

Abstract
The reaction of the old yellow enzyme and reduced flavins with organic nitrate esters has been studied. Reduced flavins have been found to react readily with glycerin trinitrate (GTN ) (nitroglycerin) and propylene dinitrate, with rate constants at pH 7.0, 25°C of 145 M−1s−1 and 5.8 M−1s−1, respectively. With GTN, the secondary nitrate was removed reductively 6 times faster than the primary nitrate, with liberation of nitrite. With propylene dinitrate, on the other hand, the primary nitrate residue was 3 times more reactive than the secondary residue. In the old yellow enzyme-catalyzed NADPH-dependent reduction of GTN and propylene dinitrate, ping-pong kinetics are displayed, as found for all other substrates of the enzyme. Rapid-reaction studies of mixing reduced enzyme with the nitrate esters show that a reduced enzyme–substrate complex is formed before oxidation of the reduced flavin. The rate constants for these reactions and the apparent Kd values of the enzyme–substrate complexes have been determined and reveal that the rate-limiting step in catalysis is reduction of the enzyme by NADPH. Analysis of the products reveal that with the enzyme-catalyzed reactions, reduction of the primary nitrate in both GTN and propylene dinitrate is favored by comparison with the free-flavin reactions. This preferential positional reactivity can be rationalized by modeling of the substrates into the known crystal structure of the enzyme. In contrast to the facile reaction of free reduced flavins with GTN, reduced 5-deazaflavins have been found to react some 4–5 orders of magnitude slower. This finding implies that the chemical mechanism of the reaction is one involving radical transfers.
Old yellow enzyme (OYE; EC 1.6.99.1), isolated from brewer's bottom yeast by Warburg and Chistian in 1933, was the first enzyme where the requirement of a prosthetic group (“cofactor”) for catalysis was demonstrated (1). OYE is a mixture of homodimers and heterodimers arising from different yeast genes, with each monomer of ≈45 kDa binding a molecule of flavin mononucleotide. Although its exact physiological function is still unknown, many tight binding ligands such as phenols and steroids have been identified, raising the possibility that this enzyme might be involved in yeast sterol metabolism (2). On binding with phenols and heteroaromatic compounds containing an ionizable hydroxyl group, OYE produces characteristic long-wavelength charge transfer complexes (3).
The crystal structure of OYE has been solved, revealing several amino acid residues around the active site of the enzyme to effect catalysis and ligand binding (4). Phenolic compounds are held parallel to the flavin by π-overlap with the isoalloxazine ring and oriented by hydrogen bonding of the phenolate oxygen with His-191 and Asn-194. The long wavelength-absorbance band arises from parallel stacking interaction between the phenolate anion and the flavin. Several mutant forms of OYE have been constructed and have confirmed that His-191, Asn-194, Tyr-196, and Thr-37 residues interact in ligand binding and catalysis (5–7). We have discovered that this enzyme catalyzes the reduction of the olefinic bond of α,β-unsaturated carbonyl compounds by using NADPH as a reductant (2, 8) and also catalyzes slow aromatization of cyclic enones like cyclohexenone by a novel dismutation reaction (8). By using R-(2H) NADPH and D2O, it was established that reduction of α,β-unsaturated carbonyl compounds proceeds via stereospecific transfer of hydride from NADPH via the flavin to the carbon atom β to the carbonyl function and transfer of a proton to the α position, resulting in trans-addition to the double bond (8). A growing number of substrates for OYE have been discovered. Recently, we showed that unsaturated nitro compounds act as substrates for OYE, being converted to the saturated nitro compounds in a two-step mechanism, first hydride transfer from the reduced flavin to form an aci-nitro intermediate, followed by proton uptake mediated by Tyr-196 (9).
During the last few years, an increasing number of proteins have been isolated from bacterial, yeast, and plant sources, which share significant sequence identity with OYE (10–18). A number of these enzymes from microbial sources have been shown to catalyze the degradation of nitrate esters (19). Nitrate esters, condensation products of alcohol and nitric acid, although not commonly found in nature, are now widely spread in the environment because of their extensive use as industrial explosives and as vasodilators in medicine. Microbes isolated from ammunition-testing sites led to the isolation of several nitrate ester-degrading enzymes such as PETN reductase (11) and the so-called xenobiotic reductases A and B (17). All these enzymes have large sequence homology with OYE, which also was reported (19) to catalyze the reduction of organic nitrate esters such as trinitroglycerine (glycerin trinitrate, GTN). We therefore undertook a detailed study of the OYE-catalyzed reaction of organic nitrate esters and report the results here. During this study, we observed that reduced flavins could reduce nitrate esters, although more slowly than enzyme catalysis by OYE. We also studied the reduction of GTN by the mutant forms of OYE 1, Y196F, and H191N. It was found, in contrast to the reduction of α,β-unsaturated carbonyl or nitro compounds, that replacement of the acid/base residue Tyr-196 by phenylalanine had no effect on catalysis of nitrate ester reduction. On the other hand, mutation of His-191 greatly reduced the enzyme activity.
Materials and Methods
Preparation of OYE 1.
