

Abstract
Estrogen action in mammary gland development and breast cancer progression is tightly linked to the GH/IGF-I axis. Although many of the effects of GH on mammary gland growth and development require IGF-I, the extent to which GH action in breast cancer depends on IGF-I is not known. We examined GH action in a panel of estrogen receptor-positive breast cancer cell lines and found that T47D cells express significant levels of GH receptor and that GH significantly enhances 17β-estradiol (E2)-stimulated proliferation in these cells. GH action in the T47D cells was independent of changes in IGF-I and IGF-I receptor (IGF-IR) expression and IGF-IR signaling, suggesting that GH can exert direct effects on breast cancer cells. Although E2-dependent proliferation required IGF-IR signaling, the combination of GH+E2 overcame inhibition of IGF-IR activity to restore proliferation. In contrast, GH required both Janus kinase 2 and epidermal growth factor receptor signaling for subsequent ERK activation and potentiation of E2-dependent proliferation. Downstream of these pathways, we identified a number of immediate early-response genes associated with proliferation that are rapidly and robustly up-regulated by GH. These findings demonstrate that GH can have important effects in breast cancer cells that are distinct from IGF-IR activity, suggesting that novel drugs or improved combination therapies targeting estrogen receptor and the GH/IGF axis may be beneficial for breast cancer patients.
Estrogens, such as 17β-estradiol (E2), are a major factor in the initiation and progression of breast cancer. Endocrine therapies targeting E2 production or estrogen receptor (ER) activity have proven effective in most patients with ER+ breast cancers. However, resistance to these therapies can occur in up to 50% of these patients. Therefore, additional therapeutic strategies are needed for women with ER+ breast tumors.
Targeting the IGF-I pathway has been of particular interest, given the tight association between ER and the IGF-I receptor (IGF-IR) signaling pathway. Cross talk between these factors can occur on many levels including ER and IGF-IR interaction (1, 2), E2 activation of the IGF-I pathway (3), and up-regulation of IGF-I signaling components by E2 (4–7). IGF-I is also implicated in breast cancer resistance to endocrine therapies (8–10) and can activate a gene signature that associates with luminal B breast tumors, a subtype of ER+ tumors with poor outcome (11). Based on this evidence, cotargeting both ER and IGF-IR seems to be a reasonable therapeutic approach for patients with ER+ breast cancer that fail to respond to endocrine therapy. In fact, inhibition of IGF-IR can block E2-stimulated proliferation in vitro (3), and IGF-IR targeted therapy improves the efficacy of antiestrogens in the breast cancer cell lines BT474 and MCF-7 (12). However, despite the success of IGF-IR-targeted therapies in in vitro and preclinical in vivo models, outcomes of clinical trials have been disappointing (13–15). For example, a randomized phase II trial found no difference in progression-free survival when an anti-IGF-IR antibody (AMG 479) was given in addition to endocrine therapy (exemestane or fulvestrant) among postmenopausal women with ER+ metastatic breast cancer (16). Furthermore, a recent study has shown that tamoxifen-resistant tumors, representing the patients most likely to be entered into a clinical trial testing an IGF-targeted therapy, actually lose IGF-IR expression and thus do not respond to such treatments (17).
Another possible reason for failure of IGF-IR therapies could be that they increase circulating GH levels (18, 19) through the disruption of a negative feedback loop between the liver and the pituitary gland (20). Several lines of emerging evidence implicate a role for GH in breast cancer. A recently published 22-year long study found cancer to be virtually nonexistent among a GH receptor (GHR)-deficient population in Ecuador (21). GH signaling was the pathway third most highly associated with breast cancer risk in a genome-wide association study (22). Furthermore, GHR expression is higher in breast tumors vs adjacent normal tissue (23), and expression of autocrine GH in mammary carcinomas was found to predict worse survival outcomes (24). Despite this evidence, the role of GH in human breast cancer has not been well studied.
Although many of GH's actions on growth and development of the rodent mammary gland are mediated by IGF-I (as reviewed in References 25 and 26), here we examined the direct role of GH in ER+ breast cancer cell lines. We found that GH is capable of potentiating the effects of E2 on proliferation in cells expressing high levels of GHR. Importantly, GH was capable of overcoming the growth-suppressive effects of IGF-IR tyrosine kinase inhibition, suggesting that GH action may, in fact, be a plausible cause of IGF-IR inhibitor failure in the clinic. In addition, we have dissected the pathways activated directly by GH in breast cancer cells and found that Janus kinase 2 (JAK2), epidermal growth factor receptor (EGFR), and ERK may all play essential roles in mediating the effects of GH on both the proliferation and expression of immediate early target genes.
Materials and Methods
Reagents
Recombinant human GH was purchased from Dr A. F. Parlow (National Hormone and Peptide Program, National Institute of Diabetes and Digestive and Kidney Diseases) and reconstituted in PBS with 0.1% BSA. E2 was purchased from Sigma (St Louis, Missouri). IGF-I was purchased from R&D Systems (Minneapolis, Minnesota). ICI 182,780 (ICI) was purchased from Tocris (Bristol, United Kingdom). NVP-AEW541 (AEW541) was generously provided by Novartis Pharmaceuticals (Basel, Switzerland). AG1478 was purchased from Cayman Chemicals (Ann Arbor, Michigan); gefitinib from LC Laboratories (Woburn, Massachusetts); U0126 from Calbiochem (EMD Millipore, Billerica, Massachusetts); and AS703026 and AZD1480 from Selleckchem (Houston, Texas). Antibodies used for Western blotting of cell extracts were as follows: phospho-ERK (Santa Cruz Biotechnology, Santa Cruz, California; sc-7383); ERK (Santa Cruz Biotechnology; sc-94); phospho-IGF-IR (Cell Signaling Technology, Danvers, Massachusetts; number 3024); IGF-IR (Cell Signaling; number 3027); phospho-signal transducer and activator of transcription (STAT)-5 (Y694) (Cell Signaling; number 9359 and number 9351); STAT5 (Cell Signaling; number 9363); ERα (NeoMarkers, Fremont, California; Ab-21 H222); EGFR (Cell Signaling; number 2646); and β-actin (Sigma; clone AC-15, number A5441). Small interfering RNA targeting JAK2 and a nontargeting negative control were purchased from Ambion (Life Technologies, Grand Island, New York).
