model begins with a single (malignant) cell which undergoes clonal expansion and eventually, if the clone survives extinction, progresses to clinically detectable cancer. In contrast to natural history models in which the preclinical state of a tumor is typically assumed to be screen-detectable, the preclinical tumor development in the MSCE model starts off with a single malignant cell which undergoes a clonal expansion and eventually, if the clone survives extinction, is detected as cancer. Mathematically, the growth of the malignant tumor is described by a stochastic birth-death (bd) process with cell division rate αM and cell death rate βM. Clinical detection of the tumor is similarly treated as a stochastic event with rate ρ per cell. This implies that a tumor of size n cells has probability nρΔt to be detected in a time interval Δt short enough for the tumor to be constant in size. We refer to this generalization of the bd process as a birth-death-observation (bdo) process. Note, all analyses reported here are with a fixed value of ρ = 10−7. The rationale for this particular value of ρ is that a typical tumor contains about 109 cells upon (symptomatic) detection and that only about 1% of the tumor volume is occupied by actively dividing tumor cells [19]. Results obtained with other values for ρ (in the range of 10−6 – 10−8) were similar and did not change our conclusions (see Table S1).
The essential stochastic components of the MSCE model are illustrated in Figure 1, including separate clonal-expansions for premalignant and malignant cells. As we will show here (see Results), the MSCE model contains an approximation (referred to as MSCE-1) which differs from the original MSCE model in two important aspects: (1) the rate at which P cells suffer a transformation event that gives rise to a detectable cancer is approximated by an ‘effective’ transformation rate, , and (2) the approximation requires a lag-time to allow or the time from first malignant cell that forms a persistent cancer clone to the time of diagnosis. Furthermore, not all of the MSCE model parameters are identifiable from incidence data — some parameters must be fixed initially in order to achieve parameter identifiability, as discussed in the Results section and in the Supplemental Information (SI).
While multistage generalizations of the models shown in Figure 1 have also been explored by others [20–24], the general impact of malignant tumor progression on the hazard function (and the age-specific incidences of the cancers modeled here) has not been fully characterized [1–4], especially in regard to the time scales of premalignant and malignant clonal expansion. However, Fakir et al. [20,21] modeled stochastic effects of lung cancer progression by augmenting a similar model with a more realistic progression model, including extinction and/or dormancy, proliferation, and invasive growth.
MSCE model hazard function
For the full version depicted in Figure 1, which includes two stochastic clonal expansions, it is straightforward to derive the probability, PMSCE(t), for a cancer diagnosis/detection to occur by time t. A general approach is to solve the Kolmogorov backward equations for the marginal probability generating functions properly conditioned on no cancer occurring before time t, as shown in the SI. This yields the MSCE survival function SMSCE(t) = 1 − PMSCE(t). The MSCE hazard function — the rate at which preclinical cancer cells collectively trigger a first clinical observation event — is given by
| (1) |
(where the dot represents a derivative with respect to t). hMSCE(t) can be computed by numerically solving a set of ordinary differential equations as described in SI.
Data
We used data from the Surveillance Epidemiology and End Results (SEER) database by the National Cancer Institute. Incidence data were for all races by single-years of age between 10 and 84 and calendar-year between 1975 and 2008 for males and females (all races) in the original nine registries (SEER-9) [5]. Incident cancers were defined using the International Classification of Diseases for Oncology, Third Edition (ICD-O-3) as follows: colorectal cancer (C18-C20); esophageal (C150-C159, 8140/3 adenocarcinoma NOS); gastric cancer (C16); and pancreatic cancer (C25). Population data were also downloaded for the nine SEER catchment areas by gender and single years of age and calendar year. Methods for adjusting the incidence for secular trends (period and cohort effects) are described in SI and the SEER-9 incidences for all four cancers are shown in Figures S3a-h, both unadjusted and adjusted for secular trends. Figure S2 shows the secular trends estimated using a modified age-period-cohort (APC) model described in the SI.
Results
We find that modeling of cancer incidence data provides new insights into the importance of clonal extinction and clonal growth rates (or doubling times) of premalignant and malignant clones in relation to three underlying time scales in carcinogenesis: the mean sojourn time for premalignant clones until occurrence of the first malignant cell (T1), the mean sojourn time for premalignant clones until the first surviving malignant clone ( ), and the mean sojourn time of persistent preclinical cancers from first malignant cell to time of diagnosis (T2). In the following we demonstrate how these time scales contribute to, and are estimable from, the age-specific incidence curves of four digestive tract cancers.
Mathematical properties of MSCE-1 Approximation
To gain insights into how tumor progression is impacting cancer incidence we begin with a mathematical dissection of the hazard function generated by the MSCE model depicted in Figure 1. This will demonstrate that the MSCE model (with a distinct clonal expansion for malignant cells) can be closely approximated by a reduced model (MSCE-1) which adjusts the rate of malignant transformation, μ2, for non-extinction and further models the outgrowth of persistent malignant clones as a constant time-lag, tlag, which in turn is approximated by the mean sojourn time of a surviving malignant clone, from its inception to detection of cancer, T2. To better understand the relationship between tlag and the mean sojourn time T2 in the MSCE model we show (proof given in SI) that the hazard function of a model with two consecutive clonal expansions for premalignant and malignant cells is mathematically equivalent to a model with a single clonal expansion of premalignant cells with a time-dependent mutation rate, i.e., replacing μ2 ↔ μ2(1 – SM (u)), where 1 – SM(u) is the unnormalized probability of detection of malignant clones a time u after the malignant clone is seeded. An exact expression for SM(u) is given in the SI for constant parameters. However, this mathematical “simplification” of the model from a double to a single clonal expansion process comes at the cost of a time-dependent (conditional) mutation rate.