Wild-type and mutant enzyme forms were prepared as described (2, 5, 7). NADPH, NADP, glucose 6-phosphate, and glucose-6-phosphate dehydrogenase (from Leuconostoc mesenteroides) were from Sigma. Glycerol and 1,2-propanediol were from Aldrich. GTN and propylene 1,2-dinitrate (PDN) were synthesized by using the reported procedure (20) as follows. A 0.92-g (10 mmol) portion of glycerol was added drop-wise to a mixture of 2 ml of fuming nitric acid and 2.5 ml of concentrated sulfuric acid stirred at −10°C. The resulting mixture was stirred at 0°C for 2 h. Then the reaction mixture was poured onto crushed ice and the yellow oily liquid was extracted with dichloromethane, washed with water, cold sodium bicarbonate solution, and brine. After evaporation of the solvent, vacuum distillation gave 2 ml of clear liquid [BP 90°C/0.25 mmHg (1 mmHg = 133 Pa)]. 1H NMR showed pure GTN. By using the same procedure, PDN was made from 1,2-propanediol (boiling point 70°C/10 mmHg).
Spectroscopic Studies.
UV-visible absorbance spectra were recorded with a Hewlett–Packard Diode Array spectrophotometer or a Cary (Varian) model 219 double-beam spectrophotometer. 1H NMR spectra were recorded with a Bruker (Billerica, MA) 500 MHz NMR instrument. Chemical shift values are reported in parts per million relative to tetramethylsilane.
Steady-State Kinetics Experiments.
To determine the steady-state kinetic parameters it is necessary to exclude oxygen from the system. We therefore made use of the capabilities of a stopped-flow spectrophotometer to mix together NADPH and nitrates and to follow anaerobically the course of the reaction. With PDN as substrate, 2.64 μM OYE 1 (all concentrations given are those after mixing) was mixed in one set of experiments with 60 μM NADPH and 70, 250, 500, and 1,000, 2,000, and 5,000 μM propylene dinitrate, and the oxidation of NADPH was followed at 340 nm. Turnover numbers were estimated from the time-interval changes during the reaction. In a second set of experiments, the concentration of propylene dinitrate was limiting (initially 50 μM) and the NADPH was in excess (213 and 320 μM). Under these conditions, the enzyme is largely in the oxidized form during turnover, and becomes reduced as the propylene dinitrate is exhausted. The reaction was followed at 340 nm and at 462 nm, and enzyme-monitored turnover traces at the latter wavelength were analyzed as described in ref. 21. For GTN, similar enzyme-monitored turnover experiments were carried out, with an initial NADPH concentration of 50 μM and GTN concentrations of 50, 200, 300, 350, 500, and 1,050 μM. All reactions were carried out in 0.05 M KPi buffer (pH 7.0) at 25°C, using a HiTech (Salisbury, U.K.) SF-61 stopped-flow spectrophotometer. Anaerobiosis was achieved as described (7).
Rapid-Reaction Studies of Oxidative Half Reaction.
OYE 1 was made anaerobic in a tonometer and reduced slowly with an NADPH-generating system as described (7). Reoxidation of the reduced enzyme was followed at 462 nm and other selected wavelengths on mixing with different concentrations of GTN or PDN that had been bubbled in syringes with O2-free argon before introduction into the stopped-flow spectrophotometer. Reaction traces were analyzed as described (7). All reactions were carried out in 0.05 M KPi buffer (pH 7.0) at 25°C.
Kinetics of Oxidation of Reduced Riboflavin with GTN and PDN.
Riboflavin was made anaerobic in a tonometer and reduced photochemically with the EDTA-induced photoreduction as described (7). Reoxidation of the reduced flavin was followed at 450 nm and other selected wavelengths on mixing with different concentrations of GTN and PDN that had been bubbled in syringes with O2-free argon before introduction into the stopped-flow spectrophotometer. All reactions were carried out in 0.05 M KPi buffer (pH 7.0) at 25°C.
Product Analysis.
To determine the regioselectivity of denitration of GTN and PDN by reaction with reduced riboflavin and by the enzyme-catalyzed reaction with OYE, products were analyzed by NMR and HPLC procedures. For example, 17 mg (0.045 mmol) of riboflavin was added to a mixture of 0. 3 M EDTA in 10 ml of 0.1 M (pH 7.0) phosphate buffer and 200 ml of water. The resulting solution was made anaerobic by repeated evacuation and flushing with argon, and then reduced by shining visible light from a commercial sun gun. Under a stream of argon, a solution of 12 mg (0.053 mmol) of GTN was added and the reaction flask closed. The reaction mixture was stirred in the dark overnight and then extracted with dichloromethane, washed with water, cold sodium bicarbonate solution, brine, and finally dried with anhydrous magnesium sulfate. The organic solvent was evaporated, and the resulting residue was analyzed by NMR and HPLC.
For the OYE-catalyzed reaction, 40 mg (0.053 mmol) of NADPH was added to 20 ml of 0.1 M (pH 7.0) phosphate buffer containing 0.6 μM OYE 1. The resulting mixture was made anaerobic by repeated evacuation and flushing with argon. Under a stream of argon, a solution of 12 mg (0.053 mmol) of GTN was added and the reaction flask closed. The reaction mixture was stirred overnight in the dark and then extracted with dichloromethane, washed, and analyzed as described previously.
HPLC Analysis.
RP-HPLC analysis was carried out with a Beckman System Gold instrument (model 126) UV detector, using a C-18 250 × 10-mm column. Elution was carried out with a linear gradient between 1% and 90% acetonitrile in ultra pure HPLC-grade water, and the eluant was monitored by UV at 217 nm. The flow rate was kept at 1 ml/min at room temperature. The system was calibrated with standard solutions of GTN.
