

Abstract
Runx (Cbfa/AML) transcription factors are critical for tissue-specific gene expression. A unique targeting signal in the C terminus directs Runx factors to discrete foci within the nucleus. Using Runx2/CBFA1/AML3 and its essential role in osteogenesis as a model, we investigated the fundamental importance of fidelity of subnuclear localization for tissue differentiating activity by deleting the intranuclear targeting signal via homologous recombination. Mice homozygous for the deletion (Runx2ΔC) do not form bone due to maturational arrest of osteoblasts. Heterozygotes do not develop clavicles, but are otherwise normal. These phenotypes are indistinguishable from those of the homozygous and heterozygous null mutants, indicating that the intranuclear targeting signal is a critical determinant for function. The expressed truncated Runx2ΔC protein enters the nucleus and retains normal DNA binding activity, but shows complete loss of intranuclear targeting. These results demonstrate that the multifunctional N-terminal region of the Runx2 protein is not sufficient for biological activity. We conclude that subnuclear localization of Runx factors in specific foci together with associated regulatory functions is essential for control of Runx-dependent genes involved in tissue differentiation during embryonic development.
Factors that mediate transcription, processing of gene transcripts, DNA replication, and DNA repair are organized as discrete domains within the nucleus (1–13). The osteogenic and hematopoietic Runx transcription factors contain a unique and conserved amino acid motif in the C terminus designated the nuclear matrix targeting signal that directs Runx proteins to subnuclear foci where gene regulatory complexes are assembled in situ (14–20). Thus the Runx transcription factors provide a paradigm for pursuing mechanisms that coordinate the compartmentalization of regulatory proteins in intranuclear sites. In this study we directly addressed the fundamental question of whether subnuclear targeting and the associated regulatory activities of Runx factors are obligatory for in vivo function.
The Runx (CBFA/AML/PEBP2α)‖ family of transcription factors are critical for cellular differentiation and organ development (21–23). Key studies have established that Runx2/CBFA1/AML3 is required for osteoblast differentiation and in vivo bone formation (24–28). Ablation of the Runx2 gene in mice results in a complete absence of intramembranous and endochondral bone that is attributed to the maturational arrest of hypertrophic chondrocytes and osteoblasts (26, 29, 30). Haplo-insufficiency of this gene results in the human syndrome cleidocranial dysplasia, a dominantly inherited developmental disorder of bone (24, 25). A basis is thereby provided for examining the biological consequences resulting from absence of the C-terminal targeting function.
Runx factors share multiple functional domains. The N- terminal runt homology DNA binding domain (RHD), which interacts with many coregulatory factors and chromatin modifying complexes (e.g., CBFβ, p300, EAR2, ALY, mSin3A, MITF, C/EBP, and steroid receptors), contributes to regulated transcription of its target genes (reviewed in refs. 22 and 23). Nuclear import is mediated by a nuclear localization signal located contiguous to the RHD (31, 32). The C-terminal domain is responsible for subnuclear trafficking, as well as transcriptional activation and repression (23).
Using the requirement of Runx2 for osteogenesis as a model, we investigated the necessity for fidelity of subnuclear localization to support regulatory activity in vivo. We deleted the C-terminal intranuclear targeting signal by homologous recombination. Our results indicate that subnuclear targeting and the associated regulatory functions are essential for control of Runx-dependent genes involved in tissue differentiation during embryonic development.
Materials and Methods
Generation of Targeted Embryonic Stem Cells and Mutant Mice.
Genomic clones containing the 3′ end of the mouse Runx2 locus were obtained from a 129/sv mouse DNA library in λ Fix II (Stratagene). The mutation introduced a premature stop codon after amino acid 376 and created a BclI restriction site (Fig. 1). The final targeting vector containing a thymidine kinase (TK) and NEO marker genes flanked by two loxP sites (33, 34) was electroporated into AB2.2 embryonic stem cells, and 300 G418-resistant clones were analyzed for homologous recombination by Southern blotting. The floxed TK-NEO cassette was specifically deleted by expressing the Cre enzyme and selecting for 1-(2-deoxy-2-fluoro-β-d-arabinofuranosyl)-5-iodouracil-resistant embryonic stem cell clones. Excision of the TK-neo cassette was confirmed by Southern blot analysis (data not shown). Two independent clones were used for blastocyst injection. Chimeric mice were bred to C57BL/6 mice to yield heterozygous mice [Runx2 wild type (WT)/ΔC] which were interbred to yield homozygous mutant animals (Runx2 ΔC/ΔC).
Figure 1.

