

Background: BMP-2-induced signal transduction requires receptor-specific Smads for osteogenesis.
Results: c-Abl regulates BMP-2-induced canonical Smad and noncanonical PI 3-kinase in osteoblasts.
Conclusion: BMP-2-stimulated c-Abl tyrosine kinase potentiates osteoblast and osteoclast differentiation.
Significance: c-Abl is essential for bone remodeling.
Keywords: Bone Morphogenetic Protein (BMP), Osteoblasts, Osteoclast, PI 3-Kinase, SMAD Transcription Factor, Colony-stimulating Factor-1 (CSF-1), c-Abl
Abstract
Skeletal remodeling consists of timely formation and resorption of bone by osteoblasts and osteoclasts in a quantitative manner. Patients with chronic myeloid leukemia receiving inhibitors of c-Abl tyrosine kinase often show reduced bone remodeling due to impaired osteoblast and osteoclast function. BMP-2 plays a significant role in bone generation and resorption by contributing to the formation of mature osteoblasts and osteoclasts. The effects of c-Abl on BMP-2-induced bone remodeling and the underlying mechanisms are not well studied. Using a pharmacological inhibitor and expression of a dominant negative mutant of c-Abl, we show an essential role of this tyrosine kinase in the development of bone nodules containing mature osteoblasts and formation of multinucleated osteoclasts in response to BMP-2. Calvarial osteoblasts prepared from c-Abl null mice showed the absolute requirement of this tyrosine kinase in maturation of osteoblasts and osteoclasts. Activation of phosphatidylinositol 3-kinase (PI 3-kinase)/Akt signaling by BMP-2 leads to osteoblast differentiation. Remarkably, inhibition of c-Abl significantly suppressed BMP-2-stimulated PI 3-kinase activity and its downstream Akt phosphorylation. Interestingly, c-Abl regulated BMP-2-induced osteoclastogenic CSF-1 expression. More importantly, we identified the requirements of c-Abl in BMP-2 autoregulation and the expressions of alkaline phosphatase and osterix that are necessary for osteoblast differentiation. c-Abl contributed to BMP receptor-specific Smad-dependent transcription of CSF-1, osterix, and BMP-2. Finally, c-Abl associates with BMP receptor IA and regulates phosphorylation of Smad in response to BMP-2. We propose that activation of c-Abl is an important step, which induces into two signaling pathways involving noncanonical PI 3-kinase and canonical Smads to integrate BMP-2-induced osteogenesis.
Introduction
Balanced activities of two principal cell populations, osteoblasts and osteoclasts with other cells in the bone matrix, contribute to bone remodeling. Many protein factors, including bone morphogenetic protein-2 (BMP-2),4 stimulate mesenchymal cells to differentiate into osteoblast precursors and induce maturation of the osteoblasts via expression of osteoblastic gene markers (1, 2). Dimeric BMP-2 contains two receptor binding domains. Wrist epitope binds with high affinity to BMP receptor I and the low affinity kunkel epitope binds receptor II (3–6). Binding of BMP-2 to type I receptor is affected by type II receptor and induces the formation of receptor oligomer (7, 8). In this complex, the constitutively active receptor II transphosphorylates the type I receptor at the GS domain located upstream of the serine-threonine kinase domain, resulting in an increase in kinase activity of the type I receptor, which determines the specificity of the intracellular signaling by BMP-2 (9). Thus BMP receptor I acts as a downstream effector of type II receptor and phosphorylates Smads 1/5/8 (R-Smads) in their C-terminal SSXS motif leading to their activation. R-Smads contain MH1 and MH2 domains joined by a linker region (10). MH1 binds to the MH2 domain in the absence of receptor activation and ensures inhibition of receptor function. Phosphorylation of R-Smads releases this interaction resulting in oligomerization with Smad4 and translocation to the nucleus. In the nucleus, the MH-1 domain binds to specific DNA sequences and recruits other nuclear proteins to stimulate or repress gene transcription (10, 11). In addition to the activation of Smads, BMP-2 activates non-Smad signaling pathways, including the MAPK family: p38, ERK1/2, and JNK (9, 12, 13). In fact, BMP-2-stimulated p38 occurs via activation of TAK1/TAB1 (14, 15). Both TAK1 and Smad5 null mice show similar phenotypes, indicating non-Smad signaling may contribute to BMP signal transduction (16). In fact, activation of p38 MAPK and JNK induces osteoblast differentiation (17).
Role of receptor and nonreceptor tyrosine kinases in osteoblast differentiation has been reported (2). Two nonreceptor tyrosine kinases Abl1 (c-Abl) and Abl-related gene (Arg) contribute to normal development and regulate cell cycle, cell survival, actin cytoskeleton, oxidative stress, and DNA damage response (18, 19). Although c-Abl is activated by TGFβ in rodent models of fibrosis, it antagonizes the epithelial mesenchymal transition and cytostatic and oncogenic actions of TGFβ (20–23). c-Abl-deficient mice exhibit low viability and defective hematopoiesis, cardiac hyperplasia, and decreased systolic blood pressure (24–27). c-Abl has an SH3-SH2-tyrosine kinase domain cassette. C-terminal to this segment, it contains a DNA binding domain. A long C-terminal stretch contains proline-rich motifs, which interact with SH3 domains of Crk, Grb2, and Nck. Also, binding sites for p53, Rb, ATM, and RNA polymerase II are also present in this segment (19). Nuclear localization and export signals have been identified in c-Abl, which confer upon its localization in the nucleus and cytoplasm (28–30). Tyrosine phosphorylation of this enzyme regulates its catalytic activity, subcellular localization of its substrates, and SH2 domain binding (31).
A recent study demonstrated no difference in number or rate of proliferation of osteoblasts from c-Abl null mice. Rather, these cells underwent senescence (32). Another study in rats treated with the c-Abl inhibitor STI 571 (imatinib, Gleevec) showed significant effects on both osteoclasts and osteoblasts in the same bone leading to dysregulated bone remodeling (33). Similarly, c-Abl inhibition in children with chronic myeloid leukemia is associated with growth retardation (34). The mechanism by which c-Abl affects bone remodeling is not known. We have shown recently that BMP-2 stimulates osteogenesis by promoting differentiation/maturation of osteoblasts, which in turn contribute to osteoclastogenesis via increased expression of CSF-1 (35–37). We implicated requirement of tyrosine kinase(s) in BMP-2-induced signal transduction during osteoblast differentiation (37). In this report, we show an essential role of c-Abl tyrosine kinase in BMP-2-stimulated osteoblast differentiation and osteoclastogenesis. We demonstrate that c-Abl is required for the expression of osteoblastic and osteoclastic genes in response to BMP-2. Finally, we establish the existence of a cross-talk between BMP-specific Smad and c-Abl.
EXPERIMENTAL PROCEDURES
Materials
Recombinant BMP-2 was obtained from Wyeth Pharmaceuticals, Cambridge, MA. Tissue culture reagents were purchased from Invitrogen. c-Abl inhibitor STI 571 (imatinib mesylate) was obtained from Selleckchem (Houston, TX). FuGENE HD was obtained from Roche Diagnostics. Anti-phosphotyrosine antibody was purchased from Upstate Biotechnology, Inc. (Lake Placid, NY). Phospho-c-Abl (Tyr-412), phospho-c-Abl (Tyr-245), c-Abl, phospho-Akt (Ser-473), Akt, phospho-Smad1/5, antibodies, and GST-Crk were purchased from Cell Signaling (Boston). Smad1/5 antibody was obtained from Santa Cruz Biotechnology. Osterix antibody was purchased from Abcam (Cambridge, MA). β-Actin antibody, acid phosphatase kit, and Fast Garnet dye for tartrate-resistant alkaline phosphatase (TRAP) activity and Alizarin Red S were obtained from Sigma. TRIzol reagent for RNA isolation was purchased from Invitrogen. Luciferase reporter assay kit was purchased from Promega (Madison, WI). ELISA kit to detect CSF-1 was obtained from R & D Systems (Minneapolis, MN). Polyvinylidene membrane for Western blotting was purchased from PerkinElmer Life Sciences. Kinase-dead c-Abl KR (pSRαMSVc-AblK(290)R-tKNeo) expression plasmid was provided by Dr. Charles Sawyers, UCLA (38). Constitutively active Smad5 containing S463D/S465D mutation was a kind gift from Dr. David Kimelman, University of Washington. BMP-2 promoter driven luciferase (BMP-2-Luc), CSF-1 promoter luciferase (CSF-1-Luc), Osterix promoter-driven luciferase (Osx-Luc) reporter plasmids were described previously (35, 37, 39, 40).
Cell Culture and Adenovirus Infection
Mouse calvarial 2T3 osteoblast cells are responsive to BMP-2 and undergo bone matrix formation in vitro. These cells have been characterized extensively (36, 37, 41–45). These cells were grown in α-MEM in the presence of 10% fetal bovine serum. The cells were infected with adenovirus vector expressing dominant negative c-Abl or green fluorescence protein (control infection) essentially as described (35, 37, 40, 46). Mouse primary calvarial osteoblasts were prepared by sequential digestion with trypsin and collagenase as described (36, 47). The c-Abl inhibitor STI 571 was dissolved in sterile PBS. Genistein was dissolved in DMSO. The cells were pretreated with the indicated concentration of inhibitors. Control cells received the vehicle.
Coculture Assay
Cocultures of 2T3 and mouse spleen cells were done as described previously (35, 39). In brief, 2T3 cells were cultured for 24 h followed by plating 106 mouse spleen cells in the presence of 10−8 m 1,25-dihydroxyvitamin D3, 10−7 m dexamethasone, and 300 ng/ml recombinant BMP-2 or bovine serum albumin (vehicle control). Medium was replaced every 2 days. At 6 days adherent cells were fixed in 10% formalin for 5 min and treated with a 1:1 mixture of ethanol and acetone for 1 min. The cultures were then dried and stained for TRAP activity using the acid phosphatase kit and Fast Garnet dye (Sigma). TRAP-positive multinucleated cells with three or more nuclei were photomicrographed using a light microscope.
Bone Marrow Assay for Mature Osteoclast Formation
Bone marrow cells were flushed from mice femur and tibia using α-MEM. 1 × 106 nonadherent cells were cultured in α-MEM containing 10% serum in 24-well tissue culture plates in the presence of 10−8 m 1,25-dihydroxyvitamin D3. Growth medium was replenished every 48 h for 7 days. TRAP staining was performed as described above. For TRAP activity assay, cells were fixed with 37% formaldehyde for 30 s, followed by washing with PBS. TRAP activity was measured at 405 nm using TRAP assay buffer, pH 4.7–5.0, containing sodium acetate, tartaric acid, glacial acetic acid, and phosphatase substrate (Sigma).
