

Abstract
Computational design of protein-ligand interfaces finds optimal amino acid sequences within a small molecule binding site of a protein for tight binding of a specific small molecule. It requires a search algorithm that can rapidly sample the vast sequence and conformational space, and a scoring function that can identify low energy designs. This review focuses on recent advances in computational design methods and their application to protein-small molecule binding sites. Strategies for increasing affinity, altering specificity, creating broad-spectrum binding, and building novel enzymes from scratch are described. Future prospects for applications in drug development are discussed, including limitations that will need to be overcome in order to achieve computational design of protein therapeutics with novel modes of action.
Introduction
Protein-based therapeutics are an important part of the current pharmacological arsenal. Proteins offer significant advantages over small molecules, including high specificity, low cross-reactivity and off-target effects, novel modes of action, and better patient tolerance [1,2]. As of 2008, >130 therapeutic proteins had been approved for use in humans for treatment of >30 different diseases [1,3]. Their functions are quite diverse, and include replacing deficient or defective proteins (e.g. insulin for treatment of diabetes); sequestering ligands (e.g. etanercept, a tumor necrosis factor-α inhibitor for treatment of various autoimmune diseases); blocking receptor interactions (e.g. anakinra, an interleukin (IL)-1 receptor antagonist for management of rheumatoid arthritis); stimulating signaling pathways (e.g. erythropoietin, a erythropoiesis stimulator for treatment of anemia); delivering other molecules to sites of action (e.g. denileukin diftitox, a fusion of IL-2 and diphtheria toxin for treatment of cutaneous T-cell lymphoma); and serving as in vivo diagnostics (e.g. capromab pendetide, an anti-prostate specific antigen antibody for prostate cancer detection) [1]). The market for clinical protein therapeutics, some $94 billion in 2010, is expected to grow to half of total prescription drug sales by 2014 [2].
Antibodies are the dominant class of biologics with >25 approved for use, including several that are blockbuster drugs and over 200 in clinical studies [4]. Their popularity partly results from their ability to bind to a wide range of protein, peptide, and small-molecule targets with both high affinity and high specificity. However, antibodies also have various disadvantages that stem from the fact that they are large, glycosylated proteins with multiple chains and disulfide linkages [5]. Consequently, there is considerable interest in designing ligand binding sites within non-immunoglobulin scaffolds for clinical applications [6] (Box 1).
Box 1. Engineered protein scaffolds as alternatives to antibody-based drugs.
A variety of scaffolds, most of which are small, soluble monomeric proteins or protein domains, have been used for designing “next generation” antibody therapeutics [69,70]. These reengineered molecules bind specific targets with high affinities and provide several practical advantages over antibodies, including high yields in microbial expression systems and the ability to fine-tune their properties in vitro. Moreover, non-immunoglobulin binding proteins are particularly well-suited for applications, such as in vivo diagnostics, because their smaller sizes allow for better tissue penetration as well as rapid clearance, which is important for reducing background in imaging. Short plasma half-life is also an advantage for creating reagents that can bind toxic molecules. A modified lipocalin that binds digoxigenin with subnanomolar affinity has been shown to completely reverse digitalis overdosing in animal models [71].
More than 10 engineered protein scaffolds are in clinical trials [72], and Kalbitor (ecallantide), a 60 amino acid Kunitz domain that inhibits plasma kallikrein, has recently been approved by the US Food and Drug Administration for the treatment of acute attacks of hereditary angioedema [73]. Thus far, such reengineered protein scaffolds have been generated by in vitro directed evolution methods. However, they represent potential design targets for computational methods.
Controlled manipulation of the physical and chemical properties of proteins is crucial for drug development. Computational protein design offers a useful strategy not only for optimizing properties of lead candidates, such as stability (Box 2), and for developing novel reagents through the design of new functions. Moreover, unlike screening methods (e.g. directed evolution), computational design provides a general approach that also tests and expands our understanding of the fundamental forces that underlie protein stability, structure, folding, and function.
Box 2. Increasing stability through computational protein design.
One factor that often limits the efficacy of protein therapeutics is their stability. Computational design offers an automated way of improving the stability of proteins, and has been recently applied to bacterial cocaine esterase (CocE), a potential candidate for treatment of cocaine overdose and addiction. CocE is the most efficient cocaine-degrading enzyme characterized thus far and provides effective protection and reversal of cocaine toxicity in mice [74]. However, CocE is unstable at physiological temperatures (in vitro half-life ~13 min. at 37°C) [75], which severely limits its development as a therapeutic agent.
CocE presents distinct challenges for computational design since it is relatively large (574 amino acids), contains 3 domains, and is an enzyme. To help identify sites that could be altered without affecting the structure or dynamics of the active site, MD simulations were performed at a high temperature [76]. They reveal conformational changes in stretch of ~30 residues adjacent to the active site that might lead to enzyme inactivation. Residues within this region have been selected for computational redesign, and 34 mutations were predicted to be stabilizing [77].
The most thermostable variant is a double mutant (L169K/G173Q) which displays an in vitro half-life of 2.9 days at 37°C and temperature inactivation at 42–48°C [78]. Pretreatment of mice with L169K/G173Q provides improved protection from cocaine-induced lethality and suppresses the reinforcing properties of cocaine [78].