Gene replacement for Runx2ΔC. (A) Generation of the mutant locus. The schematic at the top shows the functional domains of Runx proteins and Runx2 exon organization. Domains in the N terminus (N) conserved among Runx transcription factors are indicated: the runt homology/DNA binding domain (RHD) and the nuclear localization signal (NLS). The letters Q and A designate homopolymeric stretches of glutamine and alanine residues unique to Runx2. The C terminus (C) defined by exon 8 includes the nuclear matrix targeting signal (NMTS). Below this schematic is indicated the sequence of the WT (wt) and mutated (mt) allele, and ** denotes the premature stop codon. The bottom portion shows a diagram of target constructs used for introduction of the mutation at the start of exon 8. The C-terminal genomic organization of the WT and mutated alleles are also illustrated. The restriction sites (B, BamHI; E, EcoRI) in the Runx2 genomic locus and the regions of homologous recombination are indicated. Removal of the neo-thymidine kinase cassette by Cre recombinase results in the mutant allele. The loxP sites are shown by filled triangles. The Runx2 gene encodes two major isoforms with two distinct N termini (p56/Type I and p57/Type II) that are generated from two different promoters. Our strategy produces a C-terminal deletion in both isoforms. UTR, untranslated region. (B) Genotyping by PCR analysis of mouse tail genomic DNA from WT (+/+), heterozygous (+/ΔC), and homozygous mutant (ΔC/ΔC) mice. Locations of the forward and reverse primers (which span the lox P site) used as probes for genotyping are indicated by arrows. The presence of the lox P site generates a slower migrating PCR band that represents the Runx2ΔC mutant allele carrying the premature stop codon. (C) Sequence analysis of the PCR products amplified from genomic DNA of WT (+/+) and homozygous mutant (ΔC/ΔC) mice confirms in vivo incorporation of the premature stop codon (denoted by *).
Northern Blot, Reverse Transcription (RT)-PCR, and Western Analyses.
Total RNA was isolated separately from the head and lower body of eviscerated embryos at 17.5 days postcoitum (dpc) by using Trizol reagents (Life Technologies, Rockville, MD). Standard procedures were used for Northern blot and RT-PCR analyses and for hybridization with a probe specific for Runx2/Cbfa1. RT-PCR was carried out by using primers specific for exon 8. Cellular proteins were harvested from calvarial-derived cells of embryos at 17.5 dpc. After culture for 20 days (35), nuclear lysates (36) were prepared from isolated nuclei (37) and resolved by 10% SDS/PAGE. Western blot analysis was performed by using a monoclonal Runx2(PEBP2αA) antibody αA8G5 (38) or an anti X-press antibody (Invitrogen), followed by incubation with an horseradish peroxidase-tagged anti-mouse IgG secondary antibody (Molecular Probes). Bands were detected by ECL reagents (Amersham Pharmacia).
Reporter Expression Constructs and Transfection Assays.
The cytomegalovirus-driven Runx2 expression vector was obtained from James C. Neil (University of Glasgow, Glasgow, U.K.) (39), and X-press epitope tag sequences were added. The Runx2ΔC construct was generated by eliminating amino acids 377–528 and inserting a cassette containing a stop codon. HeLa cells (plated at 0.5 × 106 per ml) were transfected at 50% confluency by using Superfect reagent (Qiagen, Valencia, CA). Plasmid constructs containing the Gallus bone sialoprotein, rat osteocalcin, and rat transforming growth factor β receptor-1 promoters fused to the chloramphenicol acetyltransferase coding sequence and Rous sarcoma virus Luciferase construct (internal control) were transfected as described (14, 40–42).
Electrophoretic Mobility-Shift Analysis.
Nuclear extracts (36) from HeLa cells transfected with WT or mutant constructs were analyzed by electrophoretic mobility-shift assays as described (41). Antibody supershift experiments contained 1 μl of polyclonal antiserum directed to a Runx2 (AML3)-specific peptide (43).
Immunofluorescence Microscopy.
Procedures for cellular fixation, in situ preparation of nuclear matrix-intermediate filaments (NMIF), and data acquisition were followed as described (14). For detection and localization of endogenously synthesized WT and Runx2ΔC proteins in bone cells from the 17.5 dpc WT/WT, WT/ΔC, and ΔC/ΔC mice, ex vivo expanded calvarial cells were cultured on glass coverslips. Whole cells and NMIF preparations were incubated with a 1:100 dilution of the Runx2 (αA8G5) antibody, followed by incubation with a 1:800 dilution of FITC-conjugated secondary antibody (Molecular Probes).
Results
Subnuclear Targeting and Associated Regulatory Functions of Runx2 Are Genetically Required for Osteogenesis During Fetal Development.
To abolish subnuclear targeting of Runx2, we introduced a stop codon after amino acid 376 in the last exon (Fig. 1A). The point mutation eliminates the C terminus and also creates a BclI restriction site for detection of the mutant mRNA in vivo. Two founder lines were established and characterized, each from microinjection of an independent clone of targeted embryonic stem cells. Genotypes of offspring were routinely determined by PCR detection (Fig. 1B). Presence of the stop codon mutation in the mouse genome was confirmed by sequence analysis (Fig. 1C).
To ensure that the Runx2ΔC mutation does not cause a defect in expression of the mutant allele, we monitored Runx2ΔC mRNA and protein levels (Fig. 2). Northern blot analyses revealed the expected major Runx2 transcript in the head and lower skeletons of WT, WT/ΔC, and ΔC/ΔC embryos at 17.5 dpc (Fig. 2A). RT-PCR analysis and BclI cleavage were used to discriminate the mutant from the WT mRNA (Fig. 2B). We also confirmed the mutation in the ΔC mouse by sequence analysis of the RT-PCR product (data not shown). Western blot analysis using lysates from ex vivo cultures derived from calvaria of WT, heterozygous, and homozygous ΔC embryos (17.5 dpc) demonstrated that both the WT and truncated mutant proteins are expressed (Fig. 2C). Both proteins are present in the heterozygote at similar levels, whereas only the truncated protein is observed in the ΔC homozygote (Fig. 2C). These data indicate that stable expression of the mutant Runx2ΔC protein, rather than absence of expression, accounts for the phenotype of the Runx2ΔC mouse.
