

Background: Primate-specific melanoma antigen-A11 (MAGE-A11) increases steroid receptor transcriptional activity and enhances prostate cancer cell growth.
Results: MAGE-A11 activates E2F1 by interacting with retinoblastoma-related protein p107.
Conclusion: MAGE-A11 influences cell cycle regulatory pathways in a molecular hub for transcription.
Significance: MAGE-A11 is a proto-oncogene whose increased expression impacts multiple signaling mechanisms that contribute to prostate cancer growth and progression.
Keywords: Androgen Receptor, E2F Transcription Factor, Gene Transcription, Prostate Cancer, Retinoblastoma (Rb), MAGE-A11, Cell Cycle Regulation, Melanoma Antigen-A11, p107, Steroid Receptor Coregulator
Abstract
Melanoma antigen-A11 (MAGE-A11) is a low-abundance, primate-specific steroid receptor coregulator in normal tissues of the human reproductive tract that is expressed at higher levels in prostate cancer. Increased expression of MAGE-A11 enhances androgen receptor transcriptional activity and promotes prostate cancer cell growth. Further investigation into the mechanisms of MAGE-A11 function in prostate cancer demonstrated interactions with the retinoblastoma-related protein p107 and Rb tumor suppressor but no interaction with p130 of the Rb family. MAGE-A11 interaction with p107 was associated with transcriptional repression in cells with low MAGE-A11 and transcriptional activation in cells with higher MAGE-A11. Selective interaction of MAGE-A11 with retinoblastoma family members suggested the regulation of E2F transcription factors. MAGE-A11 stabilized p107 by inhibition of ubiquitination and linked p107 to hypophosphorylated E2F1 in association with the stabilization and activation of E2F1. The androgen receptor and MAGE-A11 modulated endogenous expression of the E2F1-regulated cyclin-dependent kinase inhibitor p27Kip1. The ability of MAGE-A11 to increase E2F1 transcriptional activity was similar to the activity of adenovirus early oncoprotein E1A and depended on MAGE-A11 interactions with p107 and p300. The immunoreactivity of p107 and MAGE-A11 was greater in advanced prostate cancer than in benign prostate, and knockdown with small inhibitory RNA showed that p107 is a transcriptional activator in prostate cancer cells. These results suggest that MAGE-A11 is a proto-oncogene whose increased expression in prostate cancer reverses retinoblastoma-related protein p107 from a transcriptional repressor to a transcriptional activator of the androgen receptor and E2F1.
Introduction
Melanoma antigen-A11 (MAGE-A11)4 is a primate-specific steroid hormone receptor coregulator that increases transcriptional activity of the human androgen receptor (AR) (1) and isoform B of the human progesterone receptor (2). The effects of MAGE-A11 are mediated by interactions with p160 coactivators and p300 acetyltransferase (3, 4). MAGE-A11 interacts with the NH2-terminal FXXLF motif region of human AR that overlaps with the androgen-dependent NH2- and carboxyl-terminal interaction important for AR transcriptional activity (1, 5–8). MAGE-A11 also interacts with an extended LXXLL motif region in the NH2-terminal region of human progesterone receptor B, which is absent in the otherwise identical shorter progesterone receptor A (2). Lower mammals, such as mice, do not have the MAGE-A11 gene. Sequence differences in the FXXLF motif region of the mouse AR inhibit an interaction with MAGE-A11 (9). These findings suggest that the evolution of MAGE-A11 in primates provides greater regulatory control of steroid receptor transcriptional activity. It was shown recently that MAGE-A11 enhances human AR transcriptional activity by bridging AR dimers in a mechanism that accounts for the dual functions of the AR FXXLF motif in the androgen-dependent AR NH2- and carboxyl-terminal interaction and binding to MAGE-A11 (10).
MAGE-A11 is a member of a family of cancer-testis antigen genes that are frequently overexpressed in cancer (11). MAGE-A11 is also expressed at low levels in normal tissues of the human male and female reproductive tracts. It was first identified as an AR-interacting protein in human testis and is present at low levels in human foreskin fibroblasts (1). MAGE-A11 expression is regulated hormonally in human endometrium during the menstrual cycle and up-regulated by cyclic AMP (12). MAGE-A11 expression is cell cycle-dependent (4), and its coregulator activity depends on Chk1, a cell cycle-dependent kinase that phosphorylates a threonine residue in the relatively conserved carboxyl-terminal MAGE homology domain that characterizes this gene family (13). MAGE-A11 mRNA can increase exponentially during prostate cancer progression to castration-recurrent growth (10, 11, 14). Inhibition of MAGE-A11 expression arrests the growth of androgen-stimulated prostate cancer cells (10).
