

Abstract
The phagocyte NADPH oxidase catalyzes the reduction of O2 to reactive oxygen species with microbicidal activity. It is composed of two membrane-spanning subunits, gp91-phox and p22-phox (encoded by CYBB and CYBA, respectively), and three cytoplasmic subunits, p40-phox, p47-phox, and p67-phox (encoded by NCF4, NCF1, and NCF2, respectively). Mutations in any of these genes can result in chronic granulomatous disease, a primary immunodeficiency characterized by recurrent infections. Using evolutionary mapping, we determined that episodes of adaptive natural selection have shaped the extracellular portion of gp91-phox during the evolution of mammals, which suggests that this region may have a function in host-pathogen interactions. On the basis of a resequencing analysis of approximately 35 kb of CYBB, CYBA, NCF2, and NCF4 in 102 ethnically diverse individuals (24 of African ancestry, 31 of European ancestry, 24 of Asian/Oceanians, and 23 US Hispanics), we show that the pattern of CYBA diversity is compatible with balancing natural selection, perhaps mediated by catalase-positive pathogens. NCF2 in Asian populations shows a pattern of diversity characterized by a differentiated haplotype structure. Our study provides insight into the role of pathogen-driven natural selection in an innate immune pathway and sheds light on the role of CYBA in endothelial, nonphagocytic NADPH oxidases, which are relevant in the pathogenesis of cardiovascular and other complex diseases.
Keywords: innate immunity, immunogenetics, chronic granulomatous disease
Introduction
The phagocyte NADPH oxidase, also known as the “respiratory burst oxidase,” is an enzymatic complex that plays a critical role in innate immunity. Phagocyte NADPH oxidase catalyzes the reduction of oxygen to 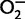 , generating reactive oxygen species (ROS) that are key components of phagocytic microbicidal activity (Heyworth et al. 2003). Phagocyte NADPH oxidase includes two membrane-spanning polypeptide subunits, gp91-phox and p22-phox (encoded by CYBB and CYBA, respectively), and a set of cytoplasmic polypeptide subunits, p40-phox, p47-phox, and p67-phox, as well as a GTPase, either Rac1 or Rac2 (encoded by NCF4, NCF1, NCF2, and RAC1 or RAC2, respectively). Upon induction, the cytoplasmic subunits bind the transmembrane components and activate the enzymatic complex, producing ROS (fig. 1; Sumimoto et al. 2005). Mutations in CYBB, CYBA, NCF1, NCF2, or NCF4 can result in chronic granulomatous disease (CGD), a primary immunodeficiency. Most CGD patients have no measurable respiratory burst, and in less than 5% of patients, low levels of ROS production are noted (Heyworth et al. 2003). Approximately 70% of CGD cases are X-linked, owing to mutations in CYBB (Heyworth et al. 2003), and there is a high degree of allelic heterogeneity in X-linked as well as in autosomal forms of CGD, except for cases due to NCF1 mutations (see the Immunodeficiency Mutations Database: http://bioinf.uta.fi/base_root/mutation_databases_list.php, last accessed July 16, 2013). NCF1 resides in a complex region of chromosome 7q11, and most CGD mutations result from gene conversion of the wild-type gene to one of several neighboring, highly paralogous pseudogenes (Chanock et al. 2000).
, generating reactive oxygen species (ROS) that are key components of phagocytic microbicidal activity (Heyworth et al. 2003). Phagocyte NADPH oxidase includes two membrane-spanning polypeptide subunits, gp91-phox and p22-phox (encoded by CYBB and CYBA, respectively), and a set of cytoplasmic polypeptide subunits, p40-phox, p47-phox, and p67-phox, as well as a GTPase, either Rac1 or Rac2 (encoded by NCF4, NCF1, NCF2, and RAC1 or RAC2, respectively). Upon induction, the cytoplasmic subunits bind the transmembrane components and activate the enzymatic complex, producing ROS (fig. 1; Sumimoto et al. 2005). Mutations in CYBB, CYBA, NCF1, NCF2, or NCF4 can result in chronic granulomatous disease (CGD), a primary immunodeficiency. Most CGD patients have no measurable respiratory burst, and in less than 5% of patients, low levels of ROS production are noted (Heyworth et al. 2003). Approximately 70% of CGD cases are X-linked, owing to mutations in CYBB (Heyworth et al. 2003), and there is a high degree of allelic heterogeneity in X-linked as well as in autosomal forms of CGD, except for cases due to NCF1 mutations (see the Immunodeficiency Mutations Database: http://bioinf.uta.fi/base_root/mutation_databases_list.php, last accessed July 16, 2013). NCF1 resides in a complex region of chromosome 7q11, and most CGD mutations result from gene conversion of the wild-type gene to one of several neighboring, highly paralogous pseudogenes (Chanock et al. 2000).
Fig. 1.

Components of the phagocyte NADPH oxidase. Representation of the inactivated (left) and activated (right) forms of the phagocyte NADPH oxidase components, reproduced from Heyworth et al. (2003). The activated form is responsible for the respiratory burst. The proteins (and genes) are gp91 (CYBB, Xp21.1), p22 (CYBA, 16q24), p67 (NCF2, 1q25), p40 (NCF4, 22q13.1), and p47 (NCF1, 7q11.23).
Several studies in animal models and in vitro have confirmed the long-standing clinical observation that the NADPH oxidase is critical for defense against catalase-positive bacteria and fungi (Buckley 2004). Association studies have suggested a role for common genetic variants in CGD genes as susceptibility alleles for tuberculosis and malaria (Bustamante et al. 2011), as well as for immune related diseases such as Crohn’s disease and lupus, as identified in genome-wide association studies (GWAS) in European populations (Rioux et al. 2007; Roberts et al. 2008; Jacob et al. 2012). Besides the phagocyte NADPH oxidase, other NADPH oxidases with different functions are expressed in a variety of nonphagocytic cells, including the endothelium, and have been implicated in cardiovascular and renal disease. Although p22-phox (encoded by CYBA) is a protein component shared by several of these NADPH oxidases (also called Nox), other more specific protein subunits are encoded by different Nox genes homologous to the genes coding for the phagocytic subunits (Sumimoto et al. 2005; San José et al. 2008). Although these nonphagocytic NADPH oxidases normally produce less 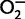 , even small imbalances in ROS levels may cause tissue damage due to oxidative stress, which is correlated with the pathogenesis of gout, chronic obstructive pulmonary disease, rheumatoid arthritis, and cardiovascular diseases (Brandes and Kreuzer 2005). Therefore, variants in NADPH oxidase genes may have pleiotropic effects across a spectrum of disorders (Santiago et al. 2012).
, even small imbalances in ROS levels may cause tissue damage due to oxidative stress, which is correlated with the pathogenesis of gout, chronic obstructive pulmonary disease, rheumatoid arthritis, and cardiovascular diseases (Brandes and Kreuzer 2005). Therefore, variants in NADPH oxidase genes may have pleiotropic effects across a spectrum of disorders (Santiago et al. 2012).