Immunoblotting, Immunoprecipitation, and PI 3-Kinase Assay
Cells were washed twice with PBS. The cells were lysed in RIPA buffer (20 mm Tris-HCl, pH 7.5, 5 mm EDTA, 150 mm NaCl, 1% Nonidet P-40, 1 mm Na3VO4, 1 mm PMSF, and 0.1% protease inhibitor mixture) for 30 min at 4 °C. The cell extracts were centrifuged at 10,000 × g for 30 min at 4 °C. Protein was estimated in the supernatant. Equal amounts of cell lysates were separated by SDS-PAGE. The separated proteins were transferred to PVDF membrane and immunoblotted using indicated antibodies. The protein bands were developed using HRP-conjugated secondary antibodies with ECL chemiluminescent reagent as described previously (35, 37, 39, 40, 45, 46, 48, 49). For PI 3-kinase activity, equal amounts of cell lysates were immunoprecipitated with anti-phosphotyrosine antibody. The immunoprecipitates were assayed for PI 3-kinase activity using phosphatidylinositol as a substrate in the presence of [γ-32P]ATP. The reaction products were separated by thin layer chromatography to detect PI 3-phosphate as described (37, 49).
c-Abl Immunocomplex Kinase Assay
The cell lysates were immunoprecipitated with c-Abl antibody as described (50). The immunoprecipitates were incubated with 1 μg of GST-Crk in the presence of 20 μCi of [γ-32P]ATP in kinase buffer (50 mm HEPES, pH 7.4, 10 mm MnCl2). The labeled protein was then separated by SDS-PAGE, dried on a filter paper, and autoradiographed.
Alkaline Phosphatase Staining and Assay
Fixed cells in 10% formalin were stained using 5-bromo-4-chloro-3-indoyl phosphate and nitro blue tetrazolium as described previously (37). The stained structures were photomicrographed (×200). Lysates of cells were assayed for alkaline phosphatase activity using p-nitrophenyl phosphate as substrate as described (37, 46, 47).
Mineralized Bone Nodule Formation
Cells were grown to confluency in 24-well plates. Growth medium was supplemented with differentiation medium containing serum-free α-MEM, 100 mg/ml ascorbic acid, 5 mm β-glycerophosphate, and 7% FBS before addition of BMP-2. The cells were cultured with a fresh medium change every 2 days for 10 more days. The cells were washed with PBS and fixed in ice-cold ethanol (70%) for 1 h at 4 °C. The cells were then washed with water and stained for 5 min with 2% solution of Alizarin Red S, pH 4.0, for calcium detection. Unbound stains were removed by washing with water (45). Plates were dried and photographed. The stains were extracted in DMSO, and absorbance was determined at 590 nm for quantification. For von Kossa (for mineral) and Van Giesen (for collagen) staining, the cells were fixed with 10% formalin, ethanol-washed, and air-dried before staining with 5% silver nitrate followed by 5% sodium thiocyanate and counterstaining with 1% acid fuchsin in picric acid (Sigma) (36).
RNA Isolation and Real Time Quantitative RT-PCR
Total RNA was prepared from cells using TRIzol reagent as described previously (39, 45, 46). 2 μg of total RNA was used to make cDNA, which was subsequently used for amplification by quantitative PCR using the ABI Prism 7300 sequence detection system and analyzed by SDS 2.1 software and SYBR Green probe method (Applied Biosystems). The primer sequences and PCR conditions for BMP-2 and CSF-1 were described previously (39, 45). The PCR condition for osterix was 35 cycles (95 °C for 15 s, 58 °C for 30 s, and 72 °C for 30 s). The primer sequences for Osx were as follows: forward, TGAGGAAGAAGCCCATTCAC; reverse, ACTTCTTCTCCCGGGTGTG.
Transfection and Luciferase Assay
Cells were transfected with the indicated plasmids using FuGENE HD reagent, and luciferase activity was determined using luciferase assay kit as per the vendor's instructions. The luciferase activity was quantified as described previously (35, 40, 45, 48).
Statistics
The significance of the data was determined by analysis of variance followed by Student-Newman-Keuls analysis as described (35, 39, 40, 45, 46, 48).
RESULTS
BMP-2-stimulated c-Abl Tyrosine Kinase Activity Is Required for Osteoclastogenesis
To systematically investigate a role for tyrosine kinase in BMP-2-induced signal transduction, we incubated 2T3 preosteoblasts with BMP-2. As expected, BMP-2 increased tyrosine phosphorylation of a panel of proteins (Fig. 1A) (37). The broad spectrum tyrosine kinase inhibitor genistein inhibited BMP-2-stimulated tyrosine phosphorylation (Fig. 1A). One of the BMP-2-stimulated tyrosine-phosphorylated proteins migrated at 120 kDa (Fig. 1A, indicated by arrow). We hypothesized that this protein may represent c-Abl tyrosine kinase. One of the best substrates of c-Abl is the SH3 domain containing adaptor protein Crk, which binds to the PXXP motif of c-Abl (51). To assess directly the c-Abl tyrosine kinase activity, we used recombinant Crk protein as substrate in vitro. Immunocomplex kinase assay of c-Abl immunoprecipitates from BMP-2-stimulated cells showed increase in tyrosine kinase activity (Fig. 1B). STI 571, a c-Abl inhibitor, abolished BMP-2-stimulated phosphorylation of Crk (Fig. 1B). Autophosphorylation (Tyr-245) of c-Abl in the linker region between SH2 and kinase domains and in the activation loop of the kinase domain (Tyr-412) is associated with its increased kinase activity (52, 53). Therefore, we tested phosphorylation of these two sites in c-Abl. BMP-2 increased phosphorylation of c-Abl at Tyr-412 and Tyr-245 (Fig. 1C), which was significantly inhibited by STI 571 (Fig. 1C).
FIGURE 1.

BMP-2-induced c-Abl tyrosine kinase activity is required for osteoclast formation. A, genistein blocks BMP-2-induced tyrosine phosphorylation. 2T3 preosteoblasts were treated with 10 μm genistein prior to incubation with 100 ng/ml BMP-2 for 5 min. The cell lysates were immunoblotted with anti-phosphotyrosine antibody. Arrow indicates the position of a 120-kDa protein. B, 2T3 cells were incubated with 25 μm STI 571 followed by treatment with 100 ng/ml BMP-2 for the indicated periods of time. The cell lysates were immunoprecipitated with c-Abl antibody. The immunoprecipitates were used in an immunocomplex kinase assay using GST-Crk fusion protein as a substrate in the presence of [γ-32P]ATP as described under “Experimental Procedures” (50). Bottom panel shows immunoblotting of the cell lysates with c-Abl antibody. Con, control. C, 2T3 cells were treated with 25 μm STI 571 and incubated with BMP-2 for 10 min. The cell lysates were immunoblotted with antibodies against phospho-c-Abl (Y412), phospho-c-Abl (Y245), and c-Abl antibodies. D, 2T3 cells were left untreated or treated with 25 μm STI 571 and cultured with mouse spleen cells in the absence and presence of 100 ng/ml BMP-2. At 7 days, the cells were fixed and stained for TRAP-positive multinucleated osteoclasts as described under “Experimental Procedures” (39). The bottom part of D shows quantification of multinucleated (MNC) TRAP-positive cells. Mean ± S.E. of triplicate wells is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. E, 2T3 cells were infected with Ad DN c-Abl or Ad GFP. The infected cells were cultured with mouse spleen cells in the presence of 100 ng/ml BMP-2. TRAP-positive multinucleated cells were detected as described (39). The bottom part of E shows quantification of multinucleated (MNC) TRAP-positive cells. Mean ± S.E. of triplicate wells is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. F, calvarial osteoblasts were prepared from wild type and c-Abl null mice as described under “Experimental Procedures.” These osteoblasts were cocultured with mouse spleen cells in the presence and absence of 100 ng/ml BMP-2. TRAP-positive multinucleated cells were detected as described (39). G and H, mouse bone marrow cells were cultured as described under “Experimental Procedures.” The cells were treated STI 571 followed by incubation with BMP-2. TRAP-positive multinucleated cells were detected as described above in F. H, TRAP activity was determined as described under “Experimental Procedures.” Mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. I and J, cultured mouse bone marrow cells were infected with Ad DN c-Abl or Ad GFP as described under “Experimental Procedures.” Infected cells were incubated with BMP-2. TRAP-positive multinucleated cells were detected as described above in F. J, TRAP activity was determined as described under “Experimental Procedures.” Mean ± S.E. of triplicate wells is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. K, bone marrow-derived cells from wild type (WT) and c-Abl null mice were cultured for 7 days. TRAP-positive multinucleated cells were detected as described above. L, TRAP activity was determined as described under “Experimental Procedures.” Mean ± S.E. of triplicate wells is shown. *, p = 0.01 versus wild type.