Determination of Nitrite.
Nitrite release was measured by using the diazo-coupling method (22). Thus, for the OYE-catalyzed reaction, 0.3 mM NADPH and 0.5 mM GTN were incubated under anaerobic conditions with 4 μM OYE in 0.1 M phosphate buffer (pH 7.0) and kept at room temperature in the dark overnight. A 100-μl aliquot then was mixed with 0.5 ml of 1% sulfanilamide in 20% (vol/vol) HCl followed by 0.5 ml of 0.02% N-(1-naphthyl) ethylenediamine dihydrochloride. After 20 min, the resulting diazo-coupling product was measured at 540 nm and the concentration of nitrite estimated from the ɛ540 value of 41,000 M−1cm−1
Results
Synthesis of GTN and PDN.
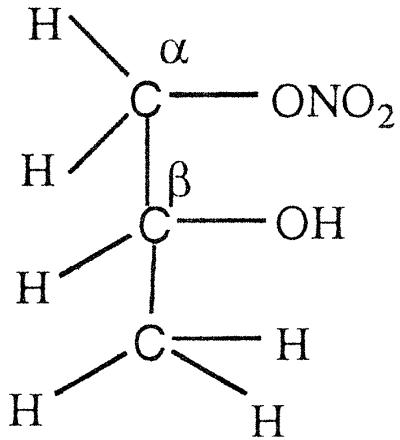
Because commercially available samples of GTN are too dilute to perform analytical experiments, we synthesized it and PDN by using a published procedure (20). The product from the reaction, after extraction from aqueous media by organic solvent, can be obtained easily in pure form by vacuum distillation. Organic nitrate esters are explosive substances and on heating at an elevated temperature, both GTN and PDN explode. However, in our hands with a relatively small sample (a couple of grams) and using very high vacuum to keep the distillation bath temperature ≈100°C, it was found to be safe to heat the compounds at this temperature. The products, clear colorless liquids, were pure GTN and PDN, respectively, as judged from the agreement of the obtained 1H NMR spectra, as shown in Table 3, with those of the published values (23). Data (500 MHz 1H NMR) for PDN: δ 1.38, 1.37 [doublet (d), 3H], 4.44–4.406 [multiplet (m), 1H], 4.64–4.61 (m, 1H), and 5.34–5.31 (m, 1H); and for GTN: 4.63–4.67 (m, 2H), 4.83–4.79 (m, 2H), and 5.52–5.49 (m, 1H). 1H NMR data also show very distinctive 1H NMR signals for secondary nitrate from that of primary nitrate, [for GTN, 5.52–5.49 (secondary 1H) vs. 4.6–4.8 (primary 1H); and for PDN, 5.34–5.31 (secondary 1H) vs. 4.6–4.4 (primary 1H)], which was used to identify the degradation products as described in a later section.
Table 3.
Proton NMR signals (ppm) of nitrate
| Proton | 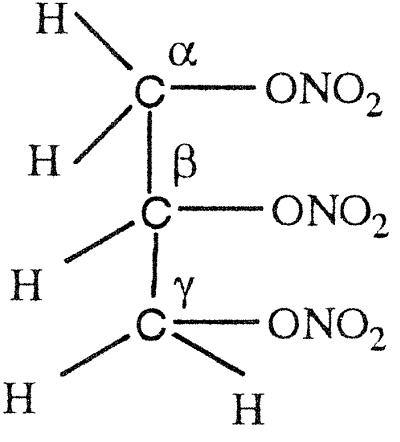 |
 |
 |
 |
 |
 |
|---|---|---|---|---|---|---|
 |
4.826–4.629 | 3.970–3.901 | 4.577–4.533 | 4.644–4.406 | 3.797–3.665 | 4.457–4.287 |
 |
5.519–5.495 | 5.357–5.327 | 4.309–4.279 | 5.340–5.307 | 5.233–5.136 | 4.124–4.110 |
 |
4.826–4.629 | 4.600–4.509 | 4.577–4.533 |
Steady-State Kinetics.
Enzyme-monitored turnover experiments were carried out by using a stopped-flow spectrophotometer, and data were analyzed as described (21). A known concentration of OYE in 50 mM NaPi (pH 7.0) was reacted with a range of concentrations of GTN and fixed concentration of NADPH (25 μM). The reaction was followed at 340 nm to monitor NADPH oxidation and at 462 nm to monitor the reduction-oxidation status of the enzyme flavin during turnover. Turnover analysis by monitoring the flavin absorbance (21) gave identical results to those obtained by following the time course of NADPH oxidation at 340 nm. Plots of 1/turnover number vs. 1/[NADPH] yielded a series of parallel lines for the varied concentrations of GTN and from a replot of intercepts against 1/[GTN], the kinetic constants were obtained as listed in Table 1. Similar results were obtained by using propylene dinitrate as substrate (Table 1). Thus, in its reaction with the nitrate esters, ping-pong kinetics were obtained, similar to the results with all other substrates of the enzyme so far studied (5).
Table 1.
Steady-state kinetics of OYE 1 with NADPH and nitrate esters
| Compound | kcat, s−1 | Km, mM | Km, NADPH, μM |
|---|---|---|---|
| GTN | 5.5 | 0.38 | 7.5 |
| PDN | 2.5 | 4.5 | Too low to measure |
Kinetics of the Oxidative Half Reaction of Reduced OYE by Organic Nitrates.
