

Abstract
Group B Streptococcus (GBS) remains the leading cause of early onset sepsis among term infants. Evasion of innate immune defences is critical to neonatal GBS disease pathogenesis. Effectors of innate immunity, as well as numerous antibiotics, frequently target the peptidoglycan layer of the Gram-positive bacterial cell wall. The intramembrane-sensing histidine kinase (IM-HK) class of two-component regulatory systems has been identified as important to the Gram-positive response to cell wall stress. We have characterized the GBS homologue of LiaR, the response regulator component of the Lia system, to determine its role in GBS pathogenesis. LiaR is expressed as part of a three-gene operon (liaFSR) with a promoter located upstream of liaF. A LiaR deletion mutant is more susceptible to cell wall-active antibiotics (vancomycin and bacitracin) as well as antimicrobial peptides (polymixin B, colistin, and nisin) compared to isogenic wild-type GBS. LiaR mutant GBS are significantly attenuated in mouse models of both GBS sepsis and pneumonia. Transcriptional profiling with DNA microarray and Northern blot demonstrated that LiaR regulates expression of genes involved in microbial defence against host antimicrobial systems including genes functioning in cell wall synthesis, pili formation and cell membrane modification. We conclude that the LiaFSR system, the first member of the IM-HK regulatory systems to be studied in GBS, is involved in sensing perturbations in the integrity of the cell wall and activates a transcriptional response that is important to the pathogenesis of GBS infection.
Introduction
Group B Streptococcus (Streptococcus agalactiae, or GBS) is a Gram-positive coccus that is a leading cause of infection in newborns, often resulting in sepsis, pneumonia and meningitis (Puopolo & Baker, 2013). In neonates, infection with GBS occurs either in utero or during passage through the birth canal of an infected mother. GBS is also an increasingly important cause of infection among elderly and immunocompromised adults (Edwards & Baker, 2005; Edwards et al., 2005). Despite the potential to cause significant morbidity and mortality, GBS is most commonly found as a harmless colonizing organism in the gastrointestinal and genito-urinary tract of healthy individuals.
Maternally derived, transplacentally acquired polysaccharide capsule-specific antibody provides the neonate the best defence against invasive GBS disease (Baker & Kasper, 1976, 1977). In the absence of specific antibody, neonatal innate immune defences provide the primary defence against GBS infection. Toll-like receptor 2 (TLR2) and MyD88 signalling in response to specific components of the GBS cell surface are central to the neonatal immune response to GBS (Henneke & Berner, 2006). Cationic antimicrobial peptides (CAMPs) are another component of the innate immune response that functions in neonatal host defence. Produced both at host skin and mucosal surfaces and by leukocytes, these small molecules provide bactericidal host defence by binding to the negatively charged bacterial cell surface, disrupting the cell wall and cell membrane, which results in microbial lysis (Jenssen et al., 2006). Disruption of cell surface integrity via targeting of peptidoglycan synthesis, degradation of the cell wall, and cell membrane compromise by pore-forming agents are also mechanisms of action for a variety of naturally occurring antibiotics (Breukink & de Kruijff, 2006; Mascher, 2006). Consequently, bacteria have evolved multiple mechanisms for both sensing and responding to cell surface stress such as that mediated by antibiotics and antimicrobial peptides (Aldridge et al., 2005; Nizet, 2006).
Multiple mechanisms have been described for bacterial evasion of CAMP-mediated killing, including the production of non-specific proteases; peptide-specific binding proteins and extrusion pumps; and genetic alterations in the cell wall that diminish CAMP-binding (Peschel & Sahl, 2006). Bacteria can also sense and respond to changes in their environment through two-component transcription systems (TCS). TCS are signal transduction systems consisting of a membrane-bound sensor histidine kinase and a cytoplasmic response regulator protein (Mascher et al., 2006a). The sensor molecule contains both transmembrane and extracellular domains and can recognize a change in the environment—such as the integrity of the cell or the presence of a specific extracellular antibiotic—and subsequently undergo autophosphorylation as well as phosphorylation of a cognate, cytoplasmic response regulator protein. The response regulator is a DNA-binding transcription factor that affects changes in the expression of downstream genes in response to the signal detected by the sensor. TCS are important in the microbial response to a multitude of naturally occurring antimicrobials such as bacitracin, vancomycin and nisin (Giammarinaro et al., 1999; Mascher et al., 2004, 2006b). There are conceptual parallels and functional overlaps between the microbial defence systems developed to combat attack from CAMPs and antibiotics that are central to the virulence of both Gram-positive and Gram-negative organisms (Peschel & Sahl, 2006).
Very little is known about GBS mechanisms of antimicrobial defence despite the predicted importance of such systems in the pathogenesis of neonatal infection. GBS contain the dlt operon, which encodes the genes necessary for d-alanylation of lipoteichoic acid (LTA) on the surface of GBS. As is true in Staphylococcus aureus, mutation of the dlt system results in alteration of surface-expressed LTA, an increase in the overall surface negative charge, and increased susceptibility of GBS to antimicrobial peptides (Poyart et al., 2003). Deletion of penicillin-binding protein 1A (product of the ponA gene) results in increased susceptibility of GBS to antimicrobial peptides and in decreased virulence in a rat model of GBS sepsis and pneumonia (Hamilton et al., 2006; Jones et al., 2007). Deletion of pilB, one of the GBS pilus-forming proteins, was shown to result in increased susceptibility to neutrophil and antimicrobial peptide killing, and decreased virulence in a mouse model of GBS sepsis (Maisey et al., 2008b). Whether global regulatory systems are involved in the GBS defence against antimicrobial agents remains unclear. Multiple TCS are predicted by whole genome analysis in GBS, but only a few such as CsrR/S (CovR/S) (Lamy et al., 2004; Jiang et al., 2005, 2008, 2012; Rajagopal et al., 2006; Lembo et al., 2010; Cumley et al., 2012; Park et al., 2012) and RgfA/C (Spellerberg et al., 2002; Al Safadi et al., 2011) have been studied in detail to date. Here we report the identification of a TCS in GBS that is a homologue of the LiaSR TCS of Bacillus subtilis, the LiaRS TCS in Streptococcus pneumoniae and Streptococcus mutans and the VraSK TCS of S. aureus, all of which are involved in the species-specific response to cell wall-active antibiotics and antimicrobial peptides (Gardete et al., 2006; Jordan et al., 2007; Kuroda et al., 2003; Mascher et al., 2004; Eldholm et al., 2010; Suntharalingam et al., 2009). We demonstrate here that the LiaR response regulator is involved in GBS response to cell wall-active antimicrobial agents, including antibiotics and antimicrobial peptides, and may regulate the response to these agents by expression of genes involved in cell wall synthesis as well as genes involved in cell membrane modifications and pili formation in GBS. Furthermore, deletion of LiaR results in a significant attenuation of virulence in GBS mouse models of sepsis and pneumonia.
Methods
Bacterial strains and growth conditions.
GBS strain A909 is a type Ia/C laboratory strain originally obtained from the Lancefield collection. GBS was grown in Todd–Hewitt broth (THB) or THY broth (THB supplemented with 5 g l−1 yeast extract) or on THY-blood agar plates (all from Difco Laboratories). Recombinant DNA manipulations were performed in E. coli strain DH5α, grown at 37 °C in Luria–Bertani (LB) broth (Difco Laboratories) or on LB agar plates. When appropriate, 100 µg ml−1 spectinomycin was added to media plus 10 µg ml−1 erythromycin for E. coli or 1 µg ml−1 for GBS. Bacitracin, cefotaxime, ampicillin, colistin (polymixin E), polymixin B and vancomycin were purchased from Sigma and diluted in sterile water. Nisin was a gift from Dr Subhabrata Biswas and was purified as follows: a commercial preparation of nisin (2.5 % w/w) containing salt and denatured milk proteins was obtained from MP Biomedicals. Ten grams of crude nisin was resuspended in 120 ml water; nisin solubility was optimized by adjusting to pH 2.0 with HCl. The solution was centrifuged at 3,000 g and the pellet discarded. Nisin was then precipitated from solution with 1.5 M NaCl, adjusting to pH 6.3 with NaOH. Nisin was collected by centrifugation and redissolved in 0.05 % (v/v) acetic acid to 3 mg ml−1 (~1 mM) final concentration by Bradford assay (Bio-Rad). The purity and integrity were verified by SDS-PAGE and silver-staining.
DNA and RNA techniques.
Genomic DNA was isolated from overnight GBS cultures using the DNeasy Tissue kit (Qiagen), after treating bacteria with mutanolysin and lysozyme in 25 % glucose-Tris-EDTA buffer to generate protoplasts. Total RNA was isolated from GBS grown to early exponential phase. Bacteria were collected and resuspended in RNA Protect Reagent (Qiagen), then pelleted and resuspended in 25 % glucose-Tris-EDTA buffer with lysozyme and mutanolysin to generate protoplasts. The protoplasts were collected, resuspended in lysis buffer and lysed by centrifugation through a QIAshredder spin column (Qiagen). RNA was then purified using the RNeasy kit (Qiagen). Contaminating DNA was removed by on-column DNase digestion using RNase-free DNase (Qiagen). The concentration of the final RNA preparation was determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies) before the addition of RNase inhibitor. Aliquots of the final RNA preparation were stored at −80 °C. Southern and Northern transfer and hybridization were performed using standard techniques; probes were labelled and signal detected using an electrochemiluminescent (ECL) nucleic acid labelling and detection kit (GE Healthcare/Amersham). Plasmid DNA was isolated from E. coli using a plasmid mini-prep kit (Qiagen); restriction digests, DNA ligation and transformation of competent E. coli were all performed using standard protocols. Electrocompetent GBS were prepared and transformed as previously described (Klinzing et al., 2009; Puopolo et al., 2001). Group B streptococci protoplast preparations used as a genomic DNA source in PCRs were prepared as previously described (Chaffin & Rubens, 1998). Reverse transcriptase PCR (RT-PCR) was performed as previously described (Puopolo et al., 2001).