We have recently shown that BMP-2 stimulates osteoblast-aided osteoclastogenesis from mouse splenocytes (39). We investigated the role of c-Abl in this process. As expected, BMP-2 enhanced the formation of multinucleated osteoclasts in this coculture assay (Fig. 1D). STI 571 significantly attenuated BMP-2-induced osteoclastogenesis (Fig. 1D, compare panel d with b). To confirm these results, we employed an adenovirus vector expressing dominant negative c-Abl. Expression of this kinase-dead c-Abl markedly decreased BMP-2-stimulated osteoclast formation (Fig. 1E). To further evaluate the contribution of c-Abl in osteoclastogenesis, we used calvarial osteoblasts as the feeder layer from c-Abl−/− mouse in coculture assay. Calvarial osteoblasts from wild type mice showed weak basal multinucleated osteoclast formation as compared with that from c-Abl null mouse (Fig. 1F, compare panel c with a). BMP-2 significantly increased the formation of mature multinucleated osteoclasts in wild type mice. However, c-Abl−/− osteoblasts showed significant impairment in supporting osteoclast formation in response to BMP-2 (Fig. 1F, compare panel d with b). These results suggest that BMP-2-stimulated c-Abl tyrosine kinase regulates osteoblast-aided osteoclast formation/maturation. To address whether c-Abl regulates osteoclastogenesis in normal bone marrow-derived cells, we examined the effect of STI 571 on osteoclast differentiation using mouse bone marrow culture in the presence of BMP-2. As shown in Fig. 1G, BMP-2 significantly increased osteoclast formation. STI 571 inhibited BMP-2-induced osteoclastogenesis from normal bone marrow-derived cells (Fig. 1G, compare panel d with b). Similarly, STI 571 significantly inhibited BMP-2-stimulated TRAP enzyme activity in mouse bone marrow-derived cultures (Fig. 1H). To confirm this observation, we used adenovirus vector expressing dominant negative c-Abl to infect normal mouse bone marrow culture. Similar to STI 571, dominant negative c-Abl attenuated osteoclast differentiation and TRAP enzyme activity from normal bone marrow-derived cells (Fig. 1, I, compare panel d with b, and J). Furthermore, bone marrow cells prepared from c-Abl null mice showed reduced osteoclast differentiation and TRAP activity (Fig. 1, K and L). These data indicate a significant role of c-Abl in osteoclast differentiation.
c-Abl Regulates BMP-2-induced Osteoblast Differentiation
BMP-2 is a potent osteogenic factor (54). We and others have previously shown that BMP-2-induced osteoblast differentiation of 2T3 cells is associated with expression of alkaline phosphatase (2, 36, 37, 55). To examine the effect of c-Abl tyrosine kinase on BMP-2-induced alkaline phosphatase expression, we treated 2T3 preosteoblasts with BMP-2 in the presence and absence of STI 571. As expected, BMP-2 increased expression of alkaline phosphatase (Fig. 2A). Inhibition of c-Abl activity by STI 571 blocked BMP-2-induced alkaline phosphatase expression (Fig. 2A, compare panel d with b). In an independent experiment, we assayed for alkaline phosphatase activity in 2T3 cell lysates. STI 571 significantly abrogated BMP-2-stimulated alkaline phosphatase activity (Fig. 2B). Similarly, expression of kinase-dead c-Abl (KR c-Abl) markedly inhibited BMP-2-induced alkaline phosphatase activity (Fig. 2, C and D). Furthermore, BMP-2 enhanced the alkaline phosphatase activity in calvarial osteoblasts isolated from wild type mice (Fig. 2E). However, the osteoblasts prepared from c-Abl null mice showed markedly impaired basal and BMP-2-responsive alkaline phosphatase activity (Fig. 2E).
FIGURE 2.

BMP-2-stimulated osteoblast differentiation requires c-Abl tyrosine kinase. A, 2T3 cells were treated with 25 μm STI 571 (STI) for 1 h prior to incubation with 100 ng/ml BMP-2 for 48 h. The cells were treated with the alkaline phosphatase substrate 5-bromo-4-chloro-3-indoyl phosphate and nitro blue tetrazolium as described under “Experimental Procedures.” The stained cells were photomicrographed. Bottom panel shows stained dishes of respective conditions. B, lysates of cells treated similarly as in A were assayed for alkaline phosphatase activity with p-nitrophenyl phosphate as a substrate. Mean ± S.E. of triplicate measurements is shown. *, p < 0.01 versus control; **, p < 0.01 versus BMP-2-stimulated. C and D, 2T3 cells were transfected with kinase-dead c-Abl (KR-c-Abl) or vector. Transfected cells were incubated with BMP-2 as described in A. C, cells were stained for alkaline phosphatase expression as in A. Bottom panel shows stained dishes of respective conditions. D, lysates of cells were assayed for alkaline phosphatase activity as described in B. Mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. E, calvarial osteoblasts prepared from wild type (+/+) and c-Abl null (−/−) mice were incubated with BMP-2. The cell lysates were used to assay alkaline phosphatase activity. Mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. F, 2T3 cells were treated with 10 μm STI 571 for 1 h prior to incubation with 100 ng/ml BMP-2 and were stained with Alizarin Red S as described under “Experimental Procedures” (45). Stained cells were photomicrographed. G, stains in F were extracted with DMSO, and absorbance was determined at 590 nm. Mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. H, 2T3 cells were infected with Ad DN c-Abl or Ad GFP as described in Fig. 1E and incubated with BMP-2. The cells were stained with Alizarin Red S as described under “Experimental Procedures” (45). I, stains in H were extracted with DMSO, and absorbance was determined at 590 nm. Mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. J, calvarial osteoblasts from wild type and c-Abl null mice were incubated with BMP-2 and stained with von Kossa and Van Giesen stain as described under “Experimental Procedures” (36, 56). Con, control.
BMP-2 induces osteoblast differentiation to form mature osteoblasts leading to mineralized bone nodules. We examined the role of c-Abl in mature osteoblast formation using a mineralization assay by staining with Alizarin Red S reagent, which stains mineralized bone nodules (45). As expected, BMP-2 enhanced mineralized nodule formation (Fig. 2F). Inhibition of c-Abl by STI 571 inhibited BMP-2-stimulated nodule formation (Fig. 2F, compare panel d with b). The Alizarin Red S stain retained in the bone nodules was measured. The quantification showed that STI 571 significantly blocked BMP-2-induced mineralized bone formation (Fig. 2G). Analogous to these results, dominant negative c-Abl showed a significant inhibition of BMP-2-stimulated nodule formation (Fig. 2, H and I). To confirm the contribution of c-Abl in bone nodule formation, we treated the calvarial osteoblasts from wild type and c-Abl null mice with BMP-2 and stained with Van Giesen and von Kossa stains that detect matrix collagen and mineral formation, respectively (36, 56). As expected, BMP-2 increased bone nodule formation in calvarial cells from wild type mice (Fig. 2J). However, BMP-2-induced bone nodule formation was significantly abrogated in calvarial cells isolated from c-Abl−/− mice (Fig. 2J, compare panel d with c). These results indicate that c-Abl regulates BMP-2-induced osteoblast differentiation and mature bone nodule formation.
c-Abl Regulates BMP-2-stimulated PI 3-Kinase/Akt Signaling
We have previously reported that BMP-2 stimulates PI 3-kinase-dependent signaling pathways to drive osteoblast differentiation and osteoblast-aided mature osteoclast formation (35, 37). We examined the role of c-Abl in this signaling. 2T3 cells were incubated with STI 571 prior to incubation with BMP-2. As expected BMP-2 increased PI 3-kinase activity in the anti-phosphotyrosine immunoprecipitates (Fig. 3A). Surprisingly, STI 571 inhibited BMP-2-stimulated PI 3-kinase activity (Fig. 3A). Expression of kinase-dead c-Abl also blocked PI 3-kinase activity in response to BMP-2 (Fig. 3B). One of the downstream targets of PI 3-kinase is Akt, and we previously showed activation of this kinase by BMP-2 (37). Inhibition of c-Abl abrogated BMP-2-induced Akt phosphorylation (Fig. 3C). Similarly, expression of kinase-dead c-Abl inhibited the Akt phosphorylation in response to BMP-2 (Fig. 3D).
FIGURE 3.

BMP-2-stimulated PI 3-kinase/Akt signaling requires c-Abl tyrosine kinase. A and C, 2T3 cells were treated with 25 μm STI 571 for 1 h prior to incubation with 100 ng/ml BMP-2 for 5 min. A, cells lysates were immunoprecipitated by anti-phosphotyrosine antibody, and the immunoprecipitates were assayed for PI 3-kinase activity as described (37, 49). C, cell lysates were immunoblotted with phospho-Akt (Ser-473) and Akt antibodies as indicated. B and D, 2T3 cells were transfected with kinase-dead c-Abl (KR c-Abl) or vector plasmid. Transfected cells were incubated with 100 ng/ml BMP-2 for 5 min. B, anti-phosphotyrosine immunoprecipitates were assayed for PI 3-kinase activity as described under “Experimental Procedures” (37, 49). D, cell lysates were immunoblotted with phospho-Akt, Akt, c-Abl, and actin antibodies.
c-Abl Regulates Osteoclastogenic and Osteoblastic Marker Expression
Expression and secretion of CSF-1 by the osteoblasts serve as a potent mediator of osteoclastogenesis. We found significantly reduced expression of CSF-1 mRNA in osteoblasts prepared from c-Abl−/− mice as compared with those from wild type mice (Fig. 4A). We have shown recently that BMP-2 stimulates CSF-1 expression in osteoblasts (39). We tested the role of c-Abl in secretion of CSF-1 from 2T3 cells. Conditioned medium from BMP-2-treated 2T3 cells was used to detect CSF-1 levels by ELISA. BMP-2-induced secretion of CSF-1 was significantly inhibited by STI 571 (Fig. 4B). Expression of dominant negative c-Abl also markedly inhibited BMP-2-induced secretion of CSF-1 (Fig. 4C). To directly examine the effect of c-Abl on CSF-1 expression, we determined the levels of mRNA. BMP-2 enhanced the CSF-1 mRNA expression (Fig. 4D). STI 571 significantly attenuated BMP-2-induced expression of CSF-1 mRNA (Fig. 4D). Similarly, expression of kinase-dead c-Abl blocked CSF-1 mRNA expression in response to BMP-2 (Fig. 4E). We have recently shown that BMP-2 increases CSF-1 expression by a transcriptional mechanism using a reporter plasmid in which the CSF-1 promoter drives the luciferase gene (39). BMP-2 increased reporter activity in 2T3 cells (Fig. 4, F and G). Inhibition of c-Abl either by STI 571 or by expression of dominant negative c-Abl significantly suppressed BMP-2-stimulated reporter activity (Fig. 4, F and G). These results suggest that c-Abl regulates osteoclastogenic CSF-1 expression by regulating its transcription.
FIGURE 4.

c-Abl regulates BMP-2-induced CSF-1 expression. A, expression of CSF-1 in calvarial osteoblasts prepared from c-Abl null mouse. Total RNAs were prepared from wild type and c-Abl null mice calvarial cells. Levels of CSF-1 mRNA were determined and corrected for GAPDH expression as described under “Experimental Procedures.” Mean ± S.E. of six measurements is shown. *, p = 0.0006 versus wild type (WT). B and D, 2T3 cells were treated with 25 μm STI 571 prior to incubation with BMP-2. B, conditioned media were tested for CSF-1 protein by ELISA as described under “Experimental Procedures” (39). D, the expression of CSF-1 mRNA was determined and corrected by GAPDH expression as described under “Experimental Procedures” (35, 48). Mean ± S.E. of triplicate measurements is shown in each panel. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated for both panels. C and E, 2T3 cells were transfected with dominant negative c-Abl (KR c-Abl) or vector plasmids. Transfected cells were incubated with BMP-2. CSF-1 protein in the conditioned medium (C) and CSF-1 mRNA were detected (E) as described above. Mean ± S.E. of triplicate measurements is shown. C, *, p < 0.01 versus control; **, p < 0.05 versus BMP-2-stimulated. E, *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. F, CSF-1 promoter-driven luciferase reporter (CSF-1-Luc) was transfected into 2T3 cells. Transfected cells were incubated with STI 571 followed by incubation with BMP-2. The cell lysates were assayed for luciferase activity as described under “Experimental Procedures” (35, 39, 48). Relative luciferase activity (RLU) is presented. Mean ± S.E. of triplicate measurements is shown. *, p < 0.05 versus control; **, p < 0.01 versus BMP-2-stimulated. G, 2T3 cells were transfected with the CSF-1-Luc and vector or dominant negative c-Abl (KR c-Abl). The cells were then incubated with BMP-2. Luciferase activity was measured in the cell lysates as described (35, 39, 48). Mean ± S.E. of triplicate measurements is shown. *, p < 0.01 versus control; **, p < 0.01 versus BMP-2-stimulated.