Although the in vitro half-life improves by 340-fold, the serum half-life of L169K/G173Q is similar to WT (2.3 vs. 2.2 hr). The pharmacokinetic properties must be improved for L169K/G173Q to become a viable therapeutic agent. The authors note that further modification of L169K/G173Q such as PEGylation [79], may help increase its half-life in vivo.
Although the ultimate goal of automated design of binding sites to any target is still largely out of reach, recent years have witnessed the successful execution of a number of proof-of-concept experiments. These include the design of metal binding sites [7,8], non-biological cofactor binding sites [9], protein-protein interactions [10,11], protein-peptide interactions [12,13], protein-DNA interactions [14], and novel enzymes [15–18] (Box 3). Here, we review the state of computational design of protein-ligand interfaces, including current capabilities, challenges in the field, and prospects for protein drug development. For this review, we consider ligands that are typically small organic molecules of ≤ 1,000 Da.
Box 3. Lessons learned from de novo enzyme design.
Four of the computational designs that catalyze the Kemp elimination of 5-nitrobenzisoxazole have been studied using mixed quantum and molecular mechanics calculations [80]. The single-step catalytic mechanism is computed to be identical to reference reactions that are catalyzed by the hydroxide ion or glutamate in water. No new intermediates are formed in the enzymatic processes. Hence, the authors have concluded that further improvement of the designs requires optimizing interactions within the active site and increasing the reactivity of the catalytic base.
Directed evolution of a computationally designed Kemp eliminase produces a >200-fold increase in catalytic efficiency through incorporation of up to eight mutations [17,81]. However, these changes also decrease thermodynamic stability and reduce activity at elevated temperatures [81]. The crystal structure of an evolved variant shows conformational changes in one of the molecules within the asymmetric unit, which suggests increased flexibility within the active site. These results illustrate the difficulty of predicting the effect of stability and dynamics on catalysis.
Kinetic studies of the most active retro-aldolase designs found that the pKa values of the catalytic lysine are shifted, as predicted, but contribute only ~10-fold to the rate accelerations [60]. Hydrophobic substrate binding interactions contribute ~500-fold to the rate acceleration, whereas an explicitly-bound water designed to aid in proton shuffling is not involved in catalysis. Tight product inhibition and lack of stereospecificity are observed, which suggest that positioning of substrate interactions and catalytic groups needs to be optimized.
MD simulations of a different retro-aldol design have been conducted, in which product inhibition is not a limiting factor [82]. Fluctuations within the active site distort its geometry and prevent proton abstraction by the designed His-Asp dyad. These results indicate that protein dynamics play an important role in the catalytic efficiency of this enzyme.
Taken together, these studies indicate that more finely-tuned geometries and a better understanding of dynamics in the active site are needed to improve catalytic efficiencies of designed enzymes. Methods for increased conformational sampling should aid design efforts, although it is unclear whether existing protein scaffolds can be easily remodeled to accommodate desired active site features. New methods for constructing large numbers of de novo backbone structures have recently been described [83], and these templates may provide better starting points for introducing novel functionalities.
Methods for computational interface design
Computational protein design is often described as an inverse-folding problem, the goal of which is to identify amino acid sequences that are compatible with a given 3D protein structure. For interface design, structures of the protein scaffold and ligand are inputted, and the design algorithm proceeds through repeated rounds of sequence-conformation searching, followed by scoring of each resultant model (Figure 1). If a given model does not meet the predetermined scoring criteria, it undergoes further perturbation by the search algorithm. The cycle continues until a model meets the scoring criteria and is outputted as a sequence and/or 3D protein-ligand model. Typically, several thousand models are outputted for iterative rounds of design and evaluation.
Figure 1.
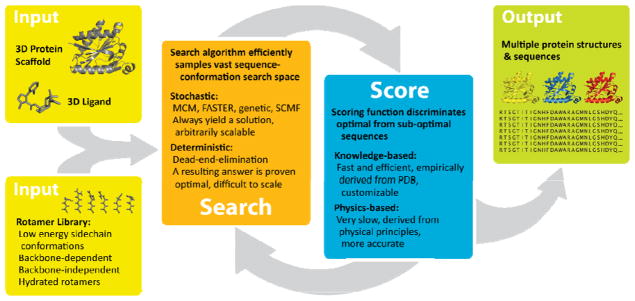
General components of an interface design algorithm, with protein and ligand structures as inputs for design. Rotamer libraries of statistically probable conformations of amino acid side chains or ligands reduce the degrees-of-freedom of the search. After multiple cycles of sequence-conformation searching followed by scoring, models are outputted that meet scoring criteria.
Design methods share two general components: a search algorithm to sample efficiently the vast sequence–conformation space, and a scoring function to discriminate optimal from sub-optimal sequences (Figure 1). Systematic or brute-force searching of all possible sequence and conformational permutations is not possible, even for relatively simple systems; therefore, several approximations are typically made. First, amino acid side chains are represented by a set of discrete conformations called rotamers. Rotamers are derived from the most frequently observed conformations seen in the Protein Data Bank (PDB) [19], and rotamer libraries can be dependent or independent of the local backbone conformation. Second, the protein backbone is often kept rigid during the actual design procedure; some protocols minimize all degrees of freedom after completion of the design procedure, or in an iterative fashion [20]. Third, iterative rounds of design with increasing resolution are used to focus the conformation and sequence search.
