

Abstract
The constitutively expressed, chromosomally-encoded β-lactamase (BlaC) is the enzyme responsible for the intrinsic resistance to β-lactam antibiotics in Mycobacterium tuberculosis. Previous studies from this laboratory have shown that the enzyme exhibits an extended-spectrum phenotype, with very high levels of penicillinase and cephalosporinase activity, as well as weak carbapenemase activity (1). In this report, we have determined the pH dependence of the kinetic parameters, revealing that the maximum velocity depends on the ionization state of two groups; a general base exhibiting a pK value of 4.5 and a general acid exhibiting a pK value of 7.8. Having defined a region where the kinetic parameters are pH-independent (pH 6.5), we determined solvent kinetic isotope effects (SKIEs) for three substrates whose kcat values differ by 4.5 orders of magnitude. Nitrocefin is a highly activated, chromogenic cephalosporin derivative that exhibits steady-state solvent kinetic isotope effects of 1.4 on both V and V/K. Cefoxitin is a slower cephalosporin derivative that exhibits a large SKIE on V of 3.9, but a small SKIE of 1.8 on V/K in steady-state experiments. Pre-steady-state, stopped-flow experiments with cefoxitin revealed a burst of β-lactam ring opening with associated SKIE values of 1.6 on the acylation step and 3.4 on the deacylation step. Meropenem is an extremely slow substrate for BlaC, and exhibits burst kinetics in the steady-state experiments. SKIE determinations with meropenem revealed large SKIEs on both the acylation and deacylation step of 3.8 and 4.0, respectively. Proton inventories in all cases were linear, indicating the participation of a single solvent-derived proton in the chemical step responsible for the SKIE. The rate limiting steps for β-lactam hydrolysis of these substrates are analyzed, and the chemical step(s) responsible for the observed SKIE are discussed.
Keywords: antibiotic resistance, beta-lactamase, solvent kinetic isotope effects, tuberculosis
INTRODUCTION
One in three people is infected with M. tuberculosis worldwide. It is the leading killer of people living with HIV causing one quarter of all deaths. Furthermore, the emergence of multidrug resistant and extensively drug resistant strains of M. tuberculosis poses a serious problem for cost effective treatment therapies (2). The intrinsic resistance of M. tuberculosis to β-lactams is attributed to the genomically encoded, Ambler class A β-lactamase (BlaC). β-lactam antibiotics irreversibly bind to the D,D-transpeptidase domains of penicillin-binding proteins (PBPs) by structurally mimicking the D-Ala-D-Ala C-terminal dipeptide of the pentapeptide of the cell wall peptidoglycan (3). In bacteria, β-lactams acylate the active site serine of PBPs, forming stable inactive acyl-enzymes (4).
The catalytic mechanism of BlaC relies on three highly conserved active site residues, Lys73 and Glu166, which are involved in the activation of the acylating nucleophile Ser70 and the activation of the active site water molecule for deacylation, respectively (5). Although the class A β-lactamase acylation mechanism is still debated (6), BlaC site-directed mutagenesis and crystallographic data with the cephalosporin cefamandole strongly suggest that Lys73 is involved in the activation of Ser70 for acylation and Glu166 in activating the conserved water molecule for deacylation. The Lys73Ala mutant form of BlaC permitted the structural identification of the Michaelis complex, but has no catalytic activity, indicating Lys73 is essential for the acylation step. The Glu166Ala mutant form of BlaC similarly has no hydrolytic activity and forms a stable acyl-enzyme complex that can be structurally characterized, suggesting that Glu166 is required for deacylation and product release (7).
Previously, BlaC has been shown to rapidly hydrolyze penicillin and cephalosporin classes of β-lactams, as well as exhibiting weak carbapenemase activity (3). While there has been decades of work in characterizing the mechanism of class A β-lactamases in Gram-negative and Gram-positive bacteria (8–12), no extensive studies have been performed to elucidate the rate-limiting steps in the reaction catalyzed by BlaC in M. tuberculosis. In this study, we have performed steady state and pre-steady state studies to measure solvent kinetic isotope effects on β-lactams that vary in catalytic efficiency. Cephalosporins, nitrocefin and cefoxitin, are rapidly hydrolyzed by BlaC, whereas the carbapenem meropenem is hydrolyzed extremely slowly. Our comparative kinetic analysis indicates that slow substrates are highly solvent sensitive for both acylation and deacylation reactions, whereas intermediate substrates have a small SKIE on acylation and a larger SKIE on deacylation and the fast substrates exhibit a small SKIE on acylation only. These findings provide greater mechanistic insight into the rate-limiting steps and chemical mechanism of the BlaC-catalyzed hydrolysis of β-lactam substrates.