Differentiation of osteoblasts is controlled by several transcription factors, including Osx (57). We and others have shown previously that Osx is a BMP-2-inducible gene (40, 57). To gain more insight for the role of c-Abl in osteoblast differentiation, we tested the effect of STI 571 on the abundance of Osx protein in 2T3 preosteoblasts. Inhibition of c-Abl attenuated BMP-2-stimulated Osx protein levels (Fig. 5A). Expression of dominant negative c-Abl also inhibited Osx protein expression by BMP-2 (Fig. 5B). Both STI 571 as well as kinase-dead c-Abl blocked BMP-2-induced expression of Osx mRNA, indicating a possible transcriptional regulation of this gene (Fig. 5, C and D). Therefore, we determined transcription of Osx using a reporter construct in which luciferase gene is driven by the Osx promoter (40). As expected, BMP-2 increased transcription of Osx. Both STI 571 and dominant negative c-Abl significantly abrogated BMP-2-stimulated Osx transcription (Fig. 5, E and F).
FIGURE 5.
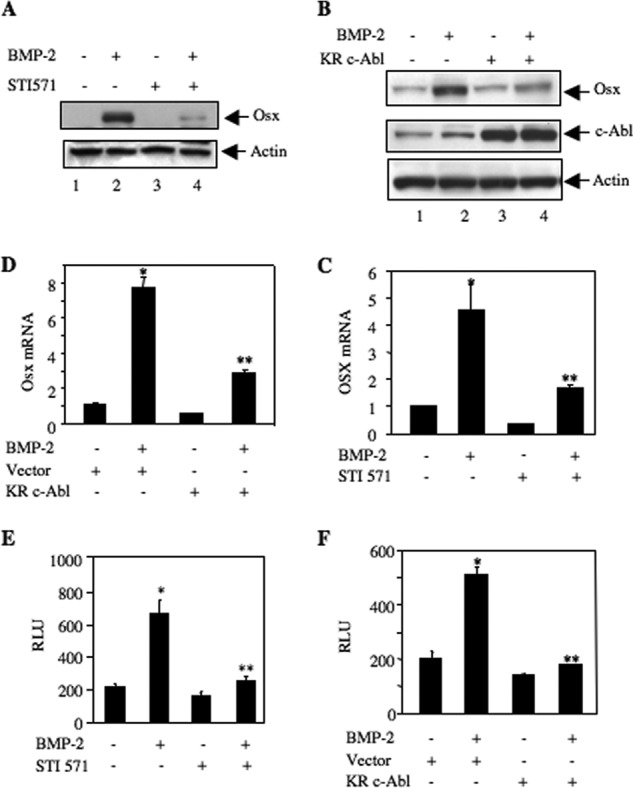
c-Abl regulates BMP-2-stimulated Osx expression. A and C, 2T3 cells were treated with 25 μm STI 571 prior to incubation with BMP-2. A, cell lysates were immunoblotted with Osx and actin antibodies. C, total RNA was used for qRT-PCR to detect Osx mRNA as described under “Experimental Procedures” (35, 48). Mean ± S.E. of six measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. B and D, 2T3 cells were transfected with dominant negative c-Abl or vector. The cells were incubated with BMP-2. B, cell lysates were immunoblotted with Osx, c-Abl, and actin antibodies. D, Osx mRNA was detected by qRT-PCR. Mean ± S.E. of six measurements is shown. *, p < 0.01 versus control; **, p < 0.05 versus BMP-2-stimulated. E, 2T3 cells were transfected with Osx promoter-driven luciferase reporter (Osx-Luc) (40). The cells were treated with 25 μm STI 571 followed by incubation with BMP-2. Luciferase activity was determined in the cell lysates. Mean ± S.E. of triplicate measurements is shown. *, p < 0.01 versus control; **, p < 0.001 versus BMP-2-stimulated. F, 2T3 cells were transfected with Osx-Luc and vector or dominant negative c-Abl. The cells were incubated with BMP-2. The cell lysates were assayed for luciferase activity. Mean ± S.E. of triplicate measurements is shown. *, p < 0.05 versus control; **, p < 0.01 versus BMP-2-stimulated. RLU, relative luciferase activity.
Autoregulation of BMP-2 is a prominent process during BMP-2-induced osteoblast differentiation (36). We have previously shown that BMP-2 increased expression of its own mRNA and protein (44, 45). Both STI 571 and dominant negative c-Abl blocked the expression of BMP-2-induced BMP-2 protein and mRNA (Fig. 6, A–D). Furthermore, using a BMP-2 promoter-driven reporter plasmid, we showed that inhibition of c-Abl by STI 571 or expression of kinase-dead c-Abl significantly abrogated BMP-2-stimulated transcription of BMP-2 (Fig. 6, E and F). These results together demonstrate a significant role of c-Abl in the expression of osteoclastogenic and osteoblastic genes during osteoblast differentiation.
FIGURE 6.

c-Abl regulates BMP-2-induced BMP-2 expression. A and C, 2T3 cells were treated with 25 μm STI 571 followed by incubation with BMP-2. A, cell lysates were immunoblotted with BMP-2 and actin antibodies. C, expression of BMP-2 mRNA was determined by qRT-PCR. Mean ± S.E. of six measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. B and D, 2T3 cells were transfected with dominant negative c-Abl (KR c-Abl) or vector. The cells were incubated with BMP-2. B, cell lysates were immunoblotted with BMP-2, c-Abl, and actin antibodies. D, BMP-2 mRNA was detected by qRT-PCR. Mean ± S.E. of six measurements is shown. *, p < 0.01 versus control; **, p < 0.01 versus BMP-2-stimulated. E, 2T3 cells were transfected with BMP-2 promoter-driven luciferase reporter (BMP-2-Luc) (37). The cells were treated with 25 μm STI 571 followed by incubation with BMP-2. Luciferase activity was determined in the cell lysates. Mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. F, 2T3 cells were transfected with BMP-2-Luc and vector or dominant negative c-Abl (KR c-Abl). The cells were incubated with BMP-2. The cell lysates were assayed for luciferase activity. Mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2-stimulated. RLU, relative luciferase activity.
c-Abl Regulates BMP Receptor-specific Smad-dependent Transcription of Target Genes
BMP-2 uses its receptor-specific Smads to induce its biological activities. Smad5 plays a significant role in induction of osteoblastic genes that are necessary for osteoclastogenesis and osteoblast differentiation (39, 40). We showed that BMP-2 increased osteoclastogenic CSF-1 expression by Smad5-dependent transcriptional mechanism (39). Using CSF-1 promoter-driven reporter plasmid, we tested the role of c-Abl in Smad5-mediated transcription of CSF-1. Both STI 571 and expression of dominant negative c-Abl significantly inhibited BMP-2-stimulated transcription of CSF-1 (Fig. 7, A and B). We have reported earlier that Smad5 also regulates expression of Osx and BMP-2 (35, 37). Therefore, using reporter plasmids, we tested the effect of c-Abl inhibition on transcription of osteoblastogenic Osx and BMP-2. Both STI 571 and dominant negative c-Abl significantly inhibited Smad5-stimulated transcription of Osx (Fig. 7, C and D). Similarly, inhibition of c-Abl markedly blocked BMP-2 transcription induced by Smad5 (Fig. 7, E and F).
FIGURE 7.

c-Abl regulates Smad-dependent transcription of CSF-1, Osx, and BMP-2. A, C, and E, 2T3 cells were transfected with CSF-1-Luc (A), Osx-Luc (C), and BMP-2-Luc (E) along with Smad5 expression plasmid. Transfected cells were incubated with 25 μm STI 571. Luciferase activity was determined in the cell lysates. A, mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus Smad5-transfected. C, mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus Smad5-transfected. E, mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.005 versus Smad5-transfected. B, D, and F, 2T3 cells were transfected with CSF-1-Luc (B), Osx-Luc (D), and BMP-2-Luc (F) along with Smad5 and dominant negative c-Abl expression plasmids. Luciferase activity was determined in the cell lysates. B, mean ± S.E. of triplicate measurements is shown. *, p < 0.01 versus control; **, p < 0.05 versus Smad5-transfected. D, mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus Smad5-transfected. F, mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus Smad5-transfected. RLU, relative luciferase activity.
c-Abl Association with BMPRIA Induces Its Tyrosine Phosphorylation
Our results above demonstrate a role of c-Abl in BMP-2-induced osteogenic signal transduction. To characterize the mechanism, we examined association of c-Abl with BMPRIA. Coimmunoprecipitation experiments using c-Abl immunoprecipitates showed increased association of BMPRIA with c-Abl in response to BMP-2 (Fig. 8A). Reciprocal coimmunoprecipitation confirmed these results (Fig. 8B). Next, we determined the role of c-Abl tyrosine kinase activity in this association. Inhibition of c-Abl blocked BMP-2-induced association of BMPRIA with c-Abl, suggesting tyrosine kinase activity of c-Abl is necessary for this association (Fig. 8C). To examine whether the association of c-Abl induces tyrosine phosphorylation of BMPRIA, anti-phosphotyrosine immunoblotting was carried out with BMPRIA immunoprecipitates. As shown in Fig. 8D, BMP-2 enhanced tyrosine phosphorylation of BMPRIA. STI 571 inhibited BMP-2-stimulated BMPRIA tyrosine phosphorylation (Fig. 8D). Similarly, expression of dominant negative c-Abl blocked tyrosine phosphorylation of BMPRIA in response to BMP-2 (Fig. 8E). These results indicate a significant role of c-Abl in tyrosine phosphorylation of BMPRIA.
FIGURE 8.
