

Abstract
Silicon wafers and glass surfaces were functionalized with facially amphiphilic antimicrobial copolymers using the “grafting from” technique. Surface initiated atom transfer radical polymerization (ATRP) was used to grow poly(butylmethacrylate)-co-poly(Boc-aminoethyl methacrylate) from the surfaces. Upon Boc-deprotection, these surfaces became highly antimicrobial and killed S. aureus and E. coli 100% in less than 5 min. The molecular weight and grafting density of the polymer were controlled by varying the polymerization time and initiator surface density. Antimicrobial studies showed that the killing efficiency of these surfaces was independent of polymer layer thickness or grafting density within the range of surfaces studied.
Keywords: Antibacterial polymers, antimicrobial surfaces, surface modification, atom transfer radical polymerization, biocidal surfaces
Introduction
Hospital acquired infections pose a major global healthcare issue. Over 2 million cases are reported annually in the USA alone, leading to 100,000 deaths and adding nearly 5 billion dollars to US healthcare costs.1, 2 Contamination of medical devices (catheters, implants, etc.) is responsible for 45% of these infections.3 Sources of infectious bacteria can be traced to surgical equipment, medical staff clothing, resident bacteria on the patient’s skin, and the ambient atmosphere of the hospital.4 In addition, Medicare recently adopted a new policy that will not reimburse hospitals for these acquired infections.5, 6
Bacterial contamination of surfaces typically begins with the initial adherence of only a few microorganisms to the surface following implantation7, but then develops into a biofilm in less than 24 hrs.8 These biofilms exhibit extreme resistance to antibiotics and the host’s own immune system. In order to reduce or even prevent these infections, an attractive strategy is to consider materials that resist the initial phase of bacterial colonization, thus preventing biofilm formation.9 Strategies to make antimicrobial materials include the addition of leachable biocides to the material.8, 10 However, these materials have disadvantages including contamination of the environment and the host, as well as short durations of antimicrobial action due to rapid leaching. Alternatively, biocides can be covalently bonded to the surface or a non-leaching biocide can be used.1N-halamines are commonly used to make biocidal materials.11-13 Quaternary ammonium compounds (QAC) are widely used as biocides, and were successfully applied as antimicrobial layers over glass14 and poly(ethylene terephthalate) surfaces.15 Polymers containing QACs have been previously covalently attached onto different materials such as glass14, 16-19, polymers20, 21, paper19 and metals.22
The killing mechanism of bacteria by those surfaces, and specially the effect of polymer length on killing efficiency, is still under debate. Klibanov and co-workers hypothesized that the killing efficiency of immobilized polycationic chains would be enhanced if the chains are sufficiently long and flexible.17 They showed that immobilized N-hexyl poly(4-vinylpyridine)17 and N-hexyl-N-methyl polyethyleneimine23 polymers with longer chain lengths (160 kDa and 25 kDa rather than 60 kDa and 2 kDa; respectively) had stronger bactericidal activity than the shorter ones. However, their “grafting onto” technique allowed polymer chain attachment to the surface at multiple points along the backbone, thus the actual free chain-length of the attached polymer is not precisely known. Ober and coworkers demonstrated that the antimicrobial properties of surfaces coated with polystyrene-b-poly(4-vinyl-N-alkylpyridiniumbromides) copolymers were related to the molecular composition and polymer organization in the top 2-3 nm of the surface. Furthermore, their results suggest that molecular weight does not seem to be the limiting factor for antimicrobial activity.24 Recently, in detailed studies, Russell and coworkers16 along with Matyjaszewski and coworkers25 used both “grafting from” and “grafting onto” techniques to prepare quaternized poly(2-dimethylamino)ethyl methacrylate immobilized on both silicon wafers and glass surfaces. In these studies, they prepared surfaces with various polymer chain lengths and surface charge density. Their results showed that the killing efficiency of these surfaces is independent of polymer chain length but is dependent on surface charge density.
Facially amphiphilic, cationic polymers belong to a class of molecules that mimics natural host defense peptides.2, 26 They contain hydrophobic and hydrophilic side chains (protonated amines) which can segregate to opposite regions, or faces, of the molecule forming a facially amphiphilic polymer. These polymers have been termed Synthetic Mimics of Antimicrobial Peptides (SMAMPs).26 This facially amphiphilic topology and polycationic nature leads to bacterial membrane insertion, followed by disruption of the membrane. An important distinction from biocidal QAC is the fact that SMAMPs can be designed to be antimicrobial yet non-toxic to mammalian cells.2
In this study, we used ATRP to graft one family of these SMAMPs, poly(butylmethacrylate)-co-poly(Boc-aminoethyl methacrylate)27 (3), from silicon wafers and glass surfaces in order to determine if their antimicrobial activity was retained. Our results showed that these surface-bound polymers retained their antibacterial properties and killed S. aureus and E. coli 100% by contact in less than 5 min. We show that the antimicrobial properties of these polymers are independent of polymer chain length and grafting density.