Generation of A909ΔliaR using allelic replacement.
PCR products containing (a) ~1300 bp of sequence upstream of the start codon for liaR through nucleotide 54 of the liaR gene, and (b) the last 67 bp of the liaR gene to approximately 900 bp downstream of the gene, were amplified using primer pairs YvqC_KO_upstream_F_BamHI+YvqC_KO_upstream_R and YvqC_KO_downstream_F+YvqC_KO_downstream_R_Xho, respectively (all primers are listed in Table S1 available in Microbiology Online). The products were used as template DNA in subsequent PCR with primers YvqC_KO_upstream_F_BamHI+YvqC_KO_downstream_R_Xho to create a fusion of the two original fragments. The fusion product was cloned into the Gram-positive cloning vector, pJRS233 using the BamHI and XhoI restriction enzyme sites. This plasmid contains a Gram-negative replicon, a temperature-sensitive Gram-positive replicon and an erythromycin resistance gene (Perez-Casal et al., 1993). Use of this construct in allelic exchange mutation results in removal of an internal 520 bp of liaR. Two-step allelic exchange mutagenesis was then performed as previously described (Puopolo et al., 2001). In brief, the pJRS233-liaR gene fusion construct was introduced into A909 by electroporation and selected for growth at 30 °C in the presence of erythromycin (erm). A single erm-resistant colony was shifted to growth at 37 °C to select for organisms in which the recombinant plasmid had integrated into the liaR gene by homologous recombination. Potential integrants were screened for gene interruption by PCR using primers YvqC_upstream_F and YvqC_downstream_R. Confirmed integrants were passaged in the absence of erm to allow for plasmid excision via a second recombination event. Erm-sensitive colonies were screened by PCR using primers YvqC_upstream_F and YvqC_downstream_R to confirm the expected deletion of the gene fragment. Deletions were confirmed by Southern blot hybridization using genomic DNA isolated from the deletion strain; and by Northern blot hybridization using RNA from the deletion strain, probed with the deleted fragment.
Construction of complementation plasmid to rescue A909ΔliaR phenotypes.
A plasmid was constructed to express the full-length liaR using the promoter from the bca gene of GBS, a strong, constitutive promoter that is not regulated by LiaR (Klinzing et al., 2009; Puopolo et al., 2001). A 246 bp fragment of the upstream region of bca (the N2 region) was cloned into Gram-positive vector pDL278, to create the expression vector pKPN2 (Klinzing et al., 2009). Beginning with the first amino acid following the ATG start codon, liaR was amplified with primers containing BamHI (5′ end) and SphI (3′ end) sites and was cloned into the BamHI and SphI sites of pKPN2, downstream of the bca promoter, to create the construct pKPN2-LiaR. Inserts and reading frame were confirmed by sequencing. pKPN2-LiaR was introduced into A909ΔLiaR by electroporation.
Colorimetric reporter construct to measure promoter activity.
An alkaline phosphatase reporter plasmid was constructed in the promoterless Gram-positive cloning vector pDL278 (Dunny et al., 1991). The phoZ gene, encoding alkaline phosphatase, was amplified by PCR from Enterococcus faecalis (strain 29212 purchased from Hardy Diagnostics) using primers containing the restriction enzyme sites, BamHI and SphI. The amplified PCR product was digested with BamHI and SphI, ligated into the multi-cloning site of pDL278, and transformed into E. coli. Recombinant colonies were identified by selection on spectinomycin-containing media; recombinant plasmid was isolated and sequenced to confirm insertion of the phoZ gene. A single confirmed clone was named pDKphoZ and used in all subsequent studies. To measure lia promoter activity, a 269 bp fragment of DNA upstream from the ATG start codon of LiaF was amplified and cloned upstream of the phoZ gene in pDKphoZ. To quantify alkaline phosphatase activity, GBS were grown to saturation overnight, then diluted 1 : 100 into 5 ml fresh THB and grown to early/mid exponential phase (OD650 ~0.3–0.35.) Bacteria were pelleted, resuspended in 5 ml MOPS buffer 50 mM NaCl, 10 mM NH4Cl, 10 mM MgCl2, 40 mM 3-(N-morpholino) propanesulfonic acid (MOPS) (K1 salt), pH 7.3, and the absorbance at 650 nm (A650) was recorded. The resuspended bacteria (100 µl) were combined with 100 µl Tris-Zinc buffer (1 M Tris pH 8.0, 0.1 mM ZnCl2) and 20 µl of 1 mg pNPP ml−1 (para-nitro phenol phosphate) in a 1.5 ml Eppendorf tube and incubated at 37 °C for 60–75 min. Bacteria were then pelleted; 100 µl of supernatant was removed to a microtitre plate well and the absorbance at 405 nm (A405) was recorded. Alkaline phosphatase activity was calculated in Miller units as [A405/(A650×bacteria added ml×time of incubation min)]×1000 (Zhang & Bremer, 1995).
Quantification of capsular polysaccharide and whole cell blots.
Capsular polysaccharide was quantified by competitive ELISA using purified type Ia capsule protein and anti-type Ia antisera as previously described (Cieslewicz et al., 2001). Whole cell blots were performed by growing bacteria on blood agar plates overnight. Bacteria were transferred to nitrocellulose by laying nitrocellulose circles briefly on agar plates. The subsequent whole cell blots were air-dried and Western blotting with GBS polysaccharide type Ia antisera was performed as previously described (Puopolo et al., 2001).
Characterization of A909ΔliaR growth under stress conditions.
Overnight cultures of A909 and A909ΔliaR were diluted 1 : 100 into 10 ml of fresh THB with antibiotics or antimicrobial peptide at the indicated concentrations. Growth was monitored in all cases by hourly measurements of the optical density at 650 nm (OD650). For temperature sensitivity testing, cultures of each strain were grown at 37 °C to OD650 ~0.35, then diluted 1 : 100 into THB media on a 96-well plate. Strains were subsequently grown at the indicated temperatures using a temperature gradient thermocycler. Growth was measured by (OD650) after 6–10 h at the indicated temperature. Results for growth at each temperature were expressed as a percentage of the growth measured at 37 °C.
UV light sensitivity assay.
Growth after exposure to shortwave UV (UV) light was assayed as previously described (Puopolo et al., 2001).
Murine models of GBS infection.
Animal experiments were performed with the permission of the Harvard Medical Area Standing Committee on Animals, experimentation protocol # 04350.
Intraperitoneal model of sepsis: Adult CD-1 female outbred mice were obtained from Charles River Laboratories. Varying concentrations of wild-type (A909) and mutant (A909ΔliaR) GBS in fresh THB (without antibiotics) were administered to mice by intraperitoneal injection (4 groups of 5 mice/group), and followed to 96 h post-injection. The LD50 was determined by the method of Reed & Muench (1938).
Pneumonia model: Wild-type (A909) and mutant (A909ΔliaR) GBS were grown at 37 °C to OD650 of approximately 0.6. The bacteria were pelleted and resuspended in 1 ml fresh THB. The average bacterial concentration was 5×108 c.f.u. ml−1. The cultures were kept on ice until ready for inoculation. Four- to 6-week-old female FVB/N mice (Harlan Sprague–Dawley Farms) were sedated by intraperitoneal injection of xylazine and ketamine; 20 µl of bacterial culture was added to each nostril. Inoculation with THB alone was performed as a control. Mouse survival was recorded at 48 h. The overall survival results shown in Table 1 are combined from two separate experiments. Three mice inoculated with A909, two with A909ΔliaR, and two with THB were sacrificed after 48 h post-inoculation. Lungs and spleens were removed using standard sterile techniques and homogenized in 200 µl of sterile PBS. The homogenates were plated on Todd–Hewitt agar plates and incubated overnight at 37 °C to determine c.f.u. per lung and spleen. Lungs and spleens from mice inoculated with THB only were found to be sterile (no bacterial colonies were identified after overnight incubation).
Table 1. Effect of LiaR deletion in murine models of GBS sepsis and pneumonia.
| Sepsis model* | Pneumonia model† | ||||
| Strain | LD50 trial 1 | LD50 trial 2 | Survival no. (%) | Lungs (c.f.u.) | Spleen (c.f.u.) |
| A909 | 1.0×104 | 7.5×104 | 4/19 (19) | 2.3×108 | 5.9×107 |
| A909ΔliaR | 6.5×106 | 9.5×106 | 14/14 (100) | 9.9×102 | 2.0×102 |
| A909ΔliaR+pLiaR | ND | 1.5×106 | ND | ND | ND |
Sepsis model LD50 expressed as bacterial c.f.u. (colony-forming units). LD50 calculated by survival 96 h post-intraperitoneal injection, using the method of Reed & Muench (1938). The difference in mean LD50 between A909 and A909ΔliaR is statistically significant (P = 0.0338 by t-test).
