

Despite the advance in treatment of acute myocardial infarction (MI) with timely reperfusion of ischemic myocardium, coronary artery disease remains a leading cause of morbidity and mortality worldwide. In patients survived acute MI, heart undergoes a remodeling process characterized by changing in size, shape, structure, and function. The progressive ventricular dilation, wall thinning, fibrosis, together with loss of contractile function lead to life-threatening heart failure and arrhythmia. Therefore understanding the process of pathological remodeling in post-MI hearts is of paramount importance.
It is well established that inflammatory response is elicited by MI and contributes significantly to cardiac remodeling. At the onset of MI injury, inflammatory leukocytes produced in bone marrow are mobilized and infiltrate the myocardium from circulation. The locally targeted leukocytes can be beneficial to wound healing by removing dead cells and matrix debris. However, prolonged inflammation may also contribute to additional cell death and scar formation due to fibrosis. Therefore inflammatory response has to be precisely controlled and timely resolved to avoid adverse remodeling in the post-infarct heart 1, 2. As a result, modulating inflammatory response has been considered a potential therapeutic approach to preserve and recover heart function after MI 3, 4. A great deal of effort has been made to understand the cellular and molecular mechanisms of inflammatory response following MI 2, 5-8.
Prostaglandins (PGs) are a group of 20-carbon lipid metabolites derived from arachidonic acids through a cascade of cyclo-oxygenases (COX-1 and COX-2) and terminal prostaglandin synthases. Prostaglandin E2 (PGE2) is one of the most abundant species with an important role in both promotion and resolution of inflammation 9-11. mPGES-1 (microsomal prostaglandin E2 synthase-1) encoded by Ptges gene is the major terminal prostaglandin synthase of PGE2. Genetic inactivation of Ptges gene in mice led to attenuated inflammation, reduced pain, less brain ischemia/reperfusion injury and atherosclerosis 12. Therefore, inhibiting PGE2 synthase and downstream receptors is viewed as a viable alternative strategy to COX-2 inhibition for inflammatory diseases.
In addition to inflammation, PGE2 can exert other effects depending on receptor subtypes, cell type, and the context of its activation 13. More relevant to heart, PGE2, among other PGs, can regulate cardiomyocytes hypertrophy 14, 15 and remodeling 16. In an earlier study by Degousee et al, systemic ablation of mouse Ptges gene promoted pathological remodeling in left ventricle after MI17. Reduced PGE2 in mPGES-1 deficient mice resulted in more severe ventricle dilation and worse cardiac function after MI. Therefore, PGE2 produced in infarct heart appears to have a beneficial effect in post-MI myocardial remodeling. In the same study, Degousee et al also found that the transient induction of the mPGES-1 protein observed in the post-MI heart was mainly produced from inflammatory cells recruited to infarct zone and peri-infarct region of the heart. Therefore, bone marrow derived inflammatory cells may be an important source of PGE2 that attenuates pathological remodeling in post-MI heart. However, this observation is correlative based on immuno-histochemistry and there is no direct evidence to support the relative contribution of PGE2 from bone marrow derived leukocytes vs. other cell types.
A report by the same group in this issue provided a more definitive answer by clarifying the crucial role of bone marrow derived leukocyte in the cardiac remodeling after MI 18. Taking advantage of a well established bone-marrow transplant approach, the authors established two chimera wildtype female mouse lines reconstituted with bone marrow from either Ptges+/+ (BM+/+) or Ptges−/− (BM−/−) male mice. By performing MI on these chimera mice followed by functional and molecular studies, the specific contribution of PGE2 produced from bone marrow derived leukocytes to post-MI myocardial remodeling was evaluated. As shown in their study, inactivation of mPGES-1 in bone marrow derived leukocytes led to a more severe pathological remodeling and worse function. This is within expectation since this phenotype is similar to what is observed in the mouse model of systemic inactivation of mPGES-117. It was quite unexpected, however, when they observed that local inflammation was enhanced in the infarct area in the BM-/- mice and an even higher level of PGE2 was detected in the myocardium post infarct, possibility due to the induction of mPGES-1 activity in cardiac fibroblasts18.
These observations have unveiled a mechanism of cardiac remodeling orchestrated by leukocytes remotely generated in bone marrow (Figure 1). Although implication of inflammatory response in cardiac remodeling is not new and there has been a growing interest for the crosswalk between bone marrow and heart failure 19, this report has added a new molecular link involving mPGES-1 from bone marrow derived leukocytes 18. It is interesting to note that even though the recruitment of bone-marrow derived leukocytes is largely confined within the infarct zone and peri-infarct area17, the impact of prostaglandin PGE2 produced from these cells on cardiac remodeling can be observed throughout the myocardium. However, the current report only demonstrated the necessary role of PGE2 production from BM derived leukocytes in myocardial remodeling18. It remains to be determined whether the PGE2 produced from these cells is sufficient to deliver a beneficial effect to post-MI heart. An obvious experiment would be to establish chimera mice of Pges-/- genotype but reconstituted with Pges+/+ bone marrow, and to determine whether post-MI myocardial remodeling can be ameliorated. Clearly, more work is needed to establish the role of prostaglandins, especially PGE2, in mediating the remote control of myocardial remodeling by bone marrow derived leukocytes.