Figure 2.

Runx2ΔC mRNA expression and protein synthesis in vivo. (A) Northern blot analysis of total cellular RNA (10 μg/lane) prepared from the heads of WT (+/+), heterozygous (+/ΔC), and homozygous (ΔC/ΔC) embryos at 17.5 dpc. The Runx2 major transcript (arrowhead) migrates above the 28S ribosomal RNA. (Lower) Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA as a control for RNA loading. (B) Detection of WT and Runx2ΔC mRNAs in +/+, +/ΔC, and ΔC/ΔC mouse embryos by RT-PCR and BclI restriction enzyme digestion. Lane M represents size markers. Schematic shows the locations of the primers (arrows) used for PCR amplification of the WT and mutant mRNAs. The BclI site present in the mutant generates the 0.2- and 0.3-kb digestion products. (C) Western blot analysis of nuclear lysates from cultured calvarial cells isolated from +/+, +/ΔC, and ΔC/ΔC Runx2 mice at 17.5 dpc and cultured for 2 wk. Lysates from ROS 17/2.8 osteoblast-like cells serve as a positive control for full-length Runx2. Standard immunoblotting was performed with the monoclonal Runx2 antibody. The expected molecular masses for Runx2 WT and ΔC proteins are indicated by arrows below the 62-kDa and above the 38-kDa markers, respectively. Relative marker positions are indicated.
When the Runx2ΔC F1 heterozygotes were mated, there were no live births of homozygote mice from 10 or more pregnancies of each line. We determined that embryonic lethality occurs after 17.5 dpc but before 18.5 dpc. Fig. 3A shows representative embryos of each genotype at 17.5 dpc. The ΔC/ΔC 17.5-dpc embryos (0.75 ± 0.1 g) are smaller (18–20%; P < 0.001) compared with WT/WT (0.99 ± 0.1 g) and WT/ΔC (0.99 ± 0.1 g). The ratio of live births of the WT (WT/WT)/heterozygous (WT/ΔC)/homozygous (ΔC/ΔC) mice is 1:2:0, but the ratio of live animals at 17.5 dpc is 1:2:1, conforming to normal Mendelian segregation. Thus, the Runx2ΔC mutation results in late embryonic lethality.
Figure 3.

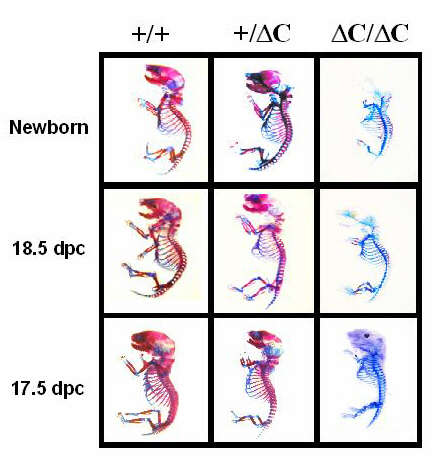
The Runx2ΔC knock-in results in embryonic lethality in homozygotes and absence of clavicles in heterozygotes. (A) Whole animal contact radiographs of 17.5 dpc mice (Faxitron X-Ray, Wheeling, IL). At this age no skeletal elements are detected by soft x-rays in the ΔC/ΔC compared with +/+ and +/ΔC mice. (B) Radiography of 6-wk mice to visualize phenotype of the heterozygote. Shown is a dorsal view of a +/+ mouse with arrow pointing to the clavicle that is missing in the +/ΔC mouse. (C) Staining of skeletons from (17.5 dpc and 18.5 dpc) embryos with Alizarin red to detect bone and Alcian blue to detect cartilage by standard procedures (60).
We established that the Runx2ΔC mutation causes a severe skeletal defect. Contact radiography revealed that neither calcified cartilage nor mineralized bone is present in the ΔC/ΔC mice at 17.5 dpc (Fig. 3A), but a few mineralized vertebrae are detectable at 18.5 dpc (data not shown). Radiography also indicated that cranial and facial bones are completely absent in the homozygous Runx2ΔC mice from 17.5 dpc to birth. Heterozygotes have normal skeletons and craniofacial features, except for missing clavicles (Fig. 3B). Most heterozygotes have a small calcified remnant of the clavicle at the acromial end, similar to that observed in the Cbfa1−/− mice (26). The clavicle is, in part, formed by intramembranous bone, but develops at 14 dpc, earlier than other intramembranous bones (e.g., cranium). These results suggest that haplo-insufficiency of Runx2 C-terminal functions at this stage of embryogenesis prevents formation of the clavicles. Body mass (weight) was recorded for several litters during postnatal development of the surviving WT and heterozygous mice. Male heterozygotes exhibit a significantly lower weight (18–20%) and weight gain than WT animals, whereas differences between female WT and heterozygous mice are not remarkable. The male-specific differences in weight may be related to the observations that Runx2 mRNA is expressed in testis (44).
To further characterize the skeletal defect, we evaluated formation of cartilaginous and osseous tissues in the Runx2 WT/WT, WT/ΔC, and ΔC/ΔC mice by analysis of skeletal stained embryos (Fig. 3C). At 17.5 dpc the cartilaginous skeleton of the ΔC homozygote is apparently normal with respect to the number and proportion of the long bones, which are formed by endochondral ossification. However, mineral-containing tissues are not detectable at 17.5 dpc, although there is evidence for initiation of osteogenesis at 18.5 dpc and in the newborns (Fig. 6, which is published as supplemental material on the PNAS web site, www.pnas.org). Strikingly, there is a complete absence of intramembranous bone formation (cranium and mandible) at all stages of development in the ΔC/ΔC mouse. Except for the missing clavicles, the skeleton of the heterozygote is almost identical to that of the WT and displays similar proportions of cartilage and mineralized bone tissues. Together, these results demonstrate that Runx2 C-terminal functions are required for ossification of the skeleton.