Survival was calculated at 48 h post-nasal injection. The difference in survival between A909 and A909ΔliaR is statistically significant with P<0.0001 by Fisher’s exact test. Bacterial c.f.u. recovered from the lungs and spleens in the pneumonia model were obtained from three different mice (A909) and two different mice (A909ΔliaR).
DNA microarray analysis.
The microarray used for LiaR transcriptional profiling was constructed at The Institute for Genomic Research (TIGR; now the J. Craig Venter Institute) and consisted of PCR amplicons representing all of the unique, annotated ORFs from GBS strains A909, 2603V/R and 515. Amino-allyl cDNA was synthesized from 2 µg of total RNA and hybridized to printed UltraGap slides (Corning), as previously described (Jiang et al., 2008). RNA was separately isolated twice resulting in two biological replicates. Technical replication consisted of (a) ≥3-fold spotted replication on a single slide, (b) hybridization of each RNA sample twice in one flip-dye replicate to control for dye-specific bias, and (c) duplicate or triplicate distinct amplicons designed per gene for a subset of genes. This level of replication resulted in ≥12 data points for each gene per condition assayed. The array image processing was performed with TIGR Spotfinder (Saeed et al., 2003). Data were normalized using iterative log mean centring with a 30K low intensity filter with TIGR Microarray Data Analysis System (MIDAS) (Saeed et al., 2003). The geometric mean ratio was calculated for all good spots across all four hybridizations. Regulation levels were set at a ratio of >2 or <0.5. Functional classification was categorized using the Sagalist server (http://genolist.pasteur.fr/SagaList/index.html) classification scheme (Moszer et al., 1995). The relative representation of regulated genes within each functional category was determined. The number of genes up- and down-regulated in A909ΔliaR within each category relative to the total number of regulated genes (119 for down-regulation, 89 for up-regulation) was compared to the total number of genes within that category relative to the total number of predicted GBS genes, using Fisher’s exact test (GraphPad Software). To account for multiple comparisons within each set of genes up- or down-regulated in A909ΔliaR, Bonferroni correction was made to set the significance level at P<0.002 for genes down-regulated in A909ΔliaR; and at P<0.0026 for genes up-regulated in A909ΔliaR.
MIC determination.
The minimum inhibitory concentration (MIC) of antimicrobial agents was determined for A909 and A909ΔliaR by the serial dilution method. Fresh cultures of bacteria were grown to OD650 ~0.30–0.35 and placed on ice. Fifteen microlitres of culture was added to the wells of a 96-well microtitre plate containing 150 µl of THB medium supplemented with twofold serial dilutions of antimicrobials. Bacterial growth after 6–24 h at 37 °C was spectrophotometrically measured by using an ELISA microtitre plate reader (model 3550; Bio-Rad Laboratories) at an absorbance of 650 nm (OD650). The compound concentration at which no growth was detectable was defined as the MIC.
Statistical analysis.
Mean values for bacterial growth under stressed conditions (heat, UV light, antimicrobials) were compared by t-test (GraphPad Scientific software available at www.graphpad.com) using a significance level of P<0.05.
Results
The GBS homologue of LiaR is expressed as part of a three-gene operon from a single promoter
The predicted DNA transcriptional regulatory protein encoded by SAK_0392 of the A909 genome sequence was identified among the GBS genes most strongly regulated by the stress-responsive TCS CiaRH (D. C. Klinzing and others, unpublished data). blast analysis reveals that the protein encoded by SAK_0392 is most closely related to the LiaR two-component regulator (originally named YvqC) found in B. subtilis. The GBS LiaR predicted protein sequence is 56 % identical (77 % conserved) to the LiaR protein in B. subtilis and 54 % identical (76 % conserved) with the VraR gene in S. aureus. In bacilli, LiaR is expressed as part of a six-gene operon (liaIHGFSR), but only identified homologues of liaFSR are found in the GBS genomes (Fig. 1a). LiaIHGFSR is expressed in bacilli from two promoters, producing transcripts containing liaIH and liaIHGFSR (Mascher et al., 2003, 2004). Northern blot analysis using liaR, liaF or liaS DNA as probes identified the same 2.3 kb message, consistent with a single transcript containing all three genes (Fig. 1b). Further confirmation that the three genes are transcribed as an operon is demonstrated by reverse-transcriptase PCR (RT-PCR) using RNA isolated from GBS cultures (Fig. 1c). Amplified fragments corresponding to the junctional regions between liaF, liaS and liaR support the expression of a single transcript containing all three genes. To confirm that the liaFSR operon is transcribed from a single promoter, DNA upstream of liaF and liaS was cloned into an alkaline phosphatase (AP) reporter plasmid. Only the sequence upstream of liaF drove detectable AP activity (Fig. 1d), consistent with a promoter located upstream of liaF.
Fig. 1.

(a) Structure of the genome region containing the proposed Lia operon. (b) Expression of LiaR in wild-type and ΔliaR mutant GBS. Ten micrograms of total RNA was isolated from GBS wild-type strain A909 and from the isogenic mutant A909ΔliaR and electrophoresed on 1 % denaturing formaldehyde agarose gel. RNA was transferred to nylon membrane and probed with a fragment of the LiaR gene deleted from the A909ΔliaR mutant. Message of ~2.3 kb was observed in A909; this is absent in the mutant strain (arrowhead). The blot was reprobed with a RecA gene fragment to ensure even loading of RNA. The size of the mRNA is consistent with a message encompassing liaFSR genes, and RNA analysis with liaF and liaS probes confirms that these genes are expressed as an operon (data not shown). (c) Operon structure determination by RT-PCR. Confirmation of the operon structure of liaFSR was done by performing reverse transcription using the indicated primer followed by PCR with primers for the indicated genes. The genes for LiaS and LiaR were found to be coexpressed when using a LiaR primer for reverse transcription. Additionally, LiaF and LiaS were coexpressed when using a LiaS reverse transcription primer. PCR products were identical in size to those found using genomic DNA as a template. Template consisting of RNA that did not undergo reverse transcription did not produce any product. (d) liaFSR Promoter Activity. DNA fragments encompassing 269 bp upstream from the ATG start codon of liaF, and 473 bp upstream of the ATG start codon of liaS were amplified and cloned upstream of the alkaline phosphatase gene phoZ in reporter construct pDKphoZ and introduced into GBS. Alkaline phosphatase activity was measured and calculated in Miller units from wild-type A909 containing no reporter construct (no vector control); A909 containing the reporter construct without experimental DNA upstream of phoZ (empty vector control); and constructs containing the liaF and liaS upstream regions. The region upstream of liaF was able to drive transcription of the phoZ gene indicating that this is the functional promoter for the liaFSR operon in GBS, while the region upstream of liaS did not drive phoZ expression. Error bars denote the mean and standard deviation from three separate sets of experiments.
Construction of A909ΔliaR and initial characterization
To examine the role of the liaFSR system in GBS disease pathogenesis, we generated an isogenic liaR deletion in GBS strain A909 by allelic exchange mutagenesis. The mutant strain A909ΔliaR contains a deletion of the 520 nt internal portion of liaR (out of 642 nt total); Northern analysis using a probe specific for the deleted segment reveals no message (Fig. 1b). Northern analysis using a probe specific for liaF or liaS demonstrates that a message is still produced in A909ΔliaR and suggests that LiaR does not autoregulate the expression of the liaFSR operon (data not shown).
LiaR and environmental stress in GBS
We measured the response of the A909ΔliaR compared to wild-type A909 in response to elevated temperature and UV light exposure (Fig. 2). A909ΔliaR grew poorly at 40 °C compared to A909 (P = 0.019 for difference between strains’ growth at 37 °C versus 40 °C). The mutant strain was also significantly more sensitive to killing by UV light exposure than wild-type (P = 0.0003 for difference in survival after 5 s of UV light exposure), suggesting a defect in the ability to repair DNA damage.
Fig. 2.