The search space algorithms can be further classified as either deterministic or stochastic (Figure 1). Deterministic methods, such as dead-end elimination, do not always arrive at a solution, however when they do, it can be mathematically proven to be the global energy minimum [21]. Stochastic search algorithms, such as Monte Carlo-Metropolis (MCM) with simulated annealing [22,23], fast and accurate side chain topology and energy refinement (FASTER) [24], genetic algorithms [25], and self-consistent mean-field optimization (SCMF) [26], will always find a solution to a search query; however, the solution is not guaranteed to be mathematically optimal.
The energy functions used to score and evaluate protein sequence-structure models represent a compromise between speed and accuracy (Figure 1). Physics-based energy potentials rely on accurate models of the basic forces that constitute the free energy of a protein [27], but are computationally expensive to be used for design. Knowledge-based energy potentials are derived through statistical analysis of structures deposited in the PDB and capture large amounts of empirically-derived data into efficient mathematical functions [28,29]. In practice, most design programs use some combination of both. Explicit modeling of individual water molecules is impractical, thus the solvent is treated implicitly as a continuum (e.g. by a desolvation penalty for burial of polar groups, as in the Rosetta algorithm [30]). Van der Waals interactions are typically described by a Lennard-Jones potential that is sometimes softened so that it is less sensitive to small atom overlaps caused by fixed rotamer sampling. Hydrogen bonds are explicitly considered because the strength of the interaction is dependent on both distance and orientation between donor and acceptor groups. The energies of designed sequences are determined with respect to a reference state, usually the unfolded protein [31]; however, most algorithms ignore the effect of mutations on the unfolded state, and instead, represent it through a constant amino acid reference energy [28]. The relative weights of the various energy terms are adjusted empirically to match experimental data.
To increase efficiency, design algorithms require pair-wise decomposable terms (i.e. no interaction involves more than two functional groups). This procedure allows determination of the interaction energy between two amino acid side chains, independent from all others. As a result, pair-wise interaction energies between all possible side chains and conformations in all positions can be pre-computed and stored in a database. The actual design simulation relies on look-up and summation of these energy terms. This strategy is impractical for higher-order interactions because their number increases exponentially.
Design protocols have difficulty modeling flexibility at binding interfaces
Traditionally, protein design has relied on methods that approximate the lock-and-key (LK) model of binding (Figure 2). Protein backbones are held fixed, and only residue side chains are allowed to change conformation. In some cases, small φ/ψ angle adjustments are allowed on the protein backbone during gradient minimization of the ligand complex to accommodate slight changes in conformation. The magnitude of these changes is small; therefore, these methods severely restrict the diversity of sequences that can be designed. These limitations in sampling can be mitigated by using expanded rotamer libraries at the cost of increased computing times [32,33].
Figure 2.
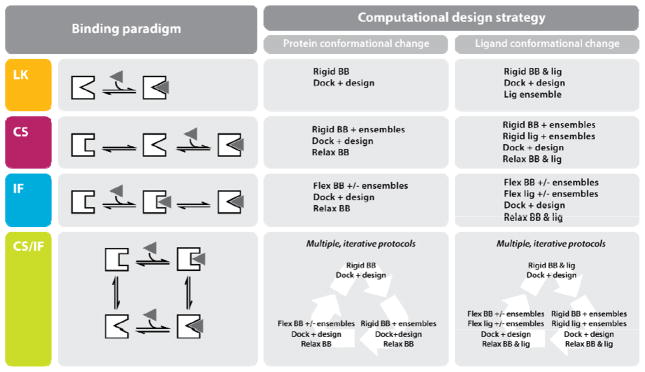
Ligand binding paradigms and corresponding computational design strategies. The four binding paradigms outlined in the text are listed in the colored badges, with schematics immediately to the right: LK (orange); CS (purple); IF (blue); hybrid CS/IF (green). Possible computational design strategies are presented for each binding mode. Abbreviations: BB, backbone; lig, ligand.
Conformational mobility is an intrinsic property of proteins that allows them to adjust upon binding. In crystal structures of protein–ligand complexes, 70–100% of the ligand is usually buried [34], which is consistent with induced fit (IF) models of binding (Figure 2). It has been shown that using a single, static receptor conformation in molecular docking algorithms results in incorrect binding poses for 50–70% of all ligands [35]. Thus, for the computational design of protein-ligand interfaces to progress, structural plasticity of the interacting partners needs to be considered. Incorporation of backbone flexibility into protein design is non-trivial because it massively increases the search space and also requires scoring functions that can discriminate between alternate backbone conformations. However, recent designs of protein-peptide, protein-protein and protein-small molecule interfaces illustrate some of the methodological advances in sampling strategies.