Histological analysis shows that deletion of the Runx2 C terminus disrupts bone formation at the stage of hypertrophic chondrocyte maturation. Limb patterning is normal in the ΔC/ΔC mouse at 17.5 dpc (Fig. 4, compare a and b); however, the limbs consist almost entirely of cartilage (compare ΔC to WT). Heterozygous mice (not shown) exhibit normal formation of endochondral bone and a medullary cavity containing hematopoietic tissue, whereas the limbs of ΔC/ΔC mice display no evidence of a marrow cavity (Fig. 4 b and d). Fig. 4e shows calcified cortical and trabecular bone matrix in the WT/WT limb. In contrast, the ΔC/ΔC tibia (Fig. 4f) has some calcified cartilage matrix as well as bone matrix on the periosteal surface at the middiaphysis. A thin layer of alkaline phosphatase-positive osteoblasts confirms the initiation of bone formation at this site (Fig. 4g), but further progression of osteoblast differentiation is inhibited in the ΔC/ΔC mice. Thus endochondral bone formation in the ΔC mutant appears to be arrested at the stage in which the diaphysis is composed almost entirely of hypertrophic chondrocytes.
Figure 4.

Absence of bone tissues in the ΔC/ΔC knock-in mutant mouse. Histologic appearance of limbs from WT and Runx2ΔC homozygous mutant embryos (ΔC/ΔC) at 17.5 dpc. Tibial bone paraffin sections stained with Safranin O-fast green (a–d) from WT (a and c) and homozygous ΔC/ΔC (b and d) mice at ×4 (a and b) and ×10 (c and d) magnifications to show growth plate zones. Cartilage (red) and bone, marrow, blood vessels (blue) are distinguished. Brackets in a and b show regions at higher magnification in c and d. Undecalcified tibial bone sections with hematoxylin and eosin/von Kossa silver staining (to detect mineral) for WT (e) and ΔC/ΔC (f) at ×10 magnification. (g) Alkaline phosphatase and (h) tartrate-resistant acid phosphatase histochemistry of the ΔC/ΔC bone (×20). (i) The central portion of the limb from ΔC/ΔC bone and indicates accumulation of blood vessels (hematoxylin and eosin stain) (×40 magnification). Arrow indicates one osteoclast (Ocl). V designates blood vessels. P shows periochondrium.
Osteoclasts are also present in the mutant bones (Fig. 4h), consistent with the presence of bone matrix and indicative of early vascular invasion. However, an aggregation of blood vessels is consistently observed in the middiaphyseal region of the ΔC/ΔC limbs (Fig. 4i). This finding suggests that vascular ingrowth, which is necessary for and precedes marrow cavity formation, has been initiated but has not progressed normally. In conclusion, there is an absence of skeletal development in the ΔC mutant mouse because hypertrophic chondrocyte maturation, vascular invasion, osteoblast differentiation, and mineral deposition are abnormal.
The Runx2ΔC Mutation That Abolishes Subnuclear Targeting Compromises Transcriptional Control.
The phenotype of the Runx2 ΔC/ΔC mouse is remarkably similar to the phenotype resulting from complete ablation of Runx2/Cbfa1. Therefore, we assessed functional activities of the ΔC mutant protein that contains the multifunctional DNA binding domain and the nuclear import signal, but lacks the subnuclear targeting signal. After transient transfection in cells which lack endogenous Runx proteins, we find Runx2ΔC forms a sequence-specific complex with DNA, similar to the full-length protein, as shown by oligonucleotide competition and gel mobility-supershift analyses (Fig. 5A). The truncated protein also retains the capability to heterodimerize with its partner protein core binding factor β, as reflected by the immunoreactivity of the WT and mutant Runx2 complexes with a core binding factor β antibody (data not shown). We also compared transcriptional activity of the WT and Runx2ΔC proteins on promoters that are either enhanced or repressed by Runx2. The mutant protein exhibits significantly reduced transactivation of both the osteocalcin and transforming growth factor β R1 promoters (Fig. 5B). In addition, repressor activity on the bone sialoprotein promoter is abrogated. Western blot analysis shows that both proteins are expressed to the same extent. Hence, transcriptional activity, but not DNA binding, is reduced by deletion of the Runx2 C terminus. The decrease in transcriptional function reflects the absence of Runx2 domains that contribute to subnuclear targeting and associated regulatory functions.
Figure 5.