Despite the involvement of the NADPH oxidase in a range of clinically relevant phenotypes, our knowledge of the sequence diversity of NADPH genes mostly derives from CGD patients. Although targeted SNP genotyping has been performed in the context of association studies for CYBA (Bedard et al. 2009) and NCF4 (Olsson et al. 2007), none of the large-scale resequencing efforts, such as Seattle SNPs (http://pga.gs.washington.edu/, last accessed July 16, 2013), Innate Immunity PGA (http://www.pharmgat.org/IIPGA2/index_html, last accessed July 16, 2013), and the Cornell–Celera initiative (Bustamante et al. 2005), have included the NADPH oxidase genes, and the coverage of these genes for the current release of the 1000 Genomes Project remains low for most of the studied individuals (1000 Genomes Project Consortium et al. 2012; average coverage and their standard deviations on May 2013 are CYBB: 4.0 ± 2.2, CYBA: 3.5 ± 2.0, NCF2: 4.9 ± 2.7, and NCF4: 4.9 ± 2.7). Although GWAS have identified common variants that contribute to complex phenotypes, a component of missing heritability of common diseases due to rare variants that are detectable only by resequencing is emerging. In this study, we analyzed the pattern of sequence diversity of four of the NADPH genes (CYBB, CYBA, NCF2, and NCF4) between mammalian species and in human populations by resequencing these genes in 102 ethnically diverse individuals. We interpreted our results in terms of evolutionary histories, by addressing the action of natural selection and focusing on two temporal scales: mammalian evolution and recent human evolution. We excluded NCF1 from our study because its high homology with its pseudogenes prevents reliable sequencing in individual samples (Chanock et al. 2000). Several studies have shown the importance of natural selection on the evolution of immunity genes at both the interspecific (Kosiol et al. 2008) and population levels (Ferrer-Admetlla et al. 2008; Barreiro et al. 2009; Barreiro and Quintana-Murci 2010). By definition, variants under natural selection are associated with different reproductive efficiencies (fitness) of their carriers and contribute to phenotype variability; therefore, they may be biomedically relevant by influencing the susceptibility to rare or common diseases. The goals of this study are as follows: 1) to determine whether the pattern of diversity of human phagocyte NADPH genes reflects the action of different types of natural selection, 2) to elucidate the evolutionary dynamics of NADPH genes at the temporal scales of mammals and humans, and 3) to understand the biomedical implications of this evolutionary process in human populations.
Results
Molecular Evolution of NADPH Genes along Mammalian Phylogeny
We examined signatures of natural selection across the coding regions of NADPH genes by analyzing sequences from the complete genomes of 29 mammals listed in the Entrez and Ensembl databases (Lindblad-Toh et al. 2011, one sequence for each species, see supplementary material, Supplementary Material online for details) and comparing the amount of nonsynonymous and synonymous substitutions (Nielsen et al. 2005). When comparing a set of homologous sequences from different species, most of the observed differences are fixed; that is, the differences are monomorphic within a species because enough time has passed for the observed variant to appear, increase its frequency and reach a frequency of 1 (Kimura 1974). We compared the number of fixed synonymous substitutions (dS, assumed to be neutral) and fixed nonsynonymous substitutions (dN, for which we test the hypothesis of natural selection) between species using the parameter ω = dN/dS, which is informative of the action of natural selection at the inter-specific level (Yang 2007a). Under neutral evolution of nonsynonymous substitutions, these substitutions fix at the same rate as synonymous substitutions, and therefore dN ≈ dS and ω ≈ 1. If nonsynonymous substitutions tend to be deleterious, purifying selection maintains the substitutions at low frequencies and prevents fixation at the same rate as synonymous substitutions, resulting in dN < dS and ω < 1. On the other hand, if episodes of positive natural selection (that raise the frequency of beneficial variants) are frequent, nonsynonymous substitutions increase in frequency and fix more rapidly than neutral synonymous substitutions, thus, dN > dS and ω > 1. We used the maximum likelihood framework developed by Yang (2007a) to estimate ω for the NADPH oxidase genes under a variety of evolutionary models, as implemented in the PAML software (Yang 2007b). This approach allows inferences about the evolution of a coding region along an interspecific phylogeny and maps the codons that have evolved under strong/weak purifying selection, neutrality, or adaptive positive selection (see supplementary material, Supplementary Material online for details).
In general, PAML evolutionary models that allow a combination of purifying selection and neutrality are reasonably realistic. These models are nested with respect to models that also incorporate positive selection at the cost of adding new parameters. We evaluated the improvements in the goodness of fit of the data using the latter model with respect to the former models by applying a likelihood ratio test (LRT). After fitting the data to the most appropriate evolutionary model, Naive Empirical Bayes (NEB) or Bayes Empirical Bayes (BEB) approaches were used to infer the ω parameter for each codon.
For the 29 species of mammals considered, we filtered based on quality control (supplementary table S1, Supplementary Material online) and analyzed 570 codons of CYBB in 26 species, 198 codons of CYBA in 16 species, 526 codons of NCF2 in 23 species, and 339 codons of NCF4 in 20 species (see supplementary material, Supplementary Material online for further details including the species and sequences used for the analyses, the parameter estimations for the different models and the LRT results). Here, we only present the results of model M3 of Yang (2007a) with three (K = 3) classes of ω (fig. 2). This model allows for different ω classes, including the possibility of positive selection, and is a reasonable way of presenting the results for the four genes under the same model. Moreover, in all cases, the data fit better with model M3 than the nested and simpler M0 or M2 models presented by Yang (2007a).
Fig. 2.

Inferred types of natural selection for codons of the NADPH genes at the evolutionary time scale of mammals. Codons are represented along the horizontal axis. For each gene, three classes of sites (black, dark gray, and light gray) are considered, and each class evolved under different inferred ω values (presented for each gene in the figure at the top of each graphic). These classes correspond to the model M3 of Yang (2007a) with three classes of sites. Given our data, this model is more likely than alternative models of evolution that assume simpler scenarios, such as a unique ω for the entire gene (see supplementary material, Supplementary Material online for details regarding methods and results using alternative models). The three classes correspond to different types and levels of natural selection, from strong purifying selection (in the lightest gray) to positive selection (ω > 1). For each codon, the probability of belonging to each of the three classes of ω corresponds to the height of the corresponding color in the vertical bar. For example, codon 173 of CYBB (indicated by a white arrow) has a 0.000 probability of belonging to the ω = 0.0095 class (light gray, a class corresponding to strong purifying selection), a 0.169 probability of belonging to the ω = 0.3085 class (dark gray), and a 0.831 probability of belonging to the ω = 1.8987 class (black, a class that suggests positive selection). In this case, reasonable evidence of positive selection on this codon exists.