Protein backbone flexibility
The conformational selection (CS) model of binding proposes that unbound receptors exist as an ensemble of conformers and that energetically preferred states are selected upon ligand binding [36] (Figure 2). Normal mode analysis has been used to create a backbone ensemble, which enables the design of more diverse BH3 peptide sequences that bind the anti-apoptotic protein Bcl-xl [37]. Structural ensembles generated from either NMR data (60 backbones) or molecular dynamics (MD) simulations (128 backbones) have been successfully used as inputs for design using a new FASTER search algorithm [38].
Sampling correlated side chain–backbone ‘backrub’ motions that are frequently seen in high-resolution crystal structures [39] has allowed for efficient approximation of local conformational changes [40,41]. Small backrub moves improve modeling of side chain order parameters obtained from NMR experiments [42]. Moreover, larger amplitude backrub moves have been used to obtain backbone ensembles of ubiquitin that are consistent with native-state dynamics measured by residual dipolar couplings [43].
A method that uses constrained backbone sampling to remodel flexible loops has been used to alter the substrate specificity of human guanine deaminase [44]. The goal is to introduce cytosine deaminase activity into a human scaffold to create a prodrug- activating enzyme with low immunogenicity, for use in suicide gene chemotherapy. Remodeling of a critical active site loop produces an enzyme that is 25,000-fold less active on guanine than the wild type (WT) enzyme and 100-fold more active on ammelide, a structural intermediate between guanine and cytosine. An X-ray structure of the designed apoprotein shows that the backbone root mean square deviation (RMSD) between the remodeled loop and the computational model is within 1Å. The authors note that further optimization of the substrate binding interface may require flexible backbone design of the surrounding regions to increase catalytic activity.
Ligand flexibility
In addition to protein flexibility, efforts have been made to develop methods for accommodating ligand flexibility in interface design [45,46]. The use of rotamer libraries for small molecules is advantageous because it aligns with the rotamer libraries that are used for sampling amino acid side chains. Although this procedure is relatively straight-forward for ligands that contain standard amino acids, Kaufmann et al. have demonstrated that modeling of ligands with more than four rotatable bonds requires splitting the ligand into multiple fragments for dense sampling [47]. The authors have created ligand rotamers using the Cambridge Structural Database (CSD) of small molecules. Statistically derived potentials similar to those for the protein design energy functions have been generated and used to populate a predefined ligand rotamer library. By combining the techniques to model ligand flexibility with those to model protein flexibility, both the IF binding model and a hybrid conformational selection/induced fit (CS/IF) binding model can be approximated [48] (Figure 2).
Interface dynamics
A recent study has highlighted the still little understood influence of protein dynamics on designing a ligand interface de novo. Mutations introduced into a thermophilic jellyroll template (1m4w) to achieve binding to a ligand altered the dynamics observed in the binding site [49] (Figure 3). A high-resolution X-ray structure of one of the designs (1m4w_6) was determined and revealed an unanticipated expansion of the binding pocket. Increased temperature factors (B-factors) in two regions on either side of the interface were also observed (Figure 3). B-factors reflect the thermal vibrations of an atom and are indicative of dynamic flexibility. Two residues of 1m4w_6 were mutated back to WT (1m4w_6w20v48), which closed the binding pocket as predicted but failed to restore the rigidity of the structure (Figure 3). Increased dynamics at the designed interface might explain the absence of high-affinity ligand binding.
Figure 3.
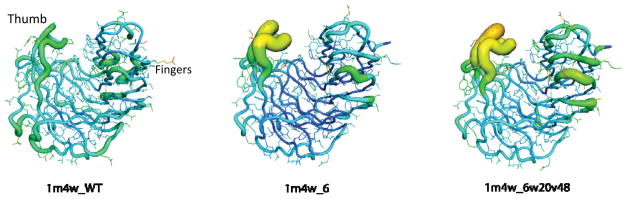
Crystallographic B-factors mapped onto X-ray structures of 1m4w designs. WT protein (1m4w_WT) and 2 design variants (1m4w_6 and 1m4w_6w20v48) are shown as ribbon diagrams, in which the width and color are proportional to the B-factor at each residue (values increase from blue to red and as ribbon gets wider). The protein resembles a hand with the binding pocket located in a cleft between the ‘thumb’ and ‘fingers’. Higher B-factors in the thumb and fingers of the designed structures indicate a shift in the dynamics of the binding pocket.
Predicting the effects of internal dynamics on computationally designed models is an inherent limitation of the design process. Computational design methods ignore changes in protein dynamics, and although sets of discrete conformations are sampled, the time scale of motions is not considered. New MD approaches help define the dynamic properties of interfaces upon binding [50–52]. Unfortunately, MD methods are typically computationally too expensive to be incorporated in sequence-conformation sampling during protein design. Present strategies use MD to generate conformational ensembles beforehand [38,53] or to analyze selected models afterwards [54]. Finding ways of integrating dynamics into ligand-interface design will be important for achieving precise placement of functional groups and improving the activity of de novo designed enzymes (Box 3).