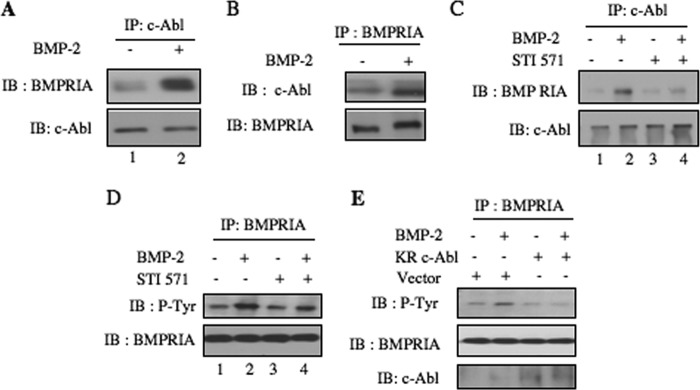
BMP-2 increases association of c-Abl with BMPRIA to induce its tyrosine phosphorylation. A, lysates of BMP-2-treated 2T3 cells were immunoprecipitated (IP) with c-Abl antibody followed by immunoblotting (IB) with BMPRIA and c-Abl antibodies as indicated. B, BMP-2-treated 2T3 cell lysates were used to immunoprecipitate BMPRIA antibody followed by immunoblotting with c-Abl and BMPRIA antibodies as indicated. C and D, 2T3 cells were treated with 25 μm STI 571 followed by incubation with BMP-2. The cell lysates were immunoprecipitated with c-Abl antibody followed by immunoblotting with BMPRIA and c-Abl antibodies (C). D, cell lysates were immunoprecipitated with BMPRIA antibody followed by immunoblotting with phosphotyrosine and BMPRIA antibodies as indicated. E, 2T3 cells were transfected with dominant negative c-Abl (KR c-Abl) or vector. The cells were incubated with BMP-2. The lysates were immunoprecipitated with BMPRIA antibody followed by immunoblotting with anti-phosphotyrosine and BMPRIA antibodies as indicated.
c-Abl Controls BMP Receptor-specific Smad Phosphorylation
Phosphorylation of receptor-specific Smad in the C-terminal sites is required for BMP-2-induced signal transduction (9, 10). We first examined the effect of STI 571 on this phosphorylation. As expected, BMP-2 increased C-terminal phosphorylation of Smad1/5 in 2T3 preosteoblasts (Fig. 9A). Surprisingly, inhibition of c-Abl abrogated BMP-2-stimulated phosphorylation of Smad1/5 (Fig. 9A). Similarly, expression of dominant negative c-Abl blocked phosphorylation of Smad1/5 in response to BMP-2 (Fig. 9B). Next, we determined association of Smad1/5 with c-Abl. Immunoblotting of c-Abl immunoprecipitates with Smad antibody showed increased association of Smad1/5 with c-Abl in the presence of BMP-2 (Fig. 9C). STI 571 inhibited this association (Fig. 9C). Interestingly, BMP-2 increased phosphorylation of Smad1/5 associated with c-Abl (Fig. 9C). STI 571 blocked phosphorylation of Smad1/5 complexed with c-Abl (Fig. 9C). To confirm the role of c-Abl in Smad1/5 phosphorylation, we used calvarial osteoblasts from c-Abl null mice. Treatment of cells prepared from wild type mice with BMP-2 increased phosphorylation of Smad1/5 (Fig. 9D). In contrast to these results, BMP-2 was unable to stimulate phosphorylation of Smad1/5 in calvarial osteoblasts derived from c-Abl−/− mice (Fig. 9D). These results conclusively demonstrate a direct role of c-Abl in BMP-2 signal transduction involving receptor-specific Smads.
FIGURE 9.
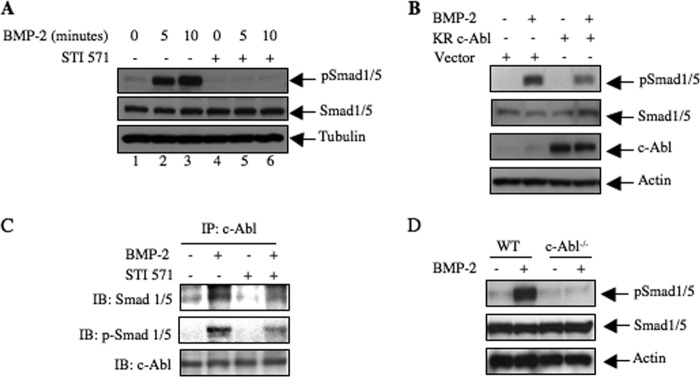
c-Abl contributes to BMP-2-stimulated Smad phosphorylation. A, 2T3 cells were treated with 25 μm STI 571 prior to incubation with BMP-2 for 5 and 10 min. The cell lysates were immunoblotted with phospho-Smad1/5, Smad1/5, and tubulin antibodies. B, 2T3 cells were transfected with dominant negative c-Abl. The transfected cells were incubated with BMP-2 for 5 min. The cell lysates were immunoblotted with phospho-Smad1/5, Smad1/5, and actin antibodies. C, 2T3 cells were treated with 25 μm STI 571 followed by incubation with BMP-2 for 5 min. The cell lysates were immunoprecipitated (IP) with c-Abl antibody followed by immunoblotting with Smad1/5, phospho-Smad1/5, and c-Abl antibodies as indicated. D, calvarial osteoblasts prepared from wild type and c-Abl null mice were incubated with BMP-2 for 5 min. The cell lysates were immunoblotted with phospho-Smad1/5, Smad1/5 and actin antibodies.
Smad5 Downstream of c-Abl Regulates BMP-2-induced Osteoclastic and Osteoblastic Marker Expression
Our results above have shown a role of c-Abl in expression of osteoclastogenic (CSF-1) and osteoblastogenic proteins (Osx and BMP-2) (Figs. 4–6). Furthermore, we show that c-Abl regulates Smad1/5 phosphorylation (Fig. 9). Therefore, using reporter transfection assays, we tested the mechanism of c-Abl-regulated expression of these proteins involving Smad5. As expected, expression of dominant negative c-Abl significantly reduced the transcription of CSF-1, Osx, and BMP-2 (Fig. 10, A–C). Interestingly, coexpression of constitutively active Smad5 significantly prevented the dominant negative c-Abl-mediated inhibition of transcription of these genes (Fig. 10, A–C). Consequently, expression of constitutively active Smad5 reversed the dominant negative c-Abl-induced suppression of BMP-2-stimulated CSF-1, Osx, and BMP-2 mRNA expression (Fig. 10, D and F). These results demonstrate a direct role of Smad5 downstream of c-Abl in induction of osteoclastic and osteoblastic gene expression.
FIGURE 10.

BMP-2-stimulated c-Abl uses Smad5 to increase CSF-1, Osx, and BMP-2 expression. A–C, 2T3 cells were transfected with CSF-1-Luc (A), Osx-Luc (B), and BMP-2-Luc (C) along with dominant negative c-Abl (KR-c-Abl) and constitutively active (CA) Smad5. The cells were treated with BMP-2. Luciferase activity was determined in the cell lysates. Mean ± S.E. of triplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-treated; @, p < 0.001 versus dominant negative-transfected and BMP-2-treated. D–F, 2T3 cells were transfected with vector or dominant negative c-Abl (KR-c-Abl) and constitutively active (CA) Smad5 as indicated. The cells were incubated with BMP-2. Total RNAs were used to detect CSF-1 (D), Osx (E), and BMP-2 (F) mRNAs. The data were corrected for GAPDH mRNA. Mean ± S.E. of quadruplicate measurements is shown. *, p < 0.001 versus control; **, p < 0.001 versus BMP-2 treated; @, p < 0.001 versus dominant negative c-Abl plus BMP-2. RLU, relative luciferase activity.
DISCUSSION
The concerted interaction between osteoblasts and osteoclasts maintains bone remodeling, which plays an important role in common bone diseases, including inflammatory osteolysis, skeletal metastasis, and osteoporosis (58–60). Many factors, including BMP-2, are produced by osteoblasts, which maintain mature osteoblast formation and stimulate osteoclastogenesis. Our results represent the first demonstration of activation of the nonreceptor tyrosine kinase c-Abl by BMP-2, which contributes to osteoblast-aided mature osteoclast formation and osteoblast differentiation. We demonstrate that c-Abl contributes to BMP-2-induced PI 3-kinase/Akt signal transduction pathways, which control osteoblast differentiation. Furthermore, our results show requirement of c-Abl for BMP-2 autoregulation and expression of CSF-1 and Osx by osteoblasts. Finally, we provide evidence that c-Abl regulates the canonical BMP-induced Smad phosphorylation, which contributes to osteogenic and osteoclastic gene expression. Thus, current data show two mechanisms, which emanate from BMP-2-stimulated c-Abl to converge on bone-specific gene expression and bone remodeling (Fig. 11).
FIGURE 11.
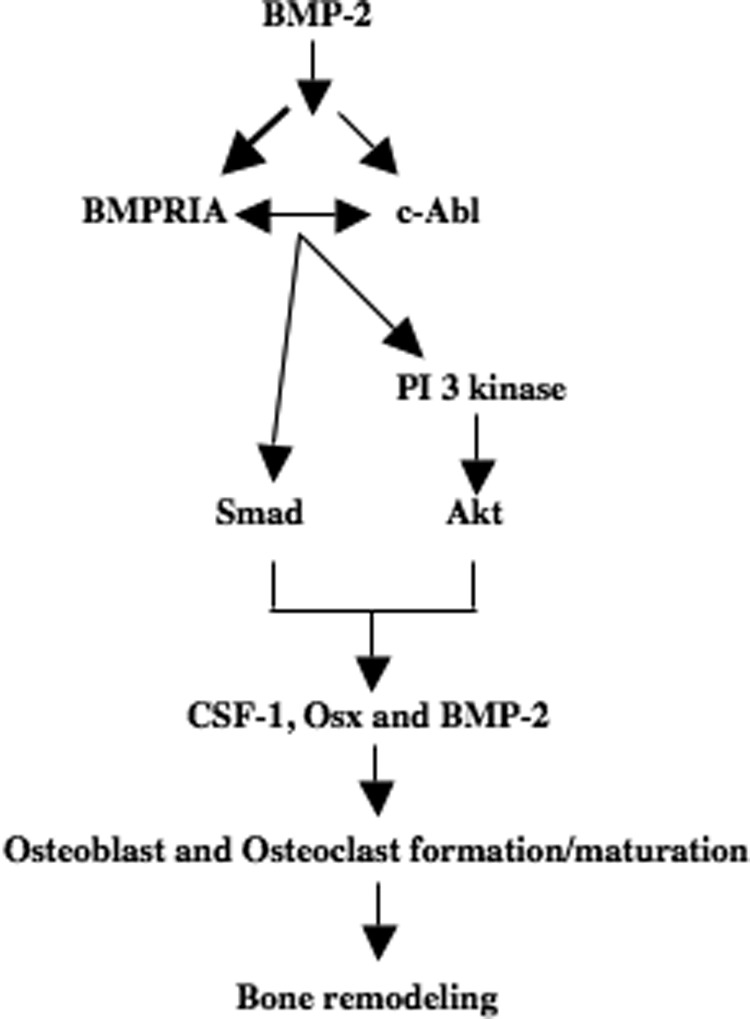
Schematic summarizing our results demonstrating BMP-2-induced signal transduction for bone remodeling.