MATERIALS & METHODS
Materials
All chemicals were of analytical or reagent grade and were used without further purification. 99.9% deuterated water was from Cambridge Isotope Laboratories. The remainder of the chemicals were purchased from Sigma-Aldrich.
Production and Purification of BlaC
The blaC gene was amplified from genomic DNA and cloned into pET28a(+) using NdeI and HindIII, as previously described (13). The expression and purification of BlaC followed the protocol previously reported (3). Based on the stoichiometric inhibition of BlaC by clavulanate (5), the active site concentration of BlaC was determined by clavulanate titration.
pH-Rate Profiles
The pH dependence of the kinetic parameters was determined by measuring initial rates at varying concentrations of cefoxitin or nitrocefin. The experiments were conducted at 25°C in 100 mM of the following buffers at the indicated pH values: sodium acetate (pH 4.0–5.5), MES (pH 5.5–6.7), HEPES (pH 6.7–7.8), and TAPS (pH 7.8–8.7). The resulting kcat data were fit to eq 1 to obtain the pKa, the negative log of the acid dissociation constant, and pKb, the negative log of the base dissociation constant, where c is the pH-independent plateau value. The kcat/Km data were fit to eq 2, which describes the pH dependence where a single group whose ionization decreases kcat/Km at high pH values.
| (1) |
| (2) |
Steady State Kinetics
The steady state rate of hydrolysis of β-lactam substrates was monitored as a decrease in the absorbance in the UV region, as previously described (14). Assays using the chromogenic substrate nitrocefin were performed at 486 nm (Δε = 20 500 M−1 cm−1). Assays using cefoxitin were performed at 270 nm ((Δε = 8 380 M−1 cm−1). All assays were performed at 25°C and 100 mM MES/NaCl (pH 6.3). Reactions were initiated by addition of enzyme at concentrations varying from 2 to 300 nM, depending on the substrate used. Initial velocity kinetic data were fit to eq 3
| (3) |
where ν is the initial velocity, V is the maximal velocity, and Km is the Michaelis constant for substrate, S.
Solvent Kinetic Isotope Effects
Steady-state solvent kinetic isotope effects were measured at varying concentrations of cefoxitin or nitrocefin and globally fit to eq 4
| (4) |
where ν is the initial velocity, V is the maximal velocity, Km is the Michaelis constant for substrate S, Ek is the SKIE on V/K (−1), Ev is the SKIE on V (−1), and Fi = 0 for H2O and Fi = 0.95 for D2O. Viscosity effects on V or V/K were evaluated by comparing rates obtained in H2O or 9% (w/w) glycerol, which mimics the viscosity increase caused by D2O (ηr = 1.24) (15). Proton inventories were determined at saturating concentrations of cefoxitin and nitrocefin at 10% volume increments of D2O from 0 to 90%. SKIEs for steady-state and proton inventory are not reported for meropenem hydrolysis due to a very low Km value, a very low kcat value, and a small extinction coefficient for hydrolysis.
Pre-steady State Kinetics
Prior to pre-steady-state kinetic studies, saturation assays were performed to confirm that sufficient amount of substrate was used to produce pseudo-first-order conditions. A large excess of substrate drives the enzyme to the Michaelis complex form by significantly increasing the rate of binding (k1[S]) so that the acylation (k2) and deacylation (k3) rates can be accurately determined.
Solvent KIEs were measured under multiple-turnover conditions. Solvent kinetic isotope effects were measured in a SX-20 (Applied Photophysics) stopped-flow, rapid-mixing spectrophotometer in the absorbance monitoring mode in either H2O or 95% D2O. Experiments were performed under pseudo-first-order conditions with at least 50-fold excess of substrate over limiting BlaC with a fixed concentration of 7 to 50 μM, depending on the substrate used. Due to the absorbance detection limit of 2 AU, assays for cefoxitin and meropenem were not measured at the wavelength of their maximal extinction coefficients but at 293 nm (Δεapp = 1 960 M−1 cm−1) and 317 nm (Δεapp = 4 700 M−1 cm−1), respectively, to obtain rates for k2 and k3. Time courses ranged from 5 to 500 s depending on the substrate in order to fully observe the burst and linear phases. Raw absorbance values were divided by the final enzyme concentration to accommodate fitting. Pre-steady state kinetic studies were not performed with nitrocefin because the kcat for hydrolysis is too high to observe a burst phase (acylation rate is faster than the 0.5 ms dead time of the spectrophotometer).