Accurate modeling of solvation and electrostatics at interfaces is critical
Electrostatic interactions, such as salt bridges and hydrogen bonds, are often essential for binding specificity and catalysis, but are difficult to model because their strength is determined by their environment (i.e. they are not pair-wise additive). Moreover, electrostatics are coupled tightly to solvation. Polar residues on the surface interact with solvent molecules; so, in order to form intermolecular contacts, the gain in energy must be sufficient to overcome the cost of desolvation. The challenge is to model solvation energies and electrostatic effects rapidly, yet accurately.
For continuum solvent models, the electrostatic potential of an amino acid residue is most accurately measured by solving the Poisson-Boltzmann (PB) equation [55]. In PB calculations, the protein is treated as a low dielectric solute that contains point charges surrounded by a high dielectric medium. The PB model captures the environment-dependent nature of electrostatic interactions, but is computationally too expensive to be used during the protein design protocol. Generalized Born (GB) models offer a fast approximation to the PB equation [56] and have been used in protein design. The unmodified physics-based CHARMM22 molecular mechanics potential energy function and GB models of solvation have been used to redesign the ligand binding site of ribose binding protein [33]. It has been found that ligand interactions with polar groups in the protein are almost exactly counterbalanced by interactions with water in the unbound protein; using a less-accurate GB model produces sequences that bind poorly to ribose. Likewise, the added computational expense of including accurate GB models has been necessary to alter the cofactor specificity of a xylose reductase [57].
Strategies employed in the redesign of antibody-antigen interactions can help guide the design of high-affinity protein-protein interfaces. An iterative computational design procedure that focuses on electrostatics has been used to introduce point mutations that improve target binding through one of two mechanisms: (i) by replacing a poorly satisfied hydrogen bond donor/acceptor with a hydrophobic residue or (ii) by introducing a charged interaction at the binding site periphery [58]. Combination of designed mutations has led to a 140-fold improvement in a lysozyme-binding antibody and a 10-fold improvement in cetuximab, a therapeutic antibody that binds to epidermal growth factor receptor, and that is used for treatment of metastatic colorectal cancer and squamous cell carcinoma. The authors also have shown that computed electrostatics alone using a PB model is a better predictor of stabilizing mutations than total free energy [58]. The identification of known affinity-enhancing mutations in the anti-fluorescein antibody 4-4-20 demonstrates the applicability to small-molecule haptens, although other mutations that are predicted to improve fluorescein binding remain to be tested.
Explicit water molecules
Continuum solvation models fail to capture the energetics of tightly bound water molecules, which can be problematic if individual water molecules are directly involved in ligand interactions or catalysis. One solution is to use solvated rotamers that include the most common positions for coordinated water atoms, as observed in the PDB [59]. Solvated rotamers are, however, limited by the fixed orientation of the water in relation to the coordinating side chain atoms. Furthermore, bridging water molecules that are bound by more than one residue cannot be accommodated. Improved modeling of explicit water molecules should aid protein-ligand design efforts. Inclusion of an explicit water molecule in the active site significantly improves the success rate in designs of retro-aldol enzymes [16], although the water molecule does not appear to contribute to catalysis [60] (Box 3).
Designing for specificity versus promiscuity
Therapeutic proteins must be able to recognize their targets in the context of crowded cellular environments. This requires high specificity in addition to stability. Positive design alone has been sufficient to achieve specific binding when the structure of the desired complex is significantly different from undesirable ones [13]. However, explicit design against competing states (i.e. negative design) is critical when the structures of the target and off-target complexes are similar [61–63]. Although most studies have designed against one or a few alternative complexes, Grigoryan et al. have conducted a large-scale experiment that has combined integer linear programming with cluster expansion to maximize the energy difference between target and off-target complexes, while minimizing losses in stability [12]. Using these methods, basic leucine zipper (bZIP)-binding peptides selective for targets over all other 19 bZIP families have been designed. Multi-state design has also been applied to large structural ensembles [38].
Computational methods have been developed that enable the design of a single protein sequence that binds to multiple targets. For example, a ‘multi-constraint’ design has been used to optimize binding of promiscuous interfaces to all known partners simultaneously [64]. These results can then be compared to interfaces that are redesigned against each interaction partner separately. These studies have revealed two different strategies for binding: shared interfaces, in which a small subset of residues form ‘hot spots’ that are used by all binding partners, and multi-faceted interfaces, in which different subsets of residues are used by each binding partner. Shared interfaces might be better small-molecule targets; as such, it might be possible to predict mutations at specific positions that alter specificity or promiscuity.
De novo enzyme design
Although the automated design of protein-ligand interfaces is ‘not a solved problem’ [65], there has been exciting progress in the computational design of new enzyme active sites. To this end, a series of papers has been published on the de novo design of enzymes that catalyze Kemp elimination [17], retro-aldol cleavage [16], and Diels-Alder reactions [18]. The basic strategy is to first build a model of the reaction transition state surrounded by suitably placed catalytic groups. For multiple step reactions, such as retrol-aldol cleavage, the active site is described by a composite of superimposed transition states and intermediates [16]. One may then search a set of protein scaffolds for potential positions that can retain the active site geometry. After grafting the active site onto a selected candidate, the protein is redesigned to optimize transition state binding affinity [66].