Experimental Section
Materials
5-Hexen-1-yl 2-bromo-2-methylpropionate (1) was prepared according to literature.28 Butylmethacrylate (BMA, 99%, Sigma-Aldrich) was vacuum-distilled and stored in an air free flask in the freezer. Karsted’s catalyst, platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex, solution in vinyl terminated polydimethylsiloxane (Alfa Aesar), N,N-diisopropylethylamine (DIEA, purified by redistillation, 99.5%, Aldrich), toluene (anhydrous, 99.8%, Aldrich), chlorodimethylsilane (97%, Alfa Aesar), n-butyldimethylchlorosilane (Gelest), CuBr (98%, Aldrich), CuBr2 (99+%, Acros), 4,4′-dinonyl-2,2′-dipyridyl (dnbpy, 97%, Aldrich), anisol (anhydrous, 99.7%, Aldrich), dichloromethane (DCM, Fisher) and hydrogen chloride solution (4.0 M in dioxane, Aldrich) were used as received. Butoxycarbonylaminoethylmethacrylate (Boc-AEMA) was prepared according to literature procedure.27 BBL™Mueller-Hinton Broth was obtained from Becton, Dickinson and Company; Sparks, MD. Silicon wafers (100 orientation, P/B doped, thickness from 450 to 575 μm and resistivity from 7.0 to 20.0 ohm cm) were obtained from International Wafer Service Inc. Glass slides were purchased from Fisher Scientific.
Instruments
1H NMR spectra were recorded on a Bruker DPX2300 spectrometer. Ellipsometric thickness measurements were made with a Rudolph Research model SL-II automatic ellipsometer with an angle of incidence of 70° from the normal. The light source was a He-Ne laser with λ= 632.8 nm. Measurements were performed on 3-5 different locations on each sample. X-ray photoelectron spectra (XPS) were recorded on a Physical Electronics Quantum 2000 spectrometer with Al Kα excitation at a spot size of 100 μm at 25 W. Spectra were obtained at 15° and 75° takeoff angles with respect to the plane of the sample surface.
Synthesis of 6-(chlorodimethylsilyl) hexyl-2-bromoisobutyrate (2)
To a dry Schlenk flask were added 1, (8.59 g, 34.5 mmol), and chlorodimethylsilane (43 mL, 387 mmol) followed by the addition of Karsted catalyst (715 μL). The mixture was stirred at room temperature under N2. Excess chlorodimethylsilane was removed under reduced pressure and the product was purified by vacuum distillation (117-122 °C/0.05 mmHg). 1H NMR (CDCl3, 300 MHz) δ: 0.40 (s, 6H, SiMe2), 0.82 (d, 2H, J = 6.6 Hz), 1.40 (m, 6H, 3CH2), 1.68 (quint, 2H, J = 6.6 Hz, OCH2CH2), 1.93 (s, 6H, 2CH3), 4.17 (t, 2H, J = 6.6 Hz, OCH2).
Initiator Immobilization on Silicon Wafer and Glass Surfaces
Silicon wafers or glass slides were cut into 1.2 × 1.2 cm pieces, rinsed with DCM and ethanol, and then dried under stream of nitrogen. The samples were then treated with oxygen plasma for 10 min at a pressure of 100 Torr using a Harrick Plasma Cleaner. Samples were placed in a custom-designed glass holder and immersed in dry toluene (10 mL) containing DIEA (275 μL, 1.58 mmol) and ATRP initiator, 6-(chlorodimethylsilyl) hexyl-2-bromoisobutyrate, pure or mixed with chlorodimethylbutylsilane at various molar ratios (1, 10, 40, 70 and 100% initiator, 1.46 mmol total). Samples were left under N2 atmosphere for 24 h and then rinsed several times with hexanes, ethanol, ethanol-water (1:1), ethanol then water followed by drying under a stream of N2.