Functional properties of the Runx2ΔC protein. (A) Runx2ΔC protein retains DNA binding activity. (Left) HeLa cell nuclear extracts (NE) were prepared 24 h after transfection with empty vector (V), WT, or Runx2ΔC mutant protein expression plasmids. Nuclear extracts from Ros 17/2.8 cells that endogenously express Runx2 provided a positive control. An oligonucleotide containing the Runx consensus sequence was used as labeled probe in all lanes. Competitor lanes (CPT) contained 100-fold excess of either WT (W) or mutant (M) Runx binding site oligonucleotides compared with control without specific competitor (−) are indicated. The arrows indicate the specific protein-DNA complexes for full-length (solid arrow) and truncated (open arrow) Runx2; NS represent a nonspecific band. (Right) Gel mobility-immunoshift analysis with (+) or without (−) the addition of 1 μl polyclonal Runx2 antibody (Ab) to detect WT and Runx2ΔC (ΔC) mutant proteins transiently expressed in HeLa cells. CON designates two control lanes for probe alone and probe plus antibody. V represents HeLa cell nuclear extracts transfected with the empty vector. The arrow indicates the supershifted complex. (B) Functional activity of the WT and ΔC Runx proteins on Runx-dependent promoters. (Left) HeLa cells were cotransfected with −1,097/+23 rat osteocalcin (OC), −1.0-kb transforming growth factor β R1, or −620/+25 bone sialoprotein (BSP) promoter-chloramphenicol acetyltransferase constructs together with either X-press-tagged WT Runx2 or Runx2ΔC expression plasmids (40–42). Promoter activities were determined 24 h posttransfection. Studies were repeated twice with n = 6 replicates with two different DNA preparations. Data were normalized to luciferase values. (Right) Western blot analysis of lysates from transfected cells from the same experiment. The X-press-tagged WT and mutant Runx proteins were detected by using α-X-press antibody from Invitrogen. Lamin B shows equal protein loading. (C) In vivo targeting of Runx2ΔC to subnuclear sites is compromised. Cells were obtained from calvarial tissue of WT and homozygous ΔC mutant mice at 17.5 dpc cultured for 2 days on glass coverslips and prepared for in situ immunohistochemistry of WT and Runx2ΔC proteins. WT cells (×100) show punctate staining of Runx2 in whole cells (WC), which is retained in the nuclear scaffold/NMIF preparations. Homozygous (ΔC/ΔC) cells (×100) also show distinct punctate foci in whole cells, but complete absence of Runx2ΔC in the NMIF preparations. The antibody control (no primary antibody) shows background fluorescence for comparison (Fig. 7, which is published as supplemental material for this data). Phase microscopy (Insets) shows cellular and nuclear morphology in WC and NMIF. Nuclei were stained with 4′,6-diamidino-2-phenylindole (0.5 μg/ml) to verify removal of DNA during the NMIF extraction procedure (see Fig. 7 for this data).
We established that the Runx2ΔC mutation has defective subnuclear targeting. We examined subnuclear localization of the Runx protein in cells harvested from the calvaria of WT and homozygous mice at 17.5 dpc to assess in situ accumulation of the WT and mutant proteins (Fig. 5C). Runx2 and Runx2ΔC each were detected in the nuclei of whole-cell preparations; thus the nuclear localization signal present in both proteins supports efficient targeting to the nucleus. However, WT protein, but not the ΔC mutant, remains associated with the nuclear scaffold (NMIF preparation). Therefore, the genetic lesion that generates a truncated C terminus and loss of the intranuclear targeting signal abrogates targeting to nuclear matrix-associated subnuclear domains.
Discussion
Regulatory and Clinical Implications.
We find that deletion of the C-terminal exon of the Runx2 gene encoding the intranuclear targeting signal results in a severe phenotype reflected by late embryonic lethality and absence of intramembranous and endochondral bone formation. Given that the Runx2ΔC mutant protein is expressed at normal levels and retains several functional properties including DNA binding, nuclear import, and interaction with its partner protein CBFβ (45, 46), the Runx2ΔC phenotype is quite remarkable because it is strikingly similar to that of mice in which the entire Runx2 gene is ablated (25, 26). One might have anticipated that the Runx2ΔC mutant mouse would display a less severe bone phenotype than the knockout mice, because the ΔC protein includes the runt homology domain required for DNA binding and the unique amino terminus (see Fig. 1) (27, 28, 39, 47, 48). The phenotype we observe in the Runx2ΔC mouse is directly attributable to reduced transcriptional activity and complete abrogation of Runx2 subnuclear targeting. Thus, the subnuclear targeting and regulatory activities encompassed in the C terminus of Runx2 are genetically required for osteogenesis and hence mediate critical regulatory functions for control of skeletal development.
Insight into the role of Runx2 in fetal development, as well as into the mechanism of Runx2 function in formation of the skeleton, is provided by comparison of the phenotypes of the ΔC mutant mouse and Runx2 null mouse. Although the skeletal phenotypes are indistinguishable, the ΔC homozygous mice die in utero rather than at birth as previously reported for the null mice. One possibility is that the Runx2 truncated protein has altered interactions with coregulatory proteins, which could result in embryonic lethality. However, it is important to note that the truncated Runx2ΔC protein does not function genetically as a dominant negative inhibitor of the WT protein, as the phenotype of the ΔC heterozygote strongly resembles and is no more severe than the phenotype of the heterozygous null mouse. The ΔC heterozygous mice exhibit absence of clavicles, which was previously attributed to haplo-insufficiency of the entire Runx2 gene product (25, 26). Hence, our data directly indicate that osteogenic regulatory functions intrinsic to the C terminus are critical for this aspect of skeletal development.
The C termini of Runx factors are responsive to several signaling pathways that participate in control of tissue-specific gene expression during development. These include bone morphogenetic protein signaling through Smads (38, 49, 50), Crk/Yes/Src signaling through WW domain-containing proteins (51, 52), and transcriptional repression by Groucho/TLE and Hes (14, 31, 53–55). Collectively the C termini of Runx factors integrate biological activities of coregulatory proteins required for formation of gene-specific complexes that are targeted to specialized subnuclear sites.