High-resolution crystal structures of active designs confirm the atomic accuracy of the design process. Still, the designed proteins are rather poor catalysts compared to naturally-occurring enzymes. Follow-up studies have helped to reveal the origins of catalytic efficiency for the computationally designed Kemp eliminases and retro- aldolases, and to identify reasons for the reduced activities (Box 3).
Concluding remarks
Although computational design holds great potential for the development of new protein-based therapeutics with novel modes of action, many challenges remain. In order to achieve de novo design of protein-ligand interfaces, technological advances are needed in: (i) accommodating backbone and ligand flexibility; (ii) developing rapid methods to accurately model electrostatics and solvation; and (iii) explicit modeling of cofactors and water molecules at the binding interface. In particular, comprehensive benchmark systems are needed to monitor methodological progress in all three areas. Modeling dynamic modes of binding partners in bound and unbound states during the design procedure remains computationally intractable for the time being. Experimental and computational analysis of the dynamics of starting scaffold and designed proteins should be conducted to build a body of data; such data will help to adjust computational design protocols to better account for protein and ligand dynamics during the design simulation. High-resolution structures of designed interfaces together with detailed characterization of both successful and unsuccessful designs will be critical for improving computational methods.
Using computational methods in conjunction with functional screening techniques may be the most effective way to design protein drugs. In silico methods can explore much larger portions of sequence space than can be accessed experimentally, and can be used to design targeted libraries that are enriched in functional sequences [64,67,68]. Directed evolution, on the other hand, allows high throughput identification of lead candidates even if the underlying mechanisms of action are not well understood. Iterating between computational and experimental techniques should also provide greater insights into structure-dynamic-activity relationships that will further inform protein therapeutic development.
References
- 1.Leader B, et al. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 2.Strohl WR, Knight DM. Discovery and development of biopharmaceuticals: current issues. Curr Opin Biotechnol. 2009;20:668–672. doi: 10.1016/j.copbio.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 3.Aggarwal S. What’s fueling the biotech engine--2008. Nat Biotechnol. 2009;27:987–993. doi: 10.1038/nbt1109-987. [DOI] [PubMed] [Google Scholar]
- 4.Reichert JM. Antibody-based therapeutics to watch in 2011. MAbs. 2011;3:76–98. doi: 10.4161/mabs.3.1.13895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Binz HK, et al. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 6.Gebauer M, Skerra A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr Opin Chem Biol. 2009;13:245–255. doi: 10.1016/j.cbpa.2009.04.627. [DOI] [PubMed] [Google Scholar]
- 7.Calhoun JR, et al. Solution NMR structure of a designed metalloprotein and complementary molecular dynamics refinement. Structure. 2008;16:210–215. doi: 10.1016/j.str.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fazelinia H, et al. OptGraft: A computational procedure for transferring a binding site onto an existing protein scaffold. Protein Sci. 2009;18:180–195. doi: 10.1002/pro.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fry HC, et al. Computational design and elaboration of a de novo heterotetrameric alpha-helical protein that selectively binds an emissive abiological (porphinato)zinc chromophore. J Am Chem Soc. 2010;132:3997–4005. doi: 10.1021/ja907407m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sammond DW, et al. Computational design of second-site suppressor mutations at protein-protein interfaces. Proteins. 2010;78:1055–1065. doi: 10.1002/prot.22631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humphris EL, Kortemme T. Design of multi-specificity in protein interfaces. PLoS computational biology. 2007;3:1591–1604. doi: 10.1371/journal.pcbi.0030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grigoryan G, et al. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yosef E, et al. Computational design of calmodulin mutants with up to 900-fold increase in binding specificity. J Mol Biol. 2009;385:1470–1480. doi: 10.1016/j.jmb.2008.09.053. [DOI] [PubMed] [Google Scholar]
- 14.Ashworth J, et al. Computational reprogramming of homing endonuclease specificity at multiple adjacent base pairs. Nucleic Acids Res. 2010;38:5601–5608. doi: 10.1093/nar/gkq283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faiella M, et al. An artificial di-iron oxo-protein with phenol oxidase activity. Nat Chem Biol. 2009;5:882–884. doi: 10.1038/nchembio.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang L, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 18.Siegel JB, et al. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science. 2010;329:309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunbrack R. Rotamer Libraries in the 21st Century. Curr Opin Struct Biol. 2002;12:431–440. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- 20.Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 21.Desmet J, et al. The dead-end elimination theorem and its use in protein side-chain positioning. Nature. 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 22.Metropolis N, et al. Equation of state calculation by fast computing machines. J Chem Phys. 1953;21:1087–1092. [Google Scholar]