The results of this analysis for CYBB, CYBA, NCF2, and NCF4 are presented in figure 2, which shows the type of natural selection (i.e., based on the estimated ω) for each codon that most likely predominated during mammalian evolution. For this temporal scale, CYBA, NCF2, and NCF4 coding regions have evolved driven by a combination of different levels of purifying natural selection. Overall, the average and standard deviation values for these genes are ωNCF2 = 0.256 ± 0.227, ωNCF4 = 0.126 ± 0.140, and ωCYBA = 0.109 ± 0.116.
Our most striking result is for CYBB, which presents a wide spectrum of mutations that account for >70% of CGD patients. Although we would predict that purifying selection on genes involved in Mendelian diseases (Blekhman et al. 2008) would yield similar results for CYBB and other NADPH components, we observed a different pattern. In general, CYBB is a conserved gene, but 6% of its codons show evidence of positive natural selection (supplementary table S2, Supplementary Material online; fig. 2). In a genome-wide survey performed by Kosiol et al. (2008), they have reported CYBB as a gene showing a signal of positive natural selection. More importantly, by evolutionary mapping, we show here for the first time that most of these positive selection events map to the small extracellular portion of this protein (fig. 3). The proximity of these inferred episodes of positive natural selection to glycosylation sites in gp91 is noteworthy considering the importance of the glycome in immunity (Marth and Grewal 2008).
Fig. 3.

Natural selection mapping across CYBB (encoding gp91) along mammalian evolution, as identified using the PAML method by Yang (2007a). The topologies of gp91 and p22 are reproduced from Taylor et al. (2004, Copyright 2004. The American Association of Immunologists, Inc. Used with permission.). Dark gray amino acids have evolved under positive selection with >80% probability. Most of these amino acids are in the extracellular portion of the protein. The upper part of the figure shows the protein alignment for nine mammals of the gp91 region indicated by the black ellipse. In this region, a high level of amino acid variation is found between species, and several codons show ω > 1. In this alignment, gray vertical bars correspond to variable amino acid sites. The protein alignment of mammals shows the following species: Hom (Homo sapiens sapiens), Pan (Pan troglodytes), Mac (Macaca mulatta), Mus (Mus musculus), Rat (Rattus norvegicus), Cri (Cricetulus griseus), Het (Heterocephalus glaber), Cav (Cavia porcellus), and Ory (Oryctolagus cuniculus). EC, extracelular environment; TM, transmembrane layer; and IC, intracellular environment.
Population Genetics of NADPH Genes
We sequenced CYBB, CYBA, NCF2, and NCF4 in a publicly available panel that includes 24 individuals of African ancestry, 31 Europeans, 24 Asian/Oceanians, and 23 admixed Latin Americans (i.e., Hispanics). This panel is a suboptimal representation of the worldwide population, a limitation that is common to most human genomic diversity projects focused on SNP genotyping or resequencing efforts. However, based on how human genetic diversity is apportioned within (>85%) and between (<15%) populations (Lewontin 1972; 1000 Genomes Project Consortium 2010), even studies using suboptimal sampling are informative about the genetic structure of human populations and serve to critically identify the role of evolutionary factors in human genetic diversity (Kimura 1974; Nielsen et al. 2005; 1000 Genomes Project Consortium 2010). All the raw results are available as supplementary material, Supplementary Material online, and at the SNP500Cancer project homepage (http://variantgps.nci.nih.gov/cgfseq/pages/snp500.do, last accessed July 16, 2013) or can be downloaded from the DIVERGENOME platform (Magalhães et al. 2012, http://www.pggenetica.icb.ufmg.br/divergenome/, last accessed July 16, 2013).
To ascertain which combination of evolutionary factors has shaped the diversity of NADPH genes, we assessed the pattern of nonsynonymous and synonymous polymorphisms, as well as intra- and interpopulation diversity for NADPH genes, and tested the null hypothesis of neutrality: that patterns of diversity may be explained by considering only the demographic history of human populations and the mutation and recombination rates of each locus.
Nonsynonymous polymorphisms are underrepresented in the human genome and usually occur at low frequencies when present, reflecting the action of purifying natural selection (1000 Genomes Project Consortium 2010). By resequencing, Tarazona-Santos et al. (2008) did not observe common nonsynonymous polymorphisms for CYBB. This result is consistent with purifying natural selection acting on X-chromosome genes due to the exposure of deleterious recessive mutations to natural selection in hemizygous males. Thus, substitutions in the coding region of CYBB should be rare in human populations and are seldom captured by studies with small sample sizes. Interestingly, the lack of CYBB nonsynonymous polymorphisms in our sample of human populations contrasts with the recurrent episodes of positive selection of the extracellular portion of gp91 during mammalian evolution, as inferred in this study. For the autosomal NADPH oxidase components (table 1 and haplotype tables online, Supplementary Material online), we observe in this study two rare and conservative nonsynonymous substitutions (i.e., involving amino acids with similar chemical properties, T85N and A304E) in NCF4. But for NCF2 and CYBA, we observed patterns of nonsynonymous substitutions that seldom occur in human genes. NCF2 has nine nonsynonymous substitutions; three of them are common (with a frequency higher than 5% in at least one of the studied population samples), and six are rare. On the other hand, two nonsynonymous substitutions in CYBA are common and ubiquitous in human populations, namely Y72H (rs4673) and V174A (rs1049254, in a position where variation among mammalian species is also observed). Moreover, the following two of the five common amino acid changes observed in the autosomal NADPH genes are predicted to be possibly damaging (i.e., radical) by the Polyphen resource (Ramensky et al. 2002); the two changes are R395W in Hispanic NCF2 (rs13306575) and Y72H (rs4673) in CYBA. In general, Polyphen accurately predicts the effect of nonsynonymous substitutions based on biochemical and evolutionary data (Williamson et al. 2005). Notably, the Immunodeficiency Mutations Database (http://bioinf.uta.fi/NCF2base/?content=pub/IDbases, last accessed July 16, 2013) reports one 395W/395W autosomal recessive CGD patient, but we and the HapMap project (www.hapmap.org, last accessed July 16, 2013) observed the W allele at frequencies between 5% and 10% in Asians and admixed Latin Americans, including one supposedly healthy 395W/395W Japanese HapMap individual. On the basis of these results, we verified whether Native Americans, who descend from an ancestral Pleistocene Asian populations that peopled the Americas by the Behring Straits more than 14,000 years ago, may have relatively higher frequencies of this variant. We genotyped the variant 395W using a Taqman assay in 558 Native Americans (see supplementary table S3, Supplementary Material online, for detailed results) and observed an allele frequency of 1.2%, being this variant always present in heterozygous individuals.
Table 1.