Cell culture
All cell lines were routinely maintained in RPMI 1640 (Invitrogen Life Technologies, Grand Island, New York) with phenol red containing 10% fetal bovine serum, as described previously (27). Growth media for MCF-7 and BT474 cells contained 6 ng/mL insulin. Cells seeded for experiments were cultured in RPMI 1640 without phenol red, containing 5% charcoal-dextran-stripped fetal bovine serum for 2–3 days prior to hormone treatment.
Proliferation assays
The methylene blue assay was carried out as described previously (28). Briefly, media were removed and cells were rinsed with PBS. Cells were then incubated for 1 hour at 37°C in methylene blue fixing and staining solution (Hanks' balanced salt solution with 1.25% glutaraldehyde and 0.6% methylene blue). After removing the solution and rinsing in distilled water, the stain was eluted for 15 minutes with gentle agitation using elution solution (49% PBS, 50% ethanol, 1% acetic acid). Absorbance was read on an automated plate reader (Synergy HT; Bio-Tek, Winooski, Vermont) at a 562-nm wavelength. Data represent relative cell numbers, presented as a percentage of control. The DNA assay was carried out as previously described (29). After hormone treatment, media were removed and cells were covered with fresh RPMI 1640 without serum or additives, and Hoechst dye (Invitrogen) was added for a final concentration of 1.25 μg/mL. Cells were incubated for 1 hour at 37°C, and fluorescence (excitation 360/40, emission 460/40) was measured. DNA content was calculated from a standard curve and is reported as a percentage of vehicle control. The 5-bromo-2′-deoxyuridine (BrdU) assay was carried out according to the manufacturer's instructions (BrdU cell proliferation assay kit; Millipore). Absorbance, reflecting incorporation of BrdU into the DNA of the cells and thus transition through the cell cycle, was read at 490 nm wavelength.
Western blot
Whole-cell extracts were prepared using M-PER (Thermo Scientific, Rockford, Illinois) and protein concentrations were measured using the bicinchoninic assay method (Thermo Scientific). Proteins were separated by SDS-PAGE (10% Tris-HCl precast gels; Bio-Rad Laboratories, Hercules, California), transferred to nitrocellulose membranes (Thermo Scientific), blocked for 1 hour in buffer containing 5% nonfat dry milk (Lab Scientific, Livingston, New Jersey) or 5% BSA, and incubated with the appropriate primary antibody overnight. The next day, secondary antibody was applied and the signal was visualized on a Molecular Imager ChemidocXRS (Bio-Rad Laboratories), using the Pierce Supersignal West Pico chemiluminescent substrate (Thermo Scientific). In the case of phosphoproteins, membranes were first probed for the phosphoprotein, stripped with Re-Blot Plus Mild antibody stripping solution (Millipore) and then reprobed for the respective total protein.
RT-quantitative PCR (QPCR)
RNA was isolated and QPCR performed using the ΔΔcycle threshold method with 36B4 as the internal control, as described previously (30). Primer sequences are listed in Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org.
Statistics
Data are presented as mean ± SEM from at least 3 independent determinations. Statistical analyses consisted of 1- or 2-way ANOVA followed by Bonferroni posttest, or t test, as appropriate.
Results
GH and E2 enhance proliferation in T47D breast cancer cells
To examine whether GH has direct effects on human breast cancer cells, we first examined GHR mRNA expression in several ER+ breast cancer cell lines. T47D cells showed the highest level of expression (Figure 1A) compared with ZR75-1, BT474, and MCF-7 cells. It should be noted that the pattern of expression of prolactin receptor (PRLR) mRNA was similar to GHR mRNA among the 4 cell lines in that both were most highly expressed in T47D cells. MCF-7 cells, on the other hand, expressed higher levels of both ERα and IGF-IR mRNA (Supplemental Figure 1). E2 stimulated proliferation in all ER+ cell lines (Figure 1B and Supplemental Figure 2). In contrast, GH action was evident only in T47D cells (Figure 1B), which may be explained by the higher relative GHR expression. In T47D cells, GH caused a slight increase in the cell number that did not consistently reach significance. Notably, however, E2+GH significantly increased cell number compared with E2 alone in T47D cells (Figure 1B). To verify that the increase in cell number was due to enhanced proliferation rather than reduced cell death, a BrdU incorporation assay was performed and confirmed that GH primarily acts to potentiate E2-dependent proliferation (Figure 1C). In addition, the ER antagonist ICI significantly reduced proliferation in response to E2 as well as E2+GH (Figure 1D). ICI did not affect the basal rate of cell proliferation because the cells are grown under nonestrogenic conditions (phenol-red free media supplemented with charcoal-dextran stripped serum). GH had no effect on ERα mRNA or protein expression in T47D cells (Supplemental Figure 3). These results suggest that GH is able to promote E2-dependent proliferation in ER+ breast cancer cells that express higher levels of GHR.
Figure 1.