The reaction kinetics of enzyme reoxidation by organic nitrates can be followed conveniently in anaerobic stopped-flow experiments by monitoring the reoxidation of reduced OYE at 462 nm. OYE was reduced slowly with an NADPH-generating system as described (7) and mixed under anaerobic conditions with different concentrations of GTN. Reoxidation of the enzyme flavin was measured at 462 nm and other wavelengths. Reciprocals of observed rate constants plotted against reciprocal of [GTN] results in the double-reciprocal plot of Fig. 1. From this plot, it can be concluded that GTN binds to reduced enzyme in a step preceding reoxidation, with a Kd of 2.9 mM and a kox of 40 s−1 for GTN (24). Experiments using PDN gave similar results, with a kox of 7 s−1 and a Kd of 12 mM for binding of PDN. These results are summarized in Table 2.
Figure 1.

Kinetics of oxidation of reduced OYE by GTN. See text for details.
Table 2.
Kinetics for the reoxidation of reduced enzymes and flavins by nitrate esters
| Compound | OYE 1 | Y196F OYE 1 | H191N OYE 1 | Flavin |
|---|---|---|---|---|
| GTN | kox 40 s−1 | kox 80 s−1 | kox 0.013 s−1 | kox 145 M−1 s−1 |
| Kd 2.7 mM | Kd 33 mM | Kd 0.5 mM | ||
| PDN | kox 7 s−1 | kox 5.8 M−1 s−1 | ||
| Kd 12 mM |
Kinetics of the Oxidative Half Reaction of Reduced OYE, H191N, and Y196F by Organic Nitrates.
The inability of Y196F to catalyze the protonation step in the reaction of α,β-unsaturated carbonyl and nitro compounds prompted us to explore its effect on the reduction of organic nitrates. Reduced Y196F OYE 1 was reoxidized by GTN, and reoxidation was monitored at 462 nm and other wavelengths in stopped-flow experiments, similar to those with wild-type enzyme (shown in Fig. 1). The reaction was twice as fast as with wild-type enzyme, with a kox of 80 s−1 and Kd of 33 mM for GTN, indicating weaker binding. These results are summarized in Table 2 also. From these results, it can be concluded that Y196F probably plays some role in substrate binding, but that the tyrosine residue is not required to protonate or deprotonate the nitrate substrate.
Similar experiments were performed by using OYE 1 H191N. With this mutant form, the reoxidation of the reduced flavin was surprisingly slow, with a limiting rate constant of 0.013 s−1 and an apparent Kd of 0.5 mM (Table 2). For all enzyme forms, the reoxidation of the reduced enzyme flavin gave the same single exponential behavior, independent of the wavelength of observation. Thus, no spectroscopically distinct intermediates such as radical species, flavin-substrate adducts, or charge-transfer complexes could be observed in the reaction.
Reaction of Reduced Flavins with PDN and GTN.
Because Tyr-196 is not required for the reduction of nitrate esters, we postulate that the OYE reaction with nitrate is probably a simple reduction reaction from the reduced flavin and hence does not require an active site acid/base catalytic residue like Tyr-196. This hypothesis was confirmed by the demonstration that free reduced flavins also could carry out the denitration reactions catalyzed by the enzyme.
Under anaerobic conditions, with a slight excess of FADH2 (formed photochemically with EDTA as photodonor) over that of GTN, complete reduction of GTN was observed in a triphasic reaction, with 2.95 ± 0.1 mol of FADH2 oxidized per mol of GTN. Thus, when 187 μM FADH2 was allowed to react anaerobically with 60 μM GTN, a total of 170 μM FADH2 was reoxidized, with approximately one-third being oxidized with t1/2 of 30 s, another third with t1/2 of ≈10 min and the remainder with t1/2 ≈140 min. From these results we can conclude that all three nitrate ester groups can be reduced by the flavin, with the first reacting ≈20 times faster than the second, which in turn is considerably more reactive than the third.
The reaction kinetics of flavin reoxidation can be followed conveniently in anaerobic stopped-flow experiments by monitoring the reoxidation of reduced riboflavin or flavin mononucleotide at 450 nm by organic nitrates at varied concentrations as performed for the OYE1 reactions described previously. In this case, the observed rate constants were directly proportional to the concentration of the nitroester, yielding the second order rate constants summarized in Table 2.
NMR Analysis of the Reaction Products.
To identify the reaction products of GTN and PDN with OYE and reduced riboflavin, the products were analyzed by NMR and HPLC. Dichloromethane extracts of the reaction media were dried and analyzed by NMR to identify the degradation products of GTN and PDN. Table 3 summarizes 1H NMR data for parent and derivative compound (23). There is a distinctive 1H NMR signal for secondary nitrate (δ 5.3–5.5) from that of the primary nitrate (δ 4.4–4.8). This distinctive nature of NMR signals was used previously by White et al. (25) to identify denitration products in the reaction with the GTN reductase from Agrobacterium radiobacter (12).
Product Analysis from Chemical Reaction Between Reduced Riboflavin and GTN.