We propose that the regulatory defect in Runx2ΔC mice is due to misdirection of the mutant Runx2 protein within the nucleus. In support of this concept, we have demonstrated that a specific domain in the C termini of Runx1 and Runx2 is responsible for targeting of these factors to distinct subnuclear sites and that loss of the subnuclear targeting signal compromises transcriptional regulation of bone-specific genes (15, 17, 18). Transgenic rescue experiments (56) aimed at restoring the hematopoietic defect of Runx1 null mice further indicate the importance of subnuclear localization. These studies (56) revealed that the Runx domain mediating repression by Groucho/TLE (i.e., VWRPY motif) (14, 31, 53) is dispensable whereas the segment spanning the subnuclear targeting signal is critical for normal fetal development.
Mutations in Runx factors that cause human disease also are associated with aberrations in subnuclear targeting (12, 57, 58). Leukemia-associated translocations in Runx1 that eliminate the C terminus result in altered transcriptional functions and subnuclear targeting (21, 59). A nonsense mutation in the human Runx2 gene analogous to our knock-in mutation in the mouse gene produces a truncated protein lacking the subnuclear targeting signal and is linked to cleidocranial displasia (38). Interestingly, the C-terminal segment encoding the subnuclear targeting signal of Runx factors overlaps with a Smad interacting region (38) as well as a transactivation domain. The extent to which these C-terminal activities are functionally independent and structurally separable remains to be established. However, the molecular convergence of these regulatory functions within the same segment of Runx2 may indicate that integration of cell signaling, subnuclear targeting, and transcriptional activation reflects inseparable components of the same developmental regulatory mechanism. The genetic requirement for subnuclear localization of Runx proteins functionally links the intranuclear organization of regulatory factors with tissue-specific transcriptional control and with competency for embryonic development.
Supplementary Material
Acknowledgments
We thank Ernesto Canalis (St. Francis Hospital, Hartford, CT) for the use of the Faxitron x-ray and Charlene Baron (Univ. of Massachusetts Medical School) and Leslie Antinnarella (Univ. of Rochester) for assistance with digital imaging and photography. We are appreciative of the editorial assistance of Elizabeth Bronstein and Judy Rask. We thank members of the laboratory for stimulating discussions and support throughout the course of these studies and John Eisman (Garvin Institute, Australia) for critical review of the manuscript and thoughtful comments. These studies were supported by National Institutes of Health Grants AR39588, AR45688, CA82834, and AR43510.
Abbreviations
- WT
wild type
- RT
reverse transcription
- dpc
days postcoitum
- NMIF
nuclear matrix-intermediate filament
Footnotes
The nomenclature committee of the Human Genome Organization has adopted the following designations for Runt-related transcription factors: RUNX1 (AML1/CBFA2/PEBP2αB), RUNX2 (AML3/CBFA1/PEBP2αA), and RUNX3 (AML2/CBFA3/PEBP2αC).
References
- 1.Blencowe B J, Nickerson J A, Issner R, Penman S, Sharp P A. J Cell Biol. 1994;127:593–607. doi: 10.1083/jcb.127.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Steensel B, Jenster G, Damm K, Brinkmann A O, van Driel R. J Cell Biochem. 1995;57:465–478. doi: 10.1002/jcb.240570312. [DOI] [PubMed] [Google Scholar]
- 3.Penman S. Proc Natl Acad Sci USA. 1995;92:5251–5257. doi: 10.1073/pnas.92.12.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamond A I, Earnshaw W C. Science. 1998;280:547–553. doi: 10.1126/science.280.5363.547. [DOI] [PubMed] [Google Scholar]
- 5.Tang Y, Getzenberg R H, Vietmeier B N, Stallcup M R, Eggert M, Renkawitz R, DeFranco D B. Mol Endocrinol. 1998;12:1420–1431. doi: 10.1210/mend.12.9.0169. [DOI] [PubMed] [Google Scholar]
- 6.Wei X, Samarabandu J, Devdhar R S, Siegel A J, Acharya R, Berezney R. Science. 1998;281:1502–1505. doi: 10.1126/science.281.5382.1502. [DOI] [PubMed] [Google Scholar]
- 7.Cook P R. Science. 1999;284:1790–1795. doi: 10.1126/science.284.5421.1790. [DOI] [PubMed] [Google Scholar]
- 8.Misteli T, Spector D L. Mol Cell. 1999;3:697–705. doi: 10.1016/s1097-2765(01)80002-2. [DOI] [PubMed] [Google Scholar]