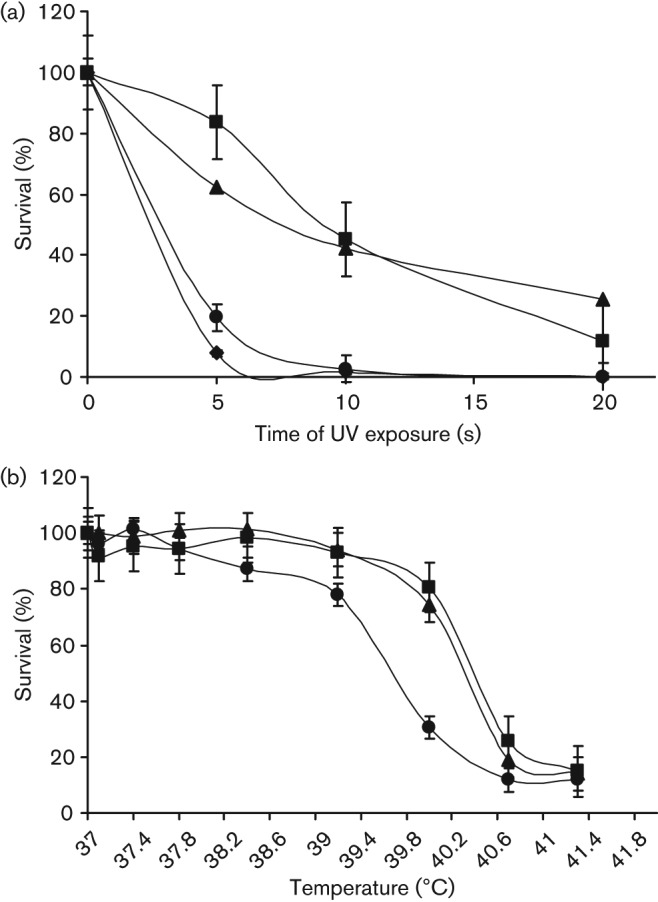
Environmental stress phenotypes of A909ΔliaR. (a) Killing by UV light. Survival of wild-type A909 (▪), A909ΔliaR (•), the complemented mutant strain A909ΔliaR+pLiaR (▴), and the DNA repair mutant, A909ΔrecA (♦) were compared following exposure to short-wave UV light for 0, 5, 10, 15 and 20 s. Results shown are mean and standard deviation from three separate experiments. (b) Growth at elevated temperature. Wild-type A909 (▪), A909ΔliaR (•), and the complemented mutant strain A909ΔliaR+pLiaR (▴), were grown at 37 °C to an OD650 ~0.35, then diluted 1 : 100 into THB media on a 96-well plate. Strains were subsequently grown at the indicated temperatures using a temperature gradient thermocycler. Growth was measured by (OD650) after 6–10 h at the indicated temperature. Results for growth at each temperature were expressed as a percentage of the growth measured at 37 °C; mean±sd of two separate experiments.
A909ΔliaR is sensitive to attack by cell wall antibiotics and antimicrobial peptides
The Lia TCS regulates responses to cell wall stress mediated by antibiotics and antimicrobial peptides in B. subtilis, and the homologous S. aureus TCS VraSK is involved in the response of that organism to vancomycin (Gardete et al., 2006; Jordan et al. 2008; Mascher et al., 2003). We examined the effect of several antibiotics and antimicrobial peptides on growth of A909ΔliaR compared to wild-type A909. The growth curve of the A909ΔliaR mutant over time is affected by vancomycin (Fig. 3a), bacitracin (Fig. 3b), colistin (polymyxin E) (Fig. 3c) and nisin (Fig. 3d) as compared to A909, demonstrating slower growth at different concentrations of antibiotic. The MICs for these compounds, as well as for the antibiotics ampicillin, cefotaxime and polymixin B, and the antimicrobial peptide LL-37 (Bergman et al., 2005; Jenssen et al., 2006; Schauber et al., 2006) were determined for A909 and A909ΔliaR (Table 2). The MIC for the mutant strain was significantly lower for colistin, nisin and polymixin B, and borderline significant for vancomycin. There was no significant difference found for cefotaxime or ampicillin. The MIC for LL-37 trended lower in the mutant strain but the difference over two experiments was not statistically significant. Interestingly, although we found a significant growth defect in the presence of bacitracin over a 6 h growth period, the MIC of the wild-type and mutant strains were identical with overnight growth. To demonstrate the specificity of these phenotypes, expression of liaR was restored to A909ΔliaR using pKPN2, a plasmid construct that drives expression of cloned DNA in GBS using the strong bca (non-LiaR-regulated) promoter (Klinzing et al., 2009). Complementation of A909ΔliaR with pKPN2-LiaR reversed the antimicrobial sensitivity phenotypes in the presence of vancomycin, nisin and colistin (Table 2 and Fig. 4). The bacitracin growth curve phenotype could not be fully complemented (data not shown), although the MIC trended higher for the complemented strain.
Fig. 3.

Antimicrobial sensitivity of wild-type GBS and ΔliaR mutant strain. The sensitivity of wild-type A909 (solid lines) and A909ΔliaR (dotted lines) was assessed by growth of each strain on increasing concentrations of antimicrobial. (a) Vancomycin (0 µg ml−1, ▪; 0.5 µg ml−1, •; 0.75 µg ml−1, ▴). (b) Bacitracin (0 µg ml−1, ▪; 1.0 µg ml−1, •; 15 µg ml−1, ▴). (c) Colistin (0 µg ml−1, ▪; 20.0 µg ml−1, •; 30.0 µg ml−1, ▴). (d) Nisin (0 µg ml−1, ▪; 0.3 µg ml−1, •; 0.9 µg ml−1, ▴). Growth was monitored hourly by OD650 measurement. Results are from three (vancomycin), or two (bacitracin, colistin and nisin) separate experiments.
Table 2. Minimal inhibitory concentrations (MIC).
| Antimicrobial | MIC (µg ml−1) | |||
| A909 | A909ΔliaR | A909ΔliaR+pLiaR | P-value* | |
| Ampicillin | 0.07 | 0.08 | 0.08 | 1.000 |
| (n = 3) | (n = 1) | (n = 1) | ||
| Bacitracin | 118.8 | 118.8 | 166.7 | 1.000 |
| (n = 8) | (n = 8) | (n = 3) | ||
| Cefotaxime | 0.06 | 0.05 | ND | 0.537 |
| (n = 3) | (n = 3) | |||
| Colistin | 66.7 | 26.7 | ND | 0.009 |
| (n = 3) | (n = 3) | |||
| LL-37 | 24.0 | 4.0 | 16.0 | 0.130 |
| (n = 2) | (n = 2) | (n = 2) | ||
| Nisin | 2.30 | 0.96 | 3.63 | 0.040 |
| (n = 4) | (n = 4) | (n = 3) | ||
| Polymixin B | 15.60 | 6.50 | ND | 0.002 |
| (n = 3) | (n = 3) | |||
| Vancomycin | 0.89 | 0.67 | 1.08 | 0.054 |
| (n = 5) | (n = 5) | (n = 2) | ||
P-values are for comparison of A909 and A909ΔliaR. The P-value for comparison between A909 and the complemented strain A909ΔliaR+pLiaR was non-significant where tested (P≥0.05).
Fig. 4.

Antimicrobial sensitivity complementation. (a) Growth of A909, solid line ▪; A909ΔliaR dotted line, ▪; A909ΔliaR +pN2GFP (control plasmid) dotted line, •; A909ΔliaR+pLiaR (dotted line, ▴) are similar in the absence of antibiotic (a). Restoration of LiaR expression in the complemented strain A909ΔliaR+pLiaR (dotted line, ▴) but not with control plasmid A909ΔliaR +pN2GFP (dotted line, •) restores growth in the presence of (b) vancomycin, 0.5 µg ml−1, and (c) nisin, 0.3 µg ml−1, and (d) colistin, 30 µg ml−1, to nearly wild-type levels.
Deletion of LiaR results in an attenuation in virulence in two models of mouse infection
The increased susceptibility of A909ΔliaR to stressful environmental conditions and to cell wall-targeted antimicrobials suggested that the Lia system may be important in vivo to GBS pathogenesis. The strain was compared to wild-type A909 in two separate murine models of GBS disease. In an intraperitoneal-injection model of GBS sepsis, A909ΔliaR had an LD50 over two orders of magnitude higher than wild-type A909 (Table 1). Expression of liaR from pKPN2-LiaR was able to partially complement the attenuation of the A909ΔliaR mutant. The lack of complete rescue of the attenuated phenotype by the strain containing pKPN2-LiaR may be due to the lack of antibiotic selection in the injected samples (fresh THB only) and loss of plasmid during treatment. In the murine model of GBS pneumonia, A909ΔliaR was highly attenuated (Table 1). Intranasal administration of the wild-type strain A909 resulted in high levels of bacterial invasion of the lung parenchyma and presumed bacteraemia, as evidenced by high levels of bacteria found in splenic tissue. Lung and spleen were not sterile in the A909ΔliaR-exposed animals, but very low numbers of bacteria were found in these organs compared to wild-type A909 (Table 1).
Encapsulation of A909ΔliaR mutants
The magnitude of attenuation observed with A909ΔliaR in the murine models of GBS infection has only been previously observed in experiments comparing wild-type GBS to GBS that lack expression of the type-specific polysaccharide capsule (acapsular mutants) (Rubens et al., 1987). Therefore, we assessed the encapsulation of the A909ΔliaR mutant strain. As qualitatively assessed by Northern analysis probing with a type IA-specific capsule locus gene (cpsA), and by immunoblot using type Ia-specific antibody; and quantitatively using type Ia-specific antibody and competitive ELISA inhibition; and confirmed by DNA microarray analysis (see below), there was no detectable difference in the level of capsular polysaccharide or the levels of capsule gene transcription between A909ΔliaR and wild-type A909 (data not shown).
GBS Lia transcriptional response to cell wall stress
The Lia operon is one of the most highly induced mRNAs in B. subtilis and S. aureus when treated with cell wall-active antibiotics (Gardete et al., 2006; Jordan et al., 2008; Mascher et al., 2003, 2004). We examined the level of lia-containing mRNA in GBS by Northern blot analysis to determine if lia operon mRNA is also induced by cell wall stress. GBS were grown to mid-exponential phase and treated with 1× and 10× the MIC of a cell-wall active antibiotic (vancomycin) or an antimicrobial peptide (nisin). In both cases, the level of liaFSR mRNA did not appear to be significantly altered when compared to an untreated culture (data not shown).