Figure 1.
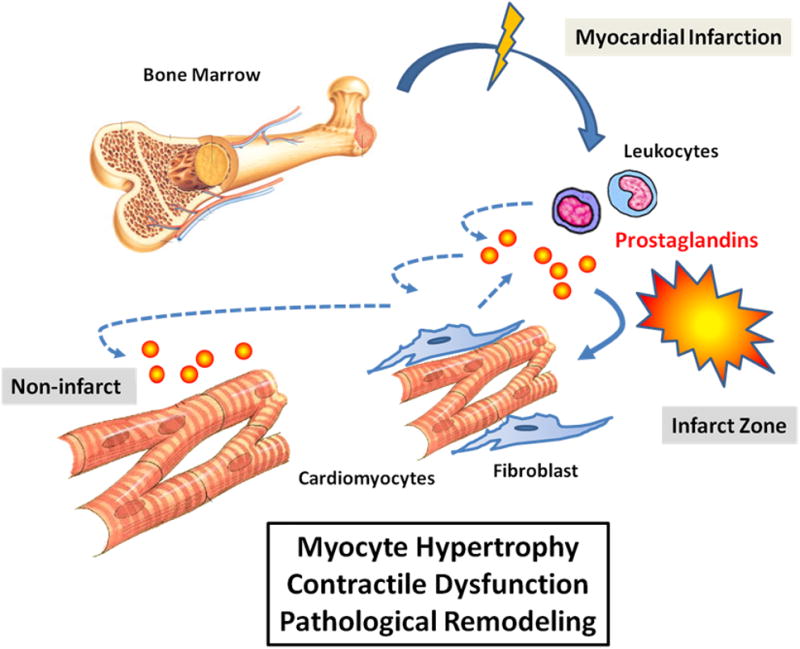
Illustration of Prostaglandin-E2 from bone marrow derived leukocytes in myocardial remodeling after myocardial infarction.
From these studies, another interesting paradox emerges: both lower and higher levels of PGE2 are associated with worse cardiac remodeling and function. PGE2 level in the remote myocardium is lower in the mPGES-1 deficient mice than that of the wildtype controls17 while higher in BM−/− mice than that of BM+/+ mice 18. Yet, under both circumstances, hearts developed more left ventricular (LV) dilation, worse LV systolic and diastolic function, and higher LV end-diastolic pressure following MI. These results highlight the confounding factors involved in the pathological remodeling in heart. One possibility is that the direct causes of such adverse myocardium remodeling are not limited to PGE2 or PGE2 downstream signaling alone in cardiomyocytes. Prostaglandins biosynthesis is not regulated in a linear fashion but rather by a convoluted network of reactions. In fact, the deficiency of mPGES-1 activity, systemically or in BM derived leukocytes, led to significant changes in the overall prostaglandin profiles in the myocardium. It is likely that the combined effects of all prostaglandin species are ultimately responsible for the myocyte hypertrophy and remodeling16. Additional animal models with targeted manipulation of PG receptors in myocytes would be needed to sort out the specific and direct contribution from different PG species.
Another interesting observation from these studies is the cross-talk between BM derived leukocytes and other different cell types in myocardium. Elevated PGE2 was observed to be produced in the resident cardiac fibroblasts in the BM-/- hearts 18. Although the functional significance of this observation is unclear, the result indicates an active interaction between BM derived cells and other cardiac resident cells beyond cardiomyocytes. As these cardiac cells such as fibroblasts also contribute significantly to myocardial remodeling, PGE2 may exert its effect though regulation on these cells. Indeed, prostaglandin mediated cross-talk may also impact on vascular remodeling, progenitor/stem cell mobilization and differentiation 20,21. The study presented in this report provides an excellent model system and outstanding opportunity to further investigate these questions.
The clinical significance of mPGES-1 function goes beyond its possible role in MI or heart failure. COX-2 inhibitors were once used mainly as anti-inflammatory and pain medicine by millions of people worldwide. However, after elevated cardiovascular risk was linked this class of non-steroidal anti-inflammatory drugs, a major effort has been shifted to target mPGES-1 as an alternative to COX-2. In light of the study reported here, however, it is clear that we also need to better understand the role of mPGES-1 and its product PGE2 in the pathological progression of heart diseases. If the critical role of mPGES-1 in the remodeling of left ventricle after MI is also implicated in human patients as suggested from these mouse studies, an mPGES-1 selective inhibitor may also have an adverse effect on myocardium remodeling and long-term mortality of the post-MI patients. It is, therefore, highly prudent to evaluate the safety profile of this class of drugs, especially for any potential risks of cardiovascular incidents.