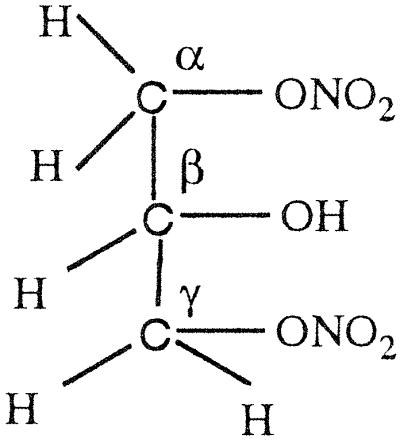
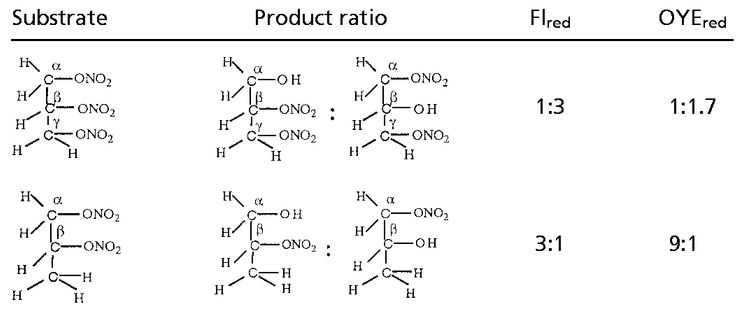
GTN (45 μmol) was added to EDTA-photoreduced riboflavin (45 μmol) in 0.01 M KPi (pH 7.0) under anaerobic conditions and kept stirring overnight. Then the sample was extracted with dichloromethane, evaporated, and dried for NMR analysis. As summarized in Table 4, a multiplet for one secondary proton of Cβ at 5.35–5.33 ppm, for CαH2OH CβHONO2CγH2ONO2, and a multiplet at 4.29- 4.27 ppm for one secondary proton of Cβ, for CαH2ONO2CβHOHCγH2ONO2, were found in the ratio of ≈1:3. When a nitro group is denitrated from the nitroester, the resulting hydrogen atom attached to the carbon atom is significantly down-shifted (corresponding to an alcoholic functional group instead of a nitroester group). Thus, a secondary nitroester hydrogen shows at 5.53–5.49 ppm and secondary alcohol at 4.85–4.68 ppm. The NMR also showed a multiplet for a secondary proton at 3.95–3.91 ppm with a corresponding multiplet at 3.79–3.67 ppm for a primary alcohol group, revealing the presence of the mononitrate, CαH2ONO2CβHOHCγH2OH. These results are in accord with the multiphasic reaction described earlier for reaction of FADH2 with GTN, and reveal the decided preference for reduced flavins to react with the secondary rather than primary nitrate ester groups of GTN.
Table 4.
Stereoselectivity in denitration reaction
 |
Fl, flavin.
Product Analysis from OYE-Catalyzed Reaction with GTN.
The NMR spectra obtained from a reaction of OYE with stoichiometric concentrations of NADPH and GTN showed a multiplet for the secondary 1H at 5.35–5.33, for CαH2OH CβHONO2CγH2ONO2, and a multiplet at 4.29–4.27 for the secondary 1H, for CαH2ONO2CβHOHCγH2ONO2, in the ratio of ≈1:1.7. In contrast to the experiment with reduced riboflavin, there was no detectable glycerin mononitrate formed during the enzyme-catalyzed reaction. It seems that the enzyme-catalyzed reduction of GTN shows a somewhat smaller preference for reaction with the secondary nitrate than in the case with free flavin. To monitor the stereochemical outcome of the OYE-catalyzed reduction in the presence of a large excess of GTN, experiments were carried out in which the isomeric denitrated products were analyzed by HPLC. In a typical experiment, 0.3 mM NADPH was treated with 6 mM of GTN and 7 nM OYE 1 in 0.1 M (pH 7.0) phosphate buffer. After the oxidation of NADPH was complete, the reaction mixture was analyzed by HPLC as described in Materials and Methods. The glycerin 1,2-dinitrate and glycerin 1,3-dinitrate products were found in the ratio of 1:1.4, showing that the comparative rates of dinitration were not significantly altered even in the presence of an excess of GTN.
Product Analysis from Chemical Reaction Between Reduced Riboflavin with PDN.
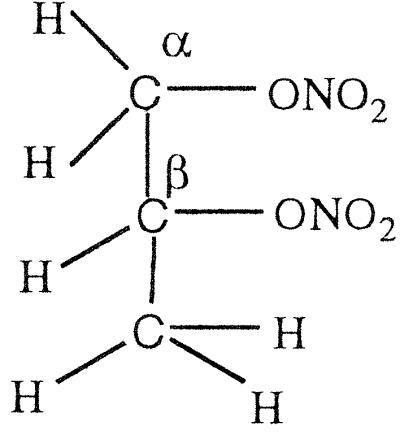
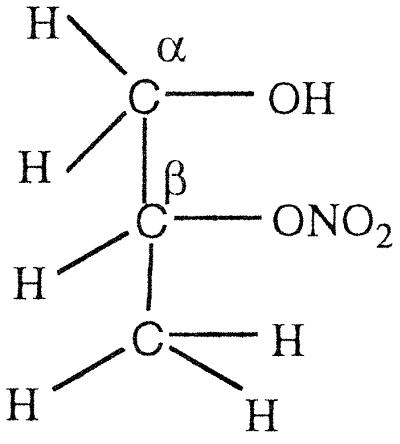
PDN (50 μmol) was added to 50 μmol of reduced flavin mononucleotide (photoreduced with EDTA) in an anaerobic flask and kept stirring overnight in the dark. NMR analysis of the dichloromethane-extracted sample revealed a multiplet corresponding to the secondary 1H at 5.35–5.33 ppm, for CH3CHONO2CH2OH, and a multiplet at 4.29–4.27 ppm, for the secondary 1H of CH3CHOHCH2ONO2, in the ratio of ≈3:1. The NMR data thus show that reduction of the secondary nitrate by reduced flavin is roughly 3 times slower than that of the primary nitrate, i.e., that in contrast to GTN, reduction of the central nitrate residue is now slower than that of the terminal residue.