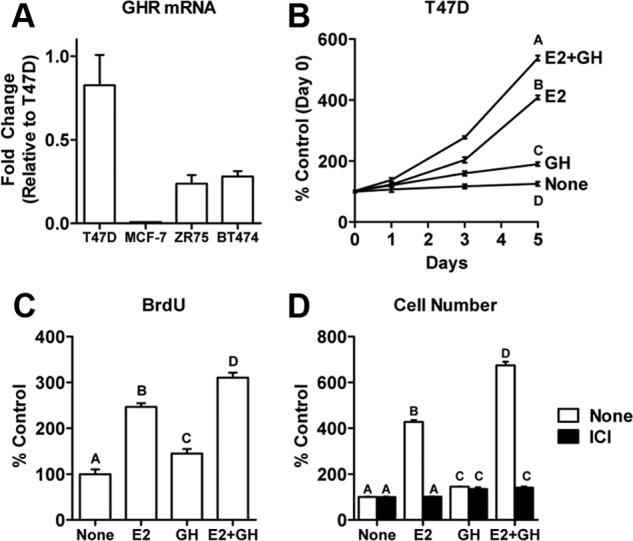
GH potentiates E2-stimulated proliferation in T47D human breast cancer cells. A, GHR mRNA expression was examined in a panel of ER+ human breast cancer cell lines by QPCR using the ΔΔcycle threshold method with 36B4 as an internal control. B, T47D cells were treated with 500 ng/mL GH, 10 nM E2, or both for 5 days. Cell numbers were determined using the methylene blue assay and are presented as percent of untreated cells on day 0. C, BrdU incorporation was assessed in T47D cells after 72 hours of treatment with GH, E2, or both. D, T47D cells were treated for 5 days as described in B in the absence or presence of 1 μM of the ER antagonist ICI 182,780. Data points or bars with different letters are significantly different (P < .05).
GH potentiates E2-dependent proliferation in a JAK2/ERK/EGFR-dependent manner but independently of IGF-IR activation
To examine whether the IGF-I pathway was involved in GH's ability to potentiate E2-dependent proliferation, IGF-I expression was measured in T47D cells. IGF-I mRNA levels were unaffected by GH, E2, or GH+E2 (Figure 2A), and secreted IGF-I protein was below detectable limits of the ELISA in all treatment groups (Figure 2B), indicating that the effect of GH on cell proliferation is not due to increased expression of IGF-I. Next, we investigated the influence of hormone treatment on IGF-IR expression and activity. Although E2 up-regulates IGF-IR expression, as expected, GH had no effect, either alone or in combination with E2 (Figure 2C). Furthermore, GH+E2 treatment does not affect phosphorylation of IGF-IR (Figure 2D). We used AEW541, a specific tyrosine kinase inhibitor that preferentially blocks activation and autophosphorylation of IGF-IR, and found that it effectively inhibits activation of IGF-IR by IGF-I treatment (Figure 2D). In proliferation assays, AEW541 reduced the effect of E2 and E2+IGF-I on cell number (Figure 2E), as suggested in previous studies (31). In contrast, AEW541 did not disrupt the ability of GH+E2 to increase the cell number compared with the effect of E2 alone (Figure 2E). Together these results indicate that the effect of GH on E2-dependent breast cancer cell proliferation is independent of both IGF-I production and IGF-IR signaling and that GH can blunt the effectiveness of IGF-IR inhibitors on cancer cell growth.
Figure 2.
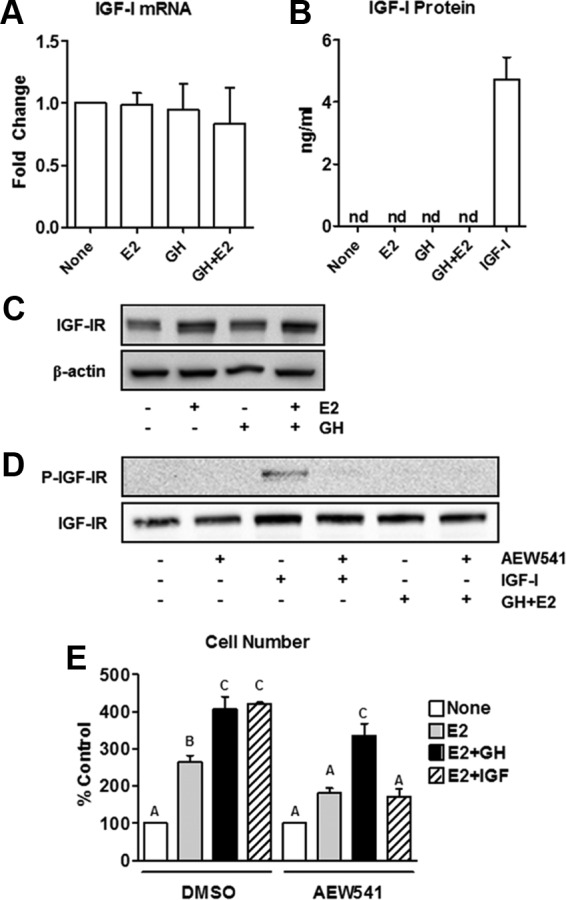
GH potentiates E2-stimulated T47D cell proliferation independently of IGF-I expression or IGF-IR activation. A, T47D cells were treated with 500 ng/mL GH for 24 hours. IGF-I mRNA expression was determined by QPCR. B, T47D cells were treated as in A, and conditioned media were collected. Levels of IGF-I protein were measured by ELISA. IGF-I (5 ng/mL) added to media served as a positive control. C, T47D cells were treated with hormones for 24 hours, after which total levels of IGF-IR protein were determined by Western blot. D, T47D cells were pretreated for 2 hours with 2 μM AEW541, an inhibitor of IGF-IR tyrosine kinase activity, followed by 30 minutes of treatment with GH+E2 or 50 ng/mL IGF-I. Phospho- and total IGF-IR levels were examined by Western blot to demonstrate the effectiveness of the inhibitor. E, T47D cells were treated with E2, E2+GH, or E2+IGF-I in the presence of 1 μM AEW541 or vehicle [dimethylsulfoxide (DMSO)] for 5 days. Proliferation was measured by the methylene blue assay. Similar results were found using the DNA assay (data not shown). Bars with different letters are significantly different (P < .05).