Data Analysis
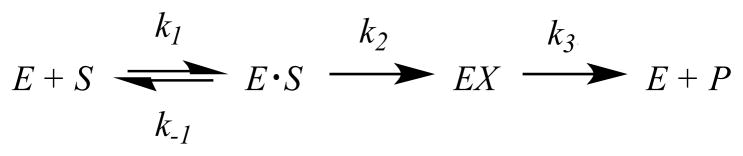
All data were fitted to the appropriate equations using the non-linear regression function of SigmaPlot 11 (SPSS, Inc.). The general mechanism of BlaC hydrolysis can be modeled as shown in Scheme 2 where k1 and k−1 represent the rates of reversible binding of substrate to and dissociation of substrate from BlaC, respectively, k2 represents the rate of acylation of enzyme BlaC by attack on the β-lactam ring, and k3 represents the rate of deacylation/hydrolysis of the BlaC—β-lactam adduct. Using this model, rate constants for k2 and k3 were determined by fitting burst kinetic traces to eq 5
Scheme 2.
| (5) |
where Ao is the burst amplitude, [E]o is the final enzyme concentration, kobs is the overall rate constant, and t is time (16) when binding step is not rate-limiting to the overall rate. The variables can be expressed in terms of k2 and k3 and substituted in eq 5 to produce eq 6 for fitting the progress curves. Estimated values for k2 and k3 from the fitting were verified by the apparent Ao and kcat values, which are comparable to steady-state turnover numbers (Table S1).
| (6) |
RESULTS AND DISCUSSION
BlaC is the chromosomally-encoded β-lactamase in Mycobacterium tuberculosis originally identified after the genome sequence was published in 1998 (17). The full-length enzyme contains an N-terminal 40 amino acid membrane insertion sequence allowing for its export into the periplasm where peptidoglycan cross-linking occurs, and anchoring in the outer aspect of the inner membrane. All mechanistic and structural studies have been performed with a soluble construct containing an N-terminal truncation of the first 40 residues, as first reported for the structure of the enzyme in 2006 (13). Using this construct, it was shown that BlaC is an extended-spectrum β-lactamase (ESBL) with extremely broad substrate specificity for penicillins, cephalosporins and even carbapenems (3), β-lactams specifically designed to be resistant to hydrolysis by ESBLs.
BlaC is included in the class A Ambler classification of β-lactamases, based on sequence similarity with others in this class (18). These enzymes all contain a conserved triad of catalytic residues, including Ser70, Lys73, and Glu166 (Ambler numbering is used throughout rather than actual sequence numbers). These are proposed to act as the nucleophiles responsible for β-lactam ring opening, and accessory general acid and base. Mechanistically, they are similar to serine proteases, and use a two-step reaction in which the enzyme is first acylated by the putative nucleophilic attack of Ser70 on the β-lactam ring with formation of a transient covalent ester bond, followed by hydrolysis of this bond by water attack on the scissile bond (deacylation).
Although the class A β-lactamases have been extensively studied, the assigned roles of the auxiliary general acid/base to either Lys73 or Glu166 have been disputed. Initially, Glu166 was thought to serve as the general base in Ser70 activation. However, an ultrahigh resolution crystallographic study of TEM-1 suggested that Glu166 was protonated at pH 8.0 and thus served the role of general acid (19). Alternatively, ab initio QM/MM calculations suggested that both Lys73 and Glu166 participate in Ser70 activation (6). Previous studies from this lab have clarified the role of each residue using site-directed mutagenesis and X-ray crystallography (7). The K73A mutant was unable to be acylated by cefamandole, and the structure of bound cefamandole in the active site revealed a pre-acylation, noncovalent Michaelis complex. The E166A mutant was crystallized as a covalent, acylated complex with bound cefamandole. Together, these results argue persuasively for the role of Lys73 as the acylation general base and for Glu166 as the deacylation base, as depicted in Scheme 3. In order to more completely characterize their respective roles in catalysis, we determined the pH dependence of the kinetic parameters for two different cephalosporin substrates.
Scheme 3.
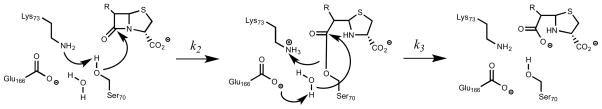
Proposed catalytic mechanism of BlaC. Activation Ser70 by Lys73 to promote β-lactam ring opening (A), formation of the covalent acyl-enzyme intermediate (B), and activation of conserved water by Glu166 for product release (C).
