

Abstract
Purpose
Ribonucleotide reductase subunit M2 (RRM2) plays an active role in tumor progression. Recently, we reported that depletion of RRM2 by systemic delivery of a nanoparticle carrying RRM2-specific siRNA suppresses head and neck tumor growth. The aim of this study is to clarify the underlying mechanism by which RRM2 depletion inhibits tumor growth.
Methods
siRNA-mediated gene silencing was performed to downregulate RRM2. Immunoblotting, RT-PCR, confocal microscopy, tissue fractionation, gene overexpression and knockdown were employed to analyze critical apoptosis signaling. Conventional immunohistochemistry (IHC) and quantum dot-based IHF were applied to detect RRM2 and Bcl2 expression and localization in tissue samples from patients and mice.
Results
Knockdown of RRM2 led to apoptosis through the intrinsic pathway in head and neck squamous cell carcinoma (HNSCC) and non-small cell lung cancer (NSCLC) cell lines. We demonstrated that Bcl-2 is a key determinant controlling apoptosis, both in vitro and in vivo and that RRM2 depletion significantly reduces Bcl-2 protein expression. We observed that RRM2 regulates Bcl-2 protein stability, with RRM2 suppression leading to increased Bcl-2 degradation, and identified their co-localization in HNSCC and NSCLC cells. In a total of 50 specimens each from HNSCC and NSCLC patients, we identified the co-localization of Bcl-2 and RRM2 and found a significant positive correlation between their expression in HNSCC (R=0.98, p<0.0001) and NSCLC (R=0.92, p<0.0001) tumor tissues.
Conclusion
Our novel findings add to the knowledge of RRM2 in regulating expression of the anti-apoptotic protein Bcl-2 and reveal a critical link between RRM2 and Bcl-2 in apoptosis signaling.
Keywords: RRM2, Bcl-2, RNAi, Apoptosis
Introduction
Ribonucleotide reductase (RR) is a potential therapeutic target for cancer because it catalyzes the conversion of ribonucleoside 5′ -diphosphates into their corresponding 2′ -deoxyribonucleoside 5′ -triphosphates that are necessary for DNA replication and repair (1). Human RR comprises two subunits, RRM1 and RRM2. The cellular RRM1 protein level is relatively stable through the entire cell cycle, whereas RRM2 is only expressed during the late G1/early S phase when DNA replication occurs (2, 3). Because the RRM2 subunit is the primary regulator of RR enzymatic activity (1), studies of its influence on the biological activities of the RR protein are highly sought. RRM2 was reported to play an active role in tumor progression (4, 5) and was identified as an indicator of poor patient outcome in several cancer types (6, 7). RRM2 is known to be involved in chemoresistance. Suppression of RRM2 can sensitize colon and pancreatic cancer cells to gemcitabine and DNA damaging agents for which RRM2 plays a role in the relevant DNA synthesis and repair mechanisms (3, 8). RRM2 serves as a prognostic biomarker of many cancer types (7, 9-11) and regulates several oncogenes that control malignant potential (12).
In recent years, several inhibitors (i.e. triapine, gemcitabine and GTI-2040) have been studied to inhibit RRM2 activity and entered in clinical trials. Shao et. al. reviewed several potential inhibitors of RR (13). Triapine and GTI-2040, potent inhibitors of RR, have been evaluated in Phase I clinical trials (14, 15). However, non-specific toxicities hamper their clinical applicability. Gemcitabine competes with RRM2 for replicating DNA, is a potential candidate for RRM2 inhibition and an FDA-approved drug for lung and pancreatic cancer treatment. However, the non-specific action of gemcitabine causes cytotoxicity (13). Overall, cancer-targeted knockdown of RRM2 is desirable. We recently explored the role of RRM2 in cancer progression by utilizing siRNA-mediated gene silencing, which has a better safety profile, and provided the first example of dose-dependent accumulation of targeted nanoparticles in human head and neck tumors to engage the RNAi machinery (16). We found that targeting RRM2 with a nanoparticle carrying RRM2-specific siRNA inhibited tumor growth in vivo. The mechanism of anti-tumor action of RRM2 inhibition merits further elucidation.
Mitochondrial outer membrane permeabilization (MOMP) integrity is highly controlled through interactions between pro-and anti-apoptotic members of the Bcl-2 protein family (17), likely providing a mechanistic basis for modulating apoptotic cell death (18, 19). Defects in the delicate balance between cell proliferation and cell death can lead to cancer development and are a hallmark of cancer (19-22). Members of the Bcl-2 protein family are critical regulators of apoptosis, contributing to prolonged cell survival through their ability to block apoptosis (23). Clinically, Bcl-2 overexpression is associated with poor prognosis, resistance to standard chemotherapy or radiation therapy and bias toward cancer cell survival (24-28). Bcl-2 activity is regulated by various mechanisms, including transcription, posttranslational modifications, and degradation. Reductions in Bcl-2 expression induces sensitivity to anti-cancer drugs (29-31) and increases survival (32). Effective mechanisms to clinically target Bcl-2 and the determination of its impact on patient outcome remain to be established.
In this study, we uncovered a novel molecular mechanism in which Bcl-2 is a key determinant controlling intrinsic apoptosis triggered by RRM2 depletion, both in vitro and in vivo. To clinically validate our novel findings, we utilized quantum dot–immunofluorescence and conventional immunohistochemistry techniques to stain RRM2 and Bcl-2 proteins in tumor tissue samples from HNSCC and NSCLC patients. We confirmed the co-localization of Bcl-2 and RRM2 and found a significant positive correlation between their expressions in these tumor tissues. Our results demonstrate a new function for RRM2 in regulating expression of the anti-apoptotic protein Bcl-2 and reveal a critical link between RRM2 and Bcl-2 in apoptosis signaling.
Materials and Methods
Cell lines
HNSCC cell lines Tu212 and Tu686 (provided by Dr. Gary L. Clayman, University of Texas M.D. Anderson Cancer Center) and NSCLC cell lines A549, H1299 and H358 were kindly provided by Dr Shi-Yong Sun (Emory University, GA). sh p53 in lentivirus vector was a generous gift from Dr. Didier Trono, Ecole Polytechnique Federale de Lausanne, Switzerland and A549-expressing sh p53 cell line was established as described (33). The dominant negative p73 plasmid generation and validation is described elsewhere (34). pWZL-NeoAKT plasmid (constitutively active) was obtained from Addgene, Cambridge, MA and pLNCX-Bcl-2 plasmid was obtained from Dr. Jackson’s lab (Case Western Reserve University, OH). Bcl-2 overexpressing Tu212 and Tu686 (pool) cell lines were generated by retroviral transduction of Bcl-2 and selection by G418. Constitutively active Akt was overexpressed in A549 and Tu686 cell lines (G418 selected pool).
Western blotting, co-immunoprecipitation and cell fractionation
Western blot was incubated with primary followed by secondary antibodies and detected using enhanced chemiluminescence system as described (35). For co-immunoprecipitation, cells were lysed in a Chaps cell extract buffer (Cell Signaling Technology, USA). Lysates were centrifuged at 15000 r.p.m. for 20 min at 4°C, and supernatants were incubated with primary antibodies for 4h and coupled to protein A/G-agarose (Pierce, USA) for 2h at 4°C. Immunoprecipitates were washed three times with Chaps buffer and subjected to immunoblotting. Cytosolic and mitochondrial fractionation was carried out using a kit (Clontech, CA, USA). Primary antibodies were anti-RRM2, anti-GAPD, anti-p53, anti-Bcl-2, anti-Bcl-XL, anti-Mcl-1, anti-Bax from Santa Cruz Biotech, CA; anti-phospho Akt, anti-caspase9, anti-caspase3, anti-PARP from Cell Signaling, MA; anti-p73 from Bethyl Laboratories, TX; and anti-β-actin from Sigma Aldrich. Secondary antibodies were from Santa Cruz Biotech. Western band quantification was performed using Image-Quant TL software (GE/Amersham Biosciences, Piscataway, NJ).
siRNA duplexes
Control siRNA (siC) was bioinformatically designed to minimize potential for targeting any human gene (Dharmacon, Lafayette, CO). Unmodified RNA duplex siR2 (RRM2 siRNA) was a gift from Calando Pharmaceuticals (Pasadena, CA). siR2-1 and siR2-2 RNA duplexes were purchased from Qiagen, California, USA. The sequences are as below:
Control siRNA: 5′-UAGCGACUAAACACAUCAAUU-3′
siR2: 5′-GCGAUUUAGCCAAGAAGUUCA-3′
siR2-1: 5′-GCGAUUUAGCCAAGAAGUUTT-3′
siR2-2: 5′-GGGAUUAAACAGUCCUUUATT-3′
Lipofectamine transfection
Cells were seeded in 6-cm plates 24 h prior to transfection in medium containing 10% fetal bovine serum (FBS), so that they reached 30~50% confluency. siRNA was complexed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions and applied to each plate. Transfection media was removed and replaced with new media after 5 hours.