GH activation of 2 major signaling pathways, JAK2/STAT5 and ERK, was detected in T47D cells treated with GH or GH+E2 but not by E2 alone (Figure 3A). Because the doses of GH and E2 used cause maximal proliferation (data not shown), and because E2 did not activate STAT5 or ERK in these cells, our findings suggest that GH activation of these pathways does not depend on E2 action. Neither pathway was significantly activated in other ER+ cell lines (data not shown), suggesting that the ability of GH to regulate these pathways correlates with its ability to potentiate proliferation. Previous studies have suggested that GH may use JAK2 and EGFR to activate ERK in various cell types, including preadipocyte and liver cells (32–35), but this has not been well studied in breast cancer cells. To examine how ERK is activated in T47D cells, specific inhibitors of JAK2 (AZD1480) and EGFR (AG1478) were used. We found that pharmacological blockade of either EGFR or JAK2 reduced the phosphorylation of both STAT5 and ERK by GH (Figure 3B), whereas the blockade of IGF-IR did not (Supplemental Figure 4). However, treatment with E2, GH, or both had no effect on EGFR expression (Supplemental Figure 5). These observations suggest that GH uses both JAK2 and EGFR to activate two of its downstream effectors, STAT5 and ERK.
Figure 3.
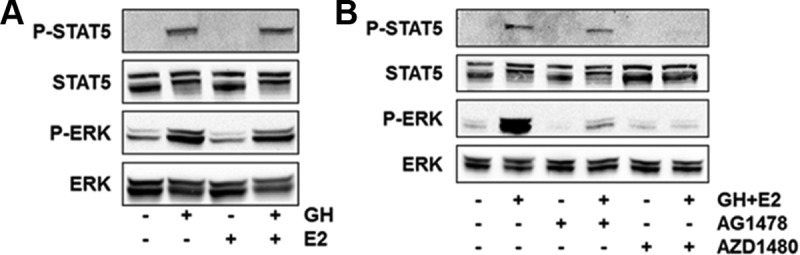
GH activates STAT5 and ERK in breast cancer cells. A, T47D cells were treated for 30 minutes with GH, E2, or both. Phospho- and total STAT5 and ERK levels were assessed by Western blot. B, T47D cells were pretreated for 2 hours with vehicle (dimethylsulfoxide) or inhibitors for EGFR (AG1478, 15 μM) or JAK2 (AZD1480, 1 μM), followed by treatment with GH+E2 for 30 minutes.
We next examined whether these pathways are required for GH-mediated cell proliferation. The JAK2 inhibitor, AZD1480, had no effect on E2 action but inhibited the ability of GH to potentiate E2-stimulated proliferation (Figure 4A), indicating that JAK2 is not required by E2 but is essential for GH to enhance E2-stimulated proliferation. Inhibition of ERK activation by U0126 (Figure 4B and Supplemental Figure 6A) or AS703026 (Supplemental Figure 6, B and C), a novel MAPK kinase (MEK) inhibitor, disrupted the ability of E2 and GH+E2 to stimulate cell proliferation. Similarly, inhibition of EGFR with AG1478 (Figure 4C) or gefitinib (data not shown) prevented the effects of E2 and GH+E2 on cell proliferation. AG1478 also abrogated STAT5 phosphorylation (Figure 3B). However, because STAT5 activation is required only for GH enhancement of E2-stimulated proliferation and not for E2 action itself (Figure 4A) and because specific MEK inhibitors dramatically impact proliferation (Figure 4B and Supplemental Figure 6), we conclude that the effects of AG1478 on proliferation most likely are due to inhibition of EGFR-mediated ERK activation. Together these findings indicate that the JAK2 pathway is essential for GH's ability to potentiate E2-dependent proliferation and that ERK and EGFR activity are essential for the proliferation induced by E2 in both the presence and absence of GH.
Figure 4.
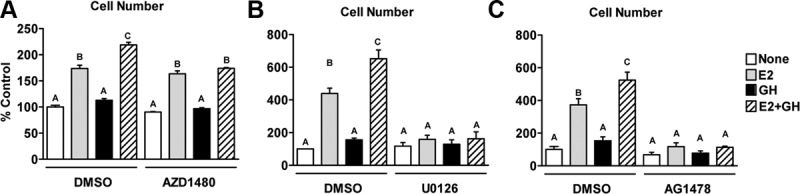
Roles of JAK2, ERK, and EGFR in E2- and E2+GH-stimulated proliferation. T47D cells were treated for 5 days with E2, GH, or both in the presence of vehicle [dimethylsulfoxide (DMSO)] or inhibitors for JAK2 (AZD1480, 1 μM) (A), MEK (U0126, 10 μM) (B), or EGFR (AG1478, 15 μM) (C). Proliferation was determined by the DNA assay. Bars with different letters are significantly different (P < .05).
GH up-regulates early proliferation genes in a JAK2-, EGFR-, and ERK-dependent manner
Activation of STAT target genes by GH is well characterized in many tissues and cell types, and STAT proteins have been associated previously with the expression of proliferation-associated genes in breast cancer (36, 37). However, it is less clear how GH uses the ERK pathway to regulate gene expression in breast cancer cells. In preadipocytes, GH has been shown to up-regulate many immediate early genes (IEGs) in an ERK-dependent manner (38). Interestingly, many of these same genes are also up-regulated by epidermal growth factor in MCF10A cells (39). Because our data indicate that GH uses EGFR signaling to activate ERK (Figure 3B), we examined the regulation of several of these IEGs by GH in T47D cells. As shown in Figure 5A, the expression of FOS, JUN, EGR1, ZFP36, and ATF3 mRNA were rapidly and robustly up-regulated by GH treatment. In contrast, E2 had little effect on these genes, either alone or in combination with GH, at these early time points (data not shown). Inhibitor studies demonstrated that GH requires JAK2, EGFR, and ERK activity to up-regulate these genes (Figure 5B). Knockdown of JAK2 using small interfering RNA had similar effects as the JAK2 inhibitor (Supplemental Figure 7). These findings suggest that GH signaling through multiple pathways may converge to up-regulate the transcriptional regulators associated with cell proliferation and thereby potentiate E2-dependent proliferation.