pH-rate profiles
Nitrocefin was developed to be an extremely active, highly chromogenic general substrate for β-lactamases (20–22). It contains a vinyl dinitrobenzene substituent attached to the C3 position of the cephalosporin nucleus, providing strong resonance electron withdrawal from the susceptible β-lactam bond. It is a very efficient substrate for BlaC, exhibiting a kcat/Km value of ~108 M−1 min−1 (Table 1), a value close to the diffusion limit (5). Determination of the pH-rate dependence allows for the identification of any ionizable groups involved in the reaction. The pH dependence of the kinetic parameters for nitrocefin are shown in Figure 1A, where the points are the experimental data and the smooth curves are the fits of the data to eq 1 (kcat; upper) or eq 2 (kcat/Km; lower). The maximum velocity changes as the result of a group that needs to be deprotonated, and exhibits a pK value of 4.5 ± 0.2, is protonated at low pH values, and as the result of a group that needs to be protonated, and exhibits a pK value of 7.8 ± 0.1, is deprotonated at high pH values. The pH dependence of kcat/Km varies only at high pH values where it deceases as a result of the deprotonation of a group exhibiting a pK value of 8.3 ± 0.1. This latter parameter includes rates for steps between the binding of nitrocefin to free enzyme and the first irreversible step, which in the case of BlaC is the acylation of the enzyme (23). The lack of any break at low pH of the kcat/Km profile suggests this parameter is not sensitive or reporting on rate-limiting chemistry that is dependent on the protonation states of enzyme groups. The decrease at high values may reflect the ionization behavior of a protonated enzyme group involved in nitrocefin binding, likely the carboxylate that is a necessary feature of all β-lactam antibiotics.
Table 1.
Solvent Kinetic Isotope Effects on BlaC
| Substrate | Nitrocefin | Cefoxitin | Meropenem |
|---|---|---|---|
| Km (μM) | 86 ± 6 | 255 ± 9 | 3.4 ± 0.7 a |
| kcat (s−1) | 38 ± 2 | 0.7 ± 0.1 | (7.7 ± 0.1) × 10−4 a |
| kcat/Km (M−1s−1) | (4.4 ± 0.1) × 105 | (2.7 ± 0.3) × 103 | (2.3 ± 0.8) × 102 a |
| k2 (s−1) | n.a. | 1.4 ± 0.01 | (1.9 ± 0.3) × 10−2 |
| k3 (s−1) | n.a. | 0.4 ± 0.001 | (4.0 ± 1.0) × 10−4 |
| DV/K | 1.4 ± 0.1 | 1.8 ± 0.1 | n.a. |
| DV | 1.4 ± 0.1 | 3.9 ± 0.1 | n.a. |
| Dk2 | n.a. | 1.6 ± 0.1 | 3.8 ± 0.1 |
| Dk3 | n.a. | 3.4 ± 0.1 | 4.0 ± 0.1 |
V/K reports on kcat/Km; V reports on kcat; k2, rate of acylation; k3, rate of deacylation; n.a., not available;
data obtained from Reference 26.
Figure 1.

pH dependence of BlaC on log kcat (■) and log kcat/Km (□) with (A) nitrocefin and (B) cefoxitin. The lines represent fits to eq 1 and 2. A mixed buffer system was utilized, with 100 mM sodium acetate, 100 mM MES, 100 mM HEPES, and 100 mM TAPS.
The bell-shaped kcat profile suggests that chemistry requires participation from both an active site base and acid. Similar bell-shaped pH profiles have been reported for the Bacillus cereus β-lactamase acting on both penicillins and cephalosporins (22). The maximum velocity includes rate contributions from steps after the formation of the enzyme-substrate complex to the final release of products, and in the context of the model for BlaC catalysis, includes both the acylation rate constant as well as the deacylation rate constant. For reasons that will be discussed further below, we believe that the acylation step in nitrocefin hydrolysis is slower than deacylation, and that the kcat pH profile is reporting on the ionization behavior of active site groups involved in this step. This suggests that the acylation base observed is Lys73, which removes a proton from the Ser70 hydroxyl that attacks the β-lactam amide linkage (Scheme 3). The extremely low pK value of 4.5 observed for this lysine likely reflects the highly perturbed active site environment after substrate binding. The observation of a general acid in the acylation reaction of nitrocefin is also unusual, since upon β-lactam ring opening, the resulting anion on the thiazole ring nitrogen is highly resonance stabilized via the conjugated vinyldinitrobenzene substituent, and would not be expected to require nitrogen protonation (Scheme 4). However, similar kcat pH profiles with this substrate were previously reported for the β-lactamase from Bacillus cereus (22).
Scheme 4.

BlaC acylation of nitrocefin, cefoxitin, and meropenem. Pyrroline ring isomerization of meropenem results in the incorporation of a solvent exchangeable proton (red) at C3.