A909ΔliaR regulates expression of cell wall synthesis, pili island 2b, cell membrane modification and stress response genes
The increased susceptibility of the A909ΔliaR strain to several antimicrobial agents suggested that LiaR may be involved in the regulation of genes involved in cell wall synthesis and maintenance, and in protection from antimicrobial agents. Transcriptional profiling by DNA microarray analysis was performed using mRNA isolated from wild-type A909 and A909ΔliaR to determine the LiaR regulon. The microarray represented all the predicted ORFs of the sequenced GBS strains A909, 2603V/R, and 515. Of the 2451 genes represented on the microarray, 8.5 % were differentially expressed in A909ΔliaR compared to A909 : 89 genes were up-regulated in A909ΔliaR g(ratio >2.0; Table S2), and 119 genes were down-regulated in A909ΔliaR (ratio <0.5; Table S3). A summary of the functional distribution of these genes is found in Table 3. Several functional categories were significantly differentially represented in the regulon compared to their representation within the GBS genome. Among genes down-regulated in A909ΔliaR, functional categories 1.2 (transport/binding proteins and lipoproteins, P<0.0001) and 6.0 (no similarity, P = 0.0005) were significantly differentially regulated; category 4.2 (detoxification, P = 0.0024) was borderline significant. Among genes up-regulated in A909ΔliaR, functional category 2.4 (metabolism of lipids, P = 0.0003) was significantly differentially regulated; category 1.2 (transport/binding proteins and lipoproteins, P = 0.0027) was borderline significant.
Table 3. Transcriptional profiling A909LiaR.
The number of genes whose expression levels were found to be significantly altered in A909ΔliaR versus wild-type GBS strain A909 is presented. Expression levels lower than 0.5 (−) and greater than 2.0 (+) were determined to be significant. Genes were classified into functional groups using the Sagalist server (http://genolist.pasteur.fr/SagaList/index.html) classification scheme (Moszer et al., 1995). The total number of genes per group is indicated, and groups in which the variation is significant by Fisher’s Exact test are indicated by the presence of the P-value. NS, not significant.
| Number of genes regulated in A909ΔLiaR | |||||
| Functional classification category | Total genes in A909 | Up-regulated | P-value | Down-regulated | P-value |
| 1.1 Cell wall | 81 | 2 | ns | 5 | ns |
| 1.2 Transport/binding proteins and lipoproteins | 255 | 21 | 0.0031 | 2 | <0.0001 |
| 1.3 Sensors (signal transduction) | 20 | 1 | ns | 4 | 0.0038 |
| 1.4 Membrane bioenergetics | 26 | 1 | ns | 3 | ns |
| 1.8 Cell surface proteins | 55 | 5 | ns | 7 | 0.0469 |
| 2.1.1 Metabolism of carbohydrates | 112 | 6 | ns | 6 | ns |
| 2.1.2 Main glycolytic pathways | 13 | 0 | ns | 1 | ns |
| 2.2 Metabolism of amino acids | 76 | 1 | ns | 8 | ns |
| 2.3 Metabolism of nucleotides/nucleic acids | 68 | 1 | ns | 1 | ns |
| 2.4 Metabolism of lipids | 37 | 8 | 0.0006 | 2 | ns |
| 2.5 Metabolism of coenzymes | 36 | 3 | ns | 1 | ns |
| 2.6 Metabolism of phosphate | 2 | 0 | ns | 0 | ns |
| 3.1 DNA replication | 28 | 1 | ns | 0 | ns |
| 3.2 DNA restriction/modification and repair | 26 | 0 | ns | 3 | ns |
| 3.3 DNA recombination | 12 | 0 | ns | 2 | 0.0164 |
| 3.4 DNA packaging and segregation | 17 | 0 | ns | 1 | ns |
| 3.5.2 Regulation | 113 | 2 | ns | 9 | ns |
| 3.6 RNA modification | 26 | 0 | ns | 0 | ns |
| 3.7.1 Ribosomal proteins | 55 | 2 | ns | 0 | ns |
| 3.7.2 Aminoacyl-tRNA synthetases | 22 | 0 | ns | 0 | ns |
| 3.7.3 Initiation | 4 | 0 | ns | 0 | ns |
| 3.8 Protein modification | 20 | 0 | ns | 5 | 0.0086 |
| 4.1 Adaptation to atypical conditions | 20 | 0 | ns | 1 | ns |
| 4.2 Detoxification | 14 | 1 | ns | 5 | 0.0024 |
| 4.3 Phage-related functions | 17 | 0 | ns | 2 | ns |
| 4.4 Transposon and IS | 46 | 2 | ns | 2 | ns |
| 4.5 Miscellaneous | 15 | 2 | ns | 1 | ns |
| 5.0 Similar to unknown proteins | 623 | 19 | 0.0063 | 46 | ns |
| 6.0 No similarity | 226 | 10 | ns | 2 | 0.0003 |
| Total | 2065 | 88 | 119 | ||
Genes down-regulated in A909ΔliaR with an attributed function that may be important to cell wall synthesis and maintenance include: (a) genes belonging to pili island PI-2b; (b) the gene for penicillin-binding protein 2b (pbp2b), a transpeptidase involved in cell wall synthesis; (c) a homologue of murN, involved in synthesis of interpeptide bridges in the peptidoglycan structure (Filipe et al., 2000; Reinscheid et al., 2002); and (d) the genes encoding a homologue of mprF/fmtC (SAK_2070 and SAK_2071), predicted to be involved in modifying the charge on the surface of the lipid membrane to act as a repellent to the charged residues of cationic antimicrobials, such as LL-37 and the defensins (Weidenmaier et al., 2005). Northern blot analysis confirmed the microarray results for each of these genes; complementation of A909ΔliaR with pKPN2liaR restored expression of each tested gene (Fig. 5a–f). Northern analysis of several other genes strongly up- or down-regulated in A909ΔliaR was performed as a general biological confirmation of the microarray results; in each case, Northern analysis was consistent with the transcriptional profile (data not shown). Finally, it is notable that the gene most affected in the LiaR regulon (SAK_2031, expression ratio 0.038) encodes a homologue of the transcriptional regulator SpxA, a global stress response regulator (Pamp et al., 2006). In B. subtilis, SpxA is involved in the response to disulphide stress via its interaction with the α-subunit of RNA polymerase (Nakano et al., 2005). This gene is expressed at high levels in GBS even in the absence of specific stress, but is undetectable by Northern analysis in A909ΔliaR.
Fig. 5.

Confirmation of transcriptional profiling results for the A909 and A909ΔliaR comparison. Ten micrograms of total RNA was prepared from A909, A909ΔliaR and the complemented strain A909ΔliaR+pLiaR and analysed by Northern blot. Probes were for the genes indicated: SAK_1439 (pili island PI-2b); SAK_0890 (penicillin-binding protein 2b); SAK_0658 (murN); SAK_2031 (spxA). In each case, expression was decreased in the A909ΔliaR strain as predicted by microarray analysis, and was restored with liaR expression from plasmid pKPN2liaR. DNA for recA (not shown to be regulated in the liaR microarray dataset) was used as control.
Discussion
GBS emerged in the United States in the 1970s as the primary bacterial cause of early-onset neonatal infection (Puopolo & Baker, 2013). The reasons for this emergence remain unclear, but GBS has proven a remarkably persistent threat to neonates (Puopolo & Baker, 2013). The pathogenesis of neonatal infection is framed as the convergence of opportunities. Approximately 20–30 % of American women are colonized with GBS at any point in time. In the absence of antibiotic prophylaxis, 50 % of neonates born to GBS-colonized mothers become colonized, and overall 1–2 % of those develop invasive GBS disease. A multitude of other maternally derived commensal bacteria have the opportunity to follow this vertical path to neonatal disease, but none do so with the frequency of GBS, suggesting that one aspect of neonatal GBS pathogenesis must lie in especially poor neonatal immune defences against GBS, or especially good strategies of GBS to avoid those defences. Relative deficiencies in neonatal phagocyte function, including poor chemotaxis, impaired intracellular killing of bacteria, and dysregulated TLR-mediated inflammatory responses have all been implicated in the pathogenesis of neonatal GBS sepsis (Henneke & Berner, 2006). Certain GBS virulence factors have been shown to specifically interfere with neonatal immune mechanisms. C5a-peptidase cleaves the C5a component of complement, interfering with both phagocyte chemotaxis and bacterial uptake (Maisey et al., 2008a). Both GBS superoxide dismutase and beta-haemolysin have been implicated in resistance to oxygen-free-radical-dependent intracellular killing by phagocytes (Maisey et al., 2008a). However, the mechanisms by which GBS regulate and coordinate responses to host immune challenges during disease pathogenesis have not been described. Here we demonstrate that the LiaFSR TCS regulates the GBS response to cell-wall active antibiotics and antimicrobial agents. Together with the significant in vivo attenuation associated with loss of LiaR, we propose that this system is needed for GBS defence against host innate immune responses.