Surface-Initiated Polymerization
A dry flask was charged with CuBr (114.7 mg, 0.8 mmol), CuBr2 (17.84 mg, 0.08 mmol), dnbpy (0.72 g, 1.76 mmol), Boc-AEMA (8.0 g, 34.9 mmol), BMA (3 mL, 18.9 mmol) and anisole (11 mL). The mixture was purged with N2 for 10 min at 80 °C (to dissolve the monomers). The surfaces with immobilized initiator were then added and the flask was sealed and kept in oil bath at 80 °C for the desired reaction time. The substrates were extracted with DCM and ethanol before drying under a stream of N2.
Activation of Polymer by Deprotection
The silicon wafers and glass surfaces were placed in a flask and covered with hydrogen chloride solution (4.0 M in dioxane) and left at RT for 12 h. They were then rinsed with ethanol and dried under a stream of N2.
Bacterial Strains and Microbiological Media
Staphylococcus aureus (ATCC 25923) and Escherichia coli (D31) (previously stored in 30% (v/v) glycerol solution at −80 °C) were grown overnight in sterile BBL™Mueller-Hinton Broth at 37 °C with rotary agitation at 200 rpm.
Antimicrobial Activity Assay
The following studies were conducted using a modified version of the Japanese Industrial Standard JIS Z 2801:2000 “Antibacterial products-Test for Antibacterial Activity and Efficacy”.1 An overnight culture of S. aureus (~109 cells/mL) was diluted to approximately 106 cells/mL. Silicon wafer test samples were placed in individual Petri plates and the bacterial suspension was then sprayed onto the silicon wafer surfaces in a fume hood using standard chromatography sprayer (VWR Scientific). Petri plates were immediately covered and transferred to a sterile laminar flow hood (Baker Company; Sanford, ME). After 3 min, 50 μL of phosphate buffered saline was added by pipette onto the sprayed area of the test surface and left for 2 additional min. The phosphate buffered saline on the test surface was mixed by pipette to ensure removal of bacteria from the test surface. An aliquot of this phosphate buffer was removed, diluted and sterilely spread on Mueller-Hinton agar plates. Plates were incubated overnight at 37 °C without agitation and then viable bacterial colonies were counted. Bacterial growth inhibition was determined as a percentage of the colonies on the control sample. Each test sample was dried after each spraying to avoid prolonged contact between bacteria, liquid, and the test surface.
Serial Subsequent Exposure Studies
Serial subsequent exposure studies were carried out using the antimicrobial surface studies described above. After the initial spraying, test samples were dried and recovered in their respective Petri plates and placed on the bench top at room temperature. The same samples were tested the following day using the identical procedure. This was carried out for 3 or 5 consecutive days.
Fluorescence Microscopy
An Olympus BX51 Reflected Fluorescence Microscope (Optical Analysis Corp. Nashua, NH) with a 100 W Mercury Lamp (Chin Technical Corp.) was used for fluorescence studies. A Live/Dead® BacLight™ Kit L-7007 (Invitrogen, Carlsbad, CA) was used as the fluorescence dye to examine S. aureus, and E. coli. The dye mixture was incubated with the bacteria at room temperature for 15 min before applying to a modified glass surface. Bacteria were viewed under a green filter (excitation/emission, 420-480 nm/520-800 nm) or a red filter (480-550 nm/5902800 nm). Viable bacterial cells appeared green whereas, cells with compromised membrane appeared red.
Results and Discussion
Polymer Synthesis and Characterization
With our goal of making non-leaching antimicrobial surfaces, we used the “grafting from” technique to attach the antimicrobial polymer 3 to silicon wafers and glass surfaces via a covalently linked initiator. Scheme 1 summarizes the synthesis of the bifunctional alkylhalide initiator 2 that contains a reactive chlorodimethylsilane, able to self-condense with silica surfaces, and an α-bromoester that can be used as an ATRP initiator. The chlorodimethylsilane group was chosen for the formation of a well-defined monolayer of the ATRP initiator via condensation with the free hydroxyl groups on the silica surface.29 Trichloroalkylsilanes and trialkoxysilanes were not used as they polymerize in the presence of trace amount of water forming a number of possible surface structures.30 The initiator 2 was synthesized by reacting 5-hexene-1-ol with 2-bromoisobutyl bromide to form the ester 1, which was hydrosililated with chlorodimethylsilane using platinum(0) as a catalyst. The chlorodimethylsilyl group of the initiator was reacted with the surface silanol groups of cleaned silicon wafers or glass in the presence of DIEA in dry toluene under N2. This resulted in the formation of a well-defined covelantly linked monolayer containing ATRP surface bound initiators. Addition of the methacrylate monomers followed by heating at 80 °C in the presence of anisole and CuBr/dnbpy generated the desired polymers. CuBr2 was used to ensure a sufficiently high concentration of deactivator, enabling better control over the polymerization by reducing the concentration of active radical end groups. The surface bound polymer was treated with 4.0 M HCl in dioxane to remove the Boc-protecting group, thus forming the active, cationically charged surfaces.