Figure 5.

GH rapidly up-regulates IEGs in a JAK2-, EGFR-, and ERK-dependent manner. A, T47D cells were treated with GH for up to 6 hours. Expression of IEGs was examined by QPCR. Data are presented as a heat map with the highest levels of expression for each gene in red and the lowest levels in green. B, GH-induced expression of IEGs was examined after pretreatment with inhibitors for EGFR (AG1478), MEK (U0126), or JAK2 (AZD1480).
Discussion
In this study, we have demonstrated that GH plays a significant role in human breast cancer cell proliferation by acting directly on human breast cancer cells to increase E2-dependent cell proliferation. Not only are these effects likely IGF-I independent because GH does not up-regulate IGF-I in the cells, nor does GH affect IGF-IR expression or activation, but GH can also overcome the growth-inhibitory effect of an IGF-IR inhibitor, indicating a potential mechanism by which therapeutics targeting IGF-IR may fail in women with ER+ breast cancer.
Although many of GH's actions on growth and development, including in the rodent mammary gland, are mediated by IGF-I (as reviewed in References 25 and 26), GH acting independently of IGF-I is not unprecedented. For example, a GHR/IGF-I double knockout stunted somatic growth more severely than either single knockout alone, a compelling indication that GH and IGF-I have both independent and overlapping functions (40). GH also has many important roles in metabolism that are largely thought to be IGF-I independent, although some uncertainty still surrounds this issue, as reviewed in Vijayakumar et al (41). Briefly, GH increases lipolysis in adipose tissue. In the liver, in which IGF-IR expression is low, GH stimulates bile acid production, hepatic glucose production, and triglyceride uptake and storage by reducing lipolysis and lipid oxidation and increases lipogenesis. GH also inhibits insulin action in both adipose tissue and the liver (41). On the other hand, GH has been shown to require IGF-IR to stimulate STAT5 activation and proliferation in osteoblasts, independently of its ability to stimulate IGF-I expression (42). Activation of ERK, however, did not require IGF-IR. In preadipocytes and pancreatic islet β-cells, GH induces complex formation of GHR, JAK2, and IGF-IR, whereas in murine osteoblasts and β-cells, GH stimulation of STAT5 depends on the presence of the IGF-IR. However, in all 3 cell types, these actions of GH occurred independently of the tyrosine kinase activity of the IGF-IR (43–45). Whether the presence of IGF-IR is also required for the effects of GH in T47D breast cancer cells requires further investigation. To our knowledge, this is the first report that GH can act independently of IGF-I production and IGF-IR tyrosine kinase activity on E2-stimulated proliferation in human breast cancer cells.
The question of how GH bypasses the requirement for IGF-IR signaling to enhance E2-stimulated proliferation remains. The GHR is likely involved because T47D cells express GHR and also respond to GH by activating signaling pathways and potentiating proliferation. However, human GH is known to also bind and activate PRLR (46–48), and a recent report of functional GHR/PRLR dimers responding to GH in T47D breast cancer cells has emerged (49). In fact, in that study antagonism of both GHR and PRLR was required to abrogate GH-stimulated STAT5 activation. Furthermore, a recent finding showed that the reduction of PRLR expression in T47D cells increases the preference of GH to bind to GHR (vs the PRLR) as well as increasing the sensitivity to GH response (50). Thus, we cannot exclude the possibility that both PRLR and GHR, perhaps in the form of heterodimers, may play a role in the GH potentiation of E2 action in breast cancer cells. Interestingly, the synergistic effects of prolactin (PRL) on E2-stimulated expression of ER target genes in T47D cells have been described (51), indicating GH and PRL may have similar roles. We have observed that GH also can potentiate E2-stimulated expression of some ER target genes in T47D cells (Felice, D. L. and J. Frasor, unreported observation) but whether GH and PRL have similar gene regulatory effects is not known.
In the present study, we find that GH activates JAK2/STAT5 and that this is required for enhanced E2-dependent proliferation. In addition, MEK/ERK signaling is activated by GH and this occurs downstream of both JAK2 and EGFR. Although links between these pathways have been established, the mechanisms of interaction appear to be complex and cell type specific. For example, GH has been shown to phosphorylate EGFR via GHR/JAK2 signaling, leading to the activation of MAPK signaling in mouse liver and Chinese hamster ovary cells (33, 34). However, in preadipocytes GH can phosphorylate EGFR via ERK, thereby delaying EGFR degradation and thus prolonging signaling (32).
In addition, the GH activation of STATs and ERK has been shown to be dependent on JAK2 in preadipocytes and hepatoma cells (35). GH and EGF can synergize in mouse preadipocytes, converging on ERK activation, whereas PRL and platelet-derived growth factor have a similar synergy in T47D cells (52). From our studies, we can conclude that in T47D breast cancer cells, GH uses both JAK2 and EGFR to activate ERK by a mechanism which does not require IGF-IR tyrosine kinase activity. It is difficult to know what role ERK and EGFR play in mediating the effect of GH on E2 action because the inhibition of either pathway prevented E2-stimulated proliferation, even in the absence of GH. Furthermore, although hormone treatments did not affect EGFR expression (Supplemental Figure 5), EGFR action is also required for ERK and STAT5 activation (Figure 3B). Nevertheless, GH activation of these pathways is likely to be important because all three (JAK2/ERK/EGFR) were essential for the GH regulation of IEGs previously associated with GH action and/or cellular proliferation. Previous studies have shown that GH activation of ERK leads to the up-regulation of IEGs encoding for transcription factor genes, including FOS, JUN, ATF3, and EGR1, in preadipocytes (53), and EGF induces expression of many of the same genes in MCF-10A cells (39).