Apoptosis assay
Cells were transfected with different concentrations of siR2 or siC and cells were collected after 72 h by trypsinzation, washed with cold 1X PBS and stained with Annexin V-phycoerythrene and 7-AAD (BD PharMingen) for 15 min at room temperature. The samples were measured using a fluorescence-activated cell sorting (FACS) caliber bench-top flow cytometer (Becton Dickinson). FlowJo software (Tree Star) was used for apoptosis analysis.
RT-PCR
RNA was collected by direct lysis in TRIzol (Invitrogen) and reverse-transcribed using SuperScriptIII reverse transcriptase. 2 μl of prepared sample cDNA was used for PCR. A Bcl-2 PCR primer set was purchased from Sigma (No. B9179). GAPDH forward primer: 5′-TGCACCACCAACTGCTTA-3′ and reverse primer: 5′-GGATGCAGGGATGATGTTC-3′ was selected. Bcl-2 levels were normalized to GAPDH levels within the same sample.
Immunohistochemistry (IHC)
To detect intracellular localization and expression levels of RRM2 and Bcl-2, we used goat anti-human R2 polyclonal antibody and mouse anti-human Bcl-2 monoclonal antibody (Santa Cruz Biotechnology) respectively, as primary antibodies, then conjugated to secondary antibody and cell nuclei were counterstained using 4,6-diamidino-2-phenylindole (DAPI, Invitrogen, Carlsbad, CA). Mouse and rabbit IgG were used as negative controls. All the tissue slides were evaluated and scored by a qualified pathologist.
Confocal microscopy
Cells were grown on glass coverslips (Lab-Tek II chamber slide, Nunc International), fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.5% Triton X-100 in PBS for 5 min. Cells were exposed consecutively to primary antibodies (anti-RRM2 and anti-Bcl-2) and rhodamine-conjugated or FITC-conjugated secondary antibodies, before mounting with ProLong Gold antifade reagent with DAPI (Invitrogen). A Zeiss LSM 510 confocal scanning microscope was used to collect the images.
Protein half-life
Tu212 cells in 100-mm dishes were transfected with siC or siR2. After 24 hrs, cells were reseeded into 60-mm dishes and allowed to attach for 24 hr. Cells were treated with 100 ug/ml cyclohexamide 5 min prior to starting the time course (0, 1, 3, 6, 9, 12H), and cell lysates were collected at the indicated time points.
Tissue specimens
This study was approved by the Institutional Review Board at Emory University and analyses were conducted using de-identified data in compliance with HIPAA. 50 HNSCC and 50 NSCLC tissues (30 adenocarcinoma, 18 squamous cell carcinoma, and 2 others) were randomly collected for the study from surgical specimens of patients who were diagnosed with HNSCC or NSCLC at Emory University Hospital and whose initial treatment was surgery without receiving prior treatment with radiation and/or chemotherapy. This was a retrospective study which used tissue samples from surgical specimens dated prior to April14, 2003.
QD-based immunohistofluorescence (QD-IHF)
QD staining was performed on paraffin-embedded formalin-fixed NSCLC and HNSCC tissue sections overnight at 4°C as described previously (36). Primary antibodies were mouse anti-human Bcl-2 at 1:50 dilution and goat anti-human R2 at 1:100 (both from Santa Cruz Biotechnology, Santa Cruz, CA) in a mixed solution. PBS alone was used as negative staining control. Tissues were incubated with QD-secondary antibody conjugates (QD 605 goat F(ab)2 anti-mouse IgG; QD 655 goat F(ab)2 anti-goat IgG, 1:50 dilution, Invitrogen, Carlsbad, CA) in a mixed solution at 37°C for 2 hours. Cell nuclei were counterstained using DAPI.
QD spectral imaging and signal quantification
QD imaging and quantification procedures were described previously (36). All cubed image files were collected from patient tissue slides at 10 nm wavelength intervals from 450-750 nm, with an auto exposure time per wavelength interval at 200× magnification. Taking the cube with a long wavelength band pass filter allowed transmission of all emission wavelengths above 450 nm. Both separated and combined QD images were obtained after establishing the QD spectral library and unmixing the image cube. For each slide, 5 cubes were taken. The background signal was removed for accurate quantification of the QD signals. Average signal of each QD signal were obtained by selecting 4-10 tumor areas in each cube for measurements by the Nuance imaging software (Caliper/PerkinElmer, Hopkinton, MA). Measurements were averaged from the 5 cubes from each slide to obtain a total average signal count on each slide for both QD signals.
Statistical analysis
All results represent the average of at least three separate experiments and are expressed as mean ± SD. Statistical analysis was performed using t-test. P<0.05 was considered statistically significant. Spearman’s correlation coefficient was estimated to measure the relationship between Bcl-2 and RRM2. A linear regression was used to plot their relationship with 95% confidence interval bound.
Results
Knockdown of RRM2 promotes apoptosis in HNSCC and NSCLC cells
Our previous observation of cell growth inhibition by RRM2-targeting siRNA (siR2) (16) prompted us to further investigate the mechanistic details of RRM2’s action. Upon knockdown of RRM2 in Tu212 (HNSCC) and A549 (NSCLC) cells (Fig. 1A and B, Inset), we observed apoptosis induction in a dose-dependent manner. After 72h, 5nM siR2 induced apoptosis (Tu212 ~45% and A549 ~60%) (Fig. 1A and B), while no significant apoptotic effects were observed at an early time (24-48h; data not shown). The target specificity of siR2 mRNA was confirmed previously (16, 37-39). Two other RRM2 siRNAs, siR2-1 and siR2-2, also effectively knocked down RRM2 and induced apoptosis in Tu212 and A549 cell lines after 72 hours (Fig. 1C and D), arguing against any non-specific effects. In all experiments, non-targeting siRNA (siC) was used as a negative control.
Figure 1.
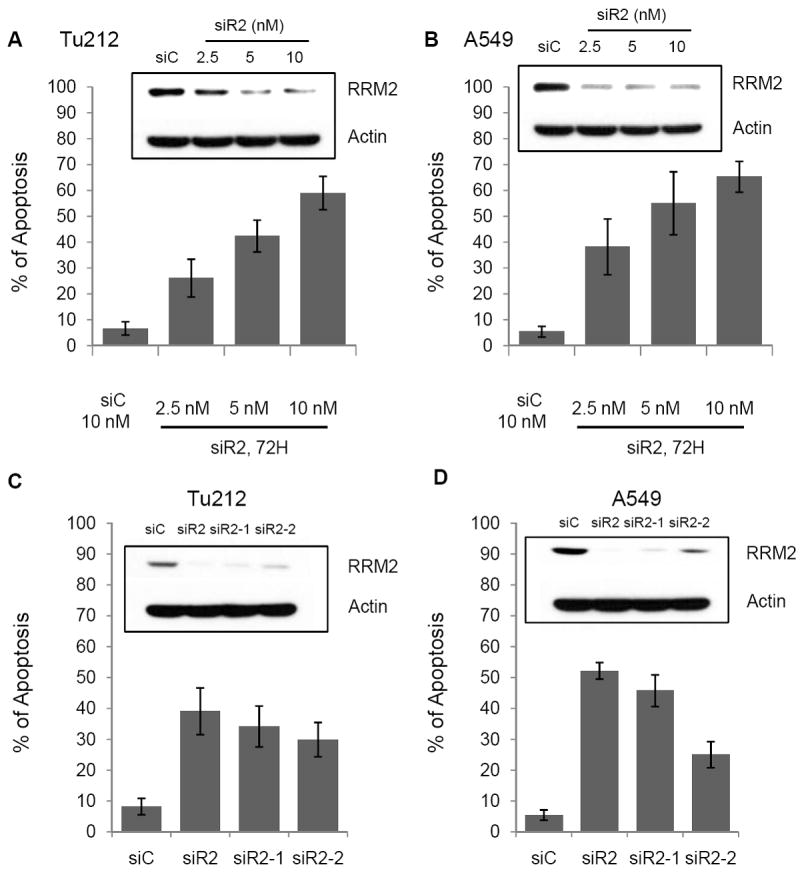
Knockdown of RRM2 induced apoptosis. Apoptosis was measured by annexin V-PE and 7-AAD staining in (A)Tu212 and (B) A459 cell lines 72h after siRNA transfection (error bars are mean ±SD from 3 independent experiments). Cell lysates were analyzed by Western blotting with the indicated antibodies after transfection (Inset). (C) Tu212 and (D) A549 cells were transfected with siC or different siRNAs against RRM2, siR2, siR2-1, siR2-2. After 72 h, apoptosis was measured by annexin V-PE and 7-AAD staining. Cell lysates were analyzed by Western blotting with the indicated antibodies after transfection (Inset).