The time-dependence of the conditional mutation rate μ2(1 – SM (u)) has two main effects: 1) it reduces the effective rate of malignant transformation, and 2) it creates a time delay for a malignant clone to grow, conditional on its non-extinction, into a detectable tumor. The latter effect is mainly due to a sharp transition of the conditional mutation rate from zero to its asymptotic value after a time which equals approximately T2, the mean sojourn time of the malignant clone to detection of cancer (see SI). Because asymptotically SM (u) → βM/αM as u → ∞, we define as the effective malignant transformation rate for the reduced (MSCE-1) model. Therefore, the approximation amounts to
| (2) |
Again, T2 is the mean sojourn time of a surviving malignant clone which avoids stochastic extinction and which, in the absence of death, would eventually be detected as cancer. The mean sojourn time of a premalignant clone to the first malignant cell (T1), the analogous mean sojourn time to the ancestor of the first persistent malignant clone ( ), and the mean sojourn time of a persistent malignant tumor (T2) to detection are functions of the cell kinetic parameters and are given by (see SI)
respectively, where SP and are survival functions defined analogously to SM (see SI).
Parameter identifiability and sensitivity
Not all of the MSCE model parameters are identifiable from incidence data — some parameters must be fixed initially in order to achieve parameter identifiability (see Heidenreich et al. [25]). Furthermore, for estimability, the exponential-then-linear character of the multistage hazard function (see SI) suggest a parametrization that involves the slope of the linear phase λ ≡ μ0Xμ1p∞ and the growth parameter of the exponential phase gP ≡ αP − βP − μ2 [1]. Note, the rates μ0 and μ1 cannot be estimated separately because the slope λ depends on their product. Analogous to premalignant growth, we introduce the malignant growth parameter gM ≡ αM − βM − ρ In order to identify μ2 and ρ we find it necessary to fix the cell division rates αP and αM. Although the product αMρ is mathematically identifiable, we were not able to obtain stable estimates and therefore also fixed the (per cell) cancer detection parameter ρ (see Materials and Methods). Otherwise, the biological model parameters were estimated using a Markov-Chain Monte Carlo (MCMC) method (see SI). Figures S1a-h show scatter-plots of the MCMC samples obtained for all four cancers studied here, separately by gender. For EAC, an additional parameter is included representing the rate of normal squamous tissue conversion to Barrett's metaplasia.
To explore the dependence of our parameter estimates on the fixed parameters αP, αM, and the cancer detection rate ρ we have also conducted a systematic sensitivity analysis. The results of this analysis (specifically, the ranges of the obtained maximum likelihood estimates for the parameters λ, gP, gM, and , assuming constant birth cohort and calendar year effects) for each fixed parameter are given in Table S1. This analysis (although limited to CRC) shows that the estimates of gM, and therefore the mean sojourn time T2, vary only slightly when αP and αM are perturbed, but may vary up to 20% as the detection rate ρ changes an order of magnitude. Therefore, the dependence of the preclinical cancer sojourn time on ρ is modest, but does not change our conclusions.
Low fitness of malignant cells
We use the above results to gain insight into the importance of clonal extinction. Figure 2 shows fits obtained with the MSCE model (solid line) to SEER incidence data for a) colorectal cancer, and b) gastric cancers. These fits include adjustments of the model-generated hazard function for secular trends (for details see SI). It is instructive to mathematically ‘dissect’ the MSCE hazard function to examine the underlying behavior of the incidence curves for the different malignant ancestors. The combined effects of extinction and time for (malignant) tumor growth on incidence can be seen by substituting the ‘full’ rate μ2 into the MSCE-1 approximation and ignoring the lag-time, i.e., tlag = 0 (dotted line in Figure 2). The higher predicted incidence sans malignant cell extinction or tumor growth shows that these processes greatly reduce and delay cancer incidence and change the shape of the incidence curve. In comparison, re-introducing the effects of extinction by replacing μ2 with (without a lag-time) restores the general shape of the incidence curve (dot-dash line) except for cancers occurring too early. Finally, re-introducing the time-lag associated with malignant tumor growth (T2) in the MSCE-1 approximation accounts for both processes (dashed line) and provides an excellent approximation to the exact incidence curve generated by the full MSCE model (solid line).
Figure 2.


Deconstruction of the MSCE hazard function: malignant clone extinction and tumor growth influence the incidence curves for a) CRC, and b) GaC. (Plots for EAC and PaC are similar, but not shown). The SEER data (adjusted for calendar-year and birth-cohort trends) are shown as circles and the overall fit using the MSCE model by the thin solid line. The dotted line on the left shows the underlying hazard for the first malignant cell, regardless of its fate. In contrast, the dash-dotted line shows the hazard for the first ancestor of a persistent (i.e. surviving) malignant clone and the dashed line represents the hazard for the first persistent malignant clone shifted to the right by the mean sojourn time of the malignant clone to cancer detection, T2 (see text).