- 23.Kirkpatrick S, et al. Optimization by Simulated Annealing. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 24.Allen BD, Mayo SL. Dramatic performance enhancements for the FASTER optimization algorithm. J Comput Chem. 2006;27:1071–1075. doi: 10.1002/jcc.20420. [DOI] [PubMed] [Google Scholar]
- 25.Desjarlais JR, Handel TM. De novo design of the hydrophobic cores of proteins. Protein Sci. 1995;4:2006–2018. doi: 10.1002/pro.5560041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koehl P. Mean-field minimization methods for biological macromolecules. Curr Opin Struct Biol. 1996;6:222–226. doi: 10.1016/s0959-440x(96)80078-9. [DOI] [PubMed] [Google Scholar]
- 27.Mackerell AD. Empirical force fields for biological macromolecules: overview and issues. J Comput Chem. 2004;25:1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 28.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci U S A. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dehouck Y, et al. A new generation of statistical potentials for proteins. Biophys J. 2006;90:4010–4007. doi: 10.1529/biophysj.105.079434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 31.Boas FE, Harbury PB. Potential energy functions for protein design. Curr Opin Struct Biol. 2007;17:199–204. doi: 10.1016/j.sbi.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Reynolds KA, et al. Computational redesign of the SHV-1 beta-lactamase/beta-lactamase inhibitor protein interface. J Mol Biol. 2008;382:1265–1275. doi: 10.1016/j.jmb.2008.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boas FE, Harbury PB. Design of protein-ligand binding based on the molecular-mechanics energy model. J Mol Biol. 2008;380:415–424. doi: 10.1016/j.jmb.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teague SJ. Implications of protein flexibility for drug discovery. Nat Rev Drug Discov. 2003;2:527–541. doi: 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- 35.Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol. 2008;18:178–184. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hammes GG, et al. Conformational selection or induced fit: a flux description of reaction mechanism. Proc Natl Acad Sci U S A. 2009;106:13737–13741. doi: 10.1073/pnas.0907195106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu X, et al. Modeling backbone flexibility to achieve sequence diversity: the design of novel alpha-helical ligands for Bcl-xL. J Mol Biol. 2007;371:1099–1117. doi: 10.1016/j.jmb.2007.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen BD, et al. Experimental library screening demonstrates the successful application of computational protein design to large structural ensembles. Proc Natl Acad Sci U S A. 2010;107:19838–19843. doi: 10.1073/pnas.1012985107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis IW, et al. The backrub motion: how protein backbone shrugs when a sidechain dances. Structure. 2006;14:265–274. doi: 10.1016/j.str.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 40.Smith CA, Kortemme T. Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J Mol Biol. 2008;380:742–756. doi: 10.1016/j.jmb.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Georgiev I, et al. Algorithm for backrub motions in protein design. Bioinformatics. 2008;24:i196–i204. doi: 10.1093/bioinformatics/btn169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friedland GD, et al. A simple model of backbone flexibility improves modeling of side-chain conformational variability. J Mol Biol. 2008;380:757–774. doi: 10.1016/j.jmb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friedland GD, et al. A correspondence between solution-state dynamics of an individual protein and the sequence and conformational diversity of its family. PLoS Comput Biol. 2009;5:1–16. doi: 10.1371/journal.pcbi.1000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murphy PM, et al. Alteration of enzyme specificity by computational loop remodeling and design. Proc Natl Acad Sci U S A. 2009;106:9215–9220. doi: 10.1073/pnas.0811070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meiler J, Baker D. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 46.Hartmann C, et al. Docking and scoring with alternative side-chain conformations. Proteins. 2009;74:712–726. doi: 10.1002/prot.22189. [DOI] [PubMed] [Google Scholar]
- 47.Kaufmann K, et al. Small Molecule Rotamers Enable Simultaneous Optimization of Small Molecule and Protein Degrees of Freedom in ROSETTALIGAND Docking. In. In: Beyer A, Schroeder M, editors. German Conference on Bioinformatics. 2008. pp. 148–157. [Google Scholar]
- 48.Chaudhury S, Gray JJ. Conformer selection and induced fit in flexible backbone protein-protein docking using computational and NMR ensembles. J Mol Biol. 2008;381:1068–1087. doi: 10.1016/j.jmb.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morin A, et al. Computational design of an endo-1,4-β-xylanase ligand binding site. Protein Eng Des Sel. 2011 doi: 10.1093/protein/gzr006. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morra G, et al. Selecting sequences that fold into a defined 3D structure: A new approach for protein design based on molecular dynamics and energetics. Biophys Chem. 2010;146:76–84. doi: 10.1016/j.bpc.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 51.Scarabelli G, et al. Predicting interaction sites from the energetics of isolated proteins: a new approach to epitope mapping. Biophys J. 2010;98:1966–1975. doi: 10.1016/j.bpj.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li L, et al. Incorporating receptor flexibility in the molecular design of protein interfaces. Protein Eng Des Sel. 2009;22:575–586. doi: 10.1093/protein/gzp042. [DOI] [PubMed] [Google Scholar]