Knockdown of RRM2 promotes apoptosis by inducing the mitochondria-mediated intrinsic apoptosis pathway in both HNSCC and NSCLC cells
We explored whether intrinsic or extrinsic apoptosis occurs following siR2-mediated depletion of RRM2 in Tu212 and A549 cell lines. Our results demonstrated the efficient triggering of the apoptotic cell signaling cascade in Tu212 and A549 cell lines following siR2 treatment (Fig. 2A). Western blot analysis revealed that suppression of RRM2 induced apoptotic signaling molecules mainly involved in intrinsic apoptosis (17, 40-42), such as cleaved caspase 9 (initiator caspase) and cleaved caspase 3 (effector caspase), followed by cleavage of substrate poly ADP-ribose polymerase (PARP). We did not detect any induction of extrinsic apoptosis signaling molecules (DR5, caspase 8) by RRM2 suppression (data not shown). We observed caspase 3 activation and PARP cleavage in both Tu212 and A549 cells following treatment with siR2-1 and siR2-2 siRNAs but less effectively than with siR2 (Fig. 2B). In the intrinsic apoptosis pathway, highly controlled mitochondrial outer membrane permeabilization is the crucial event driving initiator caspase activation and apoptosis (17). The release of cytochrome C from mitochondria has been considered a key step in the early stages of apoptosis (17, 41). We isolated mitochondrial and cytoplasmic fractions from cell lysates of Tu212 and A549 cells transfected with siR2 or siC, and observed cytochrome C release from the mitochondria to cytoplasm only in the siR2-treated group (Fig. 2C). Taken together, our results demonstrate that knockdown of RRM2 promotes apoptosis via the mitochondria-mediated intrinsic apoptosis pathway.
Figure 2.
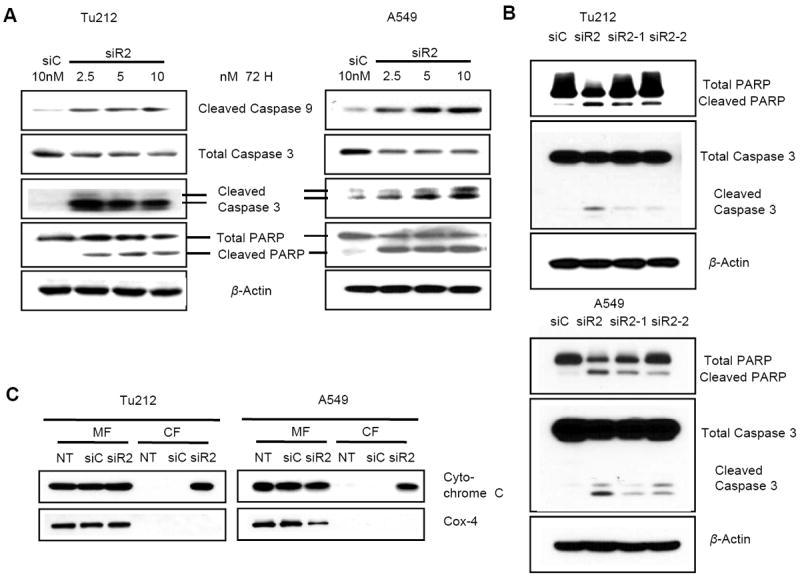
Knockdown of RRM2 induces apoptosis via the mitochondria-mediated intrinsic pathway. (A) Tu212 (left panel) and A549 (right panel) cells were transfected with siC or siR2. After 72 h, cell lysates were analyzed by Western blotting with the indicated antibodies. A single set of blots is pictured from three independent experiments. (B) Tu212 (upper panel) and A549 (lower panel) cells were transfected with siC or different siRNAs against RRM2, siR2, siR2-1, siR2-2. After 72 h, cell lysates were analyzed by Western blotting with the indicated antibodies. A single set of blots is pictured from three independent experiments. (C) Mitochondrial fraction (MF) and cytosolic fraction (CF) of Tu212 and A549 cells were isolated 72h after transfection with 5 nM siR2, 5 nM siC, or no treatment (NT). Western blotting was performed to detect Cytochrome C and Cox-4.
Depletion of RRM2 promotes apoptosis in anti-apoptotic protein Bcl-2 dependent manner
Evasion of apoptosis has proven to be critical for the development and sustained growth of cancers. Members of the Bcl-2 protein family are critical regulators of apoptosis (40). One approach to inhibiting the apoptotic pathway is through the upregulation of anti-apoptotic Bcl-2 family members (43). We determined that RRM2 suppression by siR2, siR2-1 and siR2-2 significantly reduced Bcl-2 expression in Tu212 and A549 cells (Fig. 3A, 3B), while levels of other anti-apoptotic Bcl-2 family proteins, Bcl-xL and Mcl-1 remained unchanged in both cell lines following siR2 treatment (Fig. 3A). To explore whether Bcl-2 status could affect the regulation of apoptosis by RRM2, we established a Bcl-2–overexpressing Tu212 cell line (Tu212/Bcl-2) (Supplementary Fig. S1A, upper panel). We showed significant resistance to apoptosis upon suppression of RRM2 in Tu212/Bcl-2 cell line (Fig. 3C). In addition, apoptosis marker molecules such as cleaved caspase 3 and cleaved PARP were triggered by suppressing RRM2 in Tu212 cells but not in Bcl-2–overexpressing cells (Fig. 3D). To confirm our observation, another cell line Tu686/Bcl-2 (Supplementary Fig. S1A, lower panel) was tested to determine the importance of Bcl-2 and we found similarly that RRM2-mediated apoptosis was Bcl-2-dependent (Supplementary Fig. S1B). Taken together, our results suggest that the anti-apoptotic protein Bcl-2 plays a critical role in siR2-mediated apoptosis induction.
Figure 3.
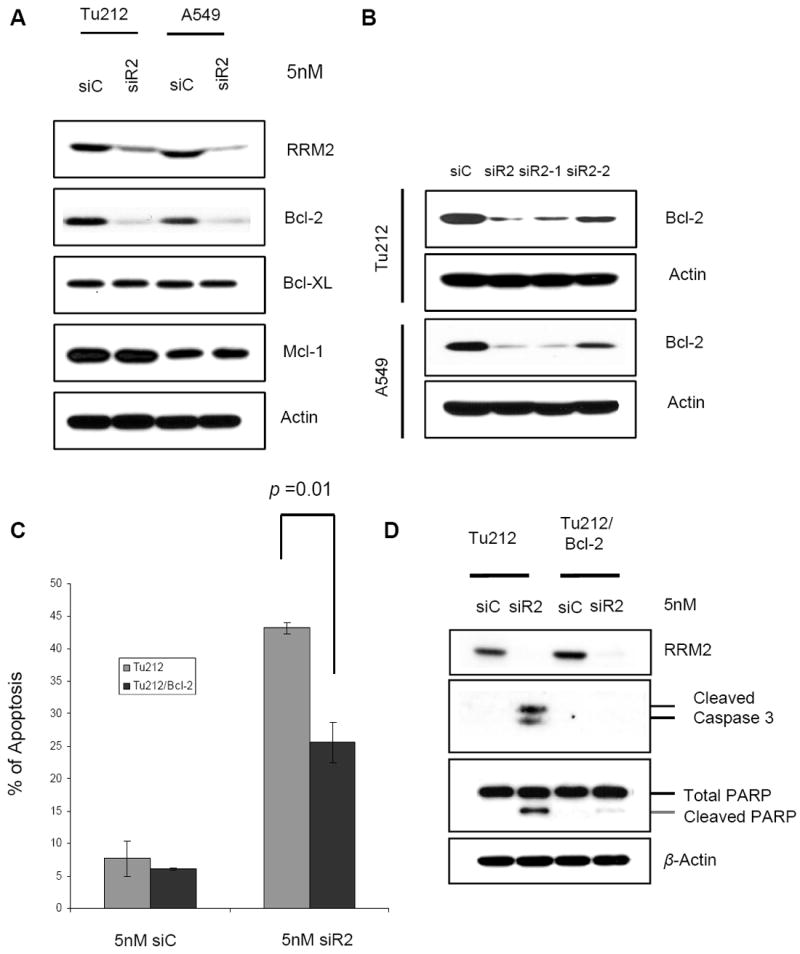
Apoptosis induction by knockdown of RRM2 is Bcl-2 dependent. (A) Cell lysates of Tu212 and A549 were collected 72h after transfection with 5 nM siC or siR2. Western blotting was performed to detect anti-apoptotic Bcl-2 family proteins. (B) Cell lysates of Tu212 and A549 were collected 72h after transfection with 5 nM siC or different siRNAs against RRM2, siR2, siR2-1, siR2-2. Western blotting was performed to detect Bcl-2 protein. (C) Apoptosis analysis (error bars are mean ±SD from 3 independent experiments). (D) Western blotting was performed with specific antibodies 72h after transfection with 5nM siC or siR2 in Tu212 and Bcl-2 overexpressing Tu212 (Tu212/Bcl-2) cell lines.