Time scales of tumor progression
The MSCE model explicitly models malignant transformations in premalignant tissues of an organ. These tissues may not be uncommon as they may arise independently from a large number of normal ancestor cells. However, our results suggest that most malignant cells and nascent malignancies undergo extinction. The time difference between the appearance of the first malignant cell in a premalignant clone, regardless of its fate, and the first ancestor cell that leads to a stable malignant clone that is bound to turn into symptomatic cancer (unless a patient dies before this happens or an intervention occurs) may be as long as 30-40 years for gastric cancer (see Table 1), as long as 20 years for CRC and EAC, or as short as 3 years, or less, in the case for pancreatic cancer. It is not clear whether these differences reflect transformation-specific differences in cell survival, exogenous factors, cell senescence, or differences in the degree of genomic instability. Whatever the origin, with the exception of pancreatic cancer, our findings suggest a generally low viability of cancer cells in spite of their aggressive and invasive behaviour.
Table 1.
MCMC-based estimates of various tumor promotion and progression time scales. See text for definitions. Estimates represent medians and 95% credibility regions of the marginal posterior distribution for each quantity listed. All units are in years.
| males | T1 (95% CI) | T1eff (95% CI) | T2 (95% CI) | Tlag(95% CI) |
|
| ||||
| CRC | 32.6 (30.4 - 36.8) | 50.6 (49.7 - 52.1) | 5.2 (3.6 - 6.2) | 5.4 (3.1 - 7.0) |
|
|
||||
| GaC | 17.9 (13.6 - 22.3) | 45.8 (43.8 - 47.9) | 12.2 (9.8 - 14.7) | 9.5 (7.7 - 12 0) |
|
|
||||
| PaC | 49.1 (40.4 - 52.6) | 52.3 (50.9 - 52.9) | 0.7 (0.4 - 2.2) | 3.0 (0.2 - 6.8) |
|
|
||||
| EAC | 15.0 (7.2 - 24.7) | 39.2 (34.2 - 44.0) | 12.0 (6.3 - 16.8) | 12.7 (3.1 - 16 5) |
|
|
||||
| females | T1 (95% CI) | T1eff (95% CI) | T2 (95% CI) | Tlag(95% CI) |
|
| ||||
| CRC | 27.5 (25.1 - 30.5) | 48.7 (47.6 - 49.9) | 6.5 (5.2 - 7.6) | 6.5 (5.1 - 8.2) |
|
|
||||
| GaC | 20.1 (16.8 - 23.4) | 58.6 (56.8 - 60.5) | 11.7 (10.1 - 13.2) | 10.6 (9.1 - 11.7) |
|
|
||||
| PaC | 53.2 (44.7 - 56.7) | 56.3 (55.2 - 57.1) | 0.6 (0.4 - 1.8) | 1.7 (0.1 -4.9) |
|
|
||||
| EAC | 15.8 (13.8 - 18.4) | 37.9 (31.8 - 45.1) | 10.6 (7.8 - 13.8) | 10.2 (7.2 - 13.2) |
In contrast, the estimated mean sojourn times T2 of persistent malignant clones vary from 10-12 years for GaC and EAC, 5-7 years for CRC, down to less than 1 year for PaC (Table 1). The latter is consistent with the observation that most pancreatic carcinoma are diagnosed at an advanced metastatic stage. Note, however, that , the estimated mean time to the appearance of the first persistent cancer clone (measured from the time the ancestral premalignant cell is born) is somewhat longer for pancreas than colon (52.3 vs 50.6 years for males, 56.3 vs 48.7 in females). This suggests that premalignant precursor lesions in pancreas, such as pancreatic intra-epithelial neoplasia (PanINs), may be present for many years before a stable malignant transformation occurs.
Table 2.
MCMC-based estimates of various tumor promotion and progression time scales. These identifiable parameters are defined as:λ = μ0 · X · μ1 ·p∞, gP = αP − βP − μ2, gM = αM − βM − ρ, . Here, we define p∞ ≈ 1 − βM/αM (see SI for more details). Estimates represent medians and 95% credibility regions of the marginal posterior distribution for each quantity listed. All units are in years.