Our findings suggest that the GH regulation of these genes in T47D breast cancer cells is ERK dependent and that the effects of JAK2/EGFR inhibition are through loss of ERK activation. However, other pathways and transcription factors may also be involved. For example, ATF3 was identified as a novel gene target of GH that is regulated in a CCAAT/enhancer-binding protein-β-dependent manner (53), and FOS also is induced by GH in preadipocytes in a JAK2-, MEK/ERK-, and CCAAT/enhancer-binding protein-β-dependent manner (38, 54–57).
It is important to note that most of the GH-regulated IEGs that we examined have the potential to play important roles in breast cancer. FOS/JUN, for example, constitute the activating protein-1 transcription factor, which regulates a number of genes involved in multiple cancer-related processes, including proliferation, differentiation, hypoxia, angiogenesis, metastasis, apoptosis, and invasion (as reviewed in Reference 58). Activating transcription factor 3 (ATF3) protein expression was found to be higher than matched normal mammary gland tissue in 48% of breast tumors examined (59), and transgenic mice overexpressing human ATF3 in the basal epithelium of the mammary gland develop mammary cancer after parity, which implicates ATF3 as an oncogene (60). Early growth response protein 1 is a zinc finger transcription factor (61, 62) that can play a role in cell proliferation, migration, and invasion (63). On the other hand, the role that GH-induced ZFP36 plays in T47D cells remains unclear. ZFP36, which was first identified as a growth-factor induced gene in mouse 3T3 cells (64), acts as a regulator of mRNA degradation (65). Considering the important roles of these IEGs, we speculate that they are essential players in GH's ability to potentiate E2-stimulated proliferation. However, it is interesting to note that GH alone, which leads to significant pathway activation and gene expression, does not have much impact on T47D cell proliferation in the absence of E2.
In conclusion, we have demonstrated that GH has direct actions on human breast cancer cells that are independent of changes in IGF-I production, IGF-IR expression, and IGF-IR activation and can overcome IGF-IR blockade, using JAK2/STAT signaling and potentially EGFR and ERK to do so. These findings are clinically relevant for women who may be directed to IGF-IR inhibitor therapies, which may elevate circulating levels of GH. The possibility of GH-regulated early genes playing a role in potentiation of E2 action is intriguing and warrants further attention. Importantly, further research will be necessary to decipher the appropriate biomarker to predict these actions of GH in breast tumors. Finally, this study highlights the importance of regarding GH as having both IGF-I-dependent and -independent functions in breast cancer biology.
Acknowledgments
We thank Dr Irida Kastrati and Bryant Marure for their technical expertise.
This work was supported in part by American Cancer Society Grant 119168-RSG-10-187--01-TBE (to J.F.) and National Institutes of Health Grant T32 HL07692--21 (to D.L.F.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AEW541
- NVP-AEW541
- ATF3
- activating transcription factor 3
- BrdU
- 5-bromo-2′-deoxyuridine
- E2
- 17β-estradiol
- EGFR
- epidermal growth factor receptor
- ER
- estrogen receptor
- GHR
- GH receptor
- ICI
- ICI 182,780
- IGF-IR
- IGF-I receptor
- JAK2
- Janus kinase 2
- MEK
- MAPK kinase
- PRL
- prolactin
- PRLR
- PRL receptor
- QPCR
- RT-quantitative PCR
- STAT
- signal transducer and activator of transcription.
References
- 1. Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C. Estrogen receptor α rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000;275:18447–18453 [DOI] [PubMed] [Google Scholar]
- 2. Lee AV, Weng CN, Jackson JG, Yee D. Activation of estrogen receptor-mediated gene transcription by IGF-I in human breast cancer cells. J Endocrinol. 1997;152:39–47 [DOI] [PubMed] [Google Scholar]
- 3. Song RX, Barnes CJ, Zhang Z, Bao Y, Kumar R, Santen RJ. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor α to the plasma membrane. Proc Natl Acad Sci USA. 2004;101:2076–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cullen KJ, Yee D, Bates SE, et al. Regulation of human breast cancer by secreted growth factors. Acta Oncol. 1989;28:835–839 [DOI] [PubMed] [Google Scholar]
- 5. Lee AV, Jackson JG, Gooch JL, et al. Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol Endocrinol. 1999;13:787–796 [DOI] [PubMed] [Google Scholar]
- 6. Yee D, Lee AV. Cross talk between the insulin-like growth factors and estrogens in breast cancer. J Mammary Gland Biol Neoplasia. 2000;5:107–115 [DOI] [PubMed] [Google Scholar]
- 7. Molloy CA, May FE, Westley BR. Insulin receptor substrate-1 expression is regulated by estrogen in the MCF-7 human breast cancer cell line. J Biol Chem. 2000;275:12565–12571 [DOI] [PubMed] [Google Scholar]
- 8. Knowlden JM, Hutcheson IR, Barrow D, Gee JM, Nicholson RI. Insulin-like growth factor-I receptor signaling in tamoxifen-resistant breast cancer: a supporting role to the epidermal growth factor receptor. Endocrinology. 2005;146:4609–4618 [DOI] [PubMed] [Google Scholar]
- 9. Massarweh S, Osborne CK, Creighton CJ, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68:826–833 [DOI] [PubMed] [Google Scholar]
- 10. Zhang Y, Moerkens M, Ramaiahgari S, et al. Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res. 2011;13:R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Creighton CJ, Casa A, Lazard Z, et al. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. 2008;26:4078–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chakraborty AK, Welsh A, Digiovanna MP. Co-targeting the insulin-like growth factor I receptor enhances growth-inhibitory and pro-apoptotic effects of anti-estrogens in human breast cancer cell lines. Breast Cancer Res Treat. 2010;120:327–335 [DOI] [PubMed] [Google Scholar]