As is true in other streptococci, the sequenced GBS genomes have few predicted alternative sigma factors that might mediate transcriptional responses to environmental challenges, but GBS contains more predicted TCSs than either group A streptococci (Streptococcus pyogenes) or Streptococcus pneumoniae (Jordan et al., 2008). Of the 20 predicted TCS in the A909 genome, four have a size and structure of the sensor component that warrants subclassification into the intramembrane-sensing histidine kinase (IM-HK) family (Jordan et al., 2008). IM-HK systems contain a relatively small (<400 aa) histidine kinase with just two transmembrane regions flanking a short (<20 aa) extracellular domain. The small size of the extracellular domain suggests that the sensing domain is either embedded in, or in close contact with, the lipid membrane of the bacterial cell (Jordan et al., 2008; Mascher, 2006). The IM-HKs have been proposed to act as ‘sentinel’ systems closely monitoring the integrity of the cell membrane and peptidoglycan cell wall (Belcheva & Golemi-Kotra, 2008), and responding to cell wall stress with a transduced transcriptional response (Mascher et al., 2004, 2006b). Analyses of the GBS LiaS sequence and predicted structure are consistent with GBS LiaFSR being a member of the IM-HK class of TCS.
The LiaSR system and its homologues have been studied in detail in few other bacterial species, most notably B. subtilis, S. pneumoniae, S. mutans and S. aureus (Gardete et al., 2006; Jordan et al., 2007; Mascher et al., 2004; Pietiäinen et al., 2005; Suntharalingam et al., 2009; Yin et al., 2006) and have been identified in the response to vancomycin and murein hydrolases expressed during competence in S. pneumoniae (Haas et al., 2005; Eldholm et al., 2010). Our studies reveal several important differences between the GBS Lia system and these homologues. The B. subtilis Lia system includes six genes (liaIHGFSR), the S. mutans Lia system includes five genes (liaFSR-ppiB-pnpB) and the S. aureus locus included four genes (orf1-vraFSR), whereas the GBS system consists only of three genes (liaFSR). The Bacillus system is transcribed from two promoters; a primary promoter located upstream of liaG, and a secondary promoter upstream of liaI. The liaIH transcript is the most highly up-regulated mRNA in response to cell wall stress in B. subtilis (Mascher et al., 2003, 2004). The B. subtilis, S. mutans and S. aureus systems are strongly autoregulated by LiaR, but we could not detect autoregulation of GBS lia by LiaR. The liaFSR genes are cotranscribed on a single message in GBS, with a primary promoter located upstream of liaF. Both Northern analysis and microarray-based transcriptional profiling demonstrate that liaF and liaS transcripts are produced at normal levels in the A909ΔliaR mutant, which contains an in-frame deletion of liaR but does not disrupt the upstream sequences. Loss of LiaR in B. subtilis and S. mutans results in increased sensitivity of the organism to bacitracin, and exposure to sublethal concentrations of bacitracin strongly induces the Lia system in both species (Mascher et al., 2003, 2004; Suntharalingam et al., 2009). Although loss of LiaR results in a similar spectrum of antimicrobial sensitivity in GBS, including variable sensitivity to bacitracin, vancomycin, nisin and the human CAMP LL-37, the GBS liaF promoter itself is not induced by these compounds. Finally, loss of the Vra system in S. aureus results in sensitivity to beta-lactam antibiotics, but has no apparent role in response to CAMPs such as LL-37, whereas loss of GBS LiaR results in the opposite effect on GBS sensitivity to LL-37 and beta-lactam antibiotics (Yin et al., 2006). These results demonstrate the unique role that the Lia system plays in GBS and suggest that other regulatory systems are operative in cell wall maintenance and antibiotic responsiveness in GBS. They also point out the importance of determining the individual role structurally homologous systems may play in the pathogenesis of disease in different Gram-positive species.
An important finding of this study is the link between the Lia system and GBS virulence. The only prior evidence for a role of the Lia system homologues in organism virulence comes from two studies in S. pneumoniae. In one study, a transposon-based signature-tagged mutagenesis (STM) library of S. pneumoniae was screened for genes essential for lung infection (Hava & Camilli, 2002). A mutant of the pneumococcal homologue of LiaF (SP0385/SPY1623) was found to be essential for lung infection, but the basis for this was not further investigated. A mutation in the LiaS histidine kinase in S. pneumoniae (HK03) revealed a role for this system in regulating pili expression. In this system, the pneumococcal pilus was substantially overexpressed in the LiaS deleted strain, and yet the mutant was attenuated in an intranasal mouse model of pneumococcal infection (Rosch et al., 2008). Similarly, expression of a second GBS pilus island, PI-1 in strain 2603V/R, has been shown to be regulated by the GBS TCS csrRS and loss of csrR activity has been shown to increase pilus expression while a csrR mutant has reduced virulence in mouse models (Jiang et al., 2005, 2008, 2012). In contrast, we observed a decrease in expression of genes encoding pili island 2b (PI-2b) in the GBS LiaR mutant and a profound attenuation in two mouse models of GBS infection.
The reasons for the decreased virulence of the LiaR mutant are likely multifactorial. It is notable that expression of genes associated with haemolysin expression (cylA-X) is increased in A909ΔliaR despite the well-documented role of haemolysis in GBS virulence (Liu & Nizet, 2004). An apparently paradoxical relationship between increased haemolysin expression and decreased virulence in animal models of GBS infection has also been noted by deletion of another GBS TCS, CsrR/S (Jiang et al., 2005), demonstrating the complexities of virulence regulation by TCS. Decreased expression from several genes in the LiaR regulon may affect the virulence of A909ΔliaR. The pili of GBS have recently been proposed to aid in antimicrobial peptide defence (Maisey et al., 2008b). At least one gene in the LiaR-regulated pili island 2b (spb1, encoded by SAK_1440) has been shown to be important for cell adhesion and invasion (Adderson et al., 2003). Pili have also been shown to be important to the transcytosis of GBS across epithelial cell layers (Pezzicoli et al., 2008), and influence resistance to phagocytosis by macrophages and neutrophils, which are a main source of LL-37 (Maisey et al., 2008b; Jiang et al., 2012). Moreover, pili appear to act as a virulence factor in a neonatal mouse model of infection (Papasergi et al., 2011). Other potential factors that may contribute to the decreased virulence of A909ΔliaR may be a loss of cell wall integrity and an inability to modify the cell surface to repel attack by antimicrobials. Microarray analysis and Northern blot confirmation demonstrate that the A909ΔliaR mutant has reduced levels of expression of penicillin-binding protein 2b (pbp2b, SAK_0890) and the peptidoglycan cross-linking peptide synthesis homologue MurN (SAK_0658), both involved in the synthesis of the cell wall. The mutant also has reduced levels of the GBS homologue of mprF (SAK_2070), the product of which modifies the charge of the cell membrane by the addition of a lysine residue to membrane phospholipids in order to repel cationic antimicrobial agents (Fig. 5) (Filipe et al., 2000; Weidenmaier et al., 2005). Similar envelope synthesis and stress response genes were found to be regulated by the Lia TCS system in S. mutans (Suntharalingam et al., 2009). Further work is required to determine if decreased pili production and/or changes in cell well integrity do occur in A909ΔliaR, and whether such changes directly contribute to the decreased virulence observed in the murine model of GBS pneumonia. In addition, as the microarray data indicate that there are additional two-component transcriptional regulators that are also down-regulated in the LiaR mutant (SAK_1992, SAK_2061 and SAK_1071), as well as the global regulator spxA, we cannot rule out the possibility that the changes in gene expression in the LiaR mutant are due to the loss of an additional downstream regulator.
The dramatic effect on virulence in the LiaR mutant does, however, suggest that this family of TCSs may be effective targets for antivirulence therapies, an evolving concept of antimicrobial development (Waldor, 2006). Disruption of a single transcriptional regulator has the effect of disrupting multiple key systems, as in the case of mutation of LiaR. High-throughput screening and structure-based design methods have been utilized to identify small molecules that specifically inhibit the action of transcriptional regulators (Bowser et al., 2007; Hung et al., 2005). Virstatin was identified as an inhibitor of ToxT in Vibrio cholerae resulting in disruption of cholera toxin and toxin co-regulated pilus production (Hung et al., 2005). Bowser et al. designed ‘broad spectrum’ inhibitors of AraC family transcriptional regulators and demonstrated effectiveness against E. coli in a urinary tract infection model (Bowser et al., 2007). A theoretical advantage of targeting regulatory molecules is that deletions are not lethal, and thus development of resistance may be less rapid than with bactericidal antibiotics.
Acknowledgements
This work was supported by Public Health Service grants from the National Institute of Child Health and Human Development, K08-HD041534 (to K. M. P.); T32-HD007466-11A1 (to D. C. K); and from the National Institute of Allergy and Infectious Diseases, R01-AI38424 (to L. C. M.); and by a Child Health Research grant from The Charles H. Hood Foundation (to K. M. P.). The authors are indebted to Dr G. Bolduc for his contribution of the mouse pneumonia model, and to Dr G. Priebe for his assistance with that model. The authors would like to thank Dr S. Kourembanas, Dr M. Wessels, and Dr D. Kasper for their support of our work; Dr M. Baron-Barshak for helpful discussions; and M. Coyne for invaluable assistance with microarray data analysis.
Abbreviations:
- CAMP
cationic antimicrobial peptide
- GBS
group B Streptococcus
- IM-HK
intramembrane-sensing histidine kinase
- LTA
lipoteichoic acid
- TCS
two-component transcription system
Footnotes
Three supplementary tables are available with the online version of this paper.