Apoptosis induction by suppression of RRM2 is p53-, p73-, Akt-independent
We then extended our study to identify any other signaling molecules involved in the regulation of siR2-mediated apoptosis, particularly p53 and p73 since these are potential candidates in apoptosis induction (44). An elevated level of p53 was observed upon RRM2 suppression in A549 cells (wild type p53), while a sustained level was seen in Tu212 cells (mutant p53 [P151S] (45)) (Fig. 4A). To explore the role of p53, we established p53-knockdown A549 cells (A549/sh p53) using short hairpin RNA (Fig. 4B). By knocking down RRM2, we found cleaved caspase 3 and PARP induction in both WT-A549 and p53 knocked-down cells (Fig 4B), suggesting that apoptosis induction by siR2 in A549 cells is p53-independent. To assess the involvement of p73, we determined the percentage of apoptosis and apoptotic marker proteins in the H1299 lung cancer cell line and in two clones of dominant negative p73-expressing H1299 cells (H1299/dN p73 Cl-7, Cl-10) (Supplementary Fig. S2A, upper panel). We found similar apoptosis induction (Fig. 4C) or PARP modification (Fig. 4D) in all cell lines upon suppression of RRM2. Another cell line H358/dNp73 (Supplementary Fig. S2A, lower panel) was also tested for apoptosis upon RRM2 knockdown and we observed a similar effect as in H1299 cells (Supplementary Fig. S2B), suggesting that apoptosis induction is also p73-independent. Akt plays a vital role in anti-apoptotic signaling (46), and we observed that Akt activation was significantly reduced upon suppression of RRM2 in both Tu212 and A549 cells (Fig. 4E). The role of Akt in RRM2-mediated apoptosis was evaluated by its overexpression in A549 cells (A549/Akt) (Supplementary Fig. S3A, upper panel), and no significant differences in apoptosis or PARP modification were seen compared to unmodified A549 cells (Fig. 4F and G). In the Tu686/Akt cell line (Supplementary Fig. S3A, lower panel), we found that knockdown of RRM2-mediated apoptosis is Akt-independent (supplementary Fig. S3B). Taken together, our results suggest that apoptosis induction by silencing of RRM2 is independent of p53, p73, and Akt.
Figure 4.
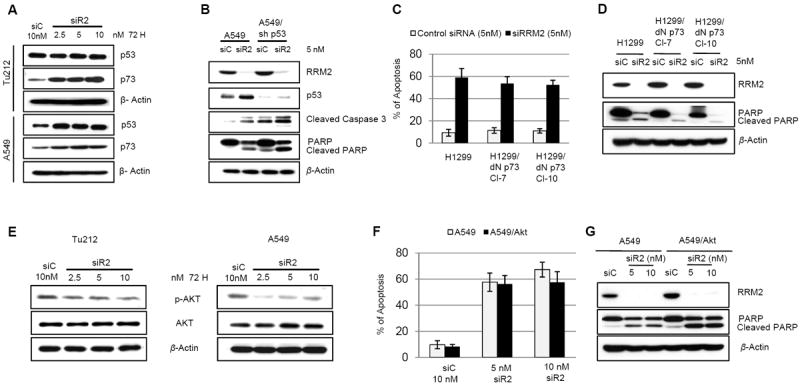
Apoptosis induction by knockdown of RRM2 is p53-, p73-, and Akt-independent. (A) Western blotting for p53 and p73 expression in Tu212 and A549 cells 72h after transfection with siC or siR2. (B) Western blotting for the indicated proteins in A549 and p53-knockdown A549 (A549/sh p53) cell lines after transfection with 5nM siC or siR2. A representative blot of three independent experiments is presented. (C) Apoptosis analysis (error bars are mean ±SD from 3 independent experiments). (D) Western blot analysis 72h after transfection with 5nM of siC or siR2 in H1299 and two clones of dominant negative p73-expressing H1299 (H1299/dN p73 Cl-7, Cl-10) cell lines. (E) Western blotting for p-Akt and Akt in Tu212 and A549 cells 72h after transfection with siC or different concentrations of siR2. (F) Apoptosis analysis. (G) Western blot of whole cell lysates from A549 and Akt-overexpressing A549 (A549/Akt) cell lines 72 h post-siRNA transfection.
RRM2 regulates Bcl-2 partially at the transcription level and the two proteins colocalize and interact with each other
To determine whether Bcl-2 is regulated by RRM2 at the transcriptional or the protein level, we transfected cells with siR2 or siC and collected RNA samples after 72 hours for RT-PCR. RRM2 levels were normalized to GAPDH levels within the same sample. The mRNA expression of Bcl-2 was suppressed ~50% (Fig. 5A) whereas the protein level was suppressed >90% (Fig. 3A) following RRM2 knockdown, indicating that RRM2 most likely regulates Bcl-2 not only at a transcriptional level but also at the post-translational level. To investigate whether these two proteins co-localize, Tu212 cells were stained with anti-RRM2 (green), anti-Bcl-2 (red) and DAPI (blue). A z-stack of 32 optical sections was created at 0.59 μm intervals using a confocal microscope (LSM 510; Carl Zeiss MicroImaging, Inc.), shown in a gallery of merged images in a single display (Supplementary Fig. S4). We found that RRM2 and Bcl-2 proteins were co-localized each other (Fig. 5B, yellow color on merge). The white box in the merged figure was magnified (Fig. 5B, inset, upper panel) and the inset lower panel shows the z-axis reconstructions along the bars indicated in the inset upper panel. To assess the impact of RRM2 depletion, Tu212 cells (Fig. 5C) and A549 cells (Fig. 5D) were transfected with siC or siR2, fixed after 48 hours and stained with anti-RRM2 (green) and anti-Bcl-2 (red). Upon silencing of RRM2, Bcl-2 protein was significantly suppressed, confirming our Western blot data showing that RRM2 regulates Bcl-2 protein stability. To demonstrate the interaction between these two proteins, co-immunoprecipitation using anti-RRM2 and anti-Bcl-2 antibodies in Tu212 and A549 cell lysates revealed that the proteins indeed existed as a complex (Fig. 5E).
Figure 5.