Scheme 1.

Reaction Scheme for the Synthesis of 3 on Surfaces.
In order to study the effect of the grafted polymer chain length on the antimicrobial activity of the surfaces, we prepared surfaces with different polymer thicknesses (5-106 nm) by varying the polymerization time. Figure 1 shows the evolution of the ellipsometric brush thickness as a function of reaction time. The observed increase in layer thickness slows down at longer reaction time, which is consistent with a loss of active chain ends during ATRP.31 Removing the Boc-protecting group with HCl led to the formation of the active polymer 3 with an expected decrease in layer thickness due to the loss of the bulky group on every repeat unit. This effect is not compensated by electrostatic repulsion of the repeat units mostly because the ellipsometry measurments were performed on dry samples. Thus active surfaces with polymer layer thicknesses between 3 nm and 70 nm were obtained (Figure 1).
Figure 1.

Evolution of the ellipsometric dry brush thickness with time for the formation of the polymer on silicon wafers before (■) and after (○) deprotection with HCl.
In order to vary the initiator density on silicon wafer surfaces, butyldimethylchlorosilane was used as a blocking agent. It was premixed with 2 in different molar ratios and allowed to react with the silicon surfaces. All of the surfaces shown in Figure 2 were polymerized in the same reaction vessel to expose them all to the identical reaction conditions. Figure 2 shows a nonlinear relationship between film thickness and initiator concentration which may be due to a facilitated radical recombination near the surface early in the polymerization for the samples with high initiator density.32 Nevertheless, films with thicknesses from 3 to 57 nm were obtained.
Figure 2.

Evolution of the ellipsometric brush thickness for the formation of the polymer as a function of initiator density.
Removal of the Boc-group and formation of the amine hydrochloride salt were monitored by XPS. The N 1s XPS spectrum (Figure 3a) showed a peak at 399.3 eV, which corresponds to the binding energy of Boc-protected amines.33 Figure 3b shows the N 1s XPS spectra for the surface bound polymer after deprotection using HCl. The peak has shifted to 401.1 eV corresponding to the protonated amine group. A small peak remains at 399.0 eV which may be due to residual Boc groups or deprotonated amines. It was observed that after extensive washing of the deprotected surface with EtOH overnight, or washing with NH4OH solution for 10 min, the peak at 401.1 eV corresponding to the protonated amine disappeared and was replaced with a peak at 399.0 eV, most likely the free amine (see Figure 3c).
Figure 3.
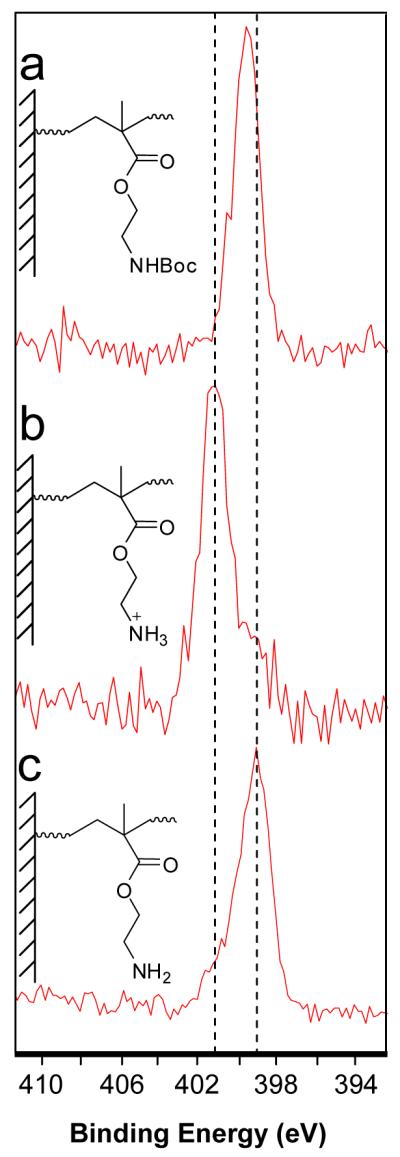
N 1s XPS spectra of surface grafted Boc-protected polymer (a). After treatment with 4.0 M HCl (b) followed by rinsing with ethanol (c).