Product Analysis from OYE-Catalyzed Reaction with PDN.
PDN (24 μmol) was mixed with a stoichiometric amount of NADPH (24.5 μmol) and a catalytic concentration of OYE 1 (0.1 ml of 10 μM) under anaerobic conditions and kept overnight. The NMR of the dichloromethane-extracted sample showed a multiplet for the secondary 1H at 5.35–5.33 ppm, for CH3CHONO2CH2OH, and a multiplet at 4.29–4.27 for the secondary 1H of CH3CHOHCH2ONO2 in the ratio of ≈9:1. Thus, the enzyme-catalyzed reaction with propylene dinitrate exhibits a marked preference for reaction with the terminal primary nitrate over the central secondary nitrate residue, even more so than that of the free flavin. Table 4 summarizes the stereoselectivity results of reductive denitration of GTN and PDN with reduced free flavin and with OYE.
Lack of Reactivity of Reduced 5-Deazaflavin with GTN.
In contrast with the ready reoxidation of reduced riboflavin and FADH2 with GTN, reduced 5-deazaflavin is only marginally reactive, if at all, with GTN. Owing to the poor solubility of 5-deazariboflavin, a water-soluble derivative, 5-deaza-10-methyl-3-sulfopropylisoalloxazine, synthesized by Blankenhorn (26) was used. A solution in 0.1 M KPi (pH 7.0) was titrated with NaBH4 until the deazaflavin was 90% reduced. Then aliquots were mixed aerobically with the same buffer, or with 2.5 mM GTN in buffer, and the course of reoxidation at 25oC followed over a period of 3 days, care being taken to exclude light, which is known to accelerate the very slow reaction with oxygen (27). The control reaction without GTN had a t1/2 of reoxidation of 4,600 min, and two reactions with GTN had t1/2 values of 3,200 and 5,200 min. From these data it can be estimated that the second order rate constant for reaction of the reduced 5-deazaflavin with GTN was at most ≈4 × 10−4 M−1s−1, some 300,000 times smaller than that with reduced riboflavin. A slightly faster reaction was obtained with another model deazaflavin, 5-deaza-3,10-dimethyl-iosalloxazine (28), where a rate constant for reaction of the reduced deazaflavin with GTN in 5% CH3CN of ≈4 × 10−3 M−1s−1 was obtained.
Discussion
That organic nitrates such as GTN or propylene dinitrate can oxidize reduced flavins under anaerobic conditions is consistent with the chemical versatility of flavins and the fact that organic nitrates are also strong oxidizing agents. Their oxidizing strength depends on the number of nitrate groups present in the compound. This property is reflected by the relative rate constants for oxidation of reduced riboflavin by GTN and PDN (Table 2).
Table 2 also reports the rate constants for oxidation of reduced OYE by GTN and propylene dinitrate. OYE acts catalytically for the NADPH-dependent denitration of organic nitrates. GTN and PDN act as true substrates, binding to the reduced enzyme to form enzyme-substrate complexes before the chemical oxidation step. In the reaction of OYE1 with other substrates, such as α,β-unsaturated aldehydes and ketones (8) or unsaturated nitro substrates like nitrocyclohexene (9), Tyr-196 acts as active-site acid, providing the second hydrogen atom involved in reduction of the substrate double bond as a proton. In such reactions, the mutant enzyme Y196F is catalytically very much impaired (6, 9). In contrast, this enzyme form is quite competent in catalysis of denitration of organic nitrate esters; indeed, as shown in Table 2, the limiting rate constant of oxidation by GTN is twice that with wild-type enzyme, although the Kd is increased by an order of magnitude. Fig. 2 shows the active site of reduced OYE 1 (Protein Data Bank ID code 1OYC) modeled with GTN positioned by hydrogen bonding of the terminal nitrate with His-191 and Asn-194 in the same way as these residues are found to bind phenolates in the crystal structure of the oxidized enzyme (4). It should be emphasized that there is considerable flexibility in the positioning of this substrate, thus the location shown may represent only a portion of those possible, a reflection of the weak binding of these substrates. Thus, it is possible that Tyr-196 can hydrogen bond to the secondary nitrate groups on GTN and PDN. Replacement of this amino acid residue by phenylalanine would disrupt its binding, accounting for the decreased binding affinity shown by GTN with the Y196F mutant of OYE. The loss of H-bonding by Tyr-196 perhaps could allow GTN to approach more favorably the reduced flavin mononucleotide, thereby increasing the oxidation rate. The efficient rate of denitration by Y196F would indicate that this reaction does not need the active site acid that the reduction of unsaturated aldehydes, ketones, and nitro compounds requires.