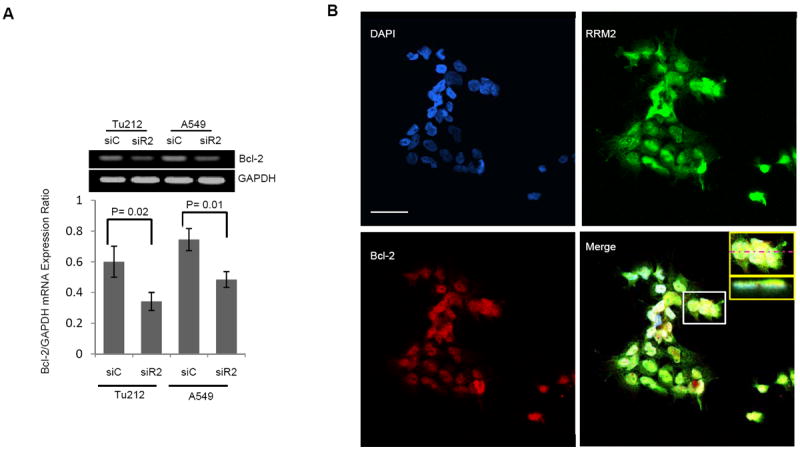
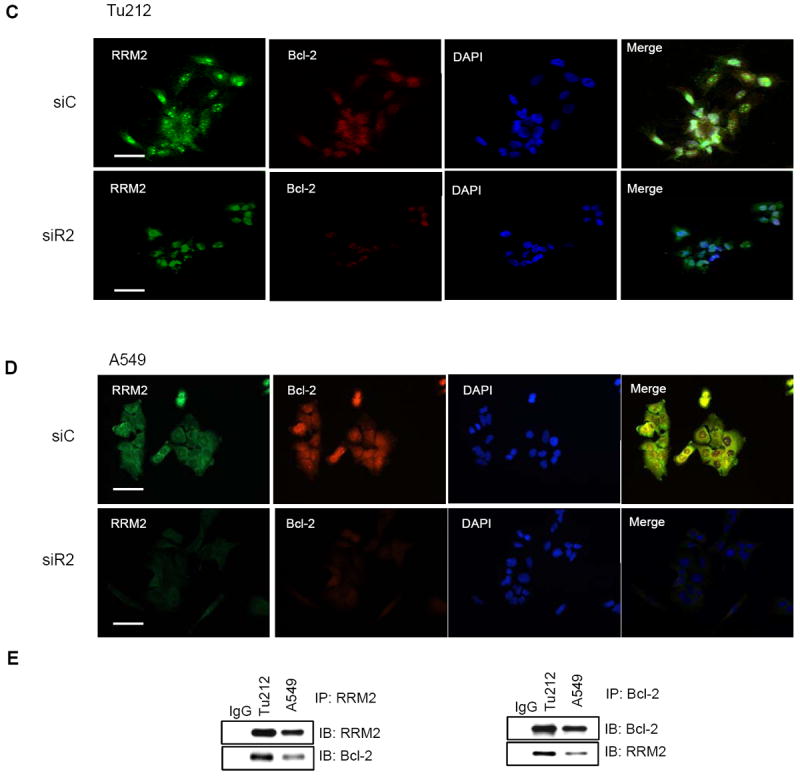
RRM2 regulates Bcl-2 through direct protein-protein interaction and partially influences Bcl-2 mRNA. (A) RT-PCR for Bcl-2 and GAPDH mRNA levels 72 h after transfection of Tu212 and A549 cells with 5nM siC or siR2. RRM2 was normalized to GAPDH levels within the same sample. (B) Tu212 cells were stained with anti-RRM2 (green), anti-Bcl-2 (red) and nuclei were counter stained with DAPI (blue). A z-stack of optical sections was created at 0.59 um intervals using a confocal microscope (LSM 510; Carl Zeiss MicroImaging, Inc.). Inset, upper panel shows magnification of white box in merged figure. Inset, lower panel shows z-axis reconstructions along the bars indicated in inset upper panel. Bar, 50μm. (C) Tu212 and (D) A549 cells were stained with anti-RRM2 (green), anti-Bcl-2 (red) and nuclei (blue) 48hr after transfection with 5 nM siC or siR2. Bar, 50μm. (E) Co-IP with anti-RRM2 (left panel) or anti-Bcl-2 (right panel) and immunoblotting with anti-Bcl-2 or anti-RRM2 in Tu212 and A549 cells.
Depletion of RRM2 regulates Bcl-2 protein expression and stability by reducing its half life
To understand the effect of RRM2 depletion on Bcl-2 protein stability, we transfected Tu212 cells with siC or siR2 for 24h. Then, cells were reseeded and allowed to attach for another 24 hr. Cells were treated with cyclohexamide to block protein synthesis, and Bcl-2 expression level was assessed at multiple time points to determine its half life. As shown in Figure 6A, knockdown of RRM2 significantly increased the rate of Bcl-2 protein degradation, reducing its half-life from ~9 hours to ~2 hours. These results demonstrate that knockdown of RRM2 causes significant reduction in Bcl-2 protein stability. It is known that Bcl-2 can be cleaved and degraded by activated caspase 3 (47, 48). Thus, the observed Bcl-2 degradation in earlier finding might be a secondary effect due to activation of caspase 3 triggered by siR2. To confirm this, we assessed caspase 3 activation in all samples shown in Figure 6A. We did not find any caspase 3 activation at early time points (Fig. 6B) of siR2 treatment, while Bcl-2 degradation was observed from one hour after blockade of de novo protein synthesis in the siR2 transfected group (Fig. 6A). This suggests that Bcl-2 degradation triggered by siR2 is not due to caspase 3 activation. To confirm the involvement of the ubiquitin-proteasome pathway in Bcl-2 degradation, we blocked proteasome function by utilizing the proteasome inhibitor MG132 in Tu212 and A549 cell lines. We observed that proteasome inhibition protected Bcl-2 degradation upon RRM2 knockdown, suggesting that RRM2 knockdown leads to Bcl-2 degradation through proteasome (Fig. 6C). We found significant reduction of Bcl-2 protein in tissue samples from mice treated with siR2-nanoparticle (CALAA-01) compared to mice treated with control siRNA-nanoparticle (siCON1) (n=8) (Fig. 6D). Taken together, our study provides functional evidence that RRM2 regulates Bcl-2 protein stability.
Figure 6.
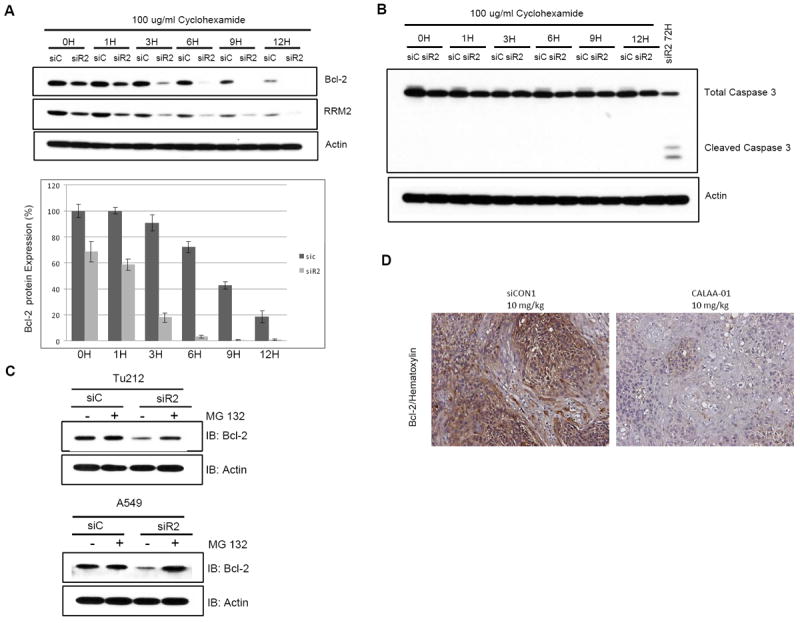
Depletion of RRM2 reduces Bcl-2 protein expression and regulates protein stability. (A, B) Tu212 cells were transfected with 5 nM siC or siR2, reseeded after 24h into 60-mm dishes and 24h later treated with cyclohexamide (100 μg/ml) for 0, 1, 3, 6, 9 and 12h. Cell lysates were collected at indicated time points and Western blotting performed with specific antibodies. (C) Tu212 and A549 cells were transfected with 5nM siC or siR2 and 24 hours later treated with 10 μmol/L MG132 for 2 hours before analyzing cell lysates by Western blotting. (D) Bcl-2 expression was detected in xenograft tumor tissue by IHC analysis. The animal study was conducted previously by treating with four doses of siCON1 (control nanoparticle) or CALAA-01 (RRM2 siRNA-nanoparticle). Representative images shown from siCON1 and CALAA-01 10 mg/kg groups (brown stain for Bcl-2 and nuclei were counter-stained by hematoxylin, blue; magnification X200).