| males | λ (95% CI) × 10-4 | gp (95% CI) | gM (95% CI) | μ2eff (95% CI) × 10-6 |
|
| ||||
| CRC | 2.13 (2.10 - 2.15) | 0.162 (0.160 - 0.164) | 2.71 (2.22 - 4.15) | 0.73 (0.55 - 0.86) |
|
|
||||
| GaC | 0.51 (0.49 - 0.53) | 0.140 (0.135 - 0.145) | 1.00 (0.80 - 1.30) | 3.21 (2.16 - 4.65) |
|
|
||||
| PaC | 0.35 (0.34 - 0.36) | 0.181 (0.177 - 0.186) | 27.7 (7.44 - 49.1) | 0.25 (0.21 - 0.32) |
|
|
||||
| EAC | 52.9 (34.8 - 99.3) | 0.163 (0.133 - 0.190) | 1.02 (0.68 - 2.19) | 4.65 (1.65 - 10.8) |
|
|
||||
| females | λ (95% CI) × 10-4 | gP (95% CI) | gM (95% CI) | μ2eff (95% CI) × 10-6 |
|
| ||||
| CRC | 1.57 (1.55 - 1.59) | 0.149 (0.147 - 0.151) | 2.12 (1.75 - 2.75) | 1.56 (1.26 - 1.87) |
|
|
||||
| GaC | 0.40 (0.36 - 0.44) | 0.100 (0.096 - 0.105) | 1.06 (0.91 - 1.25) | 2.83 (2.36 - 3.39) |
|
|
||||
| PaC | 0.34 (0.33 - 0.35) | 0.161 (0.157 - 0.165) | 30.0 (9.10 - 50.0) | 0.30 (0.26 - 0.36) |
|
|
||||
| EAC | 9.1 (2.4 - 30.9) | 0.170 (0.132 - 0.218) | 1.18 (0.86 - 1.71) | 4.65 (fixed) |
For EAC, we also estimate a (constant) tissue conversion rate, νBE, from normal esophageal tissue to the metaplastic tissue of Barrett's esophagus (BE). The age-specific prevalence of BE is therefore approximately νBE × age and appears to be subject to strong period effects [3]. Although our MCMC-based estimates for νBE and the slope parameter λ representing initiation of premalignant clones are highly anti-correlated (see Figure S1g-h), the predicted BE prevalences (about 1.5% for males and 0.5% for females at age 60 in the year 2000) are consistent with the range of epidemiological estimates obtained from studies in comparable populations [26].
Tumor growth rates
We find highly stable estimates for the net cell proliferation rate gP of premalignant cells, based on the posterior distributions of the identifiable MSCE model parameters given the observed cancer incidences in SEER (see Figure S1). The reason for this stability appears to lie in the prominence of the exponential phase of the incidence curve and the resulting linear behavior of the log-incidence (see Figure S3). Surprisingly, with the exception of gastric cancers in females, the estimated net cell proliferation rates for premalignant lesions are similar and stay within a range of 0.14 to 0.18 per year, while estimates for the net cell proliferation rate gM of the malignant lesions are much more variable and range from 1 per year in gastric and esophageal cancers to rates as high as 30 per year for pancreatic cancer (see Table 2). These values correspond to tumor volume doubling times of 250 days and 8 days, respectively for this group of cancers. While the former is consistent with clinical observations for early gastric carcinoma, which are generally slow growing [27], the latter appears too fast, but not inconsistent with tumor marker doubling times. For example, using the pancreatic tumor marker CA19-9, Nishida et al. [28] estimated doubling times from measurements in patients with inoperable pancreatic cancer in the range of 6 to 313 days. For CRC, the estimated malignant tumor volume doubling times are about 93 days for males and 119 days for females. They too appear at the lower end of the clinical spectrum, but are consistent with the determination by Bolin et al. [29] who followed 27 carcinomas radiographically in the colon and rectum, measuring a median of 130 days with a range of 53 to 1570 days. In spite of considerable uncertainty and variability of the clinical observations, the general agreement of the MSCE model predictions with sparse measurements of tumor doubling times lends support to our claim that carefully collected incidence data harbor quantitative information about the natural history of a tumor, from initiation to promotion to malignant tumor progression.
Discussion
Early models of carcinogenesis recognized the importance of rate-limiting mutations but provided only crude fits to cancer incidence and mortality [30]. Subsequent incorporation of cell proliferation made it possible to account for effects, such as the initiation/promotion effects seen in chemical carcinogenesis [31, 32] or the inverse dose-rate effect for high-LET radiation [33], that were more difficult to explain with models that did not include clonal expansion. More recently, multistage extensions of the original two-stage clonal expansion model by Moolgavkar, Venzon and Knudson [34, 35] have emerged as useful instruments to explore cancer incidence curves and isolate important secular trends that segregate with birth cohort and/or calendar year (period) from age effects driven by common underlying biological processes [3,4, 36]. While secular trends are of great interest to epidemiologists and cancer control researchers in understanding the impact of screening, potential exposures to carcinogens (e.g. tobacco smoking), infections, diet, and life-style factors on cancer incidence, in this study we focus on non-specific effects that have their origin in common cell-level processes that drive the age-effect, in particular the impact of malignant tumor progression on the age-specific incidence curve.
Incidence curves are consistent with two types of clonal expansions, slow and fast
Our MSCE model fits to the incidences of four gastrointestinal cancers (CRC, GaC, PaC, and EAC) yield parameter estimates suggesting that malignant tumor progression is preceded by a prolonged period of premalignant tumor growth characterized by a low rate of net cell proliferation (Tables 1 and 2). In contrast, malignant tumor growth is estimated to be many-fold faster than premalignant growth. The model distinguishes features of the incidence curves that relate to slow growth of premalignant lesions and fast growth of malignant lesions, and allows estimation of the time period in which tumors sojourn as slowly growing masses before becoming invasive. The effective sojourn time , i.e., the time to appearance of the first persistent malignant clone that started with a single premalignant cell, appears to be much longer than estimated from clinical data. For colon, clinical estimates range from 20-25 years [37]. However, this usually refers to the time starting with a small adenoma which must have been already present for some time. It is not known how long adenomas sojourn before they can be observed. A clue can be found in the average time to cancer among familial adenomatous polyposis (FAP) patients which can be viewed as a lower estimate for the mean sojourn time of an adenoma, since adenomas are likely to form early in life in FAP patients even though the diagnosis of polyposis may not occur until later. From the age distribution of cancer with polyposis in FAP patients (see [37]), which peaks around the age of 40, we conclude that the mean sojourn of an adenoma which has the potential to progress to cancer is likely longer than 40 years since this time generally represents the time to first diagnosis of the cancer – a first passage time in statistical parlance – and not an average time across all adenomas with neoplastic potential including some that will not turn cancerous in a person's lifetime. Our estimates of 50-55 years for the mean duration of an adenoma developing into a detectable carcinoma are therefore not inconsistent with what can be inferred from the incidence of CRC in FAP.