References
- Adderson E. E., Takahashi S., Wang Y., Armstrong J., Miller D. V., Bohnsack J. F. (2003). Subtractive hybridization identifies a novel predicted protein mediating epithelial cell invasion by virulent serotype III group B Streptococcus agalactiae. Infect Immun 71, 6857–6863. 10.1128/IAI.71.12.6857-6863.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Safadi R., Mereghetti L., Salloum M., Lartigue M. F., Virlogeux-Payant I., Quentin R., Rosenau A. (2011). Two-component system RgfA/C activates the fbsB gene encoding major fibrinogen-binding protein in highly virulent CC17 clone group B Streptococcus. PLoS ONE 6, e14658. 10.1371/journal.pone.0014658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge P. D., Gray M. A., Hirst B. H., Khan C. M. (2005). Who’s talking to whom? Epithelial-bacterial pathogen interactions. Mol Microbiol 55, 655–663. 10.1111/j.1365-2958.2004.04469.x [DOI] [PubMed] [Google Scholar]
- Baker C. J., Kasper D. L. (1976). Correlation of maternal antibody deficiency with susceptibility to neonatal group B streptococcal infection. N Engl J Med 294, 753–756. 10.1056/NEJM197604012941404 [DOI] [PubMed] [Google Scholar]
- Baker C. J., Kasper D. L. (1977). Immunological investigation of infants with septicemia or meningitis due to group B Streptococcus. J Infect Dis 136 (Suppl), S98–S104. 10.1093/infdis/136.Supplement.S98 [DOI] [PubMed] [Google Scholar]
- Belcheva A., Golemi-Kotra D. (2008). A close-up view of the VraSR two-component system. A mediator of Staphylococcus aureus response to cell wall damage. J Biol Chem 283, 12354–12364. 10.1074/jbc.M710010200 [DOI] [PubMed] [Google Scholar]
- Bergman P., Johansson L., Asp V., Plant L., Gudmundsson G. H., Jonsson A. B., Agerberth B. (2005). Neisseria gonorrhoeae downregulates expression of the human antimicrobial peptide LL-37. Cell Microbiol 7, 1009–1017. 10.1111/j.1462-5822.2005.00530.x [DOI] [PubMed] [Google Scholar]
- Bowser T. E., Bartlett V. J., Grier M. C., Verma A. K., Warchol T., Levy S. B., Alekshun M. N. (2007). Novel anti-infection agents: small-molecule inhibitors of bacterial transcription factors. Bioorg Med Chem Lett 17, 5652–5655. 10.1016/j.bmcl.2007.07.072 [DOI] [PubMed] [Google Scholar]
- Breukink E., de Kruijff B. (2006). Lipid II as a target for antibiotics. Nat Rev Drug Discov 5, 321–332. 10.1038/nrd2004 [DOI] [PubMed] [Google Scholar]
- Chaffin D. O., Rubens C. E. (1998). Blue/white screening of recombinant plasmids in Gram-positive bacteria by interruption of alkaline phosphatase gene (phoZ) expression. Gene 219, 91–99. 10.1016/S0378-1119(98)00396-5 [DOI] [PubMed] [Google Scholar]
- Cieslewicz M. J., Kasper D. L., Wang Y., Wessels M. R. (2001). Functional analysis in type Ia group B Streptococcus of a cluster of genes involved in extracellular polysaccharide production by diverse species of streptococci. J Biol Chem 276, 139–146. 10.1074/jbc.M005702200 [DOI] [PubMed] [Google Scholar]
- Cumley N. J., Smith L. M., Anthony M., May R. C. (2012). The CovS/CovR acid response regulator is required for intracellular survival of group B Streptococcus in macrophages. Infect Immun 80, 1650–1661. 10.1128/IAI.05443-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunny G. M., Lee L. N., LeBlanc D. J. (1991). Improved electroporation and cloning vector system for gram-positive bacteria. Appl Environ Microbiol 57, 1194–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M. S., Baker C. J. (2005). Group B streptococcal infections in elderly adults. Clin Infect Dis 41, 839–847. 10.1086/432804 [DOI] [PubMed] [Google Scholar]
- Edwards M. S., Rench M. A., Palazzi D. L., Baker C. J. (2005). Group B streptococcal colonization and serotype-specific immunity in healthy elderly persons. Clin Infect Dis 40, 352–357. 10.1086/426820 [DOI] [PubMed] [Google Scholar]
- Eldholm V., Gutt B., Johnsborg O., Brückner R., Maurer P., Hakenbeck R., Mascher T., Håvarstein L. S. (2010). The pneumococcal cell envelope stress-sensing system LiaFSR is activated by murein hydrolases and lipid II-interacting antibiotics. J Bacteriol 192, 1761–1773. 10.1128/JB.01489-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipe S. R., Pinho M. G., Tomasz A. (2000). Characterization of the murMN operon involved in the synthesis of branched peptidoglycan peptides in Streptococcus pneumoniae. J Biol Chem 275, 27768–27774. [DOI] [PubMed] [Google Scholar]
- Gardete S., Wu S. W., Gill S., Tomasz A. (2006). Role of VraSR in antibiotic resistance and antibiotic-induced stress response in Staphylococcus aureus. Antimicrob Agents Chemother 50, 3424–3434. 10.1128/AAC.00356-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giammarinaro P., Sicard M., Gasc A. M. (1999). Genetic and physiological studies of the CiaH-CiaR two-component signal-transducing system involved in cefotaxime resistance and competence of Streptococcus pneumoniae. Microbiology 145, 1859–1869. 10.1099/13500872-145-8-1859 [DOI] [PubMed] [Google Scholar]
- Haas W., Kaushal D., Sublett J., Obert C., Tuomanen E. I. (2005). Vancomycin stress response in a sensitive and a tolerant strain of Streptococcus pneumoniae. J Bacteriol 187, 8205–8210. 10.1128/JB.187.23.8205-8210.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A., Popham D. L., Carl D. J., Lauth X., Nizet V., Jones A. L. (2006). Penicillin-binding protein 1a promotes resistance of group B streptococcus to antimicrobial peptides. Infect Immun 74, 6179–6187. 10.1128/IAI.00895-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hava D. L., Camilli A. (2002). Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol Microbiol 45, 1389–1406. [PMC free article] [PubMed] [Google Scholar]
- Henneke P., Berner R. (2006). Interaction of neonatal phagocytes with group B streptococcus: recognition and response. Infect Immun 74, 3085–3095. 10.1128/IAI.01551-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung D. T., Shakhnovich E. A., Pierson E., Mekalanos J. J. (2005). Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310, 670–674. 10.1126/science.1116739 [DOI] [PubMed] [Google Scholar]
- Jenssen H., Hamill P., Hancock R. E. (2006). Peptide antimicrobial agents. Clin Microbiol Rev 19, 491–511. 10.1128/CMR.00056-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S. M., Cieslewicz M. J., Kasper D. L., Wessels M. R. (2005). Regulation of virulence by a two-component system in group B streptococcus. J Bacteriol 187, 1105–1113. 10.1128/JB.187.3.1105-1113.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S. M., Ishmael N., Dunning Hotopp J., Puliti M., Tissi L., Kumar N., Cieslewicz M. J., Tettelin H., Wessels M. R. (2008). Variation in the group B Streptococcus CsrRS regulon and effects on pathogenicity. J Bacteriol 190, 1956–1965. 10.1128/JB.01677-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S. M., Park S. E., Yadav P., Paoletti L. C., Wessels M. R. (2012). Regulation and function of pilus island 1 in group B streptococcus. J Bacteriol 194, 2479–2490. 10.1128/JB.00202-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. L., Mertz R. H., Carl D. J., Rubens C. E. (2007). A streptococcal penicillin-binding protein is critical for resisting innate airway defenses in the neonatal lung. J Immunol 179, 3196–3202. 10.1128/JB.00202-12 [DOI] [PubMed] [Google Scholar]
- Jordan S., Rietkötter E., Strauch M. A., Kalamorz F., Butcher B. G., Helmann J. D., Mascher T. (2007). LiaRS-dependent gene expression is embedded in transition state regulation in Bacillus subtilis. Microbiology 153, 2530–2540. 10.1099/mic.0.2007/006817-0 [DOI] [PubMed] [Google Scholar]
- Jordan S., Hutchings M. I., Mascher T. (2008). Cell envelope stress response in Gram-positive bacteria. FEMS Microbiol Rev 32, 107–146. 10.1111/j.1574-6976.2007.00091.x [DOI] [PubMed] [Google Scholar]
- Klinzing D. C., Madoff L. C., Puopolo K. M. (2009). Genomic analysis identifies a transcription-factor binding motif regulating expression of the alpha C protein in Group B Streptococcus. Microb Pathog 46, 315–320. 10.1016/j.micpath.2009.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M., Kuroda H., Oshima T., Takeuchi F., Mori H., Hiramatsu K. (2003). Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol Microbiol 49, 807–821. 10.1046/j.1365-2958.2003.03599.x [DOI] [PubMed] [Google Scholar]
- Lamy M. C., Zouine M., Fert J., Vergassola M., Couve E., Pellegrini E., Glaser P., Kunst F., Msadek T., et al. (2004). CovS/CovR of group B streptococcus: a two-component global regulatory system involved in virulence. Mol Microbiol 54, 1250–1268. 10.1111/j.1365-2958.2004.04365.x [DOI] [PubMed] [Google Scholar]