Allele Frequencies of Nonsynonymous Polymorphisms in NADPH Oxidase Genes.
| Genes | rs | Minor Allele (Amino Acid) | Polyphen Prediction | African | European | Asian | Hispanic |
|---|---|---|---|---|---|---|---|
| CYBA | |||||||
| Y72H | rs4673 | T (Y) | Possibly damaging | 0.46 | 0.32 | 0.17 | 0.22 |
| V174A | rs1049254 | T (V) | Benign | 0.17 | 0.48 | 0.48 | 0.18 |
| NCF2 | |||||||
| K181R | rs2274064 | G (R) | Benign | 0.35 | 0.37 | 0.41 | 0.48 |
| T279M | rs13306581 | T (T) | Probably damaging | 0.00 | 0.00 | 0.05 | 0.00 |
| V297A | rs35937854 | C (A) | Benign | 0.04 | 0.00 | 0.00 | 0.00 |
| T361S | Chr1:181799289 NCBI36/hg18 | T (S) | — | 0.00 | 0.00 | 0.02 | 0.00 |
| H389Q | rs17849502 | A (Q) | Benign | 0.00 | 0.05 | 0.00 | 0.07 |
| R395W | rs13306575 | T (W) | Possibly damaging | 0.00 | 0.00 | 0.00 | 0.07 |
| N419I | rs35012521 | T (I) | Probably damaging | 0.00 | 0.02 | 0.04 | 0.00 |
| P454S | rs55761650 | T (S) | — | 0.00 | 0.00 | 0.00 | 0.02 |
| L487S | Chr1:181795862 NCBI36/hg18 | C (S) | — | 0.02 | 0.00 | 0.00 | 0.00 |
| NCF4 | |||||||
| T85N | rs112306225 | A (N) | — | 0.00 | 0.00 | 0.02 | 0.00 |
| A304E | rs5995361 | A (E) | Benign | 0.04 | 0.00 | 0.00 | 0.00 |
For the four studied genes, the diversity and levels of recombination are higher in Africans than in non-Africans (table 2 and haplotype tables available as supplementary files, Supplementary Material online). This result is a consequence of the African origin of modern humans and the “out of Africa” migration that occurred 40,000–80,000 years ago after a bottleneck, leading to the peopling of other continents (Campbell and Tishkoff 2008; Laval et al. 2010). Therefore, the first divergence between continental human populations was between Africans and ancestral non-Africans. Consistently with this scenario, we observed the highest between-population differentiation for CYBB, CYBA, and NCF4 between these two groups. Interestingly, NCF2 does not match this pattern (table 3).
Table 2.
Intrapopulation Diversity Indexes in the Studied Populations for the NADPH Oxidase Genes, Obtained from Resequencing Data.a
| African | European | Asian | Hispanic | |
|---|---|---|---|---|
| Number of chromosomes | ||||
| CYBBb | 42 | 52 | 34 | 36 |
| CYBA | 48 | 62 | 48 | 46 |
| NCF2 | 48 | 62 | 48 | 46 |
| NCF4 | 48 | 62 | 48 | 46 |
| Segregating sites/singletons | ||||
| CYBB | 21/8 | 7/0 | 10/3 | 13/5 |
| CYBA | 61/22 | 33/3 | 33/5 | 34/7 |
| NCF2 | 46/13 | 33/11 | 28/16 | 37/14 |
| NCF4 | 45/12 | 26/7 | 19/1 | 30/8 |
| Haplotype structure Number of inferred haplotypesc | ||||
| CYBB | 14 | 5 | 7 | 12 |
| CYBA | 39 | 39 | 32 | 26 |
| NCF2 | 38 | 32 | 18 | 31 |
| NCF4 | 36 | 30 | 17 | 22 |
| Haplotype diversity ± SD | ||||
| CYBB | 0.88 ± 0.03 | 0.34 ± 0.08 | 0.53 ± 0.10 | 0.70 ± 0.08 |
| CYBA | 0.98 ± 0.01 | 0.98 ± 0.01 | 0.97 ± 0.00 | 0.96 ± 0.02 |
| NCF2 | 0.99 ± 0.01 | 0.96 ± 0.01 | 0.87 ± 0.04 | 0.97 ± 0.01 |
| NCF4 | 0.98 ± 0.01 | 0.93 ± 0.02 | 0.93 ± 0.02 | 0.93 ± 0.02 |
| Recombination parameter (ρ × 103, per site)c | ||||
| CYBB | 0.08 | <0.01 | <0.01 | <0.02 |
| CYBA | 2.91 | 1.48 | 0.96 | 0.88 |
| NCF2 | 0.52 | 0.38 | 0.10 | 0.58 |
| NCF4 | 1.37 | 1.03 | 0.39 | 0.22 |
| θ estimatorsd π × 103, per site | ||||
| CYBB | 0.36 | 0.12 | 0.15 | 0.28 |
| CYBA | 1.90 | 1.63 | 1.51 | 1.57 |
| NCF2 | 0.81 | 0.47 | 0.43 | 0.57 |
| NCF4 | 1.01 | 0.76 | 0.64 | 0.84 |
| θW × 103, per site | ||||
| CYBB | 0.42 | 0.13 | 0.21 | 0.27 |
| CYBA | 2.31 | 1.18 | 1.25 | 1.3 |
| NCF2 | 1.02 | 0.69 | 0.62 | 0.83 |
| NCF4 | 1.01 | 0.70 | 0.54 | 0.87 |
aMost analyses were performed using software DnaSP (Rozas 2009).
bData for CYBB (Xp21.1) are from Tarazona-Santos et al. (2008).
cHaplotypes and ρ inferred using the method by Stephens and Scheet (2005) and the software PHASE.
dπ: Tajima (1983), θW: Watterson (1975).
Table 3.
Pairwise FST Genetic Distances between Populations.
|
CYBBa |
CYBA |
|||||||
|---|---|---|---|---|---|---|---|---|
| Africa | Europe | Asia | Hispanic | Africa | Europe | Asia | Hispanic | |
| Africa | — | 0.316 | 0.264 | 0.092 | — | 0.074 | 0.083 | 0.065 |
| Europe | 0.257 | — | 0.000 | 0.107 | — | 0.002 | 0.056 | |
| Asia | 0.211 | 0.000 | — | 0.073 | — | 0.054 | ||
| Hispanic | 0.070 | 0.082 | 0.056 | — | — | |||
| NCF2 |
NCF4 |
|||||||
| Africa | — | 0.048 | 0.058 | 0.037 | — | 0.128 | 0.136 | 0.158 |
| Europe | — | 0.069 | 0.000 | — | 0.026 | 0.026 | ||
| Asia | — | 0.059 | — | 0.005 | ||||
| Hispanic | — | — | ||||||
aFor CYBB FST estimators are above the diagonal. Below the diagonal are the FST values corrected as if the effective population sizes of X chromosome genes were equal to autosomal ones.
From the four studied genes, NCF4, which encodes the regulatory protein p40-phox, shows a pattern of diversity that is typical for a gene that has evolved under neutrality. In addition to the features described in the previous paragraph, the allelic spectra of NCF4 in the studied populations are consistent with a neutral model of evolution (tables 2–4).
Table 4.