Although we previously showed a role of tyrosine kinase in BMP-2-induced signal transduction and osteoblast differentiation, the involvement of specific tyrosine kinase was not determined (37). We identified that BMP-2 stimulated c-Abl tyrosine kinase in osteoblasts (Fig. 1, A–C). Osteoblast-produced factors are necessary for mature osteoclast formation (61). Thus, coculture of osteoblasts with osteoclast precursors has the capacity to form mature osteoclasts. However, BMP-2, which is a potent osteogenic growth and differentiation factor, significantly enhances the osteoclastogenic activity of the osteoblasts (39). Although BMP receptor-specific Smad may be required for osteoclastogenesis, our results using STI 571 and dominant negative c-Abl show that this nonreceptor tyrosine kinase regulates BMP-2-induced osteoblast-aided mature osteoclast formation (Fig. 1, D and E).
A previous report described that 50% of c-Abl null mice were osteoporotic (62). This study showed no difference in osteoclast differentiation of bone marrow-derived cells when compared with wild type cells, indicating c-Abl did not contribute to osteoclastogenesis. However, we show that calvarial osteoblasts from c-Abl null mice are defective in supporting osteoclastogenesis in a coculture assay (Fig. 1F). Furthermore, we provide evidence that c-Abl regulates osteoclast differentiation of normal mouse bone marrow-derived cells in response to BMP-2 (Fig. 1, G–J).
Highly specific and ordered expression of genes regulates osteoblast differentiation. Thus, BMP-2-induced osteoblastogenesis is initiated by expression of alkaline phosphatase at an early stage (63). We showed previously that tyrosine kinase-dependent signal transduction regulates BMP-2-stimulated alkaline phosphatase expression during osteoblast differentiation (37). A study using c-Abl null mice showed significant activity of alkaline phosphatase in osteoblasts (62). However, in this study using STI 571 and expression of dominant negative c-Abl, we show complete loss of BMP-2-stimulated alkaline phosphatase activity in 2T3 preosteoblasts (Fig. 2, A–D). In conjunction with these results, in calvarial osteoblasts prepared from c-Abl null mouse, we found complete prevention of alkaline phosphatase activity in response to BMP-2 (Fig. 2E). Furthermore, our results demonstrate that inhibition of c-Abl by its inhibitor or by dominant negative c-Abl expression significantly impaired osteoblast differentiation by BMP-2 (Fig. 2, F–I). This observation was also supported by our results showing marked impairment of bone nodule formation of cells isolated from c-Abl null calvaria (Fig. 2J). Thus, our data conclusively demonstrate a requirement of c-Abl tyrosine kinase in BMP-2-induced osteoblast differentiation.
We showed that BMP-2-stimulated class IA PI 3-kinase signaling was necessary for osteoblast differentiation and osteoblast-aided mature osteoclast formation (35, 37). This group of PI 3-kinases consists of two proteins, an 85-kDa regulatory and a 110-kDa catalytic subunit (64). The regulatory subunit contains SH3 and SH2 domains; the latter is involved in association with the activated receptor and nonreceptor tyrosine kinases (64, 65). The activated mutant of c-Abl, the viral oncogene product, was previously shown to activate PI 3-kinase in fibroblasts; however, overexpression of c-Abl was not sufficient to stimulate the lipid kinase activity (66). c-Abl contains a myristoyl group at its N terminus, which interacts with its kinase domain to render an autoinhibitory state (67). Moreover, the c-Abl SH2 domain binds phosphatidylinositol 4,5-bisphosphate, which inhibits its tyrosine kinase activity (68, 69). Activated PI 3-kinase also uses phosphatidylinositol 4,5-bisphosphate as substrate to produce phosphatidylinositol 3,4,5-trisphosphate (70, 71). Thus, activation of PI 3-kinase depletes cellular phosphatidylinositol 4,5-bisphosphate levels and as a consequence may release its inhibitory action resulting in the sustained activation of c-Abl. Activated c-Abl contains multiple phosphotyrosines, which recruit several SH2/SH3 domain-containing proteins, including PI 3-kinase in hematopoietic cells; however, direct association between p85 SH2 domain and c-Abl phosphotyrosines was not detected (31, 66, 72, 73). Rather, a role for intermediate proteins that directly bind to c-Abl has been proposed to recruit PI 3-kinase (74). In conjunction with these results, current data presented here show that BMP-2 stimulates PI 3-kinase activity in anti-phosphotyrosine immune complexes in a c-Abl tyrosine kinase-dependent manner, which results in downstream Akt phosphorylation (Fig. 3).
The role of two proteins CSF-1 and RANKL in osteoclastogenesis is well established. Our results demonstrate a role of c-Abl in expression of both these factors (Fig. 4A and data not shown). We have shown recently that the expression of osteoclastogenic CSF-1 and osteoblastic Osx by the osteoblasts depend upon PI 3-kinase/Akt signaling (35, 40). Our current results demonstrate that c-Abl regulates CSF-1 and Osx expression (Figs. 4 and 5). Also, BMP-2 autoregulation is mediated by c-Abl (Fig. 6). These data suggest that c-Abl regulation of PI 3-kinase/Akt signaling may be necessary for the expression of these genes.
Along with the noncanonical PI 3-kinase/Akt signaling, BMP-2 also uses canonical receptor-specific Smad for osteoblast differentiation and osteoblast-aided osteoclast maturation (35, 39, 40). BMP receptor-specific Smad8-deficient mice did not show any overt abnormality (75). Smad1 null mice die in utero; however, Smad1 null murine embryonic fibroblasts retain BMP-2 responsiveness indicating that other Smads such as Smad5 control BMP-2 action (76). In fact, we have reported that Smad5 regulates expression of BMP-2, CSF-1, and Osx during osteoblast differentiation (37, 39, 40). In this study, we show that c-Abl contributes to Smad5-mediated expression of these genes (Fig. 7). Furthermore, we detect association between c-Abl and BMPRIA in the presence of BMP-2 and that the receptor undergoes tyrosine phosphorylation in a c-Abl-dependent manner (Fig. 8). Because we showed previously that PI 3-kinase contributes to Smad5-dependent transcription, current data demonstrate the presence of a cross-talk between BMPRIA and c-Abl, which acts upstream of PI 3-kinase to regulate Smad5 transactivation of osteogenic and osteoclastogenic genes. We have previously identified Smad-binding elements in the promoters of CSF-1 and Osx and demonstrated their interaction with BMP-2-stimulated Smad5, resulting in transcription of these genes during osteoblast differentiation (39, 40). Phosphorylation of BMP receptor-specific Smad is necessary for binding to Smad4 and translocation to the nucleus to recruit transcriptional coactivators leading to direct binding to the Smad-binding element present in the promoter region of the target genes (11). We have shown previously BMP-2 stimulated increase in phosphorylation of Smads in 2T3 preosteoblasts. In this study, we show that BMP-2-induced activation of c-Abl tyrosine kinase is necessary for BMP receptor-specific Smad phosphorylation in preosteoblasts (Fig. 9, A and B). Because c-Abl does not have any serine/threonine kinase activity, it is somewhat unlikely that this tyrosine kinase would phosphorylate Smads in their C-terminal site. For Smad phosphorylation, binding to the type I receptor is essential. TGFβ receptor-specific R-Smads are anchored to the cell membrane by binding to the cytoplasmic protein SARA (77). However, this protein does not bind BMP receptor-specific Smads. One possibility may be that BMP-2-stimulated active c-Abl may bind BMP receptor-specific Smads to facilitate the phosphorylation of Smads by the BMP receptor I. In this context, we found that c-Abl coimmunoprecipitated with BMP receptor-specific Smads in response to BMP-2 in 2T3 cells (Fig. 9C). Interestingly, inhibition of c-Abl by STI 571 suppressed this complex formation (Fig. 9C). A corollary of this observation is that Smad should be phosphorylated in this immunocomplex. In fact, we found phosphorylated Smads in the BMP-2-induced c-Abl immunoprecipitates (Fig. 9C). Interestingly, the presence of these phospho-Smads was sensitive to STI 571 (Fig. 9C), indicating c-Abl regulation of this process. Furthermore, our results demonstrate the function of BMP-specific Smad downstream of c-Abl in BMP-2-induced osteoclastic and osteoblastic gene expression (Fig. 10). Because c-Abl forms a complex with Smad in a BMP-2 inducible manner (this study) and it can be present in the nucleus (30), our results point to an interesting possibility of the presence of this tyrosine kinase in the Smad-binding element of target genes. Further studies will be necessary to examine this mode of action of c-Abl in BMP-2-induced gene expression for osteoblast differentiation.
A study of patients with chronic myeloid leukemia treated with STI 571 showed hypophosphatemia and low serum calcium levels (78). In the treated group, expression of osteoblastic and osteoclastic markers was also inhibited, leading to attenuation of bone remodeling. BMP-2 plays an important role in bone remodeling. In this study, we show three BMP-2-induced mechanisms involving c-Abl regulation of BMPRIA tyrosine phosphorylation, PI 3-kinase signaling, and Smad phosphorylation, which in concert control expression of genes necessary for osteoblast differentiation and mature osteoclast formation (Fig. 11).
Acknowledgments
We thank Drs. Brent Wagner and B. S. Kasinath for critically reading the manuscript. We thank Javier Esparza for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 AR52425 (to N. G. C.) and RO1 DK50190 (to G. G. C.). This work was also supported by Veterans Affairs Merit Review grant and CTRC P30 Cancer Center Support Grant CA 054174 (to N. G. C.) and in part by Veterans Affairs Merit Review grants (to G. G. C.).