Effect of Polymer Chain Length and Initiator Density on Antimicrobial Activity
As described above, active surfaces with high initiator density (no blocking agent) and polymer layer thicknesses between 3 nm and 70 nm were used for this study. From previous literature reports, it was expected that a trend would be observed in the surfaces’ antimicrobial activity, with the activity increasing as film thickness was increased. Surprisingly, our results shows that all surfaces killed S. aureus 100% in less than 5 min even those with thickness as low as 3 nm (Figure 4). Similarly, Figure 5 shows that surfaces with initiator densities varying from 1% to 100%, corresponding to thicknesses of 2-57 nm, were able to fully kill S. aureus regardless of the initiator density. We expected that a polymer layer thickness of 2 or 3 nm would not extend through the S. aureus cell envelope which is estimated to be about ~30-37 nm thick.34, 35 However, our results are in agreement with the findings of Russell and coworkers whose work on poly(quaternary ammonium) compounds grafted from surfaces with thicknesses as low as 10 nm killed bacteria cells.16 Similarly, Isquith and coworkers showed that a thin layer of quaternary ammonium compounds on glass had good antimicrobial properties.14
Figure 4.

Percent reduction of S. aureus growth as a function of polymer layer thickness compared to untreated silicon wafers. The modified surfaces killed 100% of the bacteria on the first exposure while decreasing efficacy was seen on subsequent exposures.
Figure 5.

Percent reduction of S. aureus growth as a function of initiator density compared to untreated silicon wafers. The modified surfaces killed 100% of bacteria on the first exposure while decreasing efficacy was seen on subsequent exposures.
In order to ensure that the killing ability of the surfaces was not due to leaching of the active polymer into the solution, the surfaces were evaluated using the Kirby–Bauer test.36 Treated and untreated silicon wafers were placed active side down onto an agar plate previously inoculated with 1 × 109 cells/mL of S. aureus. After 24 hrs incubation, treated surfaces did not exhibit a zone of inhibition around the wafer which indicated the non-leaching nature of these surfaces (Figure 6).
Figure 6.

Kirby–Bauer assay for the antimicrobial surfaces: (a) untreated silicon wafer and (b) porous scaffold loaded with 3 highlights the typical “zone of inhibition” observed in Kirby–Bauer experiments. (c) modified silicon wafer surfaces containing 70 nm of 3 shows no zone of inhibition.
Serial Subsequent Exposure Studies
All surfaces with varying polymer thicknesses and initiator densities exhibited excellent antimicrobial activities, even those with a polymer layer thickness of 3 nm or an initiator density of 1% (2 nm thickness). These surfaces were re-examined with successive bacterial challenges. After the initial spraying, test samples were dried, replaced in their respective Petri plates and stored on the bench top at room temperature. The same samples were tested the following day using the identical test procedure with no cleaning steps between successive sprays in order to examine bacterial loads. This was carried out for three to five consecutive days. As shown in Figures 4 and 5, surfaces still exhibited near quantitative bactericidal activity upon a second exposure, however, it was also observed that the third and subsequent exposures were not able to kill as many bacteria. Gorham and coworkers observed complete loss of the antimicrobial activity of polyethylene surfaces with covalently linked monolayers of quaternary ammonium compounds after the second exposure with S. aureus.37 They concluded that this loss in activity might be due to the adsorption of organic materials, such as proteins, on the positively charged membrane surface. If that was the case, removal of this material would regenerate the surface activity. We therefore washed the surface thoroughly; however, surfaces remained inactive even after cleaning with HCl/dioxane or TEA overnight, followed by sonication. The integrity of the polymer layer after that treatment was confirmed by ellipsometry. This leads to the assumption that biological components were not inhibiting the activity as such strong acids and/or bases would have been able to remove any cells, lipids or cellular debris from the surface, thus allowing viable cells to once again contact the antimicrobial polymer.38