The family of retinoblastoma proteins includes the retinoblastoma (Rb) tumor suppressor, p107 (also known as Rb-like protein 1 (pRb1)), and p130 (pRb2). Rb-like proteins suppress cell growth by restricting progression through the G1/S transition of the cell cycle by interacting through their so-called pocket regions to negatively regulate E2F transcription factors (15–17). Rb-related proteins are regulated by phosphorylation (18), and hypophosphorylated retinoblastoma proteins bind E2Fs to inhibit transcription. Phosphorylation by cyclin-dependent kinases in normally cycling cells releases bound E2Fs in a cell cycle-dependent manner (19). At least eight E2F transcription factors expressed in mammalian cells have been grouped as transcriptional activators or repressors (20).
The tumor suppressor function of Rb is often lost in late-stage cancer because of mutations in the pocket region that interfere with suppression of E2F transcriptional activity (21). In contrast, mutations in p107 have not been reported in cancer (21, 22), although p107 is important for cell cycle regulation (23, 24). Loss of Rb-related protein activity is also achieved by cancer cells through the action of viral oncogenes that target the pocket region (25, 26). One of these viral proteins, human adenovirus type 5 early region 1A (E1A), is important in cell transformation. E1A disrupts Rb-related protein complexes through competitive binding and release of transcriptionally active E2Fs that regulate genes that control the cell cycle (27–29). E1A displaces E2F transcription factors from all three Rb-related proteins and induces entry into S phase of the cell cycle.
In this report, we investigated mechanisms by which MAGE-A11 contributes to prostate cancer cell growth. We show that MAGE-A11 selectively regulates retinoblastoma family members through mechanisms similar to the adenoviral oncoprotein E1A. MAGE-A11 interacts with p107 and increases E2F1 transcriptional activity. Stabilization of p107 by MAGE-A11 correlated with increased p107 immunostaining in prostate cancer and acquisition of p107 transcriptional activator activity.
EXPERIMENTAL PROCEDURES
DNA Vectors
Human AR expression vectors included pCMV-hAR coding for 919-amino acid, full-length AR (30); pCMV-FLAG-AR (1); and pCMV-AR-(1–660) with AR NH2-terminal and DNA binding domains (31). Human MAGE-A11 expression vectors included pSG5-MAGE coding for 429-amino acid, full-length human MAGE-A11; pCMV-FLAG-MAGE; pCMV-FLAG-MAGE-(112–429) (1); and MAGE-A11 mutants in pSG5-MAGE and pSG5-HA-MAGE-(112–429), pSG5-HA-MAGE-(112–307), and pSG5-HA-MAGE-(112–298) with the human influenza HA tag (3, 4, 13). pSG5-HA-MAGE was created by PCR-amplifying pSG5-MAGE and inserting the fragment with EcoRI and SalI ends into the EcoRI and XhoI sites of pSG5-HA. Other expression vectors included pSG5-HA-p300 (4), pCMV-Rb (provided by Yue Xiong) (32), pcDNA3-p130 (33), pCMV-FLAG-ubiquitin (13), and CMX-E1A variant C (provided by Hong-Wu Chen) (34). CMV-neo-p107 (CMV-p107) expresses full-length human p107, and CMV-p107DE (CMV-p107Δ409–826) has a deletion in the pocket region (35). CMV-p107-(1–385) and CMV-p107-(385–1068) were provided by Joan Massagué (36). CMV-p107-(1–180) was constructed by cloning an EcoRI and BamHI fragment PCR-amplified from CMV-HA-p107-(1–385) into the same sites of pCMV-HA. All PCR-amplified regions were verified using DNA sequencing.
Luciferase reporter vectors included the prostate-specific antigen (PSA) enhancer luciferase PSA-Enh-Luc (4, 37) and E2F1-Luc, which contains the −728 nucleotide E2F1 promoter region in pGL2. E2F1-Luc(-E2F) is the same as E2F1-Luc, except for inactivating mutations in three E2F1 binding sites. E2F4-Luc contains a 3-kb E2F4 promoter fragment in pGL2 basic. E2F reporter vectors were provided by Joseph R. Nevins (38).