- 53.Morra G, et al. Dynamics-based discovery of allosteric inhibitors: selection of new ligands for the C-terminal domain of Hsp90. J Chem Theory Comput. 2010;6:2978–2989. doi: 10.1021/ct100334n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spiegel K, et al. Structural and dynamical properties of manganese catalase and the synthetic protein DF1 and their implication for reactivity from classical molecular dynamics calculations. Proteins. 2006;65:317–330. doi: 10.1002/prot.21113. [DOI] [PubMed] [Google Scholar]
- 55.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 56.Bashford D, Case DA. Generalized born models of macromolecular solvation effects. Annu Rev Phys Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 57.Khoury GA, et al. Computational design of Candida boidinii xylose reductase for altered cofactor specificity. Protein Sci. 2009;18:2125–2138. doi: 10.1002/pro.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lippow SM, et al. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25:1171–1176. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang L, et al. A “solvated rotamer” approach to modeling water-mediated hydrogen bonds at protein-protein interfaces. Proteins. 2005;58:893–904. doi: 10.1002/prot.20347. [DOI] [PubMed] [Google Scholar]
- 60.Lassila JK, et al. Origins of catalysis by computationally designed retroaldolase enzymes. Proc Natl Acad Sci U S A. 2010;107:4937–4942. doi: 10.1073/pnas.0913638107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Havranek JJ, Harbury PB. Automated design of specificity in molecular recognition. Nat Struct Biol. 2003;10:45–52. doi: 10.1038/nsb877. [DOI] [PubMed] [Google Scholar]
- 62.Bolon DN, et al. Specificity versus stability in computational protein design. Proc Natl Acad Sci U S A. 2005;102:12724–12729. doi: 10.1073/pnas.0506124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ashworth J, et al. Computational redesign of endonuclease DNA binding and cleavage specificity. Nature. 2006;441:656–659. doi: 10.1038/nature04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Humphris EL, Kortemme T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 2008;16:1777–1788. doi: 10.1016/j.str.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 65.Schreier B, et al. Computational design of ligand binding is not a solved problem. Proc Natl Acad Sci U S A. 2009;106:18491–18496. doi: 10.1073/pnas.0907950106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zanghellini A, et al. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006;15:2785–2794. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chica RA, et al. Generation of longer emission wavelength red fluorescent proteins using computationally designed libraries. Proc Natl Acad Sci U S A. 2010;107:20257–20262. doi: 10.1073/pnas.1013910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nannemann DP, et al. Design and directed evolution of a dideoxy purine nucleoside phosphorylase. Protein Eng Des Sel. 2010;23:607–616. doi: 10.1093/protein/gzq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Skerra A. Alternative non-antibody scaffolds for molecular recognition. Curr Opin Biotechnol. 2007;18:295–304. doi: 10.1016/j.copbio.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 70.Gronwall C, Stahl S. Engineered affinity proteins--generation and applications. J Biotechnol. 2009;140:254–269. doi: 10.1016/j.jbiotec.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 71.Schlehuber S, Skerra A. Lipocalins in drug discovery: from natural ligand-binding proteins to “anticalins”. Drug Discov Today. 2005;10:23–33. doi: 10.1016/S1359-6446(04)03294-5. [DOI] [PubMed] [Google Scholar]
- 72.Beck A, et al. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol. 2010;10:345–352. doi: 10.1038/nri2747. [DOI] [PubMed] [Google Scholar]
- 73.Zuraw B, et al. Ecallantide. Nat Rev Drug Discov. 2010;9:189–190. doi: 10.1038/nrd3125. [DOI] [PubMed] [Google Scholar]
- 74.Ko MC, et al. Cocaine esterase: interactions with cocaine and immune responses in mice. J Pharmacol Exp Ther. 2007;320:926–933. doi: 10.1124/jpet.106.114223. [DOI] [PubMed] [Google Scholar]
- 75.Cooper ZD, et al. Rapid and robust protection against cocaine-induced lethality in rats by the bacterial cocaine esterase. Mol Pharmacol. 2006;70:1885–1891. doi: 10.1124/mol.106.025999. [DOI] [PubMed] [Google Scholar]
- 76.Gao D, et al. Thermostable variants of cocaine esterase for long-time protection against cocaine toxicity. Mol Pharmacol. 2009;75:318–323. doi: 10.1124/mol.108.049486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Narasimhan D, et al. Structural analysis of thermostabilizing mutations of cocaine esterase. Protein Eng Des Sel. 2010;23:537–547. doi: 10.1093/protein/gzq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brim RL, et al. A thermally stable form of bacterial cocaine esterase: a potential therapeutic agent for treatment of cocaine abuse. Mol Pharmacol. 2010;77:593–600. doi: 10.1124/mol.109.060806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park JB, et al. PEGylation of bacterial cocaine esterase for protection against protease digestion and immunogenicity. J Control Release. 2010;142:174–179. doi: 10.1016/j.jconrel.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alexandrova AN, et al. Catalytic mechanism and performance of computationally designed enzymes for Kemp elimination. J Am Chem Soc. 2008;130:15907–15915. doi: 10.1021/ja804040s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Khersonsky O, et al. Evolutionary optimization of computationally designed enzymes: Kemp eliminases of the KE07 series. J Mol Biol. 2010;396:1025–1042. doi: 10.1016/j.jmb.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 82.Ruscio JZ, et al. The influence of protein dynamics on the success of computational enzyme design. J Am Chem Soc. 2009;131:14111–14115. doi: 10.1021/ja905396s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MacDonald JT, et al. De novo backbone scaffolds for protein design. Proteins. 2010;78:1311–1325. doi: 10.1002/prot.22651. [DOI] [PMC free article] [PubMed] [Google Scholar]