RRM2 and Bcl-2 proteins co-localize and their expression is positively correlated in tumor tissues from HNSCC and NSCLC patients
We extended the study to examine the correlation between RRM2 and Bcl-2 protein levels in tissue samples from cancer patients. A total of 50 cases each of HNSCC and NSCLC (30 adenocarcinoma, 18 squamous cell carcinoma and 2 others) were analyzed. Clinico-pathological features of the HNSCC and NSCLC patient groups are shown in Supplementary Table 1. Quantum dot (QD)-immunofluorescence (IHF) analysis allows quantification of multiple biomarkers simultaneously in the same tissue slide (36, 49). In our study, we quantified the expression levels of RRM2 and Bcl-2 in the same tissue slide by using secondary antibody-conjugated QDs with two different emission wavelengths (655 nm and 605 nm). Both separated and combined QD images were established after determining the QD spectral library and unmixing the cube. Representative QD images from HNSCC and NSCLC patients are shown in Figure 7A and 7D, respectively. RRM2 and Bcl-2 were found to be highly co-localized in both types of cancer. Average signal counts (A.U.) of each QD signal were obtained from 5 image cubes of each tissue slide using Nuance imaging software (Supplementary Table 2 and 3). QD signals were plotted by de-identified patient ID of HNSCC and NSCLC patients and are shown in Figure 7B and 7E, respectively. Spearman’s correlation coefficient demonstrated a highly significant positive correlation between RRM2 and Bcl-2 expression in both HNSCC and NSCLC patient samples (HNSCC: R=0.98, p-value <0.0001; NSCLC: R=0.92, p-value <0.0001; Fig. 7C and 7F, respectively). QD-IHF is comparatively new but promising technique and is under development in our laboratory. Therefore, we additionally applied conventional immunohistochemistry (IHC) techniques to confirm our findings. To compare the QD technique with conventional IHC, we examined 20 out of 50 (previously selected) specimens from both HNSCC and NSCLC patients. IHC signal was quantified with weighted index [WI = intensity (scored as 0-3) × percentage of positive staining] by two researchers including a pathologist. Spearman’s correlation analysis demonstrated a significant positive correlation between the RRM2 expression level quantified by IHC and QD-IHF in both HNSCC and NSCLC samples (HNSCC: R=0.59, p-value=0.0067; NSCLC: R=0.46, p-value=0.036; supplementary Fig. S5 and S6, respectively), supporting that quantification and localization of multiple biomarkers using QD-IHF is comparable but more subjective than IHC.
Figure 7.
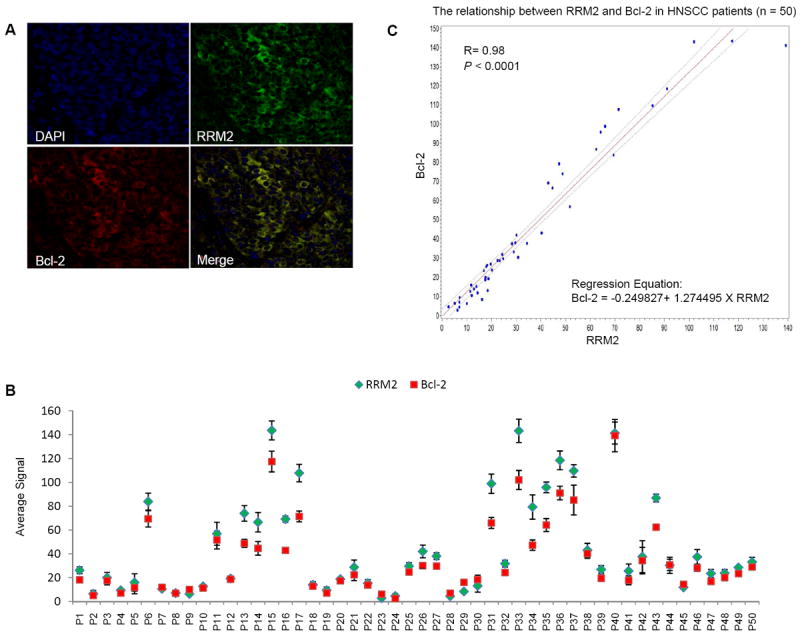
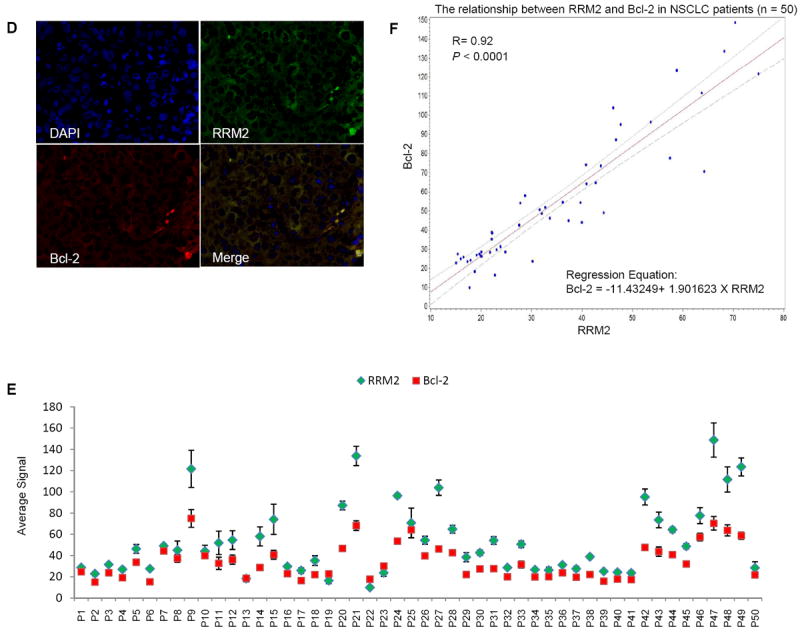
RRM2 and Bcl-2 proteins co-localize and their expression is positively correlated in tumor tissues from HNSCC and NSCLC patients. (A and D) Staining of RRM2 and Bcl-2 in paraffin-embedded formalin-fixed HNSCC (A) and NSCLC (D) tissue sections using primary antibodies with QD-secondary antibody conjugates. A representative QD-image is shown (400X magnification). (B and E) Quantification of QD signals. Average signals of RRM2 and Bcl-2 expression in each HNSCC (B) and NSCLC (E) patient sample were plotted. (C and F) Spearman’s correlation coefficient was estimated between Bcl-2 and RRM2 levels in HNSCC (C) and NSCLC (F) tumors. A linear regression was used to plot their relationship with 95% confidence interval bound.
Discussion
Elevated levels of RRM2 correlate with poor prognosis for cancer patients (50-53). In our previous report, we showed the importance of RRM2 in promoting tumor growth in HNSCC and NSCLC (16). In our current work, we unravel first time the mechanistic details linking RRM2 to apoptosis and provide vital clues for the development of novel cancer therapies. We demonstrate that RRM2 silencing by siRNA directs mitochondria-mediated intrinsic apoptosis. We revealed that RRM2 and Bcl-2 proteins exist as a complex. RRM2 was found to be significantly involved in regulating the anti-apoptotic protein Bcl-2 and a reciprocal regulation between these proteins in apoptosis signaling was observed. Clinical specimens from different types of cancer have been examined to confirm the correlation between RRM2 and Bcl-2. QD-IHF staining showed the co-localization of RRM2 and Bcl-2 expression in the same tissue slides from HNSCC and NSCLC patients. Interestingly, expression of both proteins was positively correlated in both HNSCC and NSCLC tumors.