Identifiability of a malignant progression parameter
Our mathematical analysis shows the approximate equivalency of the hazard functions generated by the MSCE model and a model with a single clonal expansion (MSCE-1) which is adjusted for clonal extinction and delayed by a lag-time representing the mean sojourn time T2 of the surviving malignancy (see Figure 1). Thus, in practice, only the time-scale associated with malignant tumor progression can be estimated from cancer incidence data but not the full malignant cell kinetics given by the rates of malignant cell division αM, cell death βM, and (per cell) detection ρ. However, assuming plausible values for the cell division rates (αM) and a (per cell) cancer detection rate ρ (see sensitivity analysis), we do obtain estimates for the net cell proliferation rate gM in malignant tumors that yield tumor volume doubling times which are consistent with clinical observations from radiographic imaging of carcinoma (see Results).
For pancreatic cancer, the estimated sojourn times T2 for male and female preclinical malignancies are very short, suggesting that the model only captures the short metastatic phase of the development but cannot identify the sojourn of the primary tumor. It is conceivable that non-invasive precursors, such as the PanINs, interact with stromal components such as myofibroblasts that facilitate invasion and metastatic colonization [38]. The resulting colonies may initially grow slowly, perhaps similar to their parental premalignant precursors, but may acquire an aggressive and expansive phenotype at a later time.
Carcinogenesis may well require more than 2 clonal expansions. However, as shown by Meza et al. (2008) [1] for CRC and PaC, the main features of the age-specific incidence curve can almost entirely be explained by the initiation and growth characteristics of premalignant tumors. Here, we posed the follow-up question: what impact does a second clonal expansion (say, representing malignant tumor growth) have on incidence curves. Our mathematical analysis shows that the impact amounts to a time-translation of the incidence curve which appears to be identifiable in the SEER incidences studied here. This is consistent with the common view that premalignant tumors and malignant tumors result from rather distinct clonal expansions which markedly different cell kinetics.
Comparison with DNA sequencing studies
For colorectal cancer Jones et al. [39] determined the time required from the founder cell of an advanced carcinoma to the appearance of the metastatic founder cell through comparative lesion sequencing in a small number of subjects. They concluded that it takes on average 2 years for the metastatic founder cell to arise in a carcinoma and an additional 3 years for the metastatic lesion to expand, thus a total of 5 years to the detection of the (metastatic) cancer after the carcinoma forms. Our model-derived estimates for T2, the mean sojourn time for preclinical CRC (5-7 years) are therefore in good agreement with the estimates for CRC using a molecular clock based on mutational data and evolutionary analysis [39].
More recently, Yachida et al. [40] undertook a similar study for pancreatic cancer sequencing the genomes of seven metastatic lesions to evaluate the clonal relationships among primary and metastatic cancers (see also [41]). They estimated 6.8 years for the length of time from the appearance of sub-clones in the primary tumor with metastatic potential to the seeding of the index metastasis and additional 2.7 years to detection. However, our T2 estimates for pancreatic cancer are inconsistent with those derived by Yachida and colleagues (see the MCMC-based posterior distributions for T2 in Fig. S1). Remarkably, we find shorter times which suggests (see discussion above), that the sub-clones found by Yachida and colleagues in the primary may already have been present in a slow growing precursor lesion. The question is therefore whether metastatic dissemination in pancreas can occur before the primary tumor undergoes a drastic transformation into a rapidly growing tumor.
Limitations
We previously conducted comparative analyses of incidence data with a variety of models: simple Markov process models without clonal expansion (e.g., the Armitage and Doll model [42, 43]), the two-stage clonal expansion (TSCE) model [44, 45], and with biologically-motivated extensions of the TSCE model [1–4]. Although the latter usually provide superior fits to cancer incidence data compared to the former [1, 2, 4], MSCE models are by no means complete descriptions of the cancer process, but should be considered biologically-motivated schemata that help to identify critical processes and time scales in carcinogenesis. The models lack many clinical and biological features that may or may not be relevant to our understanding of incidence curves. For example, secular trends may also be viewed as acting quite specifically on biological parameters, while in this study we employ a statistical approach (the age-period-cohort model [3, 4, 36]) to effectively adjust cancer incidence for secular trends. Moreover, our analyses assume that all clonal expansions give rise to (mean) exponential growth even though clinical evidence suggests that tumors may slow their growth in a Gompertzian manner due to limited nutrient/oxygen supplies as the tumor develops vasculature [46]. We also did not model effects of tumor dormancy or potential increases in tumor growth rates due to subtle selection effects in the somatic evolution of the tumor. The inferred cell kinetics does represent an average rate which may comprise passenger mutations that confer weak or no selection and possibly driver mutations that are not rate-limiting (or not requisite) but are likely to speed up the growth process, as well as spatial (niche) effects and clonal interference (as suggested by Martens et al. [48]) that have the potential to slow the tumor growth process. While modeling these processes may well improve our fits and alter certain parameter estimates, it is unlikely that such fine-tuning will alter the parameters associated with the basic two (exponential-then-linear) phases of the incidence curves in a significant way. It is remarkable that in its present form the MSCE model identifies mean sojourn times for tumors that are broadly consistent with clinical estimates in spite of the considerable uncertainties of our estimates and ambiguities in clinical observations.