Figure 2.

(A) The active site of reduced OYE (Protein Data Bank ID code 1OYC) modeled with GTN at H-bonding distance from His-191 and Asn-194. (B) Side view of A, looking down on the N-10 position of the isoalloxazine ring system.
His-191 plays a role in hydrogen bonding with the ligand oxygen atom or nitro groups, facilitating the orientation of the substrate in the active site so that the site of hydride transfer is positioned over the reactive N5 locus of the flavin (4–8). The replacement of His-191 by asparagine also has profound effects on the reaction of the reduced mutant enzyme with GTN, as shown in Table 2. The rate of oxidation of GTN by reduced OYE 1-H191N is 0.025% of that of the wild-type enzyme, although the apparent Kd for binding of GTN to OYE 1-H191N is 5 times more favorable than for the wild-type enzyme, in contrast to the 200-fold poorer binding of cyclohexenone to the same mutant form (5). Replacing His-191 with asparagine results in more space for the substrate to move into the active site, making it possible for other amino acid residues to become more actively involved in interaction with GTN. This possibility could explain the apparent decrease in Kd for GTN in the H191N mutant enzyme. The altered position of the substrate also might result in less favorable positioning for reaction, thereby accounting for the decreased oxidation rate.
The NMR results show that in the reaction of GTN with reduced riboflavin the nitrate attached to the central carbon atom is removed some 6 times faster than the primary nitrates, because, although the product ratio is 3:1, there are statistically twice as many chances to remove a primary than a secondary nitrate ester group (Table 4). This finding is readily explained by the two strong electron-withdrawing groups on either side of the central nitrate, making it a stronger oxidizing agent and hence more reactive with reduced flavin. On the other hand, the OYE-catalyzed reaction shows a somewhat smaller preference for denitration of the central residue. The hydrogen-bonding interactions of His-191 and Asn-194 with the primary nitrate would tend to negate the chemical effect of the primary nitrates on the reactivity of the secondary nitrate, possibly accounting for the decreased ratio of secondary to primary product in the enzyme-catalyzed reaction.
In the case of propylene dinitrate, despite the expected equal chemical reactivity of both nitrate residues, reaction with reduced free riboflavin shows a 3-fold preference for the terminal residue over that of the central nitrate. This result may be caused by steric interference by the methyl group on reaction with the secondary nitrate. Hydrogen bonding of the primary nitrate by His-191 and Asn-194 of OYE 1 would increase the electron density on the primary oxygen atoms, enhancing the positive character of the nitrogen, and would be expected to enhance the reactivity at the primary nitrate of the PDN substrate even more in the enzyme-catalyzed reaction. This is indeed the case with the enzyme showing a 9-fold preference for denitration of the primary nitrate group.
Mechanism of Nitrate Reduction.
The OYE-catalyzed NADPH-dependent denitration of organic nitrates GTN and PDN is a reduction reaction producing nitrite, as demonstrated previously for OYE homologs (11–12). This property was confirmed further in this study by detecting and measuring nitrite, using the diazo-coupling method (22). When OYE-catalyzed reduction of PDN was carried out, one equivalent of nitrite was produced for each equivalent of NADPH consumed. Nitric acid and nitrates have been used as strong oxidizing agents for a long time, but still the exact mechanisms of their reactions are not well understood (29).
Two possible mechanisms can be envisaged for the reductive denitration of PDN and GTN by reduced flavins, as shown in Scheme S1. In one case (Scheme S1A) the reaction would involve hydride transfer from the reduced flavin to the nitrogen of the nitrate residue. This step could be followed by or concerted with electron rearrangement, resulting in liberation of nitrite and formation of the alcohol product. Alternatively, the reaction could involve sequential electron and proton transfers, as illustrated in Scheme S1B. The failure to detect any intermediates in the reaction with either free flavins or with OYE is consistent with either mechanism, because failure to observe flavin semiquinone, an intermediate in a radical pathway, could be caused by its rate of formation being slower than its subsequent reaction. Comparison of the reactivity of GTN with reduced flavins and reduced deazaflavins does, however, give support to the radical pathway of Scheme S1B. Because of the thermodynamic properties of 5-deazaflavins (26), with redox potentials of ≈−650 mV for the oxidized/semiquinone (ox/sq) couple, ≈−300 mV for the ox/red couple, and ≈+50 mV for the sq/red couple, 5-deazaflavins generally are considered as incompetent in radical reaction pathways (30, 31). On the other hand, they are quite competent for hydride transfer, and in the case of acyl CoA dehydrogenase, the reduced 5-deazaFAD enzyme form was found to be oxidized by crotonyl CoA some 4 times faster than normal enzyme (32). Thus, the 4–5 orders of magnitude slower reaction of GTN with reduced 5-deazaflavins than with normal flavins is much more consistent with the radical pathway than with the hydride transfer mechanism.
Scheme 1.

Acknowledgments
We thank Dr. P. Andres Karplus for careful reading of the manuscript and helpful suggestions. The research was supported by U.S. Public Health Service Grant GM-11106.