It was also found that surfaces lost their antimicrobial activity completely when heated at 80 °C in sealed ampoules for 4 days, whereas they retained their activity for long times if stored at low temperature (−20 °C). This suggested that the loss of antimicrobial activity might be due to chemical modification, or rearrangement of the polymer. Armes and coworkers39 showed that a similar polymer system, poly(2-aminoethyl methacrylate) hydrochloride, underwent chemical reactions under alkaline conditions in solution. They proposed several pathways: under alkaline conditions the ammonium salt is deprotonated to give the free amine which may attack its own ester group intramolecularly to form an amide via a rearrangement. Alternatively, an interchain or intrachain condensation can occur between the primary amine and a neighboring carbonyl (see Scheme 2). According to Ikada and coworkers, poly N-[3(N,N-dimethylamino) propylacrylamide and poly 2-(dimethylamino)ethyl methacrylate grafted chains have pKa values of 9.0 and 7.0, respectively, whereas the pKa values of the corresponding monomers are 10.3 and 7.9, respectively.40 This shows that the pKa values of their polymer brushes are between 0.9 - 1.3 units lower than the pKa values of the corresponding monomers. By analogy, we assume that the pKa value of our grafted polymer will be in the range of ~ 6.8 - 7.2 based on the known pKa value of 8.1 for the monomer.41 Thus, when surfaces are sprayed with bacterial suspension in distilled water, part of the polymer will be in the free amine form, allowing polymer rearrangement to occur in pathways similar to those proposed by Armes and coworkers (Scheme 2). This rearrangement is presumably faster on the surface than in solution because of the close proximity of the polymer chains on the surface. To further test this assumption, silicon wafer surfaces with active polymer in the ammonium chloride form were covered with PBS buffer (pH 7.4) for 3 days, washed with distilled water and covered with 4.0 M HCl/dioxane for 4 h. The sample was rinsed with ethanol once and dried under a N2 stream. If the surface was just deprotonated in the buffer, treatment with HCl would regenerate it. However, the N 1s XPS spectrum (Figure 7) showed that the peak at 401.1 eV, corresponding to the protonated amine nitrogen was not present but a new peak at 399.5 eV was. This broad peak is consistent with the presence of amide nitrogens. Antimicrobial assays showed that samples treated the same way only retained 60% of their antimicrobial activity, not 100%. These results support the hypothesis that chemical rearrangement rather than contamination was responsible for the loss of the surface activity.
Scheme 2.

Possible Rearrangement Mechanisms for Polymer 3 in PBS Buffer (pH 7.4)
Figure 7.

N 1s XPS spectra of surface grafted polymer 3 (a), after treatment with PBS buffer (pH 7.4), followed by 4.0 M HCl/dioxane (b).
Fluorescent Microscopy
To confirm that this surface-grafted polymer kills bacteria by contact on the surface, we used the Live/Dead two-color fluorescence method. In this viability assay, a mixture of SYTO9 green fluorescent nucleic acid stain and propidium iodide, a red fluorescent nucleic acid stain, was used. The SYTO9 stain labels bacteria with both intact and compromised membranes. In contrast, propidium iodide penetrates only bacteria with compromised membranes. When both dyes are present, propidium iodide competes with the SYTO9 stain for nucleic acid binding sites.
The dye mixture was incubated with the bacteria (108 cell/mL) for 15 min before applying to plain glass and polymer-brush modified glass surfaces. Figures 8a and 8d show that all S. aureus cells on the glass control gave only green fluorescence, indicating all bacteria had intact cell membranes. On the other hand, Figures 8e and 8f show that almost all bacterial cells turned red after 2 min of exposure to the modified surface. It is seen that the green color faded due to replacement of SYTO9 with propidium iodide (Figures 8b and 8c). Similar results were observed when E. coli was used instead of S. aureus (Figure 9). The very fast killing effect observed using fluorescent microscopy corroborates the results in figures 4 and 5 which showed that these surfaces kill 100% S. aureus within only 5 min exposure.
Figure 8.

Fluorescence microscopy images of S. aureus: (a and d) on unmodified glass surfaces after 10 min exposure using green and red filters respectively, and (b and e) on polymer modified glass surfaces after 2 min exposure using green and red filters, respectively.
Figure 9.

Fluorescence microscopy images of E. coli: (a and c) on unmodified glass surfaces after 10 min exposure using green and red filters, respectively, and (b and d) on polymer modified glass surfaces after 2 min exposure using green and red filters, respectively.