One way to improve the MSCE model and test model assumptions is to incorporate data from screening and imaging of premalignant as well as malignant tumors. Screening for CRC provides information on the number and sizes of adenomatous polyps and screen detected carcinoma; while screening for EAC may include assessment of the presence or absence of dysplasia and/or chromosomal abnormalities in endoscopic biopsies and surveillance for early cancer. Mechanistic models such as the MSCE model may utilize these different outcomes to enhance our understanding of tumor initiation, growth, persistence and preclinical sojourn. In this study, we demonstrate that the preclinical phase of malignant tumor progression subtly influences the shape of the age-specific incidence curve, leaving a ‘footprint’ that may be identified through likelihood based analyses of incidence data after adjusting for secular trends. We identify and estimate three characteristic times scales of carcinogenesis: the mean sojourn time from premalignant cell to first malignant cell, T1; the mean sojourn time from premalignant cell to first malignant ancerstor that generates a persistent clone, ; and the mean sojourn time it takes for persistent tumors to develop from a single malignant cell to clinical cancer, T2. We conclude that malignant clone extinction and tumor sojourn times play important roles in reducing and delaying cancer incidence and influencing the shape of incidence curves for colorectal, gastric, pancreatic, and esophageal cancers.
Supplementary Material
Major Findings
Cancer incidence curves harbor information about hidden processes of tumor initiation, premalignant clonal expansion, malignant transformation, and even some limited information on tumor growth before clinical detection. Our analyses of the incidences of four digestive tract cancers show that the age-specific incidence curves –upon adjustments for secular trends and, in the case of esophageal adenocarcinoma, inclusion of an event describing the conversion of normal squamous to metaplastic Barrett's epithelium– are well approximated by a model which explicitly incorporates the stochastic growth kinetics of premalignant clones, the sporadic appearance of malignant cells within these clones, and a constant time delay corresponding to the mean sojourn time of a malignant clone. While this sojourn appears very short for pancreatic cancer (<3 years), intermediate for colorectal cancer (5-7 years), it is much longer for gastric cancer and esophageal adenocarcinoma (10-12 years). Furthermore, with the exception of pancreatic cancer, our results are consistent with the assumption of a high (>95%) probability of tumor stem cell extinction or terminal differentiation.
Quick Guide to Equations
The multistage clonal expansion model (MSCE) approximation yields the following hazard function which represents the age-specific rate at which cancers occur in a population that had no prior occurrences of that cancer:
With
Where represents the effective rate of malignant transformations that give rise to persistent tumors, and tlag is a time-lag equal to the mean sojourn time of a preclinical cancer clone from its single cell inception to clinical detection. See Figure 1 and text for a definition of the basic model parameters X (number of stem cells), αP(M) (cell division rates), βP(M) (cell death rates), μ0,1,2 (mutation rates). Furthermore,
where −pP measures approximately the net cell proliferation in premalignant clones and qP approximately the (effective) rate of malignant transformations.
Acknowledgments
We would like to thank Dr. Rafael Meza (University of Michigan) for helpful discussions.
Grant Support: This research was supported by the National Cancer Institute (NCI) under grants R01 CA107028 and UO1 CA152926 (EGL, JJ, and WDH) and by the National Science Foundation (NSF) under grant no. DGE-0718124 (KC).
Footnotes
Conflicts of interest: None.
References
- 1.Meza R, Jeon J, Moolgavkar SH, Luebeck EG. Age-specific incidence of cancer: Phases, transitions, and biological implications. Proc Natl Acad Sci U S A. 2008;105:16284–9. doi: 10.1073/pnas.0801151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luebeck EG, Moolgavkar SH. Multistage carcinogenesis and the incidence of colorectal cancer. Proc Natl Acad Sci U S A. 2002;99:15095–100. doi: 10.1073/pnas.222118199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jeon J, Luebeck EG, Moolgavkar SH. Age effects and temporal trends in adenocarcinoma of the esophagus and gastric cardia (United States) Cancer Causes Control. 2006;17:971–81. doi: 10.1007/s10552-006-0037-3. [DOI] [PubMed] [Google Scholar]
- 4.Meza R, Jeon J, Renehan AG, Luebeck EG. Colorectal cancer incidence trends in the United States and United kingdom: evidence of right- to left-sided biological gradients with implications for screening. Cancer Res. 2010;70:5419–29. doi: 10.1158/0008-5472.CAN-09-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Surveillance, Epidemiology, and End Results (SEER) Program SEER*Stat Database: Incidence -SEER 9 Regs Limited-Use, Nov 2009 Sub (1973-2007) (Katrina/Rita Population Adjustment) - Linked To County Attributes - Total U.S., 1969-2007 Counties. April 2010, based on the November 2009 submission ed: National Cancer Institute, DCCPS, Surveillance Research Program, Cancer Statistics Branch. [last accessed on 2012 Mar 13] Available from: http://seer.cancer.gov/.