These studies were repeated with cefoxitin, a significantly slower cephalosporin substrate with kcat of 72 min−1 and kcat/Km of 2.8 × 105 M−1min−1 (Table 1). The pH dependence of the kinetic parameters determined using cefoxitin appear to be similar to those exhibited by nitrocefin, and the interpretation of the kcat/Km profile is the same; a protonated group exhibiting a pK value of 6.8 ± 0.1 responsible for interacting with the carboxylic acid of cefoxitin. However, the interpretation of the pH dependence of kcat is different. The maximum velocity depends on a group that needs to be deprotonated, and exhibits a pK value of 4.3 ± 0.1, and as the result of a group that needs to be protonated, and exhibits a pK value of 7.7 ± 0.1, is deprotonated at high pH values. Cefoxitin exhibits “burst” kinetics in stopped-flow experiments (see below), thus requiring that the acylation step be faster than the deacylation step. Since kcat for this substrate is determined by the deacylation step, then the pH dependence of kcat must involve the protonation states of enzyme groups involved in that reaction. Deacylation is initiated by Glu166 abstracting a proton from a conserved water molecule in the active site that hydrolyzes the ester bond. The Ser70 alkoxide is protonated by Lys73, regenerating the protonation state of these residues to that required for the next round of catalysis. Thus, in the deacylation reaction, the function and protonation states of the two auxillary acid/base groups are reversed, with the group acting as general base being Glu166, and with the group acting as the general acid Lys73.
One final comment is worthy given the various pH environments that M. tuberculosis must confront. After ingestion by host alveolar macrophages, the organism is subjected to increasingly low pH as the phagosome is slowly acidified, and experiences intraphagosomal pH values as low as 5.5 (24). BlaC, which is located in the periplasmic space outside of the cell membrane remains remarkably active at these low pH values as a result of the low pK values of Lys73 in the acylation half-reaction and Glu166 in the deacylation half-reaction, and thus can function effectively even as acidification of the phagosome is occurring.
Solvent kinetic isotope effects
The foregoing analysis of the pH dependence of the kinetic parameters allowed us to define a broad pH window where solvent kinetic isotope effects would be unaffected by solvent isotope composition-dependent changes in the pK values of the enzyme groups involved in catalysis. Solvent kinetic isotope effects are powerful probes of proton transfer events in enzyme-catalyzed reactions and have been used to probe transition state structures for both acylation and deacylation reactions of mechanism-related proteases (25–28). This extends to the analysis of SKIEs on β-lactamases, including important contributions on penicillanic acid sulfone hydrolysis by Brenner & Knowles (29), β-lactam hydrolysis by the Bacillus cereus TEM β-lactamase by Hardy & Kirsch (22) and β-lactam hydrolysis by several related class A and C β-lactamases by Adediran & Pratt (30). We chose three substrates for this analysis with BlaC, which span nearly five orders of catalytic turnover rates. Viscosity controls showed no effect of added 9% (w/w) glycerol on rates.
Nitrocefin steady-state hydrolysis is only modestly affected by solvent isotopic composition (Figure 2A). Fits of the data points yielded equivalent, small solvent kinetic isotope effects of 1.4 ± 0.1 on V and V/K (Table 1). The small effects on nitrocefin hydrolysis by BlaC are similar to those observed previously for penicillins and cephalosporins by Hardy & Kirsch (1.53 and 1.09, respectively) and Adediran & Pratt (1.04 and 1.39, respectively) (22, 30). Since kcat/Km only reports on steps between binding of nitrocefin to the free enzyme, and the formation of the covalent acyl-enzyme, the steady-state results for nitrocefin hydrolysis revealing equivalent, small solvent KIEs on kcat/Km and kcat supports acylation as the rate-limiting step for this substrate. The proton inventory for nitrocefin hydrolysis is linear (Figure 3A), suggesting a single proton is transferred in the acylation reaction, and we assign this to the exchangeable proton on the Ser70 hydroxyl group to Lys73 (Scheme 4). Nitrocefin is too fast a substrate for stopped-flow, multiple-turnover kinetics, and thus we could not obtain independent evidence on the SKIEs on k2 (acylation) and k3 (deacylation). We then turned to both steady-state and multiple-turnover SKIE determinations of cefoxitin hydrolysis.
Figure 2.

Initial velocity pattern for (A) nitrocefin with 2 nM BlaC and (B) cefoxitin with 350 nM BlaC in H2O (●) and in D2O (○) with 100 mM MES at pH 6.3.
Figure 3.

Proton inventories of (A) nitrocefin with 2 nM BlaC and (B) cefoxitin with 350 nM BlaC measuring the initial velocity with varying % D2O in 100 mM MES at pH 6.3.