Results of Neutrality Tests for the NADPH Oxidase Genes and Their Significance.a
| African | European | Asian | Hispanic | |
|---|---|---|---|---|
| Tajima’s D | ||||
| CYBB | −0.473 | −0.274 | −0.813 | 0.084 |
| CYBA | −0.580 | 1.242 | 0.684 | 0.707 |
| NCF2 | −0.412 | −0.939 | −0.883 | −0.987 |
| NCF4 | −0.750 | 0.243 | 0.556 | −0.102 |
| Fu and Li’s D | ||||
| CYBB | −1.050 | 1.110 | 0.395 | −0.977 |
| CYBA | −1.485 | 1.734* | 1.188 | 0.138 |
| NCF2 | −0.407 | −1.105 | −1.904 | −1.398 |
| NCF4 | −0.308 | −0.443 | −1.893 | −0.266 |
| Fu and Li’s F | ||||
| CYBB | −0.980 | 0.811 | 0.114 | −0.814 |
| CYBA | −1.382 | 1.893* | 1.205 | 0.405 |
| NCF2 | −0.512 | −1.252 | −1.893 | 1.567 |
| NCF4 | −0.557 | −0.231 | 1.225 | −0.248 |
Note.—Underlined values represent significant results under the demographic model inferred by Laval et al. (2010) for human populations. See details in supplementary table S4, Supplementary Material online.
aThe McDonald–Kreitman test is nonsignificant in any of the cases.
*Significant under the Wright–Fisher model of constant population size.
Although CYBB presented the most interesting evolutionary history at the interspecific level, with repeated episodes of positive natural selection, recent human evolutionary history has resulted in interesting patterns of variation for CYBA and NCF2. CYBA encodes p22-phox, which is a transmembrane protein shared by different NADPH oxidases. In addition to harboring two common, nonsynonymous polymorphisms (V174A is also variable among different species of mammals), CYBA is the most variable and most affected by recombination among the NADPH oxidase genes (tables 1 and 2). In particular, CYBA diversity is very high in Europe: compared with 329 genes resequenced in a European sample (http://pga.gs.washington.edu/summary_stats.html, last accessed July 16, 2013), πCYBA ranks 11th (i.e., the 97th percentile). Moreover, there are contrasting proportions of the total number of polymorphisms/number of singletons between Africans (a low proportion) and Europeans (a high proportion), the latter showing an excess of common polymorphisms with respect to the neutral expectation (see the DFL test in table 4 and supplementary table S4, Supplementary Material online). This excess of common variants in Europeans is also significant when we conservatively tested it against a scenario of human evolution that incorporates the “Out of Africa” bottleneck (Laval et al. 2010) and the observed level of recombination in Europeans (ρCYBA = 8.07 for the sequenced region). Because demographic forces and recombination levels alone do not explain the high CYBA diversity and its excess of common polymorphisms, we suggest that balancing natural selection (that acts by maintaining different alleles at high frequency in a population) has contributed to shape the diversity of CYBA, at least in the European population. Indeed, figure 4a shows that the haplotype network of CYBA for the European population is consistent with the action of balancing natural selection (Bamshad and Wooding 2003), showing two well-differentiated common clades that explain the observed high diversity and the excess of common CYBA variants. A comparative genomic analysis confirms this inference; the ratio of polymorphisms to differences fixed between human and chimpanzee is not homogeneous along the gene in the different human populations (Mc Donald 1998; supplementary table S5, Supplementary Material online) as would be expected under neutral evolution. Our inference of balancing natural selection is consistent with the fact that 25–30% of the variation in levels of ROS production can be attributed to genetic factors (Lacy et al. 2000) and that ROS levels are associated with CYBA variants (Bedard et al. 2009).
Fig. 4.

Phylogenetic networks of (a) CYBA in Europeans and (b) NCF2 in Europeans (black) and Asians (yellow). The lengths of the branches are proportional to the number of mutations. Only nonsynonymous mutations are shown. The haplotype names correspond to the table of inferred haplotypes in the supplementary material, Supplementary Material online. We only show the names of haplotypes with a frequency >5%. Ancestral haplotypes (inferred as being the human haplotype or median vector most similar to the chimpanzee sequence) are indicated by an arrow. Median vectors are in red.
p67, encoded by NCF2, is a necessary cytosolic NADPH component for phagocyte ROS production. Asians show a highly differentiated NCF2 haplotype structure (see frequencies of haplotypes NCF2-D11 and NCF2-E10 in the haplotype tables online and in the network shown in fig. 4b), and the highest FST values are observed in pairwise comparisons between Asians and non-Asian populations (in particular with Europeans, table 3), and not between Africans and non-African populations, as is usually observed in the human genome. We confirmed these results by analyzing data for NCF2 from the HapMap Project (supplementary material, Supplementary Material online). Moreover, a trend toward an excess of rare polymorphisms exists in Asians that is not observed elsewhere (tables 1, 2, and 4; DFL = −1.904, FFL = −1.893). Although we cannot exclude that this pattern of diversity is compatible with the null hypotheses of neutrality and with the tested demographic history of human populations inferred by Laval et al. (2010, tables 2–4), we can speculate and envisage four additional evolutionary scenarios that may have contributed to shape the pattern of NCF2 diversity in Asians: 1) the observed trend is suggestive of a selective sweep on NCF2 standing variation: a neutral or weakly deleterious existing variant becomes beneficial and rapidly increases in frequency (together with its associated haplotypes, i.e., incomplete sweep), reducing the nucleotide diversity in the surrounding region and rendering other standing substitutions rare. During this process, new rare substitutions appear in the expanding positively selected haplotype. 2) The pattern of diversity of NCF2 in Asia may result from an incomplete selective sweep acting on ARPC5, which is located approximately 35 kb downstream of NCF2. In a genome-wide scan for recent positive selection, Voight et al. (2006) identified a strong signature of an incomplete sweep for ARPC5 in Asia (P = 0.009), characterized by a higher than expected long-range linkage disequilibrium summarized by very high iHS statistics (see Haplotter results for the HapMap II data at http://haplotter.uchicago.edu/, last accessed July 16, 2013). SNPs in NCF2 also presents high iHS statistics (P = 0.02), although values are lower than for ARPC5. 3) The differentiated pattern of diversity of NCF2 in Asia may also have been generated without the action of natural selection during the first colonization of Asia by modern humans. In a process of geographic population expansion, specifically in the front wave of the expansion, some rare alleles/haplotypes (i.e., surfing alleles) may become common by chance, mimicking the pattern of diversity generated by a selective sweep (Excoffier and Ray 2008). 4) The excess of rare variants may be an artifact of pooling individuals from different populations (Ptak and Przeworski 2002). Consistent with evolutionary scenarios 1–4 that produce similar patterns of diversity, the haplotype network of NCF2 for Eurasians (fig. 4b) shows the following: 1) a large differentiation between Asians and Europeans that is compatible with the high observed FST values and 2) a star-like shape associated with the haplotype NCF2-E10 that is common in Asia and rare elsewhere, which is compatible with the excess of rare alleles in the Asian populations.