- Lembo A., Gurney M. A., Burnside K., Banerjee A., de los Reyes M., Connelly J. E., Lin W. J., Jewell K. A., Vo A., et al. (2010). Regulation of CovR expression in Group B Streptococcus impacts blood-brain barrier penetration. Mol Microbiol 77, 431–443. 10.1111/j.1365-2958.2010.07215.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. Y., Nizet V. (2004). Extracellular virulence factors of group B Streptococci. Front Biosci 9, 1794–1802. 10.2741/1296 [DOI] [PubMed] [Google Scholar]
- Maisey H. C., Doran K. S., Nizet V. (2008a). Recent advances in understanding the molecular basis of group B Streptococcus virulence. Expert Rev Mol Med 10, e27. 10.1017/S1462399408000811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisey H. C., Quach D., Hensler M. E., Liu G. Y., Gallo R. L., Nizet V., Doran K. S. (2008b). A group B streptococcal pilus protein promotes phagocyte resistance and systemic virulence. FASEB J 22, 1715–1724. 10.1096/fj.07-093963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher T. (2006). Intramembrane-sensing histidine kinases: a new family of cell envelope stress sensors in Firmicutes bacteria. FEMS Microbiol Lett 264, 133–144. 10.1111/j.1574-6968.2006.00444.x [DOI] [PubMed] [Google Scholar]
- Mascher T., Margulis N. G., Wang T., Ye R. W., Helmann J. D. (2003). Cell wall stress responses in Bacillus subtilis: the regulatory network of the bacitracin stimulon. Mol Microbiol 50, 1591–1604. 10.1046/j.1365-2958.2003.03786.x [DOI] [PubMed] [Google Scholar]
- Mascher T., Zimmer S. L., Smith T. A., Helmann J. D. (2004). Antibiotic-inducible promoter regulated by the cell envelope stress-sensing two-component system LiaRS of Bacillus subtilis. Antimicrob Agents Chemother 48, 2888–2896. 10.1128/AAC.48.8.2888-2896.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher T., Helmann J. D., Unden G. (2006a). Stimulus perception in bacterial signal-transducing histidine kinases. Microbiol Mol Biol Rev 70, 910–938. 10.1128/MMBR.00020-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher T., Heintz M., Zähner D., Merai M., Hakenbeck R. (2006b). The CiaRH system of Streptococcus pneumoniae prevents lysis during stress induced by treatment with cell wall inhibitors and by mutations in pbp2x involved in β-lactam resistance. J Bacteriol 188, 1959–1968. 10.1128/JB.188.5.1959-1968.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moszer I., Glaser P., Danchin A. (1995). SubtiList: a relational database for the Bacillus subtilis genome. Microbiology 141, 261–268. 10.1099/13500872-141-2-261 [DOI] [PubMed] [Google Scholar]
- Nakano S., Erwin K. N., Ralle M., Zuber P. (2005). Redox-sensitive transcriptional control by a thiol/disulphide switch in the global regulator, Spx. Mol Microbiol 55, 498–510. 10.1111/j.1365-2958.2004.04395.x [DOI] [PubMed] [Google Scholar]
- Nizet V. (2006). Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr Issues Mol Biol 8, 11–26. [PubMed] [Google Scholar]
- Pamp S. J., Frees D., Engelmann S., Hecker M., Ingmer H. (2006). Spx is a global effector impacting stress tolerance and biofilm formation in Staphylococcus aureus. J Bacteriol 188, 4861–4870. 10.1128/JB.00194-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasergi S., Brega S., Mistou M. Y., Firon A., Oxaran V., Dover R., Teti G., Shai Y., Trieu-Cuot P., Dramsi S. (2011). The GBS PI-2a pilus is required for virulence in mice neonates. PLoS ONE 6, e18747. 10.1371/journal.pone.0018747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. E., Jiang S., Wessels M. R. (2012). CsrRS and environmental pH regulate group B streptococcus adherence to human epithelial cells and extracellular matrix. Infect Immun 80, 3975–3984. 10.1128/IAI.00699-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Casal J., Price J. A., Maguin E., Scott J. R. (1993). An M protein with a single C repeat prevents phagocytosis of Streptococcus pyogenes: use of a temperature-sensitive shuttle vector to deliver homologous sequences to the chromosome of S. pyogenes. Mol Microbiol 8, 809–819. 10.1111/j.1365-2958.1993.tb01628.x [DOI] [PubMed] [Google Scholar]
- Peschel A., Sahl H.-G. (2006). The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat Rev Microbiol 4, 529–536. 10.1038/nrmicro1441 [DOI] [PubMed] [Google Scholar]
- Pezzicoli A., Santi I., Lauer P., Rosini R., Rinaudo D., Grandi G., Telford J. L., Soriani M. (2008). Pilus backbone contributes to group B Streptococcus paracellular translocation through epithelial cells. J Infect Dis 198, 890–898. 10.1086/591182 [DOI] [PubMed] [Google Scholar]
- Pietiäinen M., Gardemeister M., Mecklin M., Leskelä S., Sarvas M., Kontinen V. P. (2005). Cationic antimicrobial peptides elicit a complex stress response in Bacillus subtilis that involves ECF-type sigma factors and two-component signal transduction systems. Microbiology 151, 1577–1592. 10.1099/mic.0.27761-0 [DOI] [PubMed] [Google Scholar]
- Poyart C., Pellegrini E., Marceau M., Baptista M., Jaubert F., Lamy M.-C., Trieu-Cuot P. (2003). Attenuated virulence of Streptococcus agalactiae deficient in D-alanyl-lipoteichoic acid is due to an increased susceptibility to defensins and phagocytic cells. Mol Microbiol 49, 1615–1625. 10.1046/j.1365-2958.2003.03655.x [DOI] [PubMed] [Google Scholar]
- Puopolo K. M., Hollingshead S. K., Carey V. J., Madoff L. C. (2001). Tandem repeat deletion in the alpha C protein of group B streptococcus is recA independent. Infect Immun 69, 5037–5045. 10.1128/IAI.69.8.5037-5045.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puopolo K. M., Baker C. J.(2013).Group B streptococcal infection in neonates and young infants. http://www.uptodate.com/contents/group-b-streptococcal-infection-in-neonates-and-young-infants
- Rajagopal L., Vo A., Silvestroni A., Rubens C. E. (2006). Regulation of cytotoxin expression by converging eukaryotic-type and two-component signaling mechanisms in Streptococcus agalactiae. Mol. Micro 62, 941–957. 10.1111/j.1365-2958.2006.05431.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L. J., Muench H. (1938). A simple method of estimating fifty per cent endpoints. Am J Hyg 27, 493–497. [Google Scholar]
- Reinscheid D. J., Stösser C., Ehlert K., Jack R. W., Möller K., Eikmanns B. J., Chhatwal G. S. (2002). Influence of proteins Bsp and FemH on cell shape and peptidoglycan composition in group B streptococcus. Microbiology 148, 3245–3254. [DOI] [PubMed] [Google Scholar]
- Rosch J. W., Mann B., Thornton J., Sublett J., Tuomanen E. (2008). Convergence of regulatory networks on the pilus locus of Streptococcus pneumoniae. Infect Immun 76, 3187–3196. 10.1128/IAI.00054-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubens C. E., Wessels M. R., Heggen L. M., Kasper D. L. (1987). Transposon mutagenesis of type III group B Streptococcus: correlation of capsule expression with virulence. Proc Natl Acad Sci U S A 84, 7208–7212. 10.1073/pnas.84.20.7208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed A. I., Sharov V., White J., Li J., Liang W., Bhagabati N., Braisted J., Klapa M., Currier T., et al. (2003). TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378. [DOI] [PubMed] [Google Scholar]
- Schauber J., Dorschner R. A., Yamasaki K., Brouha B., Gallo R. L. (2006). Control of the innate epithelial antimicrobial response is cell-type specific and dependent on relevant microenvironmental stimuli. Immunology 118, 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellerberg B., Rozdzinski E., Martin S., Weber-Heynemann J., Lütticken R. (2002). rgf encodes a novel two-component signal transduction system of Streptococcus agalactiae. Infect Immun 70, 2434–2440. 10.1128/IAI.70.5.2434-2440.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntharalingam P., Senadheera M. D., Mair R. W., Lévesque C. M., Cvitkovitch D. G. (2009). The LiaFSR system regulates the cell envelope stress response in Streptococcus mutans. J Bacteriol 191, 2973–2984. 10.1128/JB.01563-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldor M. K. (2006). Disarming pathogens–a new approach for antibiotic development. N Engl J Med 354, 296–297. 10.1056/NEJMcibr054591 [DOI] [PubMed] [Google Scholar]
- Weidenmaier C., Peschel A., Kempf V. A., Lucindo N., Yeaman M. R., Bayer A. S. (2005). DltABCD- and MprF-mediated cell envelope modifications of Staphylococcus aureus confer resistance to platelet microbicidal proteins and contribute to virulence in a rabbit endocarditis model. Infect Immun 73, 8033–8038. 10.1128/IAI.73.12.8033-8038.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S., Daum R. S., Boyle-Vavra S. (2006). VraSR two-component regulatory system and its role in induction of pbp2 and vraSR expression by cell wall antimicrobials in Staphylococcus aureus. Antimicrob Agents Chemother 50, 336–343. 10.1128/AAC.50.1.336-343.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Bremer H. (1995). Control of the Escherichia coli rrnB P1 promoter strength by ppGpp. J Biol Chem 270, 11181–11189. 10.1074/jbc.270.19.11181 [DOI] [PubMed] [Google Scholar]