Fits of the steady-state data for SKIEs on cefoxitin hydrolysis yielded values of D2OV/K = 1.8 ± 0.1 and D2OV = 3.9 ± 0.1 (Table 1, VFigure 2B). The much larger solvent kinetic isotope effect on suggests that deacylation is rate-limiting for this substrate. A single proton is involved in this proton transfer step as evidenced by the linear proton inventory obtained at saturating concentrations of cefoxitin (Figure 3B). We ascribe this proton transfer as being the deprotonation of the conserved hydrolytic water molecule by Glu166 (Scheme 3). The small SKIE on V/K is likely to arise from a small contribution from the acylation reaction, as observed with nitrocefin.
Under multiple-turnover conditions, using high concentrations of both BlaC and cefoxitin, a distinct burst of absorbance is observed (Figure 4A), suggesting that the on-enzyme acylation rate is faster than the slower hydrolysis of the cefoxitin-BlaC complex. When the curves are fit to an equation that describes an exponential increase followed by a linear (“steady-state”) rate, SKIEs are again observed on both the burst phase and the linear rate. By fitting progress curves to the eq 6, we can dissect the rates of acylation and deacylation and determine the SKIEs on both k2 and k3. Because k3 is not significantly slower than k2 (Table 1), we observe an initial burst phase (Ao) equivalent to approximately 70% of the initial active enzyme concentration used to initiate the reaction. The solvent kinetic isotope effect on k2, D2Ok2 was 1.6 ± 0.1 and that on k3, D2Ok3 was 3.4 ± 0.1 (Table 1). The near equivalence of the values for D2OV and D2Ok3 argues persuasively for the isotope effect on deacylation being the source of the solvent kinetic isotope effect on V. The combination of the burst and the SKIEs on each phase of the reaction demonstrates that deacylation is rate-limiting. Viscosity controls using 9% (w/w) glycerol for cefoxitin and meropenem hydrolysis exhibited a slight increase in the linear steady-state rate, as observed previously with P99 β-lactamase by Adediran & Pratt (30). The presence of glycerol in the active site may act similar to methanol by accelerating the solvolysis of the slow step (deacylation), and thereby enhances kcat (see Supplementary Fig. S2).
Figure 4.

Burst kinetic isotope effects on (A) cefoxitin and (B) meropenem in H2O (black) and D2O (gray) with fits of experimental traces to the k2 (acylation) and k3 (deacylation) values in Table 1 (red). Cefoxitin studies consisted of 1800 μM cefoxitin mixed with 27 μM BlaC. Meropenem studies consisted of 875 μM meropenem mixed with 13 μM BlaC.
The very low Km value for meropenem, coupled with its weak extinction coefficient and extremely slow turnover rate (0.08 min−1), made the determination of steady-state SKIEs both difficult and unreliable. Rather, we measured SKIEs at a single high concentration of both BlaC and meropenem. As seen in Figure 4B, there is an easily observable burst followed by a linear “steady-state” rate of hydrolysis. Here, we observe a stoichiometric burst phase with the initial enzyme concentration since deacylation rate is significantly slower than the acylation rate. When the experiment is repeated in D2O, both rates are quite significantly affected, with calculated values of D2Ok2 equal to 3.8 ± 0.1 and of D2Ok3 equal to 4.0 ± 0.1 (Table 1). The SKIE on deacylation corresponds almost exactly to that observed with cefoxitin, and has the same source, e.g. deprotonation of the water molecule by Glu166 leading to hydrolysis of the ester-linked substrate. The source of the equally large SKIE on k2 is less obvious and unique to this carbapenem substrate. During the acylation reaction of carbapenems catalyzed by BlaC, the ring opening of the β-lactam yields an initial Δ2-pyrroline ring, which subsequently isomerizes to a Δ1-pyrroline ring with stereospecific protonation at C3 to generate the final, stable acylated adduct (Scheme 4). The structure of the meropenem complex with BlaC shows that C3 is clearly sp3 hybridized (3), and structures determined for the related carbapenems, ertapenem, and doripenem, reveal similar isomerizations (23). This reaction is not, however, concerted as the Δ2-pyrroline ring form is observed crystallographically at very short soaking times with ertapenem. We propose that it is this solvent-mediated C3 protonation accompanying isomerization that is the source of the solvent KIE on k2, the acylation rate constant.
In conclusion, the M. tuberculosis blaC-encoded β-lactamase exhibits a very broad pH dependence that allows it to act effectively at a range of pH values that the organism encounters, including the low pH levels that it encounters intracellularly after macrophage capture. The pH dependence of kcat is reporting on the required ionization states of Glu166 and Lys73 for effective acylation of the enzyme (for nitrocefin) or for deacylation of the covalent enzyme-β-lactam complex (for the slower cefoxitin and meropenem). This interpretation is confirmed by the solvent kinetic isotope effect analysis with relatively slow substrates, such as cefoxitin, where large SKIEs are observed on k3, the deacylation rate constant, and not k2, the acylation rate constant. However, using meropenem, an extremely slow substrate, large SKIEs are observed on both acylation and deacylation, the former being the result of a slow isomerization/protonation step accompanying β-lactam ring opening of the carbapenem nucleus. This interpretation has support from structural studies of the Δ2- and Δ1-pyrroline forms of the enzyme bound carbapenem (7).