Discussion
By analyzing 29 mammalian genomes and four human populations, we show in this study that natural selection has acted in different ways over time to shape the pattern of diversity of the phagocyte NADPH oxidase genes. At the temporal scale of the evolution of mammals, we have inferred recurrent episodes of positive selection acting on the extracellular portion of gp-91 that have been important to shape the pattern of interspecific diversity of this gene. Our interspecific analyses did not show a similar pattern of natural selection in any of the other phagocyte NADPH oxidase genes. Even if current knowledge on the biology of NADPH does not allow us to interpret our results in terms of function, we propose that the extracellular region of gp-91 is functionally relevant. Our results also imply that this region is highly differentiated among mammals at the protein level, and this variability should be considered when mammals models are used to study the structure and function of phagocyte NADPH components.
In the time scale of human evolution, our analyses of the NADPH oxidase genes suggest that CYBA has been a target of balancing natural selection. Because we do not have evidence of population-specific variants that faced selective pressure, the inferred natural selection may have acted on a standing variation in ancestral populations. This implies that the selective pressure began after the appearance of the variant and, possibly, acted in a specific geographic region (Barret and Schluter 2008). The signatures of natural selection acting on a new mutation and on standing variation differ. In the case of selective sweeps, episodes of natural selection on standing variation are associated to a larger variance in the allelic spectrum with respect to natural selection on a new mutation. Also, selection on standing variation may produce an excess of alleles at intermediate frequencies that is not associated with high nucleotide diversity (Przeworski et al. 2005; Peter et al. 2012). This pattern contrasts with the effect of balancing natural selection, which produces an excess of common alleles associated with high genetic diversity. Thus, the observed pattern of CYBA diversity in Europeans is not consistent with a selective sweep on a standing variation, but it is consistent with a scenario of balancing selection acting on standing variation.
If we consider for CYBA that heterozygote advantage may be the mechanisms of balancing selection, we can speculate that the biological basis for this mechanism may be the following: considering that p22-phox is not exclusive of the phagocyte NADPH oxidase, but it is also part of Nox complexes expressed in other tissues, the dependence of ROS production on CYBA variants has to be finely regulated. If CYBA variants induce high levels of ROS, these variants may favor a phagocyte-dependent efficient response to pathogens but may damage other endothelial tissues. Alternatively, tissue oxidative damage does not occur if ROS production is low, but this response may be associated with a weaker phagocyte respiratory burst against pathogens. In this context, heterozygote individuals with a CYBA-dependent intermediate level of ROS production may have been favored by natural selection.
Our results contribute to the discussion regarding the relevance of balancing selection in shaping the diversity of innate immunity genes (Ferrer-Admetlla et al. 2008; Barreiro et al. 2009). Ferrer-Admetlla et al. (2008) have associated the recurrent signatures of balancing selection on inflammatory genes with the need for fine regulation. CYBA is the only phagocytic NADPH oxidase gene that also encodes nonphagocyte Nox components; thus, CYBA has a role in ROS cell signaling, a potentially dangerous process due to its capability to produce oxidative damage to tissues. Genes with these characteristics likely need even tighter regulation. The interplay between pathogen-driven selective pressure on innate immunity genes and their concomitant nonimmunological functions is complex. In addition to CYBA, other interesting examples of this interplay can be found among the 10 human toll-like receptors (TLRs) that show a variety of signatures of natural selection, TLR8, which shows the strongest signature of purifying selection, is also involved in neuronal development (Barreiro et al. 2009), and it is difficult to discriminate the role of each function in determining the observed signature of natural selection.
With few exceptions, the pathogens responsible for natural selection on immune genes are difficult to specify. In the case of NADPH oxidase, we can infer, based on the spectrum of infections in CGD patients, that catalase-positive bacteria and fungi, such as Staphylococci, Salmonella, Candida, Aspergillus, and M. tuberculosis, may be the selective pathogens. Interactions between the host and pathogens also include mechanisms of the latter to impair the respiratory burst of the former. For example, Leishmania donovani blocks the assembly of NADPH oxidase at the phagosome membrane (Lodge et al. 2006). These mechanisms may constitute selective pressures imposed by pathogens.
The associations reported in GWAS between rs4821544 in NCF4 and Crohn’s disease (an idiopathic inflammatory bowel disease that predominantly involves the ileum and colon, Rioux et al. 2007) and between rs10911363 in NCF2 and systemic lupus erythematosus (Cunninghame Graham et al. 2011) confirm the involvement of NADPH genes in the pathogenesis of inflammatory-related common diseases. Our claim that natural selection acted on CYBA (and maybe in NCF2) is relevant for biomedical studies because combining evidence of natural selection with association analyses in immune genes increases the statistical power to detect disease-associated variants (Ayodo et al. 2007). As a new generation of association studies focusing on rare variation is emerging, the combination of genes deemed interesting from GWAS and populations with an excess of rare variants in these genes, such as NCF2 in Asians, are particularly interesting as a source of rare variants with clinical relevance. Finally, by determining through molecular evolution mapping that the extracellular portion of gp91 (encoded by CYBB in the X-chromosome) has been subject to recurrent episodes of positive selection at the scale of mammals evolution, we posit the hypothesis that this portion of the NADPH oxidase is relevant for currently unknown biological processes that, once revealed by structural and functional investigations, will contribute to understanding the role of NADPH oxidase in infectious, autoimmune, and cardiovascular diseases.
Materials and Methods
Molecular Evolution Analysis of NADPH Genes
We used the maximum likelihood framework developed by Yang (2007a) to estimate ω for the NADPH oxidase genes under a variety of evolutionary models, as implemented in the PAML software (Yang 2007b). This approach allows inferences about the evolution of a coding region along an inter-specific phylogeny, mapping the codons that have evolved under strong/weak purifying selection, neutrality, or adaptive positive selection. Further details about these analyses are available as supplementary material, Supplementary Material online.
Human Population Genetics of NADPH Genes
For human population genetics analyses, we conducted bidirectional Sanger sequencing of CYBB, CYBA, NCF2, and NCF4 for a total of 35,242 bp for each of 102 healthy individuals as part of the SNP500 Cancer project (Packer et al. 2006; see supplementary fig. S1, Supplementary Material online, for details). Human population resequencing data for CYBB were published in Tarazona-Santos et al. (2008). The resequencing experiments were performed as in Packer et al. (2006). These 102 unrelated individuals include the following: 24 of African ancestry (15 African Americans from the United States and 9 Pygmies), 23 admixed Latin Americans (from Mexico, Puerto Rico, and South America), 31 Europeans (from the CEPH/UTAH pedigree and the NIEHS Environmental Genome Project), and 24 Asians–Oceanians (from Melanesia, Pakistan, China, Cambodia, Japan, and Taiwan).
