

Abstract
K2P (KCNK) potassium channels generate “leak” potassium currents that strongly influence cellular excitability and contribute to pain, somatosensation, anesthesia, and mood. Despite their physiological importance, K2Ps lack specific pharmacology. Addressing this issue has been complicated by the challenges that the leak nature of K2P currents poses for electrophysiology-based high-throughput screening strategies. Here, we present a yeast-based high-throughput screening assay that avoids this problem. Using a simple growth-based functional readout, we screened a library of 106,281 small molecules and identified two new inhibitors and three new activators of the mammalian K2P channel K2P2.1 (KCNK2, TREK-1). By combining biophysical, structure–activity, and mechanistic analysis, we developed a dihydroacridine analogue, ML67-33, that acts as a low micromolar, selective activator of temperature- and mechano-sensitive K2P channels. Biophysical studies show that ML67-33 reversibly increases channel currents by activating the extracellular selectivity filter-based C-type gate that forms the core gating apparatus on which a variety of diverse modulatory inputs converge. The new K2P modulators presented here, together with the yeast-based assay, should enable both mechanistic and physiological studies of K2P activity and facilitate the discovery and development of other K2P small molecule modulators.
K2P channels regulate electrical activity in various tissues through generation of a plasma membrane “leak” potassium conductance.1,2 Channels from this family function in excitable and nonexcitable cells and are implicated in vasodilation, respiratory control, nociception, neuroprotection, anesthesia, and antidepressant responses.1−3 Due to their involvement in pain, ischemia, and migraine, K2Ps are proposed as targets for a range of cardiovascular and neurological disorders;3 however, despite this considerable interest, the K2P family is poorly responsive to classic potassium channel blockers4 and remains practically pharmacologically orphaned.2,3 Development of specific K2P pharmacology has been hindered by the scarcity of facile methods to detect potassium flux in cells and by the fact that the channels produce a voltage-independent “leak” current that is a challenge for conventional electrophysiological screening assays. Thus, there is a need to develop new screening strategies for identifying K2P modulators.
Here, we report the development and implementation of a high-throughput yeast-based screening assay for small molecule modulators of the polymodal K2P, K2P2.1 (KCNK2, TREK-1).5−7 This channel is regulated by heat,8 mechanical force,9 general anesthetics,9,10 and G-protein coupled receptors6 and is involved in pain,11,12 general anesthetic responses,13 neuroprotection from ischemia,13 and depression.14 Although a variety of pharmacologic agents affect K2P2.1 (TREK-1) function, such as millimolar concentrations of volatile halogenated4,9,10,15,16 and gaseous general anesthetics,4,15,17 the neuroprotective agent riluzole (10–100 μM),4,18 and the antidepressant fluoxetine (Prozac) (IC50 ∼10 μM),4,19,20 these compounds have other molecular targets.15 Hence, we set out to develop small molecules that could control K2P2.1 (TREK-1) activity selectively. Such molecules should serve as tools to dissect the unconventional gating apparatus that controls K2P2.1 (TREK-1) function21−23 and may also provide lead compounds for novel anesthetics, neuroprotectants, and drugs against mood disorders.
Using a yeast-based screen, coupled with electrophysiological analysis, we discovered K2P2.1 (TREK-1) inhibitors and activators in a single 106,281 small molecule screening campaign. These modulators comprise different chemical classes and reversibly affect K2P2.1 (TREK-1). Beginning with a carbazole-based scaffold present in the activator ML67, we developed ML67-33, an activator that rapidly and reversibly affects K2P2.1 (TREK-1) with an EC50 in the low micromolar range and by acting on the extracellular C-type gate though a novel mechanism. This dihydroacridine analogue also activates the two temperature- and mechano-sensitive K2P channels most closely related to K2P2.1 (TREK-1), K2P10.1 (TREK-2), and K2P4.1 (TRAAK) but has no effects on more distantly related K2Ps. Thus, ML67-33 represents a novel K2P activator that has specificity within the K2P family. Our success with this yeast-based assay establishes a new means for discovering K2P small molecule modulators.
Results and Discussion
High-Throughput Yeast Screen Identifies K2P2.1 (TREK-1) Activators and Inhibitors
We desired to address the dearth of K2P channel pharmacology by developing a high-throughput screen (HTS) for small molecule K2P modulators. Prior studies have established that growth of the potassium-uptake-deficient yeast strain SGY152824 could be rescued in solid-media assays by ectopic expression of diverse potassium channels,25−27 including K2P2.1 (TREK-1).22 Although this platform has been used to screen libraries of <10,000 compounds against the inward rectifier Kir2.128,29 and has proven advantageous for studying blocker interactions with potassium channels,27,30 the solid media format limits the scalability for screening large libraries of compounds. Hence, we focused on developing a means to monitor rescue of growth under limiting potassium concentrations by heterologously expressed potassium channels, such as K2P2.1 (TREK-1), in a liquid media HTS format.
We measured the signal generated by the vital dye resazurin (Alamar Blue), which live cells convert to a fluorescent form,31 to quantify the abundance of living SGY1528 grown in liquid culture in 386-well plates for 24 h in media containing a range of potassium concentrations (Figure 1). When cultured in media containing 0–50 mM KCl, cells expressing the yeast potassium transporter Trk1p32 exhibited similar levels of resazurin fluorescence signals indicative of robust growth (fluorescence intensity range: 116 ± 3.2–110 ± 3.3, arbitrary units, AU, mean ± SE; p = 0.18, t test, Figure 1A). In contrast, yeast bearing a plasmid for a nonfunctional channel26 showed little growth in potassium-limited conditions, 0–2 mM KCl (30 ± 0.1–32 ± 0.2 AU) and only propagated with 50 mM KCl (87 ± 1.5 AU, Figure 1A). Yeast expressing K2P2.1 (TREK-1) that were grown in potassium-limited conditions, 0–2 mM KCl, had resazurin fluorescence signals that were substantially larger than the negative control and that indicated rescue by a functional channel (67 ± 0.5 vs 30 ± 0.1 AU, respectively, p < 0.001, t test, Figure 1A). Interestingly, in nonlimiting 50 mM KCl media, K2P2.1 (TREK-1)-expressing cells exhibited reduced growth compared to potassium-limited conditions, 1.2 or 2 mM KCl, (84 ± 1.2 AU, p < 0.001 vs 1.2 or 2 mM KCl, t test) that was comparable to that of the negative control (Figure 1A). This effect was not observed for Trk1p (Figure 1A) and is reminiscent of prior studies where activation of a heterologously expressed potassium channel caused yeast growth inhibition.33 Together, these experiments show that K2P2.1 (TREK-1) supports viability of SGY1528 in liquid media under potassium-limiting conditions, a result that agrees with solid media studies.22 Importantly, this liquid-based, 384-well format was suited to automated plate reader analysis. Hence, we next sought to define conditions suitable for a HTS screen for regulators of K2P2.1 (TREK-1) activity.
Figure 1.

Yeast screen identifies K2P2.1 (TREK-1) small molecule modulators. (A) Resazurin (Alamar blue) measurement of potassium concentration growth effects on SGY1528 yeast expressing the indicated constructs. Error bars show ± SE, n = 16. For some points, error bars are smaller than symbols. (B) Exemplar scatter plot showing growth inhibition score distribution from a 384-well screening plate. Each point represents end-point normalized resazurin fluorescence. Error bars show ± SD. (C, D) Dose–response for (C) ML67 and (D) ML45 on growth inhibition of yeast expressing K2P2.1 (TREK-1) (black circles) or Trk1p (blue triangles). Compound structures are shown.
Resazurin assessment of the effects of 1% dimethyl sulfoxide (DMSO), the test compound carrier, and 0.1% sodium dodecyl sulfate, SDS, a growth inhibition control, established two important assay properties. First, DMSO did not inhibit growth of K2P2.1 (TREK-1)-expressing yeast in potassium-limiting conditions (2 mM KCl) where the active channel is required for survival, whereas SDS was lethal. Second, measurement of the Z′ value, a widely used HTS assay metric for determining the separation between negative and positive controls where Z′ > 0.5 indicates a robust screen,34 yielded a favorable value, Z′ = 0.76 (Figure 1B). Hence, we proceeded with a screening campaign to identify candidate K2P2.1 (TREK-1) modulators.
We screened a library of 106,281 small molecules at 10 μM each for their ability to inhibit growth of K2P2.1 (TREK-1)-expressing yeast (Figure 1B, Supplementary Figure S1, Supplementary Table S1). Each plate included wells for 1% DMSO and 0.1% SDS, which served as the respective 0% and 100% growth inhibition controls for calculating the degree of compound-induced growth inhibition (Supplementary Figure S1). From the initial screen, we chose 320 compounds for further evaluation from the set that inhibited growth in the range of 44–92% (a range representing 1.25–3σ above the mean inhibition from the screen and including an upper limit chosen to reduce toxic compound identification, cf. Supplementary Methods). To distinguish generally toxic compounds in this set from those that caused K2P2.1 (TREK-1)-specific effects, we tested each of the 320 compounds over a range of 0.4–50 μM in dose–response screen against yeast expressing K2P2.1 (TREK-1) or Trk1p (Figure 1C,D). These tests identified 81 compounds having at least a 2-fold difference in the apparent IC50 required to prevent growth K2P2.1 (TREK-1) versus Trk1p expressing yeast, e.g., ML45 and ML67 (Figure 1C,D). From these K2P2.1 (TREK-1)-specific compounds, we were able to purchase 61 in quantities sufficient for electrophysiological analysis.
Twenty-five of the 61 candidate compounds were soluble in aqueous solution at a concentration range suited for initial electrophysiological assays (100–750 μM) and were tested by two-electrode voltage clamp for activity against K2P2.1 (TREK-1) expressed in Xenopus oocytes. Electrophysiological characterization identified five compounds that affected K2P2.1 (TREK-1). Two acted as inhibitors: a pyrimidine (ML45) and a thiophene (ML58) (Supplementary Figure S2). Three activated the channel: a thiazolidine (ML12), an amantadine derivative (ML42), and a carbazole (ML67) (Supplementary Figure S3). Dose–response studies showed that ML45 reversibly inhibited K2P2.1 (TREK-1) by ∼70% at the highest concentration tested (IC50 ∼21 μM, Figure 2A,C,E). In contrast, ML67 reversibly activated K2P2.1 (TREK-1), increasing currents by up to ∼11-fold (EC50 213.0 ± 1.2 μM, Figure 2 B,D,E and Table 1). Because K2P2.1 (TREK-1) activators could provide a path to novel anesthetics, analgesics, and neuroprotectants3 and because there were readily available derivatives, we chose to focus on the activator ML67.
Figure 2.

ML45 and ML67 reversibly modulate K2P activity in Xenopus oocytes. (A, B) Exemplar two-electrode voltage clamp I–V curves for application of 100 μM (A) ML45 or (B) ML67 measured using a −150 to 50 mV ramp from a −80 mV holding potential in 2 mM [K+]o. (C, D) Exemplar K2P2.1 (TREK-1) responses to 100 μM (C) ML45 or (D) ML67 measured at 20 mV and 0 mV for ML45 and ML67, respectively. (E) ML45 and ML67 dose–response for K2P2.1 (TREK-1). “Cpd” denotes tested compound. Data were normalized to basal activity and fit with the Hill equation. (F) Dose–responses measured by two-electrode voltage clamp for ML67 against K2P2.1 (TREK-1), black; K2P10.1 (TREK-2), red; K2P3.1 (TASK-1), green; and Kv7.2 (KCNQ2), blue. Error bars show SE, n ≥ 6, N ≥ 2, where n and N is the number of oocytes or independent oocyte batches, respectively.
Table 1. Effects of Activator Compounds on K2P Channels and Mutantsa.
| channel | compound | EC50 (μM) | Hb | Emax (fold) |
|---|---|---|---|---|
| K2P2.1 (TREK-1) | ML67 | 213.0 ± 1.2 | 2.1 ± 0.6 | ∼11 |
| ML67-2 | ND | na | 1.3 ± 0.1 at 500 μM | |
| ML67-13 | 177.4 ± 1.1 | 2.2 ± 0.5 | ∼20 | |
| ML67-15 | ND | 4.7 ± 0.9 at 200 μM | ||
| ML67-17 | 162.2 ± 1.2 | 2.4 ± 1.1 | ∼14 | |
| ML67-18 | 124.8 ± 1.2 | 1.2 ± 0.2 | ∼18 | |
| ML67-29 | 250.6 ± 2.0 | 1.9 ± 1.4 | ∼18 | |
| ML67-33 | 36.3 ± 1.0 | 3.6 ± 0.4 | 11.1 ± 0.4 | |
| 9.7 ± 1.2c | 2.3 ± 0.7 | 11.4 ± 1.1 | ||
| ML67-137 | >40 | na | 9.2 ± 1.4d | |
| K2P2.1 (TREK-1) G137I | ML67-33 | ND | na | 0.9 ± 0.1 at 150 μM |
| K2P2.1 (TREK-1) W275S | ML67-33 | 21.8 ± 1.3 | 1.6 ± 0.7 | 5.1 ± 0.6 |
| K2P2.1 (TREK-1)-3G | ML67-33 | 49.4 ± 1.1 | 2.2 ± 0.4 | 12.9 ± 1.0 |
| K2P10.1 (TREK-2) | ML67 | ∼250 | 1.0 ± 0.4 | 10.1 ± 1.1 at 500 μM |
| ML67-33 | 30.2 ± 1.4 | 1.6 ± 0.6 | 11.4 ± 1.8 | |
| K2P4.1 (TRAAK) | ML67-33 | 27.3 ± 1.2 | 1.8 ± 0.4 | 14.7 ± 1.1d |
| K2P9.1 (TASK-3) | ML67-33 | ND | na | 2.1 ± 0.4 at 150 μM |
| K2P5.1 (TASK-2) | ML67-33 | ND | na | 1.7 ± 0.3d |
| K2P3.1 (TASK-1) | ML67 | ND | na | 1.2 ± 0.0 at 500 μM |
| ML67-33 | ND | na | 1.1 ± 0.1d | |
| K2P18.1 (TRESK) | ML67-33 | ND | na | 0.9 ± 0.1d |
| Kv7.2 (KCNQ2) | ML67 | ND | na | 1.8 ± 0.0 at 500 μM |
ND = not determined; na = not applicable.
Cooperativity coefficient from the Hill equation.
Measurements from HEK293 cells.
Value determined at 100 μM compound.
We first addressed whether ML67 was a selective or general potassium channel opener. Two electrode voltage clamp studies showed that ML67 activated the closest K2P2.1 (TREK-1) homologue (Supplementary Figure S4) K2P10.1 (TREK-2) (EC50 ∼250 μM) but not the more distantly related K2P3.1 (TASK-1) (Figure 2F). Further, ML67 had no effect on the voltage-gated potassium channel Kv7.2 (KCNQ2) (Figure 2F) for which small molecule openers have been described.35 Having established that ML67 had some selectivity among diverse potassium channels, we sought to characterize its structure–activity relationships with respect to channel activation and improve upon its properties.
ML67 Derivatives Improve Potency for K2P2.1 (TREK-1)
Investigation of substitution on the ML67 central carbazole ring showed that the hydrophobic halogen atoms were important. Removal of the chloro substituents at the carbazole ring 3- and 6-positions abolished activity (ML67-2, Figure 3A, Table 1), whereas the 3,6-dibromo congener ML67-13 showed a slightly increased potency (EC50 177.4 ± 1.1 μM) and a dramatically improved maximum response (efficacy) (Emax ∼20 fold) (Figure 3A, Table 1). Thus, hydrophobic groups at the 3- and 6-positions are vital for function in the ML67 series.
Figure 3.

Studies to improve ML67 potency. Effects of changes to ML67 (A) halogen positions, (B) linker region, and (C) acidic group measured against K2P2.1 (TREK-1) by a −150 to 50 mV ramp from a −80 mV holding potential using two-electrode voltage clamp in Xenopus oocytes in 2 mM [K+]o. “Cpd” denotes tested compound. Data (mean ± SE, n ≥ 6, N ≥ 2) from 0 mV were normalized to basal activity and fitted to the Hill equation. EC50 values are ML67-13, 177.4 ± 1.1 μM; ML67-17, 162.2 ± 1.2 μM; ML67-29, 250.6 ± 2.0 μM μM; ML67-18, 124.8 ± 1.2 μM; ML67-33, 36.3 ± 1.0 μM. Error bars show SE, n ≥ 6 and N ≥ 2 except for ML67-2 and ML67-15 where n = 4 and N = 2. Compound structures are shown.
With regard to the N-alkyl side chain, we found that the carboxylate function was critical for activity, as the nitrile congener (ML67-1) (Figure 3B) showed no activity, even at 500 μM. Extending the alkyl chain by one methylene (ML67-15) did not improve activity (Figure 3B). Rigidification of the alkyl chain by a cyclobutane group (ML67-17 and ML67-29, cis:trans stereoisomer ratio 15:85 and 95:5, respectively) had a favorable effect compared to ML67-15: EC50 162.2 ± 1.2 and 250 ± 2 μM for ML67-17 and ML67-29, respectively (Figure 3B, Table 1). The relative positioning of carbazole ring and carboxylate function will be quite different in these two stereoisomeric analogues. Therefore, their similar EC50 values suggest that precise positioning of the carboxylate is not essential. Consistent with this notion, replacement of the carboxylate with a bioisosteric and anionic tetrazole ring (ML67-18) was well tolerated and improved potency almost 2-fold (EC50 124.8 ± 1.2, Emax ∼18-fold, Figure 3C, Table 1). Having established the importance of the halogen substituents and anionic tetrazole, we turned to modification of the core tricyclic ring system.
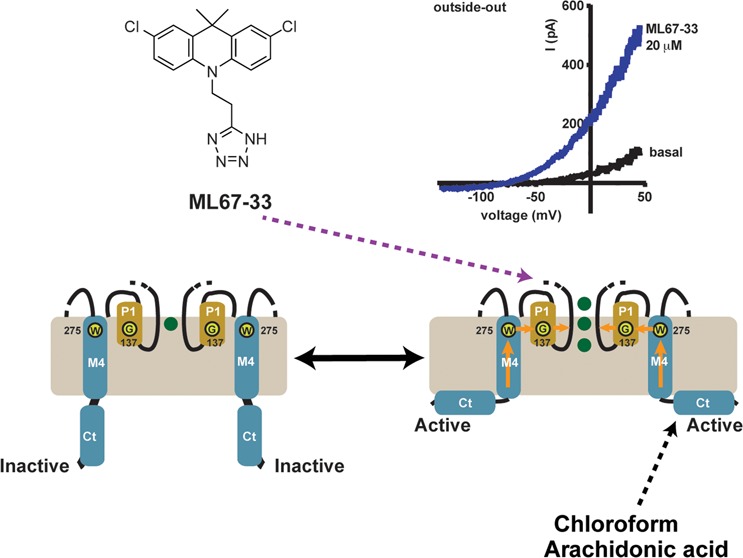
We explored a number of halogenated tricyclic ring systems bearing anionic side chains. These yielded varying degrees of success at improving K2P2.1 (TREK-1) activation. The most effective had substitution of the carbazole tricycle for 9,9-dimethyl-9,10-dihydroacridine. Analogue ML67-33 exhibited a 5-fold improved potency compared to that of ML67-18, but somewhat reduced efficacy (EC50 36.3 ± 1.0 μM, Emax 11.1 ± 0.4 fold, Figure 3C, Table 1). The corresponding dibromo congener ML67-137 was no more potent than ML67-18 (Figure 3C, Table 1). As ML67-33 (2,7-dichloro-9,9-dimethyl-10-[2-(1H-tetrazol-5-yl)-ethyl]-9,10-dihydro-acridine) was the most potent compound and had favorable solubility properties (clog P = 4.84, clog D = 3.29 at pH 7.4, Supplementary Table S2), we pursued a series of experiments designed to test its mechanism of action.
ML67-33 Activates the K2P2.1 (TREK-1) C-Type Gate
We examined how ML67-33 affected K2P2.1 (TREK-1) expressed in two widely used experimental systems, Xenopus oocytes and mammalian HEK-293T cells. ML67-33 had similar potencies and efficacies on K2P2.1 (TREK-1) expressed in both systems (Figure 4A and B) (EC50 36.3 ± 1.1 μM and 9.7 ± 1.2 μM and Emax 11.1 ± 0.4 and 11.4 ± 1.1 for oocytes and HEK cells, respectively, Figure 4C, Table 1), demonstrating that the compound acts independently of cellular context (Figure 4A–C). Further, the effects of ML-67-33 were fast, occurring within seconds (half-maximal activation time, 4.1 ± 0.5 s, mean ± SE, n = 7, N = 2) and were reversible (Figure 4D). ML67-33 application to excised membrane patched from HEK cells expressing K2P2.1 (TREK-1) showed that ML67-33 activated K2P2.1 (TREK-1) in both the outside-out (Figure 5A) and inside-out (Figure 5B) configurations. The effectiveness in both contexts strongly suggests that ML67-33 acts directly on the channel and does not require soluble cytosolic factors. The times of half-maximal activation and return to baseline following compound washout (t1/2act and t1/2wash, respectively) in the outside-out configuration were indistinguishable from those measured in the whole-cell configuration (t1/2act: t1/2wash mean ± SE, 4.1 ± 0.5 s: 3.9 ± 0.5 s and 4.6 ± 0.9 s,: 3.1 ± 0.7 s for whole-cell and outside-out, respectively, Figure 5C and D). Both t1/2 values slowed substantially when the compound was applied to the inside-out configuration. Notably, there was a larger effect on washout from inside-out patches (t1/2act: t1/2wash, 10.8 ± 1.3 s: 22.0 ± 5.1 s, Figure 5C and D). Taken together, these data indicate that although ML67-33 appears to be membrane-permeable, its site of action on K2P2.1 (TREK-1) is more readily accessible from the extracellular side.
Figure 4.

ML67-33 reversibly activates K2P2.1 (TREK-1) independent of expression system. (A, B) Exemplar I–V curves showing the effect of ML67-33 on K2P2.1 (TREK-1) activity in (A) Xenopus oocytes (two-electrode voltage clamp) or (B) HEK-293T cells (whole cell patch clamp). In both, the external solution contained 2 mM [K+]o, pH 7.4. Currents were elicited by a −150 to 50 mV voltage ramp from a −80 mV (oocytes) or −40 mV (HEK-293T) holding potential. (C) Quantification of the effect of ML67-33 on the indicated channels. Data (mean ± SE, n ≥ 6, N ≥ 2) were normalized to basal channel activity and fit with the Hill equation. EC50 36.3 ± 1.0 μM, 9.7 ± 1.2 μM and Emax at 100 μM 11.1 ± 0.4, 11.4 ± 1.1 for oocytes and HEK cells, respectively. (D) Exemplar reversible activation of K2P2.1 (TREK-1) by ML67-33 measured at 0 mV in HEK-293T cells.
Figure 5.

ML67-33 activates K2P2.1 (TREK-1) in excised membrane patches. (A, B) Exemplar I–V curves showing ML67-33 effects on K2P2.1 (TREK-1) in (A) outside-out and (B) inside-out excised patches from HEK-293T cells. Currents were elicited by a −100 to 50 mV ramp from a −40 mV holding potential. (C) Exemplar responses to ML67-33 measured at 0 mV in the indicated configurations. Gray indicates presence of 20 μM ML67-33. (D) Time to half-maximal activation following ML67-33 application and recovery from activation (wash) following ML67-33 removal, measured in HEK-293T cells at 0 mV. Error bars: mean ± SE n ≥ 6, N ≥ 2. ** p ≤ 0.01; N.S. indicates not significant (p ≥ 0.05) as determined by t test.
A selectivity filter-based C-type-like gate mediates K2P2.1 (TREK-1) activation from diverse inputs that include basic extracellular pH,22,36 intracellular acidosis,23 temperature,22 mechanical force,22 and intracellular C-terminal domain, Ct, phosphorylation.21 The C-type gate active conformation can be stabilized by high concentrations of extracellular potassium, [K+]o,21−23,36 or by mutations in the P1 pore helix, G137I,21 or the M4 transmembrane helix, W275S22 (Figure 6A). We tested how each affected the response to ML67-33. The data show that all three manipulations reduced the response of K2P2.1 (TREK-1) to ML67-33 (Figure 6B–G). Indeed, the change causing the most potent C-type gate stabilization, G137I,21 made the channels completely resistant to ML67-33 activation. In contrast, use of a triple glycine mutation, K2P2.1-3G21 (Figure 6A) that uncouples the pore from Ct, which acts as a sensor for temperature8,21,22 and mechanical stimulation,9,22 resulted in channels that could be readily activated by ML67-33. In this case, both the ML67-33 potency and efficacy were similar to that of wild-type channels (EC50 49.4 ± 0.1 μM, Emax12.9 ± 1.0, Figure 6F and G). The observation that activation of the C-type gate renders the channels resistant to ML67-33 whereas loss of coupling to Ct does not affect ML67-33 activation indicates that ML67-33 acts directly on the components comprising the C-type gate. Ct is central to K2P2.1 (TREK-1) activation by the two most effective activators previously reported, chloroform4,9 and arachidonic acid,4,9 and is crucial for channel inhibition by fluoxetine (Prozac).20 The lack of involvement of Ct in ML67-33 activation together with evidence for the direct action of ML67-33 on the C-type gate indicates that ML67-33 activates the channel by a novel mechanism (Figure 6H).
Figure 6.

ML67-33 activates the K2P2.1 (TREK-1) extracellular C-type gate. (A) K2P2.1 (TREK-1) subunit cartoon diagram. Key residue positions, transmembrane segments (M1–M4), and pore helices (P1 and P2) are indicated. First and second pore-forming regions are tan and blue, respectively. (B–G) Exemplar two-electrode voltage clamp I–V curves in Xenopus oocytes and dose response curves showing ML67-33 responses in channels having perturbed gating elements. (B) C-type gate stabilization by 90 mM [K+]o. (C) ML67-33 dose response at +40 and −40 mV in 90 mM [K+]o (90K) and 0 mV in 2 mM [K+]o (2K). (D, E) C-type gate stabilization by (D) G137I and (E) W275S. (F) Uncoupling Ct from the pore by the K2P2.1 (TREK-1)-3G mutant. (G) ML67-33 dose responses for the indicated channels at +40 mV (90K) or 0 mV (2K), normalized to basal channel activity and fit with the Hill equation. Error bars indicate SE, n ≥ 6, N ≥ 2. (H) Model of K2P2.1 (TREK-1) activation after ref (21). Green spheres indicate potassium ions. Positions of Gly137 and Trp275 are indicated. Orange arrows indicate pathway for coupling Ct activation to the C-type gate. Elements involved in activation by ML67-33, chloroform,9 and arachidonic acid9 are indicated. I–V curves were measured in (B) 90 mM [K+]o or (D–F) 2 mM [K+]o. Currents were elicited by a −100 to 50 mV ramp from a 0 mV holding potential (90K) or by a −150 to 50 mV ramp, from a −80 mV holding potential (2K).
ML67-33 Activates Temperature- and Mechano-Sensitive K2P Channels
To examine the ML67-33 specificity within the K2P family, we used heterologous expression in Xenopus oocytes of representatives from different K2P subtypes6 (Supplementary Figure S4). These included the two temperature- and mechano-sensitive K2Ps most closely related to K2P2.1 (TREK-1), K2P10.1 (TREK-2) and K2P4.1 (TRAAK); a representative from the neighboring subgroup, the TALK subgroup, K2P5.1 (TASK-2); and representatives from two divergent branches of the K2P family, the TASK subgroup, K2P3.1 (TASK-1) and K2P9.1 (TASK-3); and K2P18.1 (TRESK). ML67-33 exhibited substantial activation of K2P10.1 (TREK-2) (Figure 7A, Supplementary Figure S5A) (EC50 30.2 ± 1.4, Emax 11.4 ± 1.8 fold, Table 1) and K2P4.1 (TRAAK) (Figure 7B, Supplementary Figure S5B) (EC50 27.3 ± 1.18 μM, Emax 14.7 ± 1.12 fold, Table 1). In stark contrast, ML67-33 showed little or no activity against channels from the TALK group, K2P5.1 (TASK-2) (Figure 7C and G, Table 1); TASK group, K2P3.1 (TASK-1) (Figure 7D and G, Table 1) and K2P9.1 (TASK-3) (Figure 7E and G, Table 1); and K2P18.1 (TRESK) (Figure 7F and G, Table 1) even when applied at 100 μM, a concentration at which K2P2.1 (TREK-1) shows a maximal response. Similar to the parent compound ML67 (Figure 2F), ML67-33 at 100 μM showed no effect on Kv7.2 (KCNQ2) (Supplementary Figure S6). Together, these data establish that ML67-33 is a selective activator of channels from the K2P2.1 (TREK-1) subfamily of temperature- and mechano-sensitive channels.
Figure 7.

ML67-33 is a selective activator of temperature- and mechanosensitive K2P channels. (A–F) Exemplar I–V curves showing ML67-33 effects on (A) K2P10.1 (TREK-2), (B) K2P4.1 (TRAAK), (C) K2P5.1 (TASK-2), (D) K2P3.1 (TASK-1), (E) K2P9.1 (TASK-3), and (F) K2P18.1 (TRESK) measured in Xenopus oocytes using a −150 to 50 mV ramp from a −80 mV holding potential in 2 mM [K+]o. (G) ML67-33 dose responses for the indicated channels. Data (mean ± SE, n ≥ 6, N ≥ 2) were normalized to basal activity and fit with the Hill equation. EC50 values: K2P2.1 (TREK-1) 36.3 ± 1.0 μM, K2P10.1 (TREK-2) 30.2 ± 1.4 μM, and K2P4.1 (TRAAK) 27.3 ± 1.2 μM. Emax values at 100 μM are K2P2.1 (TREK-1) 11.1 ± 0.4, K2P10.1 (TREK-2) 11.4 ± 1.8, K2P4.1 (TRAAK) 14.7 ± 1.1, K2P5.1 (TASK-2) 2.0 ± 0.1, K2P9.1 (TASK-3) 1.7 ± 0.3, K2P3.1 (TASK-1) 1.1 ± 0.0, K2P18.1 (TRESK) 0.9 ± 0.1. Error bars indicate SE, n ≥ 6, N ≥ 2.
Discussion
K2P channels are the most diverse potassium channel class37 and function in both excitable and nonexcitable cells.1 The fact that this channel family responds poorly to classic potassium channel blockers4 and remains largely pharmacologically orphaned3,4 limits the ability to probe its function. Additionally, because K2Ps produce voltage-independent leak current, they present difficult targets for modulator discovery by conventional electrophysiological screening techniques. Our studies demonstrate that it is possible to use a solution-based yeast screening platform built upon rescue of potassium uptake by a functional K2P channel to identify both inhibitors and activators of K2Ps. This assay provides a substantial advantage in terms of scalability and quantification over solid-based media assays used previously to screen small libraries against other potassium channels.28,29
We identified a set of novel K2P2.1 (TREK-1) inhibitors and activators in a single screening campaign covering 106,281 compounds. Because all of the identified compounds inhibited K2P2.1 (TREK-1)-dependent yeast growth, our discovery of molecules that proved to be activators was unexpected. Examination of the potassium dependency of yeast rescue in solution showed that, unlike the potassium transporter Trk1p, K2P2.1 (TREK-1) conferred a bell-shaped dependence on growth rescue as a function of potassium (Figure 1A), an effect not seen previously in solid media assays.22 Prior identification of gain-of-function mutants of the yeast channel YKC1 (TOK1) has shown that hyperactive potassium channels can negatively impact yeast growth.33 Although the exact mechanism by which K2P2.1 (TREK-1) hyper-activation causes growth inhibition remains unclear, the results from the YKC1 (TOK1) studies suggest a rationale for why our high-throughput screen identified inhibitors and activators of K2P2.1 (TREK-1) in a single screening campaign. Because inhibitors and activators of K2P2.1 (TREK-1) are desirable for both physiological studies and as leads for therapeutic applications,3 this unexpected benefit substantially expands the potential of this assay to identify K2P modulators from diverse chemical libraries.
The two inhibitors and three activators that we identified produced fast, reversible changes in K2P2.1 (TREK-1) function that occurred within seconds of compound application and removal (Supplementary Figures S2 and S3). This is notable because the yeast HTS assay time scale is hours and could favor the identification of slow-acting compounds that have indirect effects on channel function by affecting factors such as channel biogenesis, assembly, or trafficking. Although such compounds could in principle be identified, the fact that we found diverse compounds that appear to act directly and immediately on the channel indicates that there is no strong bias for slow-acting effectors and underscores the potential of this assay as a discovery platform for fast-acting K2P channel modulators.
By combining biophysical characterization and chemical synthesis, we improved upon the properties of a lead activator, ML67 (Figure 3, Table 1), to create a dihydroacridine derivative that reversibly activated K2P2.1 (TREK-1) (Figure 4) with an EC50 in the low-micromolar range and an Emax of ∼11 (Table 1). The observation that ML67-33 activates K2P2.1 (TREK-1) in excised membrane patches (Figure 5) demonstrates that the compound does not act by perturbing channel trafficking or via a mechanism that involves cytosolic proteins and suggests that ML67-33 acts directly on the channel.
Diverse gating signals that include protons, temperature, mechanical force, and phosphorylation control K2P2.1 (TREK-1) function. Although many of these are sensed by the intracellular cytoplasmic domain, Ct,8,21,38−40 their actions converge on a common C-type selectivity filter-based gate located on the extracellular side of the membrane.21−23,36,41 A variety of manipulations that stabilize this C-type gate, such as high concentrations of extracellular potassium36 and mutations in two elements central to C-type gate activation, the P1 pore helix and M4 transmembrane helix,22 reduced or eliminated the activating effects of ML67-33 (Figure 6B–E and G). By contrast, decoupling Ct from the C-type gate failed to affect channel sensitivity to ML67-33 (Figure 6F and G). This result eliminates this region as the target of ML67-33 action and is striking because in addition to sensing physiological inputs, Ct is thought to be central to K2P2.1 (TREK-1) modulation by compounds such as chloroform,9 arachidonic acid,9 and fluoxetine (Prozac).20 Further, we found that ML67-33 acts quickly and reversibly when applied to channels in whole cells and outside-out patches but displays slower on and off rates when applied to channels in the inside-out patch configuration (Figure 5C and D). Together, these observations strongly support the idea that ML67-33 acts directly on the extracellular C-type gate and indicate that ML67-33 has a mechanism that is different from other K2P2.1 (TREK-1) modulators and that targets the core machinery that controls channel gating (Figure 6H).
ML67-33 activates two, closely related temperature- and mechano-sensitive K2P channels, K2P10.1 (TREK-2) and K2P4.1 (TRAAK), with an EC50 in the low-micromolar range (Figure 7, Table 1). In contrast, ML67-33 was ineffective against more the distantly related members of the K2P family K2P5.1 (TASK-2), K2P3.1 (TASK-1), K2P9.1 (TASK-3), and K2P18.1 (TRESK) and against the voltage-gated channel Kv7.2 (KCNQ2) (Figure 7, Supplementary Figure S6). The C-type, selectivity filter-based gating mechanism acts in channels that respond to ML67-33, K2P2.1 (TREK-1),22,23,36 and K2P10.1 (TREK-2),22 as well as those that were resistant to the compound, K2P3.1 (TASK-1)42−44 and K2P5.1 (TASK-2).45 Thus, the presence of a C-type gate is necessary but not sufficient for activation by ML67-33. Further, the selectivity profile of the compound suggests that key targets of ML67-33 action must lie in elements that are common to the K2P2.1 (TREK-1) subfamily.
A number of compounds modulate K2P2.1 (TREK-1) activity.4 Many are drugs having numerous molecular targets or metabolites involved in multiple pathways such as local46,47 and general9,10,17 anesthetics, antidepressants,9,19 neuroprotectants,18,48 phospholipids,38,49 protons,36,39,50 and heavy metal ions.17 Most of these K2P2.1 (TREK-1) modulators act at >100 μM, have limited effects on current amplitude,4 and current enhancements of <2-fold.10,17,47,51 The largest reported activations are for chloroform (5.5-fold at 1.6 mM)9 and arachidonic acid (3–12-fold at 10–20 μM),9,47 a polyunsaturated fatty acid with multiple biological functions. ML67-33 acts at a lower concentration (9.7–36.3 μM) and has a larger stimulatory effect (Emax ∼11-fold) than most previously reported activators.4 Although ML67-33 activation matches that of the most effective but unspecific activator, arachidonic acid,9,47 ML67-33 stimulation of K2P2.1 (TREK-1) does not require Ct (Figure 6F), a channel element that is central to the action of chloroform,4,9 arachidonic acid,4,9 and other gating inputs.8,21,38−40 Instead, our data indicate that the potent activation caused by ML67-33 involves direct action on the C-type gate that forms the core gating apparatus of the channel21−23,41 (Figure 6H). These properties, together with the fact that ML67-33 acts within seconds, suggest that ML67-33 has a novel mechanism of action and focuses attention on the C-type gate for future structure-based development of K2P modulators.
ML67-33 displays marked specificity within the K2P group and thus should provide a key step in the development of selective regulators of the K2P2.1 (TREK-1) subfamily. Such compounds may afford new entry points for neuroprotective and cardioprotective molecules that could be useful for the treatment of ischemia or pain control. Finally, the demonstrated ability of our high-throughput yeast-based screening assay to identify both activators and inhibitors of K2P2.1 (TREK-1) suggests that this platform can be adapted to screen for regulators of other K2Ps. Our findings should enable new mechanistic and physiological investigations of K2P activity as well as the further discovery of other K2P small molecule modulators.
Methods
Molecular Biology
Murine K2P channels were cloned into pGEMHE/pMO,22 IRES-GFP (Invitrogen), or pYES2-MET25 (high copy 2 μ, URA3)26 for expression in oocytes, HEK-293T cells, and yeast, respectively, using standard molecular biology procedures, and verified by DNA sequencing.
Yeast, Media, Compounds, and High-Throughput Screening
Saccharomyces cerevisiae strain SGY1528 was transformed with previously described plasmids.22,26 Resazurin (Alamar Blue, Invitrogen) signals were quantified using an automated plate reader using 560 nm excitation/590 nm emission settings. Details are found in the Supporting Information.
Library compounds were assembled at the Small Molecule Discovery Center from commercial sources. Individual compounds were purchased or synthesized.
Electrophysiology
Two electrode voltage clamp was done as previously described.21,22 HEK293T whole cell and patch clamp recording was done following established protocols.9 Data were fit with a modified Hill equation: I = Imin + (Imax – Imin)/(1 + 10(Log EC50 – Log[C])*H); Imax and Imin are maximal and minimal current values, respectively, EC50 is a half-maximal effective concentration, and H is the Hill coefficient. Detailed procedures are found in the Supporting Information.
Statistical Analysis
Results are mean ± SD or SEM from at least two independent experiments (denoted as N). Statistical analyses used the two-tailed Student’s t test; significance defined as p ≤ 0.05.
Chemical Synthesis
ML67, ML67-2, and ML67-13 were obtained from commercial sources. Syntheses of ML67-33, ML67-137, ML67-18, ML67-15, ML67-17, and ML67-29 are described in Supporting Information.
Acknowledgments
This work was supported by grants to D.L.M. from NIH R01-MH093603 and American Heart Association 0740019N and to S.N.B. from the Life Sciences Research Foundation. D.L.M. is an AHA Established Investigator. We thank E. Gracheva and Minor lab members for comments on the manuscript. S.N.B. is a Genentech Fellow of the Life Sciences Research Foundation.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# Department of Cellular and Molecular Physiology Yale University School of Medicine, New Haven, CT 06510.
Author Contributions
S.N.B. and D.L.M. conceived the study and designed the experiments. S.N.B. performed the experiments and analyzed data. S.N.B and K.K.-H.A. performed the chemical screen. A.G.G. performed chemical synthesis. S.N.B. and K.A.C. performed molecular cloning. M.R.A. supervised the chemical screen and analyzed data. A.R.R. designed new analogues and supervised chemical synthesis. D.L.M. analyzed data and provided guidance and support throughout. S.N.B. and D.L.M. wrote the paper.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Enyedi P.; Czirjak G. (2010) Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol. Rev. 90, 559–605. [DOI] [PubMed] [Google Scholar]
- Lesage F.; Barhanin J. (2011) Molecular physiology of pH-sensitive background K(2P) channels. Physiology 26, 424–437. [DOI] [PubMed] [Google Scholar]
- Es-Salah-Lamoureux Z.; Steele D. F.; Fedida D. (2010) Research into the therapeutic roles of two-pore-domain potassium channels. Trends Pharmacol. Sci. 31, 587–595. [DOI] [PubMed] [Google Scholar]
- Lotshaw D. P. (2007) Biophysical, pharmacological, and functional characteristics of cloned and native mammalian two-pore domain K+ channels. Cell Biochem. Biophys. 47, 209–256. [DOI] [PubMed] [Google Scholar]
- Fink M.; Duprat F.; Lesage F.; Reyes R.; Romey G.; Heurteaux C.; Lazdunski M. (1996) Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J. 15, 6854–6862. [PMC free article] [PubMed] [Google Scholar]
- Noel J.; Sandoz G.; Lesage F. (2011) Molecular regulations governing TREK and TRAAK channel functions. Channels (Austin) 5, 402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore E. (2007) The neuronal background K2P channels: focus on TREK1. Nat. Rev. Neurosci. 8, 251–261. [DOI] [PubMed] [Google Scholar]
- Maingret F.; Lauritzen I.; Patel A. J.; Heurteaux C.; Reyes R.; Lesage F.; Lazdunski M.; Honore E. (2000) TREK-1 is a heat-activated background K(+) channel. EMBO J. 19, 2483–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A. J.; Honore E.; Maingret F.; Lesage F.; Fink M.; Duprat F.; Lazdunski M. (1998) A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 17, 4283–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A. J.; Honore E.; Lesage F.; Fink M.; Romey G.; Lazdunski M. (1999) Inhalational anesthetics activate two-pore-domain background K+ channels. Nat. Neurosci. 2, 422–426. [DOI] [PubMed] [Google Scholar]
- Alloui A.; Zimmermann K.; Mamet J.; Duprat F.; Noel J.; Chemin J.; Guy N.; Blondeau N.; Voilley N.; Rubat-Coudert C.; Borsotto M.; Romey G.; Heurteaux C.; Reeh P.; Eschalier A.; Lazdunski M. (2006) TREK-1, a K+ channel involved in polymodal pain perception. EMBO J. 25, 2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel J.; Zimmermann K.; Busserolles J.; Deval E.; Alloui A.; Diochot S.; Guy N.; Borsotto M.; Reeh P.; Eschalier A.; Lazdunski M. (2009) The mechano-activated K+ channels TRAAK and TREK-1 control both warm and cold perception. EMBO J. 28, 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurteaux C.; Guy N.; Laigle C.; Blondeau N.; Duprat F.; Mazzuca M.; Lang-Lazdunski L.; Widmann C.; Zanzouri M.; Romey G.; Lazdunski M. (2004) TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. 23, 2684–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurteaux C.; Lucas G.; Guy N.; El Yacoubi M.; Thummler S.; Peng X. D.; Noble F.; Blondeau N.; Widmann C.; Borsotto M.; Gobbi G.; Vaugeois J. M.; Debonnel G.; Lazdunski M. (2006) Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat. Neurosci. 9, 1134–1141. [DOI] [PubMed] [Google Scholar]
- Solt K.; Forman S. A. (2007) Correlating the clinical actions and molecular mechanisms of general anesthetics. Curr. Opin. Anaesthesiol. 20, 300–306. [DOI] [PubMed] [Google Scholar]
- Lesage F.; Terrenoire C.; Romey G.; Lazdunski M. (2000) Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J. Biol. Chem. 275, 28398–28405. [DOI] [PubMed] [Google Scholar]
- Gruss M.; Bushell T. J.; Bright D. P.; Lieb W. R.; Mathie A.; Franks N. P. (2004) Two-pore-domain K+ channels are a novel target for the anesthetic gases xenon, nitrous oxide, and cyclopropane. Mol. Pharmacol. 65, 443–452. [DOI] [PubMed] [Google Scholar]
- Duprat F.; Lesage F.; Patel A. J.; Fink M.; Romey G.; Lazdunski M. (2000) The neuroprotective agent riluzole activates the two P domain K(+) channels TREK-1 and TRAAK. Mol. Pharmacol. 57, 906–912. [PubMed] [Google Scholar]
- Kennard L. E.; Chumbley J. R.; Ranatunga K. M.; Armstrong S. J.; Veale E. L.; Mathie A. (2005) Inhibition of the human two-pore domain potassium channel, TREK-1, by fluoxetine and its metabolite norfluoxetine. Br. J. Pharmacol. 144, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoz G.; Bell S. C.; Isacoff E. Y. (2011) Optical probing of a dynamic membrane interaction that regulates the TREK1 channel. Proc. Natl. Acad. Sci. U.S.A. 108, 2605–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev S. N.; Clark K. A.; Minor D. L. Jr. (2012) Metabolic and thermal stimuli control K(2P)2.1 (TREK-1) through modular sensory and gating domains. EMBO J. 31, 3297–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev S. N.; Peyronnet R.; Clark K. A.; Honore E.; Minor D. L. Jr. (2011) Multiple modalities converge on a common gate to control K2P channel function. EMBO J. 30, 3594–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechotta P. L.; Rapedius M.; Stansfeld P. J.; Bollepalli M. K.; Ehrlich G.; Andres-Enguix I.; Fritzenschaft H.; Decher N.; Sansom M. S.; Tucker S. J.; Baukrowitz T. (2011) The pore structure and gating mechanism of K2P channels. EMBO J. 30, 3607–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko C. H.; Gaber R. F. (1991) TRK1 and TRK2 encode structurally related K+ transporters. Mol. Cell. Biol. 11, 4266–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.; Ruknudin A.; Yang W.; Shaw S.; Knickerbocker A.; Kurtz S. (1995) Functional expression of a vertebrate inwardly rectifying K+ channel in yeast. Mol. Biol. Cell 6, 1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor D. L. Jr.; Masseling S. J.; Jan Y. N.; Jan L. Y. (1999) Transmembrane structure of an inwardly rectifying potassium channel. Cell 96, 879–891. [DOI] [PubMed] [Google Scholar]
- Chatelain F. C.; Gazzarrini S.; Fujiwara Y.; Arrigoni C.; Domigan C.; Ferrara G.; Pantoja C.; Thiel G.; Moroni A.; Minor D. L. Jr. (2009) Selection of inhibitor-resistant viral potassium channels identifies a selectivity filter site that affects barium and amantadine block. PloS One 4, e7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaks-Makhina E.; Kim Y.; Aizenman E.; Levitan E. S. (2004) Novel neuroprotective K+ channel inhibitor identified by high-throughput screening in yeast. Mol. Pharmacol. 65, 214–219. [DOI] [PubMed] [Google Scholar]
- Zaks-Makhina E.; Li H.; Grishin A.; Salvador-Recatala V.; Levitan E. S. (2009) Specific and slow inhibition of the kir2.1 K+ channel by gambogic acid. J. Biol. Chem. 284, 15432–15438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelain F. C.; Alagem N.; Xu Q.; Pancaroglu R.; Reuveny E.; Minor D. L. Jr. (2005) The pore helix dipole has a minor role in inward rectifier channel function. Neuron 47, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama G. R.; Caton M. C.; Nova M. P.; Parandoosh Z. (1997) Assessment of the Alamar Blue assay for cellular growth and viability in vitro. J. Immunol. Methods 204, 205–208. [DOI] [PubMed] [Google Scholar]
- Gaber R. F.; Styles C. A.; Fink G. R. (1988) TRK1 encodes a plasma membrane protein required for high-affinity potassium transport in Saccharomyces cerevisiae. Mol. Cell. Biol. 8, 2848–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukin S. H.; Vaillant B.; Zhou X. L.; Spalding E. P.; Kung C.; Saimi Y. (1997) Random mutagenesis reveals a region important for gating of the yeast K+ channel Ykc1. EMBO J. 16, 4817–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. H.; Chung T. D.; Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screening 4, 67–73. [DOI] [PubMed] [Google Scholar]
- Xiong Q.; Gao Z.; Wang W.; Li M. (2008) Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol. Sci. 29, 99–107. [DOI] [PubMed] [Google Scholar]
- Cohen A.; Ben-Abu Y.; Hen S.; Zilberberg N. (2008) A novel mechanism for human K2P2.1 channel gating. Facilitation of C-type gating by protonation of extracellular histidine residues. J. Biol. Chem. 283, 19448–19455. [DOI] [PubMed] [Google Scholar]
- Goldstein S. A.; Bayliss D. A.; Kim D.; Lesage F.; Plant L. D.; Rajan S. (2005) International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev 57, 527–540. [DOI] [PubMed] [Google Scholar]
- Chemin J.; Patel A. J.; Duprat F.; Lauritzen I.; Lazdunski M.; Honore E. (2005) A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J. 24, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore E.; Maingret F.; Lazdunski M.; Patel A. J. (2002) An intracellular proton sensor commands lipid- and mechano-gating of the K(+) channel TREK-1. EMBO J. 21, 2968–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal-Hayoun Y.; Cohen A.; Zilberberg N. (2010) Molecular mechanisms underlying membrane-potential-mediated regulation of neuronal K2P2.1 channels. Mol. Cell. Neurosci. 43, 117–126. [DOI] [PubMed] [Google Scholar]
- Rapedius M.; Schmidt M. R.; Sharma C.; Stansfeld P. J.; Sansom M. S.; Baukrowitz T.; Tucker S. J. (2012) State-independent intracellular access of quaternary ammonium blockers to the pore of TREK-1. Channels (Austin) 6, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes C. M.; Gallagher P. G.; Buck M. E.; Butler M. H.; Goldstein S. A. (2000) Proton block and voltage gating are potassium-dependent in the cardiac leak channel Kcnk3. J. Biol. Chem. 275, 16969–16978. [DOI] [PubMed] [Google Scholar]
- Lopes C. M.; Zilberberg N.; Goldstein S. A. (2001) Block of Kcnk3 by protons. Evidence that 2-P-domain potassium channel subunits function as homodimers. J. Biol. Chem. 276, 24449–24452. [DOI] [PubMed] [Google Scholar]
- Yuill K. H.; Stansfeld P. J.; Ashmole I.; Sutcliffe M. J.; Stanfield P. R. (2007) The selectivity, voltage-dependence and acid sensitivity of the tandem pore potassium channel TASK-1: contributions of the pore domains. Pflugers Arch. 455, 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer M. I.; Cid L. P.; Pena-Munzenmayer G.; Sepulveda F. V. (2010) Separate Gating Mechanisms Mediate the Regulation of K2P Potassium Channel TASK-2 by Intra- and Extracellular pH. J. Biol. Chem. 285, 16467–16475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak T. K.; Harinath S.; Nama S.; Somasundaram K.; Sikdar S. K. (2009) Inhibition of human two-pore domain K+ channel TREK1 by local anesthetic lidocaine: negative cooperativity and half-of-sites saturation kinetics. Mol. Pharmacol. 76, 903–917. [DOI] [PubMed] [Google Scholar]
- Takahira M.; Sakurai M.; Sakurada N.; Sugiyama K. (2005) Fenamates and diltiazem modulate lipid-sensitive mechano-gated 2P domain K(+) channels. Pflugers Arch. 451, 474–478. [DOI] [PubMed] [Google Scholar]
- Cadaveira-Mosquera A.; Ribeiro S. J.; Reboreda A.; Perez M.; Lamas J. A. (2011) Activation of TREK currents by the neuroprotective agent riluzole in mouse sympathetic neurons. J. Neurosci. 31, 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes C. M.; Rohacs T.; Czirjak G.; Balla T.; Enyedi P.; Logothetis D. E. (2005) PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J. Physiol. 564, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoz G.; Douguet D.; Chatelain F.; Lazdunski M.; Lesage F. (2009) Extracellular acidification exerts opposite actions on TREK1 and TREK2 potassium channels via a single conserved histidine residue. Proc. Natl. Acad. Sci. U.S.A. 106, 14628–14633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tertyshnikova S.; Knox R. J.; Plym M. J.; Thalody G.; Griffin C.; Neelands T.; Harden D. G.; Signor L.; Weaver D.; Myers R. A.; Lodge N. J. (2005) BL-1249 [(5,6,7,8-tetrahydro-naphthalen-1-yl)-[2-(1H-tetrazol-5-yl)-phenyl]-amine]: a putative potassium channel opener with bladder-relaxant properties. J. Pharmacol. Exp. Ther. 313, 250–259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.