- 6.Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999;9:M57–60. [PubMed] [Google Scholar]
- 7.Moffett BF, Baban D, Bao L, Tarin D. Fate of clonal lineages during neoplasia and metastasis studied with an incorporated genetic marker. Cancer Res. 1992;52:1737–43. [PubMed] [Google Scholar]
- 8.Beerenwinkel N, Antal T, Dingli D, Traulsen A, Kinzler KW, Velculescu VE, et al. Genetic progression and the waiting time to cancer. PLoS Computational Biology. 2007;3:e225. doi: 10.1371/journal.pcbi.0030225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–7. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 10.Karaman A, Kabalar ME, Binici DN, Oztürk C, Pirim I. Genetic alterations in gastric precancerous lesions. Genet Couns. 2010;21:439–50. [PubMed] [Google Scholar]
- 11.Norfadzilah MY, Pailoor J, Retneswari M, Chinna K, Noor LM. P53 expression in invasive pancreatic adenocarcinoma and precursor lesions. Malays J Pathol. 2011;33:89–94. [PubMed] [Google Scholar]
- 12.Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant P53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107:246–51. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 14.Ramel S, Reid BJ, Sanchez CA, Blount PL, Levine DS, Seshat K, et al. Evaluation of p53 protein expression in Barrett's esophagus by two-parameter flow cytometry. Gastroenterology. 1992;102:1220–8. [PubMed] [Google Scholar]
- 15.Moore JH, Lesser EJ, Erdody DH, Natale RB, Orringer MB, Beer DG. Intestinal differentiation and p53 gene alterations in Barrett's esophagus and esophageal adenocarcinoma. Int J Cancer. 1994;56:487–93. doi: 10.1002/ijc.2910560406. [DOI] [PubMed] [Google Scholar]
- 16.Galipeau PC, Prevo LJ, Sanchez CA, Longton GM, Reid BJ. Clonal expansion and loss of heterozygosity at chromosomes 9p and 17p in premalignant esophageal (Barrett's) tissue. J Natl Cancer Inst. 1999;91:2087–95. doi: 10.1093/jnci/91.24.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong DJ, Paulson TG, Prevo LJ, Galipeau PC, Longton G, Blount PL, et al. p16(INK4a) lesions are common, early abnormalities that undergo clonal expansion in Barrett's metaplastic epithelium. Cancer Res. 2001;61:8284–9. [PubMed] [Google Scholar]
- 18.Harris TE. The Theory of Branching Processes. New York: Dover Publications, Inc; 1989. [Google Scholar]
- 19.O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 20.Fakir H, Tan WY, Hlatky L, Hahnfeldt P, Sachs RK. Stochastic Population Dynamic Effects for Lung Cancer Progression. Radiat Res. 2009;172:383–93. doi: 10.1667/RR1621.1. [DOI] [PubMed] [Google Scholar]
- 21.Fakir H, Hofmann W, Sachs RK. Modeling progression in radiation-induced lung adenocarcinomas. Radiat Environ Biophys. 2010;49:169–76. doi: 10.1007/s00411-009-0264-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dewanji A, Moolgavkar SH, Luebeck EG. Two-mutation model for carcinogenesis: joint analysis of premalignant and malignant lesions. Math Biosci. 1991;104:97–109. doi: 10.1016/0025-5564(91)90032-e. [DOI] [PubMed] [Google Scholar]
- 23.Little MP. Are two mutations sufficient to cause cancer? Some generalizations of the two-mutation model of carcinogenesis of Moolgavkar, Venzon, and Knudson, and of the multistage model of Armitage and Doll. Biometrics. 1995;51:1278–91. [PubMed] [Google Scholar]
- 24.Little MP, Heidenreich WF, Li G. Parameter identifiability and redundancy in a general class of stochastic carcinogenesis models. PLoS One. 2009;4:e8520. doi: 10.1371/journal.pone.0008520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heidenreich WF, Luebeck EG, Moolgavkar SH. Some Properties of the Hazard Function of the Two-Mutation Clonal Expansion Model. Risk Anal. 1997;17:391–9. doi: 10.1111/j.1539-6924.1997.tb00878.x. [DOI] [PubMed] [Google Scholar]
- 26.Ronkainen J, Aro P, Storskrubb T, Johansson S, Lind T, Bolling-Sternevald E, et al. Prevalence of Barrett's esophagus in the general population: an endoscopic study. Gastroenterology. 2005;129:1825–1831. doi: 10.1053/j.gastro.2005.08.053. [DOI] [PubMed] [Google Scholar]
- 27.Teixeira CR, Haruma K, Teshima H, Yoshihara M, Sumii K, Kajiyama G. Endoscopic therapy for gastric cancer in patients more than 80 years old. Am J Gastroenterol. 1991;86:725–8. [PubMed] [Google Scholar]