Footnotes
Conflict of Interest Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liehn EA, Postea O, Curaj A, Marx N. Repair after myocardial infarction, between fantasy and reality: The role of chemokines. J Am Coll Cardiol. 2011;58:2357–2362. doi: 10.1016/j.jacc.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 3.Fraccarollo D, Galuppo P, Bauersachs J. Novel therapeutic approaches to post-infarction remodelling. Cardiovasc Res. 2012;94:293–303. doi: 10.1093/cvr/cvs109. [DOI] [PubMed] [Google Scholar]
- 4.Frantz S, Bauersachs J, Ertl G. Post-infarct remodelling: Contribution of wound healing and inflammation. Cardiovasc Res. 2009;81:474–481. doi: 10.1093/cvr/cvn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8:292–300. doi: 10.1038/nrcardio.2011.38. [DOI] [PubMed] [Google Scholar]
- 6.Kwon JS, Kim YS, Cho AS, Cho HH, Kim JS, Hong MH, Jeong SY, Jeong MH, Cho JG, Park JC, Kang JC, Ahn Y. The novel role of mast cells in the microenvironment of acute myocardial infarction. J Mol Cell Cardiol. 2011;50:814–825. doi: 10.1016/j.yjmcc.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 7.van den Borne SWM, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–37. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 8.Sun Y. Myocardial repair/remodelling following infarction: Roles of local factors. Cardiovasc Res. 2009;81:482–490. doi: 10.1093/cvr/cvn333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scher JU, Pillinger MH. The anti-inflammatory effects of prostaglandins. J Investig Med. 2009;57:703–708. doi: 10.2310/JIM.0b013e31819aaa76. 710.231/JIM.700b013e31819aaa31876. [DOI] [PubMed] [Google Scholar]
- 11.Brenneis C, Coste O, Altenrath K, Angioni C, Schmidt H, Schuh C-D, Zhang DD, Henke M, Weigert A, Brüne B, Rubin B, Nusing R, Scholich K, Geisslinger G. Anti-inflammatory role of microsomal prostaglandin e synthase-1 in a model of neuroinflammation. J Biol Chem. 2011;286:2331–2342. doi: 10.1074/jbc.M110.157362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin e synthase-1: A novel therapeutic target. Pharmacol Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 13.Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: Multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Frias MA, Rebsamen MC, Gerber-Wicht C, Lang U. Prostaglandin e2 activates stat3 in neonatal rat ventricular cardiomyocytes: A role in cardiac hypertrophy. Cardiovasc Res. 2007;73:57–65. doi: 10.1016/j.cardiores.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Xiao CY, Yuhki K, Hara A, Fujino T, Kuriyama S, Yamada T, Takayama K, Takahata O, Karibe H, Taniguchi T, Narumiya S, Ushikubi F. Prostaglandin e2 protects the heart from ischemia-reperfusion injury via its receptor subtype ep4. Circulation. 2004;109:2462–2468. doi: 10.1161/01.CIR.0000128046.54681.97. [DOI] [PubMed] [Google Scholar]
- 16.Harding P, Murray DB. The contribution of prostaglandins versus prostacyclin in ventricular remodeling during heart failure. Life Sci. 2011;89:671–676. doi: 10.1016/j.lfs.2011.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Degousee N, Fazel S, Angoulvant D, Stefanski E, Pawelzik S-C, Korotkova M, Arab S, Liu P, Lindsay TF, Zhuo S, Butany J, Li R-K, Audoly L, Schmidt R, Angioni C, Geisslinger G, Jakobsson P-J, Rubin BB. Microsomal prostaglandin e2 synthase-1 deletion leads to adverse left ventricular remodeling after myocardial infarction. Circulation. 2008;117:1701–1710. doi: 10.1161/CIRCULATIONAHA.107.749739. [DOI] [PubMed] [Google Scholar]
- 18.Degousee N, Simpson J, Fazel S, Scholich K, Angoulvant D, Angioni C, Schmidt H, Korotkova M, Stefanski E, Wang XH, Lindsay TF, Ofek E, Pierre S, Butany J, Jakobsson PJ, Keating A, Li RK, Nahrendorf M, Geisslinger G, Backx PH, Rubin BB. Lack of microsomal prostaglandin e2 synthase-1 in bone marrow derived myeloid cells impairs left ventricular function and increases mortality after acute myocardial infarction. Circulation. 2012;125 doi: 10.1161/CIRCULATIONAHA.112.099754. XXX-XXX. [DOI] [PubMed] [Google Scholar]
- 19.Maltais S, Perrault LP, Ly HQ. The bone marrow–cardiac axis: Role of endothelial progenitor cells in heart failure. Eur J Cardiothorac Surg. 2011;39:368–374. doi: 10.1016/j.ejcts.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 20.Dore-Duffy P, LaManna JC. Physiologic angiodynamics in the brain. Antioxid Redox Signal. 2007;9:1363–1371. doi: 10.1089/ars.2007.1713. [DOI] [PubMed] [Google Scholar]
- 21.Pelus LM, Hoggatt J. Pleiotropic effects of prostaglandin e2 in hematopoiesis; prostaglandin e2 and other eicosanoids regulate hematopoietic stem and progenitor cell function. Prostaglandins Other Lipid Mediat. 2011;96:3–9. doi: 10.1016/j.prostaglandins.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]