- BMP-2
- bone morphogenetic protein-2
- PI 3-kinase
- phosphatidylinositol 3-kinase
- CSF-1
- colony stimulating factor-1
- Osx
- osterix
- TRAP
- tartrate resistant acid phosphatase
- Ad
- adenoviral vector
- WT
- wild type
- DN
- dominant negative
- SH
- Src homology
- α-MEM
- α-minimal essential medium
- qRT
- quantitative PCR.
REFERENCES
- 1. Teti A. (2011) Bone development: overview of bone cells and signaling. Curr. Osteoporos. Rep. 9, 264–273 [DOI] [PubMed] [Google Scholar]
- 2. Marie P. J., Debiais F., Haÿ E. (2002) Regulation of human cranial osteoblast phenotype by FGF-2, FGFR-2, and BMP-2 signaling. Histol. Histopathol. 17, 877–885 [DOI] [PubMed] [Google Scholar]
- 3. Kirsch T., Nickel J., Sebald W. (2000) BMP-2 antagonists emerge from alterations in the low-affinity binding epitope for receptor BMPR-II. EMBO J. 19, 3314–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Allendorph G. P., Vale W. W., Choe S. (2006) Structure of the ternary signaling complex of a TGF-β superfamily member. Proc. Natl. Acad. Sci. U.S.A. 103, 7643–7648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kirsch T., Sebald W., Dreyer M. K. (2000) Crystal structure of the BMP-2-BRIA ectodomain complex. Nat. Struct. Biol. 7, 492–496 [DOI] [PubMed] [Google Scholar]
- 6. Kirsch T., Nickel J., Sebald W. (2000) Isolation of recombinant BMP receptor IA ectodomain and its 2:1 complex with BMP-2. FEBS Lett. 468, 215–219 [DOI] [PubMed] [Google Scholar]
- 7. Yu P. B., Beppu H., Kawai N., Li E., Bloch K. D. (2005) Bone morphogenetic protein (BMP) type II receptor deletion reveals BMP ligand-specific gain of signaling in pulmonary artery smooth muscle cells. J. Biol. Chem. 280, 24443–24450 [DOI] [PubMed] [Google Scholar]
- 8. ten Dijke P., Yamashita H., Sampath T. K., Reddi A. H., Estevez M., Riddle D. L., Ichijo H., Heldin C. H., Miyazono K. (1994) Identification of type I receptors for osteogenic protein-1 and bone morphogenetic protein-4. J. Biol. Chem. 269, 16985–16988 [PubMed] [Google Scholar]
- 9. Wan M., Cao X. (2005) BMP signaling in skeletal development. Biochem. Biophys. Res. Commun. 328, 651–657 [DOI] [PubMed] [Google Scholar]
- 10. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 11. Miyazono K., Kamiya Y., Morikawa M. (2010) Bone morphogenetic protein receptors and signal transduction. J. Biochem. 147, 35–51 [DOI] [PubMed] [Google Scholar]
- 12. Nohe A., Keating E., Knaus P., Petersen N. O. (2004) Signal transduction of bone morphogenetic protein receptors. Cell. Signal. 16, 291–299 [DOI] [PubMed] [Google Scholar]
- 13. Nakamura K., Shirai T., Morishita S., Uchida S., Saeki-Miura K., Makishima F. (1999) p38 mitogen-activated protein kinase functionally contributes to chondrogenesis induced by growth/differentiation factor-5 in ATDC5 cells. Exp. Cell Res. 250, 351–363 [DOI] [PubMed] [Google Scholar]
- 14. Yamaguchi K., Shirakabe K., Shibuya H., Irie K., Oishi I., Ueno N., Taniguchi T., Nishida E., Matsumoto K. (1995) Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science 270, 2008–2011 [DOI] [PubMed] [Google Scholar]
- 15. Shibuya H., Iwata H., Masuyama N., Gotoh Y., Yamaguchi K., Irie K., Matsumoto K., Nishida E., Ueno N. (1998) Role of TAK1 and TAB1 in BMP signaling in early Xenopus development. EMBO J. 17, 1019–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jadrich J. L., O'Connor M. B., Coucouvanis E. (2006) The TGF-β-activated kinase TAK1 regulates vascular development in vivo. Development 133, 1529–1541 [DOI] [PubMed] [Google Scholar]
- 17. Guicheux J., Lemonnier J., Ghayor C., Suzuki A., Palmer G., Caverzasio J. (2003) Activation of p38 mitogen-activated protein kinase and c-Jun NH2-terminal kinase by BMP-2 and their implication in the stimulation of osteoblastic cell differentiation. J. Bone Miner. Res. 18, 2060–2068 [DOI] [PubMed] [Google Scholar]
- 18. Schlatterer S. D., Acker C. M., Davies P. (2011) c-Abl in neurodegenerative disease. J. Mol. Neurosci. 45, 445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hantschel O., Superti-Furga G. (2004) Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 5, 33–44 [DOI] [PubMed] [Google Scholar]
- 20. Allington T. M., Schiemann W. P. (2011) The Cain and Abl of epithelial-mesenchymal transition and transforming growth factor-β in mammary epithelial cells. Cells Tissues Organs 193, 98–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Daniels C. E., Wilkes M. C., Edens M., Kottom T. J., Murphy S. J., Limper A. H., Leof E. B. (2004) Imatinib mesylate inhibits the profibrogenic activity of TGF-β and prevents bleomycin-mediated lung fibrosis. J. Clin. Invest. 114, 1308–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang S., Wilkes M. C., Leof E. B., Hirschberg R. (2010) Noncanonical TGF-β pathways, mTORC1 and Abl, in renal interstitial fibrogenesis. Am. J. Physiol. Renal Physiol. 298, F142–F149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Atfi A., Abécassis L., Bourgeade M. F. (2005) Bcr-Abl activates the AKT/Fox O3 signalling pathway to restrict transforming growth factor-β-mediated cytostatic signals. EMBO Rep. 6, 985–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schwartzberg P. L., Stall A. M., Hardin J. D., Bowdish K. S., Humaran T., Boast S., Harbison M. L., Robertson E. J., Goff S. P. (1991) Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell 65, 1165–1175 [DOI] [PubMed] [Google Scholar]
- 25. Tybulewicz V. L., Crawford C. E., Jackson P. K., Bronson R. T., Mulligan R. C. (1991) Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell 65, 1153–1163 [DOI] [PubMed] [Google Scholar]
- 26. Chen S., Wang R., Li Q. F., Tang D. D. (2009) Abl knockout differentially affects p130 Crk-associated substrate, vinculin, and paxillin in blood vessels of mice. Am. J. Physiol. Heart Circ. Physiol. 297, H533–H539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qiu Z., Cang Y., Goff S. P. (2010) c-Abl tyrosine kinase regulates cardiac growth and development. Proc. Natl. Acad. Sci. U.S.A. 107, 1136–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Etten R. A., Jackson P., Baltimore D. (1989) The mouse type IV c-abl gene product is a nuclear protein, and activation of transforming ability is associated with cytoplasmic localization. Cell 58, 669–678 [DOI] [PubMed] [Google Scholar]
- 29. Taagepera S., McDonald D., Loeb J. E., Whitaker L. L., McElroy A. K., Wang J. Y., Hope T. J. (1998) Nuclear-cytoplasmic shuttling of C-ABL tyrosine kinase. Proc. Natl. Acad. Sci. U.S.A. 95, 7457–7462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wen S. T., Jackson P. K., Van Etten R. A. (1996) The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products. EMBO J. 15, 1583–1595 [PMC free article] [PubMed] [Google Scholar]
- 31. Colicelli J. (2010) ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci. Signal. 3, re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kua H. Y., Liu H., Leong W. F., Li L., Jia D., Ma G., Hu Y., Wang X., Chau J. F., Chen Y. G., Mishina Y., Boast S., Yeh J., Xia L., Chen G. Q., He L., Goff S. P., Li B. (2012) c-Abl promotes osteoblast expansion by differentially regulating canonical and noncanonical BMP pathways and p16INK4a expression. Nat. Cell Biol. 14, 727–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nurmio M., Joki H., Kallio J., Määttä J. A., Väänänen H. K., Toppari J., Jahnukainen K., Laitala-Leinonen T. (2011) Receptor tyrosine kinase inhibition causes simultaneous bone loss and excess bone formation within growing bone in rats. Toxicol. Appl. Pharmacol. 254, 267–279 [DOI] [PubMed] [Google Scholar]
- 34. Rastogi M. V., Stork L., Druker B., Blasdel C., Nguyen T., Boston B. A. (2012) Imatinib mesylate causes growth deceleration in pediatric patients with chronic myelogenous leukemia. Pediatr. Blood Cancer 59, 840–845 [DOI] [PubMed] [Google Scholar]
- 35. Mandal C. C., Ghosh Choudhury G., Ghosh-Choudhury N. (2009) Phosphatidylinositol 3-kinase/Akt signal relay cooperates with Smad in bone morphogenetic protein-2-induced colony stimulating factor-1 (CSF-1) expression and osteoclast differentiation. Endocrinology 150, 4989–4998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ghosh-Choudhury N., Windle J. J., Koop B. A., Harris M. A., Guerrero D. L., Wozney J. M., Mundy G. R., Harris S. E. (1996) Immortalized murine osteoblasts derived from BMP 2-T-antigen expressing transgenic mice. Endocrinology 137, 331–339 [DOI] [PubMed] [Google Scholar]
- 37. Ghosh-Choudhury N., Abboud S. L., Nishimura R., Celeste A., Mahimainathan L., Choudhury G. G. (2002) Requirement of BMP-2-induced phosphatidylinositol 3-kinase and Akt serine/threonine kinase in osteoblast differentiation and Smad-dependent BMP-2 gene transcription. J. Biol. Chem. 277, 33361–33368 [DOI] [PubMed] [Google Scholar]
- 38. Sawyers C. L., McLaughlin J., Goga A., Havlik M., Witte O. (1994) The nuclear tyrosine kinase c-Abl negatively regulates cell growth. Cell 77, 121–131 [DOI] [PubMed] [Google Scholar]
- 39. Ghosh-Choudhury N., Singha P. K., Woodruff K., St Clair P., Bsoul S., Werner S. L., Choudhury G. G. (2006) Concerted action of Smad and CREB-binding protein regulates bone morphogenetic protein-2-stimulated osteoblastic colony-stimulating factor-1 expression. J. Biol. Chem. 281, 20160–20170 [DOI] [PubMed] [Google Scholar]