Conclusions
Silicon wafers and glass surfaces were functionalized with polymer 3 using “grafting from” techniques. ATRP allowed for control of the polymer layer thickness. Surfaces with varying grafting density and polymer thicknesses from 2 to 70 nm were prepared. This polymer is known to be antimicrobial in solution with molecular weights as low as 1.3 kDa.27 The mechanism of bactericidal action in solution is believed to be through insertion of the polymer into the bacterial phospholipid membrane. Our results showed that the killing ability of this polymer when linked to surfaces is independent of polymer layer thickness and density. These surfaces were able to kill S. aureus 100% in 5 min. A high density polymer layer thickness of 3 nm would not have polymer chains long enough to extend through S. aureus cell envelope which is estimated to be about ~ 30-37 nm thick. This means that the mechanism for bacterial killing may be more complicated than some of the models involving direct action on the membrane presented in the literature, or that solution and surface mechanisms are independent. Alternatively, even though the cell envelope is known to be ~30-37 nm thick, for gram-positive bacteria, there might be access to the plasma membrane when these cells contact hard surfaces. The microscope results are definite that membrane disruption occurs within 2 min but do not indicate how this occurs. An alternative mechanism was proposed by Kügler based on the release of Ca2+ and Mg2+ from the bacterial phospholipid membrane and the electrostatic compensation of the negative charge of the phospholipids membrane by the surface cationic charge.18 Regardless of mechanism, surfaces modified with SMAMPs led to rapid cell death. The lack of killing in serial bacterial loading experiments is related to the chemical rearrangement of 3 and alternative polymers that eliminate these side reactions are easily envisaged. Surfaces that do not support bacterial growth will be more important as “super-bugs” like MRSA become more common. This paper, and the other work in this area, will be critical for meeting this challenge.
Acknowledgment
We thank Jacob Hirsch for his help with the X-Ray Photoelectron Spectroscopy. NIH (RO1-GM-65803), the ONR (N00014-07-1-0520), and PolyMedix Inc. are greatly acknowledged for financial support.
References
- (1).Madkour AE, Tew GN. Polymer International. 2008;57:6–10. [Google Scholar]
- (2).Gabriel GJ, Som A, Madkour AE, Eren T, Tew GN. Mater. Sci. Eng., R. 2007;R57:28–64. doi: 10.1016/j.mser.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Stamm WE. Ann. Intern. Med. 1978;89:764–9. doi: 10.7326/0003-4819-89-5-764. [DOI] [PubMed] [Google Scholar]
- (4).An YH, Friedman RJ. J. Hosp. Infect. 1996;33:93–108. doi: 10.1016/s0195-6701(96)90094-8. [DOI] [PubMed] [Google Scholar]
- (5).Wald HL, Kramer AM. JAMA. 2007;298:2782–2784. doi: 10.1001/jama.298.23.2782. [DOI] [PubMed] [Google Scholar]
- (6).Department of Health and Human Services. Centers for Medicare & Medicaid Services. Fed Regist. 2008;Vol. 73:23528–23938. [Google Scholar]
- (7).von Eiff C, Jansen B, Kohnen W, Becker K. Drugs. 2005;65:179–214. doi: 10.2165/00003495-200565020-00003. [DOI] [PubMed] [Google Scholar]
- (8).Hetrick EM, Schoenfisch MH. Chem. Soc. Rev. 2006;35:780–789. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]
- (9).Williams JF, Worley SD. J. Endourol. 2000;14:395–400. doi: 10.1089/end.2000.14.395. [DOI] [PubMed] [Google Scholar]
- (10).Danese PN. Chem. Biol. 2002;9:873–880. doi: 10.1016/s1074-5521(02)00192-8. [DOI] [PubMed] [Google Scholar]
- (11).Kocer HB, Akdag A, Ren X, Broughton RM, Worley SD, Huang TS. Ind. Eng. Chem. Res. 2008;47:7558–7563. [Google Scholar]
- (12).Makal U, Wood L, Ohman DE, Wynne KJ. Biomaterials. 2006;27:1316–1326. doi: 10.1016/j.biomaterials.2005.08.038. [DOI] [PubMed] [Google Scholar]