Supplementary Material
Scheme 1.

Structures of benzylpenicillin, nitrocefin, cefoxitin, and meropenem.
Acknowledgments
This work was supported by the National Institutes of Health Grants AI33696 and AI60899 to J.S.B.
We thank Dr. Clarissa Czekster for her guidance with performing the stopped-flow experiments.
ABBREVIATIONS
- AU
absorbance units
- BlaC
β-lactamase
- ESBL
extended-spectrum β-lactamase
- HEPES
N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)
- MES
2-(N-morpholino)ethanesulfonic acid
- SKIE
solvent kinetic isotope effect
- TAPS
N-[tris(hydroxymethyl)methyl]-3-aminopropanesulfonic acid
Footnotes
SUPPORTING INFORMATION AVAILABLE
Burst SKIEs in 9% glycerol, saturation curve for cefoxitin burst kinetics, and pre-steady state fittings for kinetic parameters are summarized. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tremblay LW, Hugonnet JE, Blanchard JS. Structure of the covalent adduct formed between Mycobacterium tuberculosis beta-lactamase and clavulanate. Biochemistry. 2008;47:5312–5316. doi: 10.1021/bi8001055. [DOI] [PubMed] [Google Scholar]
- 2.Hugonnet JE, Blanchard JS. Irreversible inhibition of the Mycobacterium tuberculosis beta-lactamase by clavulanate. Biochemistry. 2007;46:11998–12004. doi: 10.1021/bi701506h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.TB Alliance. The 2010 Annual Report for TB Drug Development (online) 2010 http://www.tballiance.org/
- 4.Kurz SG, Bonomo RA. Reappraising the use of β-lactams to treat tuberculosis. Expert Rev Anti Infect Ther. 2012;10:999–1006. doi: 10.1586/eri.12.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matagne A, Lamotte-Brasseur J, Frère JM. Catalytic properties of class A beta-lactamases: efficiency and diversity. Biochem J. 1998;330:581–598. doi: 10.1042/bj3300581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tremblay LW, Xu H, Blanchard JS. Structures of the Michaelis complex (1.2 Å) and the covalent acyl intermediate (2.0 Å) of cefamandole bound in the active sites of the Mycobacterium tuberculosis β-lactamase K73A and E166A mutants. Biochemistry. 2010;49:9685–9687. doi: 10.1021/bi1015088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meroueh SO, Fisher JF, Schlegel HB, Mobashery S. Ab initio QM/MM study of class A β-lactamase acylation:3 Dual participation of Glu166 and Lys73 in a concerted base promotion of Ser70. J Am Chem Soc. 2005;127:15397–15407. doi: 10.1021/ja051592u. [DOI] [PubMed] [Google Scholar]
- 8.Anderson EG, Pratt RF. Pre-steady state beta-lactamase kinetics. The trapping of a covalent intermediate and the interpretation of pH rate profiles. J Biol Chem. 1983;258:13120–13126. [PubMed] [Google Scholar]
- 9.Bicknell R, Waley SG. Single-turnover and steady-state kinetics of hydrolysis of cephalosporins by beta-lactamase I from Bacillus cereus. Biochem J. 1985;231:83–88. doi: 10.1042/bj2310083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lietz EJ, Truher H, Kahn D, Hokenson MJ, Fink AL. Lysine-73 is involved in the acylation and deacylation of beta-lactamase. Biochemistry. 2000;39:4971–4981. doi: 10.1021/bi992681k. [DOI] [PubMed] [Google Scholar]
- 11.Kumar S, Adediran SA, Nukaga M, Pratt RF. Kinetics of turnover of cefotaxime by the Enterobacter cloacae P99 and GCl beta-lactamases: two free enzyme forms of the P99 beta-lactamase detected by a combination of pre- and post-steady state kinetics. Biochemistry. 2004;43:2664–2672. doi: 10.1021/bi030212j. [DOI] [PubMed] [Google Scholar]
- 12.Fisher JF, Mobashery S. Three decades of the class A beta-lactamase acyl-enzyme. Curr Protein Pept Sci. 2009;10:401–407. doi: 10.2174/138920309789351967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F, Cassidy C, Sacchettini JC. Crystal structure and activity studies of the Mycobacterium tuberculosis β-lactamase reveal its critical role in resistance to β-lactam antibiotics. Antimicrob Agents Chemother. 2006;50:2762–2771. doi: 10.1128/AAC.00320-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heritier C, Poirel L, Aubert D, Nordmann P. Genetic and Functional Analysis of the Chromosome-Encoded Carbapenem-Hydrolyzing Oxacillinase OXA-40 of Acinetobacter baumannii. Antimicrob Agents Chemother. 2003;47:268–273. doi: 10.1128/AAC.47.1.268-273.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheely ML. Glycerol Viscosity Tables. Ind Eng Chem. 1932;24:1060–1064. [Google Scholar]