- 28.Nishida K, Kaneko T, Yoneda M, Nakagawa S, Ishikawa T, Yamane E, et al. Doubling time of serum CA 19-9 in the clinical course of patients with pancreatic cancer and its significant association with prognosis. J Surg Oncol. 1999;71:140–6. doi: 10.1002/(sici)1096-9098(199907)71:3<140::aid-jso2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 29.Bolin S, Nilsson E, Sjödahl R. Carcinoma of the colon and rectum–growth rate. Ann Surg. 1983;198:151–8. doi: 10.1097/00000658-198308000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Renan MJ. How many mutations are required for tumorigenesis? Implications from human cancer data. Mol Carcinog. 1993;7:139–46. doi: 10.1002/mc.2940070303. [DOI] [PubMed] [Google Scholar]
- 31.Luebeck EG, Buchmann A, Stinchcombe S, Moolgavkar SH, Schwarz M. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on initiation and promotion of GST-P-positive foci in rat liver: A quantitative analysis of experimental data using a stochastic model. Toxicol Appl Pharmacol. 2000;167:63–73. doi: 10.1006/taap.2000.8980. [DOI] [PubMed] [Google Scholar]
- 32.Moolgavkar SH, Luebeck EG, de Gunst M, Port RE, Schwarz M. Quantitative analysis of enzyme-altered foci in rat hepatocarcinogenesis experiments–I. Single agent regimen Carcinogenesis. 1990;11:1271–8. doi: 10.1093/carcin/11.8.1271. [DOI] [PubMed] [Google Scholar]
- 33.Luebeck EG, Heidenreich WF, Hazelton WD, Paretzke HG, Moolgavkar SH. Biologically based analysis of the data for the Colorado uranium miners cohort: age, dose and dose-rate effects. Radiat Res. 1999 Oct;152(4):339–51. Toxicol Appl Pharmacol. 2000;167:63-73. [PubMed] [Google Scholar]
- 34.Moolgavkar SH, Knudson AG. Mutation and cancer: a model for human carcinogenesis. Journal of the National Cancer Institute. 1981;66:1037–52. doi: 10.1093/jnci/66.6.1037. [DOI] [PubMed] [Google Scholar]
- 35.Moolgavkar SH, Venzon DJ. Two-event models for carcinogenesis: incidence curves for childhood and adult tumors. Mathematical Biosciences. 1979;47:55–77. [Google Scholar]
- 36.Holford TR, Zhang Z, McKay LA. Estimating age, period and cohort effects using the multistage model for cancer. Stat Med. 1994;13:23–41. doi: 10.1002/sim.4780130105. [DOI] [PubMed] [Google Scholar]
- 37.Muto T, Bussey HJ, Morson BC. The evolution of cancer of the colon and rectum. Cancer. 1975;36:2251–70. doi: 10.1002/cncr.2820360944. [DOI] [PubMed] [Google Scholar]
- 38.Brentnall TA, Lai LA, Coleman J, Bronner MP, Pan S, Chen R. Arousal of cancer-associated stroma: overexpression of palladin activates fibroblasts to promote tumor invasion. PLoS One. 2012;7:e30219. doi: 10.1371/journal.pone.0030219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105:4283–8. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yachida S, Zhong Y, Patrascu R, Davis MB, Morsberger LA, Griffin CA, et al. Establishment and characterization of a new cell line, A99, from a primary small cell carcinoma of the pancreas. Pancreas. 2011;40:905–10. doi: 10.1097/MPA.0b013e3182207a58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luebeck EG. Cancer: Genomic evolution of metastasis. Nature. 2010;467:1053–5. doi: 10.1038/4671053a. [DOI] [PubMed] [Google Scholar]
- 42.Moolgavkar SH. Carcinogenesis models: an overview. Basic Life Sci. 1991;58:387–99. doi: 10.1007/978-1-4684-7627-9_14. [DOI] [PubMed] [Google Scholar]
- 43.Moolgavkar SH, Luebeck EG. Multistage carcinogenesis: population-based model for colon cancer. J Natl Cancer Inst. 1992;84:610–8. doi: 10.1093/jnci/84.8.610. [DOI] [PubMed] [Google Scholar]
- 44.Heidenreich WF, Luebeck EG, Moolgavkar SH. Effects of exposure uncertainties in the TSCE model and application to the Colorado miners data. Radiat Res. 2004;161:72–81. doi: 10.1667/rr3089. [DOI] [PubMed] [Google Scholar]
- 45.Meza R, Hazelton WD, Colditz GA, Moolgavkar SH. Analysis of lung cancer incidence in the Nurses' Health and the Health Professionals' Follow-Up Studies using a multistage carcinogenesis model. Cancer Causes Control. 2008;19:317–28. doi: 10.1007/s10552-007-9094-5. [DOI] [PubMed] [Google Scholar]
- 46.Xu XL. The biological foundation of the Gompertz model. Int J Biomed Comput. 1987;20:35–9. doi: 10.1016/0020-7101(87)90012-2. [DOI] [PubMed] [Google Scholar]
- 47.Brosnan JA, Iacobuzio-Donahue CA. A new branch on the tree: next-generation sequencing in the study of cancer evolution. Semin Cell Dev Biol. 2012;23:237–42. doi: 10.1016/j.semcdb.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martens EA, Kostadinov R, Maley CC, Hallatschek Spatial structure increases the waiting time for cancer. New J Phys. 2011;13:1–22. doi: 10.1088/1367-2630/13/11/115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.