- (13).Chen ZB, Sun YY. Ind. Eng. Chem. Res. 2006;45:2634–2640. doi: 10.1021/ie060088a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Isquith AJ, Abbott EA, Walters PA. Appl. Microbiol. 1972;24:859–863. doi: 10.1128/am.24.6.859-863.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).El Ola SMA, Kotek R, White WC, Reeve JA, Hauser P, Kim JH. Polymer. 2004;45:3215–3225. [Google Scholar]
- (16).Murata H, Koepsel RR, Matyjaszewski K, Russell AJ. Biomaterials. 2007;28:4870–4879. doi: 10.1016/j.biomaterials.2007.06.012. [DOI] [PubMed] [Google Scholar]
- (17).Tiller JC, Liao C-J, Lewis K, Klibanov AM. Proc. Natl. Acad. Sci. 2001;98:5981–5985. doi: 10.1073/pnas.111143098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kügler R, Bouloussa O, Rondelez F. Microbiology. 2005;151:1341–8. doi: 10.1099/mic.0.27526-0. [DOI] [PubMed] [Google Scholar]
- (19).Lee SB, Koepsel RR, Morley SW, Matyjaszewski K, Sun Y, Russell AJ. Biomacromolecules. 2004;5:877–882. doi: 10.1021/bm034352k. [DOI] [PubMed] [Google Scholar]
- (20).Tiller JC, Lee SB, Lewis K, Klibanov AM. Biotechnol. Bioeng. 2002;79:465–471. doi: 10.1002/bit.10299. [DOI] [PubMed] [Google Scholar]
- (21).Cen L, Neoh KG, Kang ET. Langmuir. 2003;19:10295–10303. [Google Scholar]
- (22).Ignatova M, Voccia S, Gilbert B, Markova N, Mercuri PS, Galleni M, Sciannamea V, Lenoir S, Cossement D, Gouttebaron R, Jérôme R, Jérôme C. Langmuir. 2004;20:10718–10726. doi: 10.1021/la048347t. [DOI] [PubMed] [Google Scholar]
- (23).Lin J, Qiu S, Lewis K, Klibanov AM. Biotechnol. Bioeng. 2003;83:168–172. doi: 10.1002/bit.10651. [DOI] [PubMed] [Google Scholar]
- (24).Krishnan S, Ward RJ, Hexemer A, Sohn KE, Lee KL, Angert ER, Fischer DA, Kramer EJ, Ober CK. Langmuir. 2006;22:11255–11266. doi: 10.1021/la061384v. [DOI] [PubMed] [Google Scholar]
- (25).Huang J, Koepsel RR, Murata H, Wu W, Lee SB, Kowalewski T, Russell AJ, Matyjaszewski K. Langmuir. 2008;24:6785–6795. doi: 10.1021/la8003933. [DOI] [PubMed] [Google Scholar]
- (26).Lienkamp K, Madkour AE, Musante A, Nelson CF, Nüsslein K, Tew GN. J. Am. Chem. Soc. 2008;130:9836–9843. doi: 10.1021/ja801662y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kuroda K, DeGrado WF. J. Am. Chem. Soc. 2005;127:4128–4129. doi: 10.1021/ja044205+. [DOI] [PubMed] [Google Scholar]
- (28).Theato P, Kim KJ, Yoon DY. Phys. Chem. Chem. Phys. 2004;6:1458–1462. [Google Scholar]
- (29).Fadeev AY, McCarthy TJ. Langmuir. 1999;15:3759–3766. [Google Scholar]
- (30).Fadeev AY, McCarthy TJ. Langmuir. 2000;16:7268–7274. [Google Scholar]
- (31).Ramakrishnan A, Dhamodharan R, Rühe J. Macromol. Rapid Commun. 2002;23:612–616. [Google Scholar]
- (32).Bao Z, Bruening ML, Baker GL. Macromolecules. 2006;39:5251–5258. [Google Scholar]
- (33).Strother T, Hamers RJ, Smith LM. Nucleic Acids Res. 2000;28:3535–3541. doi: 10.1093/nar/28.18.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Friedrich CL, Moyles D, Beveridge TJ, Hancock REW. Antimicrob. Agents Chemother. 2000;44:2086–2092. doi: 10.1128/aac.44.8.2086-2092.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Matias VRF, Beveridge TJ. J. Bacteriol. 2006;188:1011–1021. doi: 10.1128/JB.188.3.1011-1021.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bauer AW, Kirby WM, Sherris JC, Turck M. Am. J. Clin. Pathol. 1966;45:493–6. [PubMed] [Google Scholar]
- (37).McCubbin PJ, Forbes E, Gow MM, Gorham SD. J. Appl. Polym. Sci. 2006;100:381–389. [Google Scholar]
- (38).Nylander T, Samoshina Y, Lindman B. Adv. Colloid Interface Sci. 2006:123–126. 105–123. doi: 10.1016/j.cis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- (39).He L, Read ES, Armes SP, Adams DJ. Macromolecules. 2007;40:4429–4438. [Google Scholar]
- (40).Uchida E, Uyama Y, Ikada Y. Langmuir. 1993;9:11211124. [Google Scholar]
- (41).Geurts JM, Gottgens CM, Van Graefschepe MAI, Welland RWA, Van Es JJGS, German AL. J. Appl. Polym. Sci. 2001;80:1401–1415. [Google Scholar]