After controlling for multiple tests, we confirmed that all SNPs fit the Hardy–Weinberg equilibrium by the Guo and Thompson (1992) test, which was implemented in the software Arlequin 3.0 (Excoffier et al. 2007). Insertion-deletions (INDELs) were excluded from population genetics analyses. To assess intrapopulation diversity, we used two statistics: π, the per-site mean number of pairwise differences between sequences (Tajima 1983), and θw, based on the number of segregating sites (S) (Watterson 1975). We measured pairwise between-populations diversity by using the FST statistics calculated using the software DnaSP (Rozas 2009).
Haplotypes and the recombination parameter ρ were inferred using the PHASE software (Stephens and Scheet 2005), and diversity indexes calculations (tables 2 and 3) as well as neutrality statistics (table 4) were estimated using DnaSP software. We applied two kinds of neutrality tests: 1) tests based on the allelic spectrum, which is the distribution of polymorphisms across different classes of frequencies, namely, Tajima’s DT (Tajima 1989) and Fu–Li’s DFL and FFL (Fu and Li 1993) and 2) tests based on comparisons between the number of polymorphisms in human populations and fixed differences with the chimpanzee (i.e., outgroup), namely, the McDonald and Kreitman (1991) test and the adapted Kolmogorov–Smirnoff test by McDonald (1998). For the first set of tests, we used as null hypotheses both the classic Wright–Fisher model of neutrality with a constant population size, as well as the more realistic evolutionary scenario for human populations inferred by Laval et al. (2010). In the case of the scenario of Laval et al. (2010), we ignored intercontinental gene flow within the Old World because these rare gene flow events likely does not affect the level of significance of the neutrality tests given its very low inferred values (1.3 × 10−5). Null distributions used to test the significance of the neutrality tests under these evolutionary scenarios were generated using coalescent simulations and a significance level of 0.05 (Hudson 2002). The Kolmogorov–Smirnoff test of neutrality adapted by McDonald (1998) was performed using Slider software available at http://udel.edu/∼mcdonald/aboutdnaslider.html (last accessed July 16, 2013). We performed coalescent simulations using ms software (Hudson 2002). Further methodological details are available as supplementary material, Supplementary Material online. We constructed the CYBA and NCF2 networks using all SNP variants and applying the Median joining algorithm and the maximum parsimony option calculations as implemented in the software Network 4.6 (Bandelt et al. 1999).
Supplementary Material
Supplementary tables S1–S5 and figure S1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
S.J.C. conceived the study; E.T.-S., M.M., F.L., R.C., A.C., C.F., L.P., and B.S. generated the data/analyzed the sequences; L.B., A.C., W.C.S.M., and M.Y. provided tools for data generation and analyses; E.T.-S., M.M., F.L., W.C.S.M., and R.R. performed population genetics analyses; E.T.-S. and M.M. prepared tables and figures; E.T.-S. and S.J.C. wrote the manuscript. All the authors contributed with discussions about the results. The authors are grateful to Silvia Fuselli for discussions of our results, to Fernando Levi Soares for technical assistance with the figures and the Sequencing Group of the Core Genotyping Facility (National Cancer Institute) for their technical assistance. This work was supported by the Intramural Research Program of the National Cancer Institute (NCI) and National Institutes of Health (NIH). C.F. was supported by the University of Bologna. E.T-S., M.M., W.C.S.M., and F.L. were supported by the Fogarty International Center and National Cancer Institute (1R01TW007894), Conselho Nacional de Desenvolvimento Científico e Tecnológico (Brazil, Grant 307556/2012-3), Fundação de Amparo a Pesquisa de Minas Gerais (Brazil, CBB PPM 00265/12) and the Brazilian Ministry of Education (CAPES Agency, grant PNPD 02567/09-1).
References
- Ayodo G, Price AL, Keinan A, Ajwang A, Otieno MF, Orago AS, Patterson N, Reich D. Combining evidence of natural selection with association analysis increases power to detect malaria-resistance variants. Am J Hum Genet. 2007;81(2):234–242. doi: 10.1086/519221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad M, Wooding SP. Signatures of natural selection in the human genome. Nat Rev Genet. 2003;4(2):99–111. doi: 10.1038/nrg999. [DOI] [PubMed] [Google Scholar]
- Bandelt H-J, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Barreiro LB, Ben-Ali M, Quach H, et al. (17 co-authors) Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet. 2009;5(7):e1000562. doi: 10.1371/journal.pgen.1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro LB, Quintana-Murci L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet. 2010;11(1):17–30. doi: 10.1038/nrg2698. [DOI] [PubMed] [Google Scholar]
- Barrett RD, Schluter D. Adaptation from standing genetic variation. Trends Ecol Evol. 2008;23(1):38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Bedard K, Attar H, Bonnefont J, Jaquet V, Borel C, Plastre O, Stasia MJ, Antonarakis SE, Krause KH. Three common polymorphisms in the CYBA gene form a haplotype associated with decreased ROS generation. Hum Mutat. 2009;30(7):1123–1133. doi: 10.1002/humu.21029. [DOI] [PubMed] [Google Scholar]
- Blekhman R, Man O, Herrmann L, Boyko AR, Indap A, Kosiol C, Bustamante CD, Teshima KM, Przeworski M. Natural selection on genes that underlie human disease susceptibility. Curr Biol. 2008;18(12):883–889. doi: 10.1016/j.cub.2008.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovasc Res. 2005;65(1):16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Buckley RH. Pulmonary complications of primary immunodeficiencies. Paediatr Respir Rev. 2004;5(Suppl A):S225–S233. doi: 10.1016/s1526-0542(04)90043-7. [DOI] [PubMed] [Google Scholar]
- Bustamante CD, Fledel-Alon A, Williamson S, et al. (13 co-authors) Natural selection on protein-coding genes in the human genome. Nature. 2005;437(7062):1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- Bustamante J, Arias AA, Vogt G, et al. (29 co-authors) Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. 2011;12(3):213–221. doi: 10.1038/ni.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet. 2008;9:403–433. doi: 10.1146/annurev.genom.9.081307.164258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanock SJ, Roesler J, Zhan S, Hopkins P, Lee P, Barrett DT, Christensen BL, Curnutte JT, Görlach A. Genomic structure of the human p47-phox (NCF1) gene. Blood Cells Mol Dis. 2000;26(1):37–46. doi: 10.1006/bcmd.2000.0274. [DOI] [PubMed] [Google Scholar]
- Cunninghame Graham DS, Morris DL, Bhangale TR, Criswell LA, Syvänen AC, Rönnblom L, Behrens TW, Graham RR, Vyse TJ. Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet. 2011;7(10):e1002341. doi: 10.1371/journal.pgen.1002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online. 2007;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Ray N. Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol Evol. 2008;23(7):347–351. doi: 10.1016/j.tree.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Ferrer-Admetlla A, Bosch E, Sikora M, Marquès-Bonet T, Ramírez-Soriano A, Muntasell A, Navarro A, Lazarus R, Calafell F, Bertranpetit J, Casals F. Balancing selection is the main force shaping the evolution of innate immunity genes. J Immunol. 2008;181(2):1315–1322. doi: 10.4049/jimmunol.181.2.1315. [DOI] [PubMed] [Google Scholar]
- Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133(3):693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. 2010.A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491(7422):56–65. [DOI] [PMC free article] [PubMed]
- Guo SW, Thompson EA. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48(2):361–372. [PubMed] [Google Scholar]
- Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol. 2003;15(5):578–584. doi: 10.1016/s0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18(2):337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Jacob CO, Eisenstein M, Dinauer MC, et al. (21 co-authors) Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci U S A. 2012;109(2):E59–E67. doi: 10.1073/pnas.1113251108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge: Cambridge University Press; 1974. [Google Scholar]
- Kosiol C, Vinar T, da Fonseca RR, Hubisz MJ, Bustamante CD, Nielsen R, Siepel A. Patterns of positive selection in six mammalian genomes. PLoS Genet. 2008;4(8):e1000144. doi: 10.1371/journal.pgen.1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy F, Kailasam MT, O’Connor DT, Schmid-Schönbein GW, Parmer RJ. Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity. Hypertension. 2000;36(5):878–884. doi: 10.1161/01.hyp.36.5.878. [DOI] [PubMed] [Google Scholar]
- Laval G, Patin E, Barreiro LB, Quintana-Murci L. Formulating a historical and demographic model of recent human evolution based on resequencing data from noncoding regions. PLoS One. 2010;5(4):e10284. doi: 10.1371/journal.pone.0010284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin R. The apportionment of human diversity. Evol Biol. 1972;6:391–398. [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, et al. (88 co-authors) A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478(7370):476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge R, Diallo TO, Descoteaux A. Leishmania donovani lipophosphoglycan blocks NADPH oxidase assembly at the phagosome membrane. Cell Microbiol. 2006;8(12):1922–1931. doi: 10.1111/j.1462-5822.2006.00758.x. [DOI] [PubMed] [Google Scholar]
- Magalhães WC, Rodrigues MR, Silva D, Soares-Souza G, Iannini ML, Cerqueira GC, Faria-Campos AC, Tarazona-Santos E. DIVERGENOME: a bioinformatics platform to assist population genetics and genetic epidemiology studies. Genet Epidemiol. 2012;36(4):360–367. doi: 10.1002/gepi.21629. [DOI] [PubMed] [Google Scholar]
- Marth JD, Grewal PK. Mammalian glycosylation in immunity. Nat Rev Immunol. 2008;8(11):874–887. doi: 10.1038/nri2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH. Improved tests for heterogeneity across a region of DNA sequence in the ratio of polymorphism to divergence. Mol Biol Evol. 1998;15:377–384. doi: 10.1093/oxfordjournals.molbev.a025934. [DOI] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351(6328):652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Bustamante C, Clark AG, et al. (12 co-authors) A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005;3(6):e170. doi: 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson LM, Lindqvist AK, Källberg H, Padyukov L, Burkhardt H, Alfredsson L, Klareskog L, Holmdahl R. A case-control study of rheumatoid arthritis identifies an associated single nucleotide polymorphism in the NCF4 gene, supporting a role for the NADPH-oxidase complex in autoimmunity. Arthritis Res Ther. 2007;9(5):R98. doi: 10.1186/ar2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer BR, Yeager M, Burdett L, et al. (13 co-authors) SNP500Cancer: a public resource for sequence validation, assay development, and frequency analysis for genetic variation in candidate genes. Nucleic Acids Res. 2006;34(Database issue):D617–D621. doi: 10.1093/nar/gkj151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BM, Huerta-Sanchez E, Nielsen R. Distinguishing between selective sweeps from standing variation and from a de novo mutation. PLoS Genet. 2012;8(10):e1003011. doi: 10.1371/journal.pgen.1003011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M, Coop G, Wall JD. The signature of positive selection on standing genetic variation. Evolution. 2005;59(11):2312–2323. [PubMed] [Google Scholar]
- Ptak SE, Przeworski M. Evidence for population growth in humans is confounded by fine-scale population structure. Trends Genet. 2002;18(11):559–563. doi: 10.1016/s0168-9525(02)02781-6. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux JD, Xavier RJ, Taylor KD, et al. (24 co-authors) Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39(5):596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR. Confirmation of association of IRGM and NCF4 with ileal Crohn’s disease in a population-based cohort. Genes Immun. 2008;9(6):561–565. doi: 10.1038/gene.2008.49. [DOI] [PubMed] [Google Scholar]
- Rozas J. DNA sequence polymorphism analysis using DnaSP. Methods Mol Biol. 2009;537:337–350. doi: 10.1007/978-1-59745-251-9_17. [DOI] [PubMed] [Google Scholar]
- San José G, Fortuño A, Beloqui O, Díez J, Zalba G. NADPH oxidase CYBA polymorphisms, oxidative stress and cardiovascular diseases. Clin Sci (Lond). 2008;114(3):173–182. doi: 10.1042/CS20070130. [DOI] [PubMed] [Google Scholar]
- Santiago HC, Gonzalez Lombana CZ, Macedo JP, et al. (12 co-authors) NADPH phagocyte oxidase knockout mice control Trypanosoma cruzi proliferation, but develop circulatory collapse and succumb to infection. PLoS Negl Trop Dis. 2012;6(2):e1492. doi: 10.1371/journal.pntd.0001492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet. 2005;76(3):449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumimoto H, Miyano K, Takeya R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun. 2005;338(1):677–686. doi: 10.1016/j.bbrc.2005.08.210. [DOI] [PubMed] [Google Scholar]
- Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105(2):437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazona-Santos E, Bernig T, Burdett L, Magalhaes WC, Fabbri C, Liao J, Redondo RA, Welch R, Yeager M, Chanock SJ. CYBB, an NADPH-oxidase gene: restricted diversity in humans and evidence for differential long-term purifying selection on transmembrane and cytosolic domains. Hum Mutat. 2008;29(5):623–632. doi: 10.1002/humu.20667. [DOI] [PubMed] [Google Scholar]
- Taylor RM, Burritt JB, Baniulis D, Foubert TR, Lord CI, Dinauer MC, Parkos CA, Jesaitis AJ. Site-specific inhibitors of NADPH oxidase activity and structural probes of flavocytochrome b: characterization of six monoclonal antibodies to the p22phox subunit. J Immunol. 2004;173(12):7349–7357. doi: 10.4049/jimmunol.173.12.7349. [DOI] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4(3):e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975;7(2): 256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Williamson SH, Hernandez R, Fledel-Alon A, Zhu L, Nielsen R, Bustamante CD. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc Natl Acad Sci U S A. 2005;102(22):7882–7887. doi: 10.1073/pnas.0502300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Adaptive molecular evolution. In: Balding DJ, Bishop M, Cannings C, editors. Handbook of statistical genetics. 3rd ed. Vol. 1. Susex (United Kingdom): John Wiley & Sons; 2007a. pp. 377–406. [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007b;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.