- 16.Tremblay LW, Fan F, Blanchard JS. Biochemical and Structural Characterization of Mycobacterium tuberculosis β-Lactamase with the Carbapenems Ertapenem and Doripenem. Biochemistry. 2010;49:3766–3773. doi: 10.1021/bi100232q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson KA. Kinetic Analysis of Macromolecules. Oxford University Press; New York: 2003. Kinetics of a pre-steady-state burst; pp. 10–12. [Google Scholar]
- 18.Cole ST, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 19.Ambler RP. The structure of beta-lactamases. Philos Trans R Soc Lond B Biol Sci. 1980;289:321–331. doi: 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- 20.Minasov G, Wang X, Shiochet BK. An Ultrahigh Resolution Structure of TEM-1 β-Lactamase Suggests a Role of Glu166 as the General Base in Acylation. J Am Chem Soc. 2002;124:5333–5340. doi: 10.1021/ja0259640. [DOI] [PubMed] [Google Scholar]
- 21.Matthew M, Harris AM. Identification of beta-lactamases by analytical isoelectric focusing: correlation with bacterial taxonomy. J Gen Microbiol. 1976;94:55–67. doi: 10.1099/00221287-94-1-55. [DOI] [PubMed] [Google Scholar]
- 22.O’Callaghan CH, Morris A, Kirby SM, Shingler AH. Novel method for detection of beta-lactamases by using a chromogenic cephalosporin substrate. Antimicrob Agents Chemother. 1972;1:283–288. doi: 10.1128/aac.1.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardy LW, Kirsch JF. pH dependence and solvent deuterium oxide kinetic isotope effects on Bacillus cereus beta-lactamase I catalyzed reactions. Biochemistry. 1984;23:1282–1287. doi: 10.1021/bi00301a041. [DOI] [PubMed] [Google Scholar]
- 24.Hackam DJ, Rotstein OD, Zhang W, Gruenheid S, Gros P, Grinstein S. Host resistance to intracellular infection: mutation of natural resistance-associated macrophage protein 1 (Nramp1) impairs phagosomal acidification. J Exp Med. 1998;188:351–364. doi: 10.1084/jem.188.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bender ML, Clement GE, Kezdy FJ, Heck HD. The Correlation of the pH (pD) Dependence and the Stepwise Mechanism of a-Chymotrypsin-Catalyzed Reactions. J Am Chem Soc. 1964;86:3680–3690. [Google Scholar]
- 26.Quinn DM, Sutton LD. In: Enzyme Mechanism from Isotope Effects. Cook PF, editor. Chapter 3. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- 27.Stein RL, Strimpler AM, Hori H, Powers JC. Catalysis by human leukocyte elastase: proton inventory as a mechanistic probe. Biochemistry. 1987;26:1305–1314. doi: 10.1021/bi00379a016. [DOI] [PubMed] [Google Scholar]
- 28.Hyland LJ, Tomaszek TA, Jr, Meek TD. Human immunodeficiency virus-1 protease. 2 Use of pH rate studies and solvent kinetic isotope effects to elucidate details of chemical mechanism. Biochemistry. 1991;30:8454–8463. doi: 10.1021/bi00098a024. [DOI] [PubMed] [Google Scholar]
- 29.Brenner DG, Knowles JR. Penicillanic acid sulfone: an unexpected isotope effect in the interaction of 6 alpha- and 6 beta-monodeuterio and of 6,6-dideuterio derivatives with RTEM beta-lactamase from. Escherichia coli, Biochemistry. 1981;20:33680–3687. doi: 10.1021/bi00516a003. [DOI] [PubMed] [Google Scholar]
- 30.Adediran SA, Deraniyagala SA, Xu Y, Pratt RF. Beta-secondary and solvent deuterium kinetic isotope effects on beta-lactamase catalysis. Biochemistry. 1996;35:3604–3613. doi: 10.1021/bi952107i. [DOI] [PubMed] [Google Scholar]
- 31.Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE, 3rd, Blanchard JS. Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science. 2009;323:1215–1218. doi: 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.