- 9.Stenoien D L, Mancini M G, Patel K, Allegretto E A, Smith C L, Mancini M A. Mol Endocrinol. 2000;14:518–534. doi: 10.1210/mend.14.4.0436. [DOI] [PubMed] [Google Scholar]
- 10.McNally J G, Muller W G, Walker D, Wolford R, Hager G L. Science. 2000;287:1262–1265. doi: 10.1126/science.287.5456.1262. [DOI] [PubMed] [Google Scholar]
- 11.Leonhardt H, Rahn H P, Weinzierl P, Sporbert A, Cremer T, Zink D, Cardoso M C. J Cell Biol. 2000;149:271–280. doi: 10.1083/jcb.149.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stein G S, Montecino M, van Wijnen A J, Stein J L, Lian J B. Cancer Res. 2000;60:2067–2076. [PubMed] [Google Scholar]
- 13.Nickerson J A. J Cell Sci. 2001;114:463–474. doi: 10.1242/jcs.114.3.463. [DOI] [PubMed] [Google Scholar]
- 14.Javed A, Guo B, Hiebert S, Choi J-Y, Green J, Zhao S-C, Osborne M A, Stifani S, Stein J L, Lian J B, et al. J Cell Sci. 2000;113:2221–2231. doi: 10.1242/jcs.113.12.2221. [DOI] [PubMed] [Google Scholar]
- 15.Zaidi, S. K., Javed, A., Choi, J.-Y., van Wijnen, A. J., Stein, J. L., Lian, J. B. & Stein, G. S. (2001) J. Cell Sci., in press. [DOI] [PubMed]
- 16.Tang L, Guo B, Javed A, Choi J-Y, Hiebert S, Lian J B, van Wijnen A J, Stein J L, Stein G S, Zhou G W. J Biol Chem. 1999;274:33580–33586. doi: 10.1074/jbc.274.47.33580. [DOI] [PubMed] [Google Scholar]
- 17.Zeng C, McNeil S, Pockwinse S, Nickerson J A, Shopland L, Lawrence J B, Penman S, Hiebert S W, Lian J B, van Wijnen A J, et al. Proc Natl Acad Sci USA. 1998;95:1585–1589. doi: 10.1073/pnas.95.4.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng C, van Wijnen A J, Stein J L, Meyers S, Sun W, Shopland L, Lawrence J B, Penman S, Lian J B, Stein G S, et al. Proc Natl Acad Sci USA. 1997;94:6746–6751. doi: 10.1073/pnas.94.13.6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merriman H L, van Wijnen A J, Hiebert S, Bidwell J P, Fey E, Lian J, Stein J, Stein G S. Biochemistry. 1995;34:13125–13132. doi: 10.1021/bi00040a025. [DOI] [PubMed] [Google Scholar]
- 20.Bidwell J P, van Wijnen A J, Fey E G, Dworetzky S, Penman S, Stein J L, Lian J B, Stein G S. Proc Natl Acad Sci USA. 1993;90:3162–3166. doi: 10.1073/pnas.90.8.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Speck N A, Stacy T, Wang Q, North T, Gu T L, Miller J, Binder M, Marin-Padilla M. Cancer Res. 1999;59:1789s–1793s. [PubMed] [Google Scholar]
- 22.Ito Y. Genes Cells. 1999;4:685–696. doi: 10.1046/j.1365-2443.1999.00298.x. [DOI] [PubMed] [Google Scholar]
- 23.Westendorf J J, Hiebert S W. J Cell Biochem. 1999;Suppl., 32–33:51–58. doi: 10.1002/(sici)1097-4644(1999)75:32+<51::aid-jcb7>3.3.co;2-j. [DOI] [PubMed] [Google Scholar]
- 24.Mundlos S, Otto F, Mundlos C, Mulliken J B, Aylsworth A S, Albright S, Lindhout D, Cole W G, Henn W, Knoll J H M, et al. Cell. 1997;89:773–779. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- 25.Otto F, Thornell A P, Crompton T, Denzel A, Gilmour K C, Rosewell I R, Stamp G W H, Beddington R S P, Mundlos S, Olsen B R, et al. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 26.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson R T, Gao Y-H, Inada M, et al. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 27.Banerjee C, McCabe L R, Choi J-Y, Hiebert S W, Stein J L, Stein G S, Lian J B. J Cell Biochem. 1997;66:1–8. doi: 10.1002/(sici)1097-4644(19970701)66:1<1::aid-jcb1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 28.Ducy P, Zhang R, Geoffroy V, Ridall A L, Karsenty G. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 29.Kim I S, Otto F, Zabel B, Mundlos S. Mech Dev. 1999;80:159–170. doi: 10.1016/s0925-4773(98)00210-x. [DOI] [PubMed] [Google Scholar]
- 30.Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, Sato M, Yamagiwa H, Kimura T, Yasui N, et al. Dev Dyn. 1999;214:279–290. doi: 10.1002/(SICI)1097-0177(199904)214:4<279::AID-AJA1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 31.Thirunavukkarasu K, Mahajan M, McLarren K W, Stifani S, Karsenty G. Mol Cell Biol. 1998;18:4197–4208. doi: 10.1128/mcb.18.7.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu J, Maruyama M, Satake M, Bae S C, Ogawa E, Kagoshima H, Shigesada K, Ito Y. Mol Cell Biol. 1995;15:1651–1661. doi: 10.1128/mcb.15.3.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones S N, Roe A E, Donehower L A, Bradley A. Nature (London) 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 34.Ramirez-Solis R, Liu P, Bradley A. Nature (London) 1995;378:720–724. doi: 10.1038/378720a0. [DOI] [PubMed] [Google Scholar]