- 13. Sachdev D, Yee D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol Cancer Ther. 2007;6:1–12 [DOI] [PubMed] [Google Scholar]
- 14. Lee AV, Yee D. Targeting IGF-1R: at a crossroad. Oncology (Williston Park). 2011;25:535–536; discussion 551 [PMC free article] [PubMed] [Google Scholar]
- 15. Yee D. Insulin-like growth factor receptor inhibitors: baby or the bathwater? J Natl Cancer Inst. 2012;104:975–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaufman P, Ferrero J, Bourgeois H, et al. Abstract S1–4 : a randomized, double-blind, placebo-controlled, phase 2 study of AMG 479 with exemestane (E) or fulvestrant (F) in postmenopausal women with hormone-receptor positive (HR+) metastatic (M) or locally advanced (LA) breast cancer. Cancer Res. 2010;70:S1–S4 [Google Scholar]
- 17. Fagan DH, Uselman RR, Sachdev D, Yee D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: implications for breast cancer treatment. Cancer Res. 2012;72:3372–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weroha SJ, Haluska P. IGF-1 receptor inhibitors in clinical trials—early lessons. J Mammary Gland Biol Neoplasia. 2008;13:471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tolcher AW, Sarantopoulos J, Patnaik A, et al. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J Clin Oncol. 2009;27:5800–5807 [DOI] [PubMed] [Google Scholar]
- 20. Berelowitz M, Szabo M, Frohman LA, Firestone S, Chu L, Hintz RL. Somatomedin-C mediates growth hormone negative feedback by effects on both the hypothalamus and the pituitary. Science. 1981;212:1279–1281 [DOI] [PubMed] [Google Scholar]
- 21. Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3:70ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Menashe I, Maeder D, Garcia-Closas M, et al. Pathway analysis of breast cancer genome-wide association study highlights three pathways and one canonical signaling cascade. Cancer Res. 2010;70:4453–4459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gebre-Medhin M, Kindblom LG, Wennbo H, Tornell J, Meis-Kindblom JM. Growth hormone receptor is expressed in human breast cancer. Am J Pathol. 2001;158:1217–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu ZS, Yang K, Wan Y, et al. Tumor expression of human growth hormone and human prolactin predict a worse survival outcome in patients with mammary or endometrial carcinoma. J Clin Endocrinol Metab. 2011;96:E1619–E1629 [DOI] [PubMed] [Google Scholar]
- 25. Kleinberg DL, Ruan W. IGF-I, GH, and sex steroid effects in normal mammary gland development. J Mammary Gland Biol Neoplasia. 2008;13:353–360 [DOI] [PubMed] [Google Scholar]
- 26. Kleinberg DL, Wood TL, Furth PA, Lee AV. Growth hormone and insulin-like growth factor-I in the transition from normal mammary development to preneoplastic mammary lesions. Endocr Rev. 2009;30:51–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin X, Yu Y, Zhao H, Zhang Y, Manela J, Tonetti DA. Overexpression of PKCα is required to impart estradiol inhibition and tamoxifen-resistance in a T47D human breast cancer tumor model. Carcinogenesis. 2006;27:1538–1546 [DOI] [PubMed] [Google Scholar]
- 28. Felice D, Sun J, Liu R. A modified methylene blue assay for accurate cell counting. J Funct Foods. 2009;1:109–118 [Google Scholar]
- 29. Labarca C, Paigen K. A simple, rapid, and sensitive DNA assay procedure. Anal Biochem. 1980;102:344–352 [DOI] [PubMed] [Google Scholar]
- 30. Frasor J, Weaver A, Pradhan M, et al. Positive cross-talk between estrogen receptor and NF-κB in breast cancer. Cancer Res. 2009;69:8918–8925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lisztwan J, Pornon A, Chen B, Chen S, Evans DB. The aromatase inhibitor letrozole and inhibitors of insulin-like growth factor I receptor synergistically induce apoptosis in in vitro models of estrogen-dependent breast cancer. Breast Cancer Res. 2008;10:R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang Y, Kim SO, Jiang J, Frank SJ. Growth hormone-induced phosphorylation of epidermal growth factor (EGF) receptor in 3T3–F442A cells. Modulation of EGF-induced trafficking and signaling. J Biol Chem. 2003;278:18902–18913 [DOI] [PubMed] [Google Scholar]
- 33. Yamauchi T, Ueki K, Tamemoto H, et al. Tyrosine phosphorylation of the EGF receptor by the kinase Jak2 is induced by growth hormone. Nature. 1997;390:91–96 [DOI] [PubMed] [Google Scholar]
- 34. Yamauchi T, Ueki K, Tobe K, et al. Growth hormone-induced tyrosine phosphorylation of EGF receptor as an essential element leading to MAP kinase activation and gene expression. Endocr J. 1998;45(suppl):S27–S31 [DOI] [PubMed] [Google Scholar]
- 35. Jin H, Lanning NJ, Carter-Su C. JAK2, but not Src family kinases, is required for STAT, ERK, and Akt signaling in response to growth hormone in preadipocytes and hepatoma cells. Mol Endocrinol. 2008;22:1825–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alvarez JV, Frank DA. Genome-wide analysis of STAT target genes: elucidating the mechanism of STAT-mediated oncogenesis. Cancer Biol Ther. 2004;3:1045–1050 [DOI] [PubMed] [Google Scholar]