- 40. Mandal C. C., Drissi H., Choudhury G. G., Ghosh-Choudhury N. (2010) Integration of phosphatidylinositol 3-kinase, Akt kinase, and Smad signaling pathway in BMP-2-induced osterix expression. Calcif. Tissue Int. 87, 533–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen D., Ji X., Harris M. A., Feng J. Q., Karsenty G., Celeste A. J., Rosen V., Mundy G. R., Harris S. E. (1998) Differential roles for bone morphogenetic protein (BMP) receptor type IB and IA in differentiation and specification of mesenchymal precursor cells to osteoblast and adipocyte lineages. J. Cell Biol. 142, 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mundy G., Garrett R., Harris S., Chan J., Chen D., Rossini G., Boyce B., Zhao M., Gutierrez G. (1999) Stimulation of bone formation in vitro and in rodents by statins. Science 286, 1946–1949 [DOI] [PubMed] [Google Scholar]
- 43. Yoshida Y., Tanaka S., Umemori H., Minowa O., Usui M., Ikematsu N., Hosoda E., Imamura T., Kuno J., Yamashita T., Miyazono K., Noda M., Noda T., Yamamoto T. (2000) Negative regulation of BMP/Smad signaling by Tob in osteoblasts. Cell 103, 1085–1097 [DOI] [PubMed] [Google Scholar]
- 44. Ghosh-Choudhury N., Choudhury G. G., Harris M. A., Wozney J., Mundy G. R., Abboud S. L., Harris S. E. (2001) Autoregulation of mouse BMP-2 gene transcription is directed by the proximal promoter element. Biochem. Biophys. Res. Commun. 286, 101–108 [DOI] [PubMed] [Google Scholar]
- 45. Mandal C. C., Ganapathy S., Gorin Y., Mahadev K., Block K., Abboud H. E., Harris S. E., Ghosh-Choudhury G., Ghosh-Choudhury N. (2011) Reactive oxygen species derived from Nox4 mediate BMP2 gene transcription and osteoblast differentiation. Biochem. J. 433, 393–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ghosh-Choudhury N., Mandal C. C., Choudhury G. G. (2007) Statin-induced Ras activation integrates the phosphatidylinositol 3-kinase signal to Akt and MAPK for bone morphogenetic protein-2 expression in osteoblast differentiation. J. Biol. Chem. 282, 4983–4993 [DOI] [PubMed] [Google Scholar]
- 47. Ghosh-Choudhury N., Harris M. A., Wozney J., Mundy G. R., Harris S. E. (1997) Clonal osteoblastic cell lines from p53 null mouse calvariae are immortalized and dependent on bone morphogenetic protein 2 for mature osteoblastic phenotype. Biochem. Biophys. Res. Commun. 231, 196–202 [DOI] [PubMed] [Google Scholar]
- 48. Mandal C. C., Ghosh-Choudhury N., Yoneda T., Choudhury G. G. (2011) Simvastatin prevents skeletal metastasis of breast cancer by an antagonistic interplay between p53 and CD44. J. Biol. Chem. 286, 11314–11327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ghosh-Choudhury N., Abboud S. L., Mahimainathan L., Chandrasekar B., Choudhury G. G. (2003) Phosphatidylinositol 3-kinase regulates bone morphogenetic protein-2 (BMP-2)-induced myocyte enhancer factor 2A-dependent transcription of BMP-2 gene in cardiomyocyte precursor cells. J. Biol. Chem. 278, 21998–22005 [DOI] [PubMed] [Google Scholar]
- 50. Choudhury G. G., Ghosh-Choudhury N., Abboud H. E. (1998) Association and direct activation of signal transducer and activator of transcription 1α by platelet-derived growth factor receptor. J. Clin. Invest. 101, 2751–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Antoku S., Saksela K., Rivera G. M., Mayer B. J. (2008) A crucial role in cell spreading for the interaction of Abl PXXP motifs with Crk and Nck adaptors. J. Cell Sci. 121, 3071–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brasher B. B., Van Etten R. A. (2000) c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J. Biol. Chem. 275, 35631–35637 [DOI] [PubMed] [Google Scholar]
- 53. Tanis K. Q., Veach D., Duewel H. S., Bornmann W. G., Koleske A. J. (2003) Two distinct phosphorylation pathways have additive effects on Abl family kinase activation. Mol. Cell. Biol. 23, 3884–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wozney J. M. (1992) The bone morphogenetic protein family and osteogenesis. Mol. Reprod. Dev. 32, 160–167 [DOI] [PubMed] [Google Scholar]
- 55. Ghosh-Choudhury N., Harris M. A., Feng J. Q., Mundy G. R., Harris S. E. (1994) Expression of the BMP 2 gene during bone cell differentiation. Crit. Rev. Eukaryot. Gene Expr. 4, 345–355 [DOI] [PubMed] [Google Scholar]
- 56. Harris S. E., Bonewald L. F., Harris M. A., Sabatini M., Dallas S., Feng J. Q., Ghosh-Choudhury N., Wozney J., Mundy G. R. (1994) Effects of transforming growth factor β on bone nodule formation and expression of bone morphogenetic protein 2, osteocalcin, osteopontin, alkaline phosphatase, and type I collagen mRNA in long-term cultures of fetal rat calvarial osteoblasts. J. Bone Miner. Res. 9, 855–863 [DOI] [PubMed] [Google Scholar]
- 57. Nakashima K., Zhou X., Kunkel G., Zhang Z., Deng J. M., Behringer R. R., de Crombrugghe B. (2002) The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17–29 [DOI] [PubMed] [Google Scholar]
- 58. Kaplan M. S., Huguet N., Newsom J. T., McFarland B. H., Lindsay J. (2003) Prevalence and correlates of overweight and obesity among older adults: findings from the Canadian National Population Health Survey. J. Gerontol. A Biol. Sci. Med. Sci. 58, 1018–1030 [DOI] [PubMed] [Google Scholar]
- 59. Manolagas S. C. (2000) Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 21, 115–137 [DOI] [PubMed] [Google Scholar]
- 60. Nguyen D. X., Bos P. D., Massagué J. (2009) Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer 9, 274–284 [DOI] [PubMed] [Google Scholar]
- 61. Feng X., McDonald J. M. (2011) Disorders of bone remodeling. Annu. Rev. Pathol. 6, 121–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li B., Boast S., de los Santos K., Schieren I., Quiroz M., Teitelbaum S. L., Tondravi M. M., Goff S. P. (2000) Mice deficient in Abl are osteoporotic and have defects in osteoblast maturation. Nat. Genet. 24, 304–308 [DOI] [PubMed] [Google Scholar]
- 63. Stein G. S., Lian J. B., Owen T. A. (1990) Relationship of cell growth to the regulation of tissue-specific gene expression during osteoblast differentiation. FASEB J. 4, 3111–3123 [DOI] [PubMed] [Google Scholar]
- 64. Wymann M. P., Marone R. (2005) Phosphoinositide 3-kinase in disease: timing, location, and scaffolding. Curr. Opin. Cell Biol. 17, 141–149 [DOI] [PubMed] [Google Scholar]
- 65. Zhao J. J., Roberts T. M. (2006) PI3 kinases in cancer: from oncogene artifact to leading cancer target. Sci. STKE 2006, pe52. [DOI] [PubMed] [Google Scholar]
- 66. Varticovski L., Daley G. Q., Jackson P., Baltimore D., Cantley L. C. (1991) Activation of phosphatidylinositol 3-kinase in cells expressing abl oncogene variants. Mol. Cell. Biol. 11, 1107–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hantschel O., Nagar B., Guettler S., Kretzschmar J., Dorey K., Kuriyan J., Superti-Furga G. (2003) A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 112, 845–857 [DOI] [PubMed] [Google Scholar]
- 68. Plattner R., Koleske A. J., Kazlauskas A., Pendergast A. M. (2004) Bidirectional signaling links the Abelson kinases to the platelet-derived growth factor receptor. Mol. Cell. Biol. 24, 2573–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tokonzaba E., Capelluto D. G., Kutateladze T. G., Overduin M. (2006) Phosphoinositide, phosphopeptide, and pyridone interactions of the Abl SH2 domain. Chem. Biol. Drug Des. 67, 230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Whitman M., Downes C. P., Keeler M., Keller T., Cantley L. (1988) Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 332, 644–646 [DOI] [PubMed] [Google Scholar]
- 71. Carpenter C. L., Cantley L. C. (1990) Phosphoinositide kinases. Biochemistry 29, 11147–11156 [DOI] [PubMed] [Google Scholar]
- 72. Sattler M., Salgia R., Okuda K., Uemura N., Durstin M. A., Pisick E., Xu G., Li J. L., Prasad K. V., Griffin J. D. (1996) The proto-oncogene product p120CBL and the adaptor proteins CRKL and c-CRK link c-ABL, p190BCR/ABL and p210BCR/ABL to the phosphatidylinositol-3′ kinase pathway. Oncogene 12, 839–846 [PubMed] [Google Scholar]
- 73. Jain S. K., Susa M., Keeler M. L., Carlesso N., Druker B., Varticovski L. (1996) PI 3-kinase activation in BCR/abl-transformed hematopoietic cells does not require interaction of p85 SH2 domains with p210 BCR/abl. Blood 88, 1542–1550 [PubMed] [Google Scholar]
- 74. Ren S. Y., Xue F., Feng J., Skorski T. (2005) Intrinsic regulation of the interactions between the SH3 domain of p85 subunit of phosphatidylinositol-3 kinase and the protein network of BCR/ABL oncogenic tyrosine kinase. Exp. Hematol. 33, 1222–1228 [DOI] [PubMed] [Google Scholar]
- 75. Arnold S. J., Maretto S., Islam A., Bikoff E. K., Robertson E. J. (2006) Dose-dependent Smad1, Smad5, and Smad8 signaling in the early mouse embryo. Dev. Biol. 296, 104–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lechleider R. J., Ryan J. L., Garrett L., Eng C., Deng C., Wynshaw-Boris A., Roberts A. B. (2001) Targeted mutagenesis of Smad1 reveals an essential role in chorioallantoic fusion. Dev. Biol. 240, 157–167 [DOI] [PubMed] [Google Scholar]
- 77. Tsukazaki T., Chiang T. A., Davison A. F., Attisano L., Wrana J. L. (1998) SARA, a FYVE domain protein that recruits Smad2 to the TGFβ receptor. Cell 95, 779–791 [DOI] [PubMed] [Google Scholar]
- 78. Berman E., Nicolaides M., Maki R. G., Fleisher M., Chanel S., Scheu K., Wilson B. A., Heller G., Sauter N. P. (2006) Altered bone and mineral metabolism in patients receiving imatinib mesylate. N. Engl. J. Med. 354, 2006–2013 [DOI] [PubMed] [Google Scholar]