- 35.Owen T A, Aronow M, Shalhoub V, Barone L M, Wilming L, Tassinari M S, Kennedy M B, Pockwinse S, Lian J B, Stein G S. J Cell Physiol. 1990;143:420–430. doi: 10.1002/jcp.1041430304. [DOI] [PubMed] [Google Scholar]
- 36.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1989. [Google Scholar]
- 37.Montecino M, Pockwinse S, Lian J, Stein G, Stein J. Biochemistry. 1994;33:348–353. doi: 10.1021/bi00167a045. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y W, Yasui N, Ito K, Huang G, Fujii M, Hanai J, Nogami H, Ochi T, Miyazono K, Ito Y. Proc Natl Acad Sci USA. 2000;97:10549–10554. doi: 10.1073/pnas.180309597. . (First Published August 29, 2000; 10.1073/pnas.180309597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stewart M, Terry A, Hu M, O'Hara M, Blyth K, Baxter E, Cameron E, Onions D E, Neil J C. Proc Natl Acad Sci USA. 1997;94:8646–8651. doi: 10.1073/pnas.94.16.8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang R, Gerstenfeld L C. J Cell Biochem. 1997;64:77–93. doi: 10.1002/(sici)1097-4644(199701)64:1<77::aid-jcb11>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 41.Javed A, Barnes G L, Jassanya B O, Stein J L, Gerstenfeld L, Lian J B, Stein G S. Mol Cell Biol. 2001;21:2891–2905. doi: 10.1128/MCB.21.8.2891-2905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ji C, Casinghino S, Chang D J, Chen Y, Javed A, Ito Y, Hiebert S W, Lian J B, Stein G S, McCarthy T L, et al. J Cell Biochem. 1998;69:353–363. [PubMed] [Google Scholar]
- 43.Meyers S, Lenny N, Sun W-H, Hiebert S W. Oncogene. 1996;13:303–312. [PubMed] [Google Scholar]
- 44.Ogawa S, Harada H, Fujiwara M, Tagashira S, Katsumata T, Takada H. DNA Res. 2000;7:181–185. doi: 10.1093/dnares/7.3.181. [DOI] [PubMed] [Google Scholar]
- 45.Kagoshima H, Akamatsu Y, Ito Y, Shigesada K. J Biol Chem. 1996;271:33074–33082. doi: 10.1074/jbc.271.51.33074. [DOI] [PubMed] [Google Scholar]
- 46.Yergeau D A, Hetherington C J, Wang Q, Zhang P, Sharpe A H, Binder M, Marin-Padilla M, Tenen D G, Speck N A, Zhang D E. Nat Genet. 1997;15:303–306. doi: 10.1038/ng0397-303. [DOI] [PubMed] [Google Scholar]
- 47.Xiao Z S, Hinson T K, Quarles L D. J Cell Biochem. 1999;74:596–605. [PubMed] [Google Scholar]
- 48.Harada H, Tagashira S, Fujiwara M, Ogawa S, Katsumata T, Yamaguchi A, Komori T, Nakatsuka M. J Biol Chem. 1999;274:6972–6978. doi: 10.1074/jbc.274.11.6972. [DOI] [PubMed] [Google Scholar]
- 49.Attisano L, Wrana J L. Curr Opin Cell Biol. 2000;12:235–243. doi: 10.1016/s0955-0674(99)00081-2. [DOI] [PubMed] [Google Scholar]
- 50.Hanai J, Chen L F, Kanno T, Ohtani-Fujita N, Kim W Y, Guo W H, Imamura T, Ishidou Y, Fukuchi M, Shi M J, et al. J Biol Chem. 1999;274:31577–31582. doi: 10.1074/jbc.274.44.31577. [DOI] [PubMed] [Google Scholar]
- 51.Yagi R, Chen L F, Shigesada K, Murakami Y, Ito Y. EMBO J. 1999;18:2551–2562. doi: 10.1093/emboj/18.9.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen H I, Sudol M. Proc Natl Acad Sci USA. 1995;92:7819–7823. doi: 10.1073/pnas.92.17.7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levanon D, Goldstein R E, Bernstein Y, Tang H, Goldenberg D, Stifani S, Paroush Z, Groner Y. Proc Natl Acad Sci USA. 1998;95:11590–11595. doi: 10.1073/pnas.95.20.11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen G, Courey A J. Gene. 2000;249:1–16. doi: 10.1016/s0378-1119(00)00161-x. [DOI] [PubMed] [Google Scholar]
- 55.McLarren K W, Lo R, Grbavec D, Thirunavukkarasu K, Karsenty G, Stifani S. J Biol Chem. 2000;275:530–538. doi: 10.1074/jbc.275.1.530. [DOI] [PubMed] [Google Scholar]
- 56.Okuda T, Takeda K, Fujita Y, Nishimura M, Yagyu S, Yoshida M, Akira S, Downing J R, Abe T. Mol Cell Biol. 2000;20:319–328. doi: 10.1128/mcb.20.1.319-328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McNeil S, Zeng C, Harrington K S, Hiebert S, Lian J B, Stein J L, van Wijnen A J, Stein G S. Proc Natl Acad Sci USA. 1999;96:14882–14887. doi: 10.1073/pnas.96.26.14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meyers S, Hiebert S W. J Cell Biochem. 2000;35:93–98. doi: 10.1002/1097-4644(2000)79:35+<93::aid-jcb1131>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 59.McNeil S, Javed A, Harrington K S, Lian J B, Stein J L, van Wijnen A J, Stein G S. J Cell Biochem. 2000;79:103–112. [PubMed] [Google Scholar]
- 60.Lufkin T, Mark M, Hart C P, Dolle P, LeMeur M, Chambom P. Nature (London) 1992;359:835–841. doi: 10.1038/359835a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}