Abbreviations
- OYE
old yellow enzyme
- GTN
glycerin trinitrate
- PDN
propylene 1,2-dinitrate
References
- 1.Theorell H. Biochem. 1935. Z. 275, 344–346. [Google Scholar]
- 2.Stott K, Saito K, Thiele D J, Massey V. J Biol Chem. 1993;268:6097–6106. [PubMed] [Google Scholar]
- 3.Abramovitz A S, Massey V. J Biol Chem. 1976;251:5321–5326. [PubMed] [Google Scholar]
- 4.Fox K M, Karplus P A. Structure (London) 1994;2:1089–1105. [PubMed] [Google Scholar]
- 5.Brown B J, Deng Z, Karplus P A, Massey V. J Biol Chem. 1998;273:32753–32762. doi: 10.1074/jbc.273.49.32753. [DOI] [PubMed] [Google Scholar]
- 6.Kohli R M, Massey V. J Biol Chem. 1998;273:32763–32770. doi: 10.1074/jbc.273.49.32763. [DOI] [PubMed] [Google Scholar]
- 7.Xu D, Kohli R M, Massey V. Proc Natl Acad Sci USA. 1999;96:3556–3561. doi: 10.1073/pnas.96.7.3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaz A D N, Chakraborty S, Massey V. Biochemistry. 1995;34:4246–4256. doi: 10.1021/bi00013a014. [DOI] [PubMed] [Google Scholar]
- 9.Meah Y, Massey V. Proc Natl Acad Sci USA. 2000;97:10733–10738. doi: 10.1073/pnas.190345597. . (First Published September 19, 2000; 10.1073/pnas.190345597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.French C E, Bruce N C. Biochem J. 1995;312:671–678. doi: 10.1042/bj3120671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christopher E F, Nicklin S, Bruice N C. J Bacteriol. 1996;178:6623–6627. doi: 10.1128/jb.178.22.6623-6627.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snape J R, Walkley J A, Morby A P, Nicklin S, White G F. J Bacteriol. 1977;179:7796–7802. doi: 10.1128/jb.179.24.7796-7802.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miura R, Tomioka Y, Suzuki H, Monezawa M, Hishinura T, Mizugake M. Biol Pharm Bull. 1997;20:110–112. doi: 10.1248/bpb.20.110. [DOI] [PubMed] [Google Scholar]
- 14.Schaller F, Weiler E W. J Biol Chem. 1997;272:28066–28072. doi: 10.1074/jbc.272.44.28066. [DOI] [PubMed] [Google Scholar]
- 15.Buckman J, Miller S M. Biochemistry. 1988;37:14326–14336. doi: 10.1021/bi981106y. [DOI] [PubMed] [Google Scholar]
- 16.Rohde B H, Schmid R, Ullrich M S. J Bacteriol. 1999;181:814–822. doi: 10.1128/jb.181.3.814-822.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blehert D S, Fox B G, Chambliss G H. J Bacteriol. 1999;181:6254–6263. doi: 10.1128/jb.181.20.6254-6263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strassner J, Furholz A, Macheroux P, Amrhein N, Schaller A. J Biol Chem. 1999;274:35067–35073. doi: 10.1074/jbc.274.49.35067. [DOI] [PubMed] [Google Scholar]
- 19.Williams R E, Rathbone D, Bruce N C, Scrutton N S, Moody P C E, Nicklin S. In: Flavins and Flavoproteins. Ghisla S, Kroneck P, Macheroux P, Sund H, editors. Berlin: Weber Scientific; 1999. pp. 663–666. [Google Scholar]
- 20.di Carlo F J. Drug Metab Rev. 1975;4:1–38. doi: 10.3109/03602537508993747. [DOI] [PubMed] [Google Scholar]
- 21.Gibson Q H, Swoboda B E P, Massey V. J Biol Chem. 1964;239:3927–3934. [PubMed] [Google Scholar]
- 22.Asaoka K, Takahashi K. J Biochem. 1983;94:1685–1688. [PubMed] [Google Scholar]
- 23.Pouchert C J, editor. The Aldrich Library of NMR Spectra. 2nd. Ed. Milwaukee, WI: Aldrich; 1983. [Google Scholar]
- 24.Strickland S, Palmer G, Massey V. J Biol Chem. 1975;250:4048–4052. [PubMed] [Google Scholar]
- 25.White F G, Snape J R, Nicklin S. Appl Environ Microbiol. 1996;62:637–642. doi: 10.1128/aem.62.2.637-642.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blankenhorn G. Eur J Biochem. 1976;67:67–80. doi: 10.1111/j.1432-1033.1976.tb10634.x. [DOI] [PubMed] [Google Scholar]
- 27.Spencer R, Fisher J, Walsh C T. Biochemistry. 1977;16:1043–1053. doi: 10.1021/bi00650a015. [DOI] [PubMed] [Google Scholar]
- 28.Massey V, Hemmerich P. Biochemistry. 1978;17:9–17. doi: 10.1021/bi00594a002. [DOI] [PubMed] [Google Scholar]
- 29.Cotton F A, Wilkinson G. Advanced Inorganic Chemistry. New York: Wiley; 1980. pp. 430–431. [Google Scholar]
- 30.Walsh C. Acc Chem Res. 1980;13:148–155. [Google Scholar]
- 31.Ghisla S, Massey V. Biochem J. 1984;239:1–12. doi: 10.1042/bj2390001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghisla S, Thorpe C, Massey V. Biochemistry. 1984;23:3154–3161. doi: 10.1021/bi00309a008. [DOI] [PubMed] [Google Scholar]