Expression Studies
Cells were grown in medium containing penicillin, streptomycin, and 2 mm l-glutamine (Invitrogen). Human cervical carcinoma HeLa and HeLa-AR3A-PSA-ARE4 cells that stably express human AR (39) were propagated in minimal essential medium containing 10% fetal bovine serum. Monkey kidney CV1 and COS1 cells were maintained in Dulbecco's modified essential medium with 10% bovine calf serum and 20 mm Hepes (pH 7.2). LAPC-4 human prostate cancer cells were grown in RPMI 1640 medium with 10% fetal bovine serum, 1 mm sodium pyruvate, and 1 nm methyltrienolone R1881, a synthetic androgen. HEK293 cells were maintained in Eagle's minimum essential medium with 10% fetal bovine serum.
HeLa (5 × 104 cells/well) and CV1 cells (104 cells/well) in 12-well plates were transfected with expression and luciferase reporter DNA using X-tremeGENE 9 or FuGENE 6 (Roche Applied Science) (10). After 24 h, cells were transferred to serum-free, phenol red-free medium with or without 1 nm dihydrotestosterone (DHT). siRNAs for E2F1 and p107 (Dharmacon RNA Technologies) were expressed in LAPC-4 (3.8 × 105 cells/well) or COS1 cells (4.5 × 105 cells/well) in 6-well plates in 1 ml of medium without antibiotics using Lipofectamine 2000 (Invitrogen) (3, 4). Cells in 6- and 12-well plates were harvested in 0.25 ml of 1% Triton X-100, 2 mm EDTA, and 25 mm Tris phosphate (pH 7.8). Luciferase activity (mean ± S.D.) was measured in 0.1-ml aliquots using an automated Lumistar Galaxy luminometer. Data are representative of at least three independent experiments.
Immunoblots of extracts from COS1 cells (2 × 106 cells/10-cm dish, 7 × 105 cells/6-cm dish) transfected using DEAE-dextran were prepared in immunoblot lysis buffer containing 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate, 0.15 m NaCl, 2 mm EDTA, 50 mm NaF, 2 mm sodium vanadate, 50 mm Tris-HCl (pH 7.5), 1 mm phenylmethylsulfonyl fluoride, 1 mm dithiothreitol, and protease inhibitors (Roche Applied Science). Immunoprecipitation of endogenous and expressed proteins was performed by transfecting pCMV-FLAG vectors in 10-cm dishes containing 2 × 106 COS1 or 1.5 × 107 HEK293 cells using DEAE-dextran (40, 41) or 7 × 106 LAPC-4 cells using X-tremeGENE 9. After 24 h in serum-free medium, cells were harvested in phosphate-buffered saline. Cell pellets were extracted in immunoprecipitation lysis buffer containing 1% Triton X-100, 0.15 m NaCl, 50 mm NaF, 2 mm sodium vanadate, 2 mm EDTA, 50 mm Tris (pH 7.6), 1 mm phenylmethylsulfonyl fluoride, 1 mm dithiothreitol, and complete protease inhibitors (Roche Applied Science) with or without 0.5% deoxycholate or 10% glycerol. Samples containing deoxycholate were diluted 4-fold with lysis buffer without deoxycholate. Samples were precleared for 15 min at 4 °C using Sepharose CL-4B (Sigma) and immunoprecipitated for 2 h at 4 °C using anti-FLAG M2-agarose affinity resin (9). For phosphorylation experiments, 30 μg of cell extract protein in 40 μl of immunoprecipitation buffer without NaF, sodium vanadate, EDTA, or deoxycholate was incubated for 1 h at 4 °C with or without 4000 IU λ protein phosphatase (New England Biolabs), according to the instructions of the manufacturer (3).
Endogenous expression of AR and MAGE-A11 was inhibited in LAPC-4 cells using lentivirus shRNA prepared using the Open Biosystems TRC1 shRNA library. HEK293 cell medium (0.15–0.3 ml) containing ∼106 lentivirus particles/ml was added to LAPC-4 cells (107/10-cm dish) plated the day before in 6 ml of growth medium. Cells were incubated for 48 h at 37 °C with lentivirus for MAGE-A11 shRNA-947, 