- 37. Fox EM, Bernaciak TM, Wen J, Weaver AM, Shupnik MA, Silva CM. Signal transducer and activator of transcription 5b, c-Src, and epidermal growth factor receptor signaling play integral roles in estrogen-stimulated proliferation of estrogen receptor-positive breast cancer cells. Mol Endocrinol. 2008;22:1781–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cui TX, Lin G, LaPensee CR, et al. C/EBPβ mediates growth hormone-regulated expression of multiple target genes. Mol Endocrinol. 2011;25:681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Avraham R, Sas-Chen A, Manor O, et al. EGF decreases the abundance of microRNAs that restrain oncogenic transcription factors. Sci Signal. 2010;3:ra43. [DOI] [PubMed] [Google Scholar]
- 40. Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol. 2001;229:141–162 [DOI] [PubMed] [Google Scholar]
- 41. Vijayakumar A, Novosyadlyy R, Wu Y, Yakar S, LeRoith D. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm IGF Res. 2010;20:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. DiGirolamo DJ, Mukherjee A, Fulzele K, et al. Mode of growth hormone action in osteoblasts. J Biol Chem. 2007;282:31666–31674 [DOI] [PubMed] [Google Scholar]
- 43. Huang Y, Kim SO, Yang N, Jiang J, Frank SJ. Physical and functional interaction of growth hormone and insulin-like growth factor-I signaling elements. Mol Endocrinol. 2004;18:1471–1485 [DOI] [PubMed] [Google Scholar]
- 44. Gan Y, Zhang Y, Digirolamo DJ, et al. Deletion of IGF-I receptor (IGF-IR) in primary osteoblasts reduces GH-induced STAT5 signaling. Mol Endocrinol. 2010;24:644–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ma F, Wei Z, Shi C, et al. Signaling cross talk between growth hormone (GH) and insulin-like growth factor-I (IGF-I) in pancreatic islet β-cells. Mol Endocrinol. 2011;25:2119–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hughes JP, Friesen HG. The nature and regulation of the receptors for pituitary growth hormone. Annu Rev Physiol. 1985;47:469–482 [DOI] [PubMed] [Google Scholar]
- 47. Cunningham BC, Bass S, Fuh G, Wells JA. Zinc mediation of the binding of human growth hormone to the human prolactin receptor. Science. 1990;250:1709–1712 [DOI] [PubMed] [Google Scholar]
- 48. Somers W, Ultsch M, De Vos AM, Kossiakoff AA. The X-ray structure of a growth hormone-prolactin receptor complex. Nature. 1994;372:478–481 [DOI] [PubMed] [Google Scholar]
- 49. Xu J, Zhang Y, Berry PA, et al. Growth hormone signaling in human T47D breast cancer cells: potential role for a growth hormone receptor-prolactin receptor complex. Mol Endocrinol. 2011;25:597–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu J, Sun D, Jiang J, et al. The role of prolactin receptor in GH signaling in breast cancer cells. Mol Endocrinol. 2013;27(2):266–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rasmussen LM, Frederiksen KS, Din N, et al. Prolactin and oestrogen synergistically regulate gene expression and proliferation of breast cancer cells. Endocr Relat Cancer. 2010;17:809–822 [DOI] [PubMed] [Google Scholar]
- 52. Li X, Huang Y, Jiang J, Frank SJ. Synergy in ERK activation by cytokine receptors and tyrosine kinase growth factor receptors. Cell Signal. 2011;23:417–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huo JS, McEachin RC, Cui TX, et al. Profiles of growth hormone (GH)-regulated genes reveal time-dependent responses and identify a mechanism for regulation of activating transcription factor 3 by GH. J Biol Chem. 2006;281:4132–4141 [DOI] [PubMed] [Google Scholar]
- 54. Gong TW, Meyer DJ, Liao J, et al. Regulation of glucose transport and c-fos and egr-1 expression in cells with mutated or endogenous growth hormone receptors. Endocrinology. 1998;139:1863–1871 [DOI] [PubMed] [Google Scholar]
- 55. Hodge C, Liao J, Stofega M, Guan K, Carter-Su C, Schwartz J. Growth hormone stimulates phosphorylation and activation of elk-1 and expression of c-fos, egr-1, and junB through activation of extracellular signal-regulated kinases 1 and 2. J Biol Chem. 1998;273:31327–31336 [DOI] [PubMed] [Google Scholar]
- 56. Cui TX, Piwien-Pilipuk G, Huo JS, Kaplani J, Kwok R, Schwartz J. Endogenous CCAAT/enhancer binding protein β and p300 are both regulated by growth hormone to mediate transcriptional activation. Mol Endocrinol. 2005;19:2175–2186 [DOI] [PubMed] [Google Scholar]
- 57. Cui TX, Kwok R, Schwartz J. Cooperative regulation of endogenous cAMP-response element binding protein and CCAAT/enhancer-binding protein β in GH-stimulated c-fos expression. J Endocrinol. 2008;196:89–100 [DOI] [PubMed] [Google Scholar]
- 58. Milde-Langosch K. The Fos family of transcription factors and their role in tumourigenesis. Eur J Cancer. 2005;41:2449–2461 [DOI] [PubMed] [Google Scholar]
- 59. Yin X, Dewille JW, Hai T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene. 2008;27:2118–2127 [DOI] [PubMed] [Google Scholar]
- 60. Wang A, Arantes S, Yan L, et al. The transcription factor ATF3 acts as an oncogene in mouse mammary tumorigenesis. BMC Cancer. 2008;8:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Madden SL, Rauscher FJ., 3rd Positive and negative regulation of transcription and cell growth mediated by the EGR family of zinc-finger gene products. Ann NY Acad Sci. 1993;684:75–84 [DOI] [PubMed] [Google Scholar]
- 62. Gashler A, Sukhatme VP. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 1995;50:191–224 [DOI] [PubMed] [Google Scholar]
- 63. Mitchell A, Dass CR, Sun LQ, Khachigian LM. Inhibition of human breast carcinoma proliferation, migration, chemoinvasion and solid tumour growth by DNAzymes targeting the zinc finger transcription factor EGR-1. Nucleic Acids Res. 2004;32:3065–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. DuBois RN, McLane MW, Ryder K, Lau LF, Nathans D. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J Biol Chem. 1990;265:19185–19191 [PubMed] [Google Scholar]
- 65. Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin. Science. 1998;281:1001–1005 [DOI] [PubMed] [Google Scholar]