Since p53 is one of the most commonly mutated genes in cancer and inhibition of RRM2 is known to activate DNA damage (4, 8), we chose cell lines containing wild-type p53 (A549), mutant p53 (Tu212, Tu686), and no p53 (H1299, H358). These cell lines also have variable expression levels of p-AKT and Bcl-2 (54). The Bcl-2 family proteins regulate cell fate through the control of mitochondrial outer membrane potential. Dysregulation of Bcl-2 proteins is common in cancer. The upregulation of anti-apoptotic Bcl-2 proteins serves to inhibit pro-apoptotic Bcl-2 proteins to ensure the survival of abnormal cells. Thus, many malignant cells are primed for cell death and will undergo apoptosis if the function of anti-apoptotic proteins is therapeutically impaired (55). Overexpression of Bcl-2 is a marker of resistance to chemo- or radio-therapy in squamous cell carcinoma and other types of cancer (24, 26, 27). Targeting cellular processes that regulate Bcl-2 at posttranslational levels is crucial. Our novel findings suggest that targeting RRM2 is a promising approach to reduce Bcl-2 expression and to initiate apoptosis. Only Bcl-2 was identified as a key determinant controlling intrinsic apoptosis triggered by RRM2; we did not observe an increased rate of degradation of other anti-apoptotic Bcl-2 family member proteins. Bcl-2 is known to be regulated through posttranslational modifications (56). Here, we show a new molecular regulatory pathway wherein RRM2 contributes to the accumulation and stabilization of Bcl-2. Knockdown of RRM2 significantly reduced the half-life of Bcl-2. We observed that depletion of RRM2 reduced Bcl-2 expression and eventually initiated apoptosis. Bcl-2 protein expression was assessed at multiple timepoints upon RRM2 knockdown (Fig. 6A). We found that RRM2 knockdown significantly increased Bcl-2 protein degradation (Fig. 6A) but failed to induce caspase-3 activation at those time points but activation was observed only at 72 hours (Fig. 6B). Moreover, we confirmed that RRM2 knockdown leads to Bcl-2 degradation through the proteasome pathway. These observations clearly suggest that Bcl-2 downregulation by RRM2 siRNA precedes caspase-3 activation. In contrast, overexpression of Bcl-2 protected cells from apoptosis triggered by RRM2 knockdown. Our results have broader implications, suggesting that suppression of Bcl-2 by targeting RRM2 could provide an effective therapeutic strategy for Bcl-2-mediated apoptosis-resistant cancer types. Given the correlation between RRM2 and Bcl-2 expression levels, it is most likely that the expression level of RRM2 could also predict the Bcl-2 level that is involved in cancer cell survival and resistance to chemotherapy. This finding provides a strong framework for translation of this approach to the clinic.
To determine the correlation between RRM2 and Bcl-2, we employed two different techniques, QD-IHF and conventional IHC, for staining proteins in tumor specimens from HNSCC and NSCLC patients. QD-IHF allows multiple biomarker analysis in the same tissue slide (36, 49), facilitating observation of the colocalization of multiple proteins and also correlations between their expression levels. We found that RRM2 and Bcl-2 were highly co-localized and a positive correlation was observed by both QD-IHF and conventional IHC techniques.
Knockdown of RRM2 induces DNA damage (1, 4, 8) that could be the cause of p53 and p73 induction. RRM2 knockdown initially inhibits cellular growth, which might be the cause of downregulation of Akt activation. However, we excluded the possibility that activation of p53 or p73 and inhibition of p-AKT are involved in apoptosis using appropriate genetic approaches. Pathways mediating cell death in the absence of p53 are of interest for therapeutic intervention because p53 is mutated in one half of human cancers and inactivated by indirect mechanisms in a large percentage of the remainder (34, 57). Therefore, one particularly interesting indication of our study, that RRM2 siRNA may suppress cancer cell proliferation regardless of p53 status, may be relevant for RRM2-targeted cancer therapy, especially for cancers with mutant p53.
In summary, our data demonstrate that alteration of RRM2 induces apoptosis by modulating Bcl-2 expression. RRM2 suppression contributes to the instability of Bcl-2 or leaves Bcl-2 unprotected from degradation. We present RRM2 as an attractive interventional target to downregulate Bcl-2, resulting in the induction of mitochondria-mediated intrinsic apoptosis. Moreover, we confirmed the therapeutic potential of siRNA targeting RRM2 in HNSCC and NSCLC. We have provided insights into the molecular understanding of tumor progression by RRM2 that could be fundamental in developing rational therapeutic approaches against cancer. Further clinical studies are needed to elucidate the clinicopathologic significance of RRM2 and Bcl-2 expression in HNSCC and NSCLC.
Supplementary Material
Statement of Translational Relevance.
Elevated levels of RRM2 correlate with poor prognosis for cancer patients. In this study, we unravel the mechanistic details linking RRM2 to apoptosis and provide vital clues for the development of novel cancer therapies. We show a new molecular regulatory pathway wherein RRM2 contributes to the accumulation and stabilization of Bcl-2. RRM2 was found to be significantly involved in regulating Bcl-2 and a reciprocal regulation between these proteins in apoptosis signaling was observed. Furthermore, expression of both proteins was positively correlated in tumors from both HNSCC and NSCLC patients. Our results have broader implications, suggesting that suppression of Bcl-2 by targeting RRM2 could provide an effective therapeutic strategy, especially for Bcl-2-mediated apoptosis-resistant cancer types. Given the correlation between RRM2 and Bcl-2 expression levels, the expression level of RRM2 may serve as a predictive marker of Bcl-2 level. We present RRM2 as an attractive interventional target to downregulate Bcl-2, resulting in the induction of mitochondria-mediated intrinsic apoptosis. This finding provides a strong framework for translation of this approach to the clinic.
Acknowledgments
We thank Dr. Mark Davis and Dr. Yun Yen for supplying reagents. We thank Dr. Anthea Hammond for her critical and editorial review of this article.
Grant Support
This work was supported by NIH/NCI grant U54 CA119338-04, P50 CA128613 (Head and Neck Cancer SPORE). Drs. DM Shin and Z(G) Chen are recipients of Georgia Cancer Coalition (GCC) Distinguished Scholar Awards.
Footnotes
Disclosure of Potential Conflicts of Interest
There is no potential conflict of interest to authors.
References
- 1.D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, et al. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149:1023–34. doi: 10.1016/j.cell.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engstrom Y, Eriksson S, Jildevik I, Skog S, Thelander L, Tribukait B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J Biol Chem. 1985;260:9114–6. [PubMed] [Google Scholar]
- 3.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine. Oncogene. 2004;23:1539–48. doi: 10.1038/sj.onc.1207272. [DOI] [PubMed] [Google Scholar]
- 4.Zhang YW, Jones TL, Martin SE, Caplen NJ, Pommier Y. Implication of checkpoint kinase-dependent up-regulation of ribonucleotide reductase R2 in DNA damage response. J Biol Chem. 2009;284:18085–95. doi: 10.1074/jbc.M109.003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furuta E, Okuda H, Kobayashi A, Watabe K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim Biophys Acta. 2010;1805:141–52. doi: 10.1016/j.bbcan.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Souglakos J, Boukovinas I, Taron M, Mendez P, Mavroudis D, Tripaki M, et al. Ribonucleotide reductase subunits M1 and M2 mRNA expression levels and clinical outcome of lung adenocarcinoma patients treated with docetaxel/gemcitabine. Br J Cancer. 2008;98:1710–5. doi: 10.1038/sj.bjc.6604344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morikawa T, Maeda D, Kume H, Homma Y, Fukayama M. Ribonucleotide reductase M2 subunit is a novel diagnostic marker and a potential therapeutic target in bladder cancer. Histopathology. 2010;57:885–92. doi: 10.1111/j.1365-2559.2010.03725.x. [DOI] [PubMed] [Google Scholar]
- 8.Lin ZP, Belcourt MF, Cory JG, Sartorelli AC. Stable suppression of the R2 subunit of ribonucleotide reductase by R2-targeted short interference RNA sensitizes p53(-/-) HCT-116 colon cancer cells to DNA-damaging agents and ribonucleotide reductase inhibitors. J Biol Chem. 2004;279:27030–8. doi: 10.1074/jbc.M402056200. [DOI] [PubMed] [Google Scholar]
- 9.Liu X, Zhang H, Lai L, Wang X, Loera S, Xue L, et al. Ribonucleotide reductase small subunit M2 serves as a prognostic biomarker and predicts poor survival of colorectal cancers. Clinical science. 2013;124:567–78. doi: 10.1042/CS20120240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones DT, Lechertier T, Mitter R, Herbert JM, Bicknell R, Jones JL, et al. Gene expression analysis in human breast cancer associated blood vessels. PloS one. 2012;7:e44294. doi: 10.1371/journal.pone.0044294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujita H, Ohuchida K, Mizumoto K, Itaba S, Ito T, Nakata K, et al. Gene expression levels as predictive markers of outcome in pancreatic cancer after gemcitabine-based adjuvant chemotherapy. Neoplasia. 2010;12:807–17. doi: 10.1593/neo.10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan H, Villegas C, Wright JA. Ribonucleotide reductase R2 component is a novel malignancy determinant that cooperates with activated oncogenes to determine transformation and malignant potential. Proc Natl Acad Sci U S A. 1996;93:14036–40. doi: 10.1073/pnas.93.24.14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shao J, Zhou B, Chu B, Yen Y. Ribonucleotide reductase inhibitors and future drug design. Curr Cancer Drug Targets. 2006;6:409–31. doi: 10.2174/156800906777723949. [DOI] [PubMed] [Google Scholar]
- 14.Wadler S, Makower D, Clairmont C, Lambert P, Fehn K, Sznol M. Phase I and pharmacokinetic study of the ribonucleotide reductase inhibitor, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, administered by 96-hour intravenous continuous infusion. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2004;22:1553–63. doi: 10.1200/JCO.2004.07.158. [DOI] [PubMed] [Google Scholar]
- 15.Desai AA, Schilsky RL, Young A, Janisch L, Stadler WM, Vogelzang NJ, et al. A phase I study of antisense oligonucleotide GTI-2040 given by continuous intravenous infusion in patients with advanced solid tumors. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2005;16:958–65. doi: 10.1093/annonc/mdi178. [DOI] [PubMed] [Google Scholar]
- 16.Rahman MA, Amin AR, Wang X, Zuckerman JE, Choi CH, Zhou B, et al. Systemic delivery of siRNA nanoparticles targeting RRM2 suppresses head and neck tumor growth. J Control Release. 2012;159:384–92. doi: 10.1016/j.jconrel.2012.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–32. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 18.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 19.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 20.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 21.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–62. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 22.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–83. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011;18:1414–24. doi: 10.1038/cdd.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khor LY, Moughan J, Al-Saleem T, Hammond EH, Venkatesan V, Rosenthal SA, et al. Bcl-2 and Bax expression predict prostate cancer outcome in men treated with androgen deprivation and radiotherapy on radiation therapy oncology group protocol 92-02. Clin Cancer Res. 2007;13:3585–90. doi: 10.1158/1078-0432.CCR-06-2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Contu PC, Contu SS, Moreira LF. Bcl-2 expression in rectal cancer. Arq Gastroenterol. 2006;43:284–7. doi: 10.1590/s0004-28032006000400008. [DOI] [PubMed] [Google Scholar]
- 26.Condon LT, Ashman JN, Ell SR, Stafford ND, Greenman J, Cawkwell L. Overexpression of Bcl-2 in squamous cell carcinoma of the larynx: a marker of radioresistance. Int J Cancer. 2002;100:472–5. doi: 10.1002/ijc.10503. [DOI] [PubMed] [Google Scholar]
- 27.Gallo O, Boddi V, Calzolari A, Simonetti L, Trovati M, Bianchi S. bcl-2 protein expression correlates with recurrence and survival in early stage head and neck cancer treated by radiotherapy. Clin Cancer Res. 1996;2:261–7. [PubMed] [Google Scholar]
- 28.Nix P, Cawkwell L, Patmore H, Greenman J, Stafford N. Bcl-2 expression predicts radiotherapy failure in laryngeal cancer. Br J Cancer. 2005;92:2185–9. doi: 10.1038/sj.bjc.6602647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reed JC. Promise and problems of Bcl-2 antisense therapy. J Natl Cancer Inst. 1997;89:988–90. doi: 10.1093/jnci/89.14.988. [DOI] [PubMed] [Google Scholar]
- 30.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 31.Li R, Zang Y, Li C, Patel NS, Grandis JR, Johnson DE. ABT-737 synergizes with chemotherapy to kill head and neck squamous cell carcinoma cells via a Noxa-mediated pathway. Mol Pharmacol. 2009;75:1231–9. doi: 10.1124/mol.108.052969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–9. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Amin AR, Khuri FR, Chen ZG, Shin DM. Synergistic growth inhibition of squamous cell carcinoma of the head and neck by erlotinib and epigallocatechin-3-gallate: the role of p53-dependent inhibition of nuclear factor-kappaB. Cancer Prev Res (Phila Pa) 2009;2:538–45. doi: 10.1158/1940-6207.CAPR-09-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amin AR, Thakur VS, Paul RK, Feng GS, Qu CK, Mukhtar H, et al. SHP-2 tyrosine phosphatase inhibits p73-dependent apoptosis and expression of a subset of p53 target genes induced by EGCG. Proc Natl Acad Sci U S A. 2007;104:5419–24. doi: 10.1073/pnas.0700642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahman MA, Senga T, Ito S, Hyodo T, Hasegawa H, Hamaguchi M. S-nitrosylation at cysteine 498 of c-Src tyrosine kinase regulates nitric oxide-mediated cell invasion. J Biol Chem. 2010;285:3806–14. doi: 10.1074/jbc.M109.059782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang DH, Su L, Peng XH, Zhang H, Khuri FR, Shin DM, et al. Quantum dot-based quantification revealed differences in subcellular localization of EGFR and E-cadherin between EGFR-TKI sensitive and insensitive cancer cells. Nanotechnology. 2009;20:225102. doi: 10.1088/0957-4484/20/22/225102. [DOI] [PubMed] [Google Scholar]
- 37.Heidel JD, Liu JY, Yen Y, Zhou B, Heale BS, Rossi JJ, et al. Potent siRNA inhibitors of ribonucleotide reductase subunit RRM2 reduce cell proliferation in vitro and in vivo. Clin Cancer Res. 2007;13:2207–15. doi: 10.1158/1078-0432.CCR-06-2218. [DOI] [PubMed] [Google Scholar]
- 38.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–70. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6:659–68. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 40.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 41.Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5:876–85. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- 42.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–13. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 43.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 44.Stiewe T. The p53 family in differentiation and tumorigenesis. Nat Rev Cancer. 2007;7:165–8. doi: 10.1038/nrc2072. [DOI] [PubMed] [Google Scholar]
- 45.Henderson YC, Wang E, Clayman GL. Genotypic analysis of tumor suppressor genes PTEN/MMAC1 and p53 in head and neck squamous cell carcinomas. Laryngoscope. 1998;108:1553–6. doi: 10.1097/00005537-199810000-00024. [DOI] [PubMed] [Google Scholar]
- 46.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 47.Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, et al. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–8. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- 48.Grandgirard D, Studer E, Monney L, Belser T, Fellay I, Borner C, et al. Alphaviruses induce apoptosis in Bcl-2-overexpressing cells: evidence for a caspase-mediated, proteolytic inactivation of Bcl-2. EMBO J. 1998;17:1268–78. doi: 10.1093/emboj/17.5.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu J, Muller S, Nannapaneni S, Pan L, Wang Y, Peng X, et al. Comparison of quantum dot technology with conventional immunohistochemistry in examining aldehyde dehydrogenase 1A1 as a potential biomarker for lymph node metastasis of head and neck cancer. Eur J Cancer. 2012;48:1682–91. doi: 10.1016/j.ejca.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferrandina G, Mey V, Nannizzi S, Ricciardi S, Petrillo M, Ferlini C, et al. Expression of nucleoside transporters, deoxycitidine kinase, ribonucleotide reductase regulatory subunits, and gemcitabine catabolic enzymes in primary ovarian cancer. Cancer Chemother Pharmacol. 2010;65:679–86. doi: 10.1007/s00280-009-1073-y. [DOI] [PubMed] [Google Scholar]
- 51.Grade M, Hummon AB, Camps J, Emons G, Spitzner M, Gaedcke J, et al. A genomic strategy for the functional validation of colorectal cancer genes identifies potential therapeutic targets. Int J Cancer. 2011;128:1069–79. doi: 10.1002/ijc.25453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones RJ, Baladandayuthapani V, Neelapu S, Fayad LE, Romaguera JE, Wang M, et al. HDM-2 inhibition suppresses expression of ribonucleotide reductase subunit M2, and synergistically enhances gemcitabine-induced cytotoxicity in mantle cell lymphoma. Blood. 2011;118:4140–9. doi: 10.1182/blood-2011-03-340323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kretschmer C, Sterner-Kock A, Siedentopf F, Schoenegg W, Schlag PM, Kemmner W. Identification of early molecular markers for breast cancer. Mol Cancer. 2011;10:15. doi: 10.1186/1476-4598-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amin AR, Wang D, Zhang H, Peng S, Shin HJ, Brandes JC, et al. Enhanced anti-tumor activity by the combination of the natural compounds (-)-epigallocatechin-3-gallate and luteolin: potential role of p53. J Biol Chem. 2010;285:34557–65. doi: 10.1074/jbc.M110.141135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev. 2011;21:12–20. doi: 10.1016/j.gde.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choi BH, Feng L, Yoon HS. FKBP38 protects Bcl-2 from caspase-dependent degradation. J Biol Chem. 2010;285:9770–9. doi: 10.1074/jbc.M109.032466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agarwal ML, Taylor WR, Chernov MV, Chernova OB, Stark GR. The p53 network. J Biol Chem. 1998;273:1–4. doi: 10.1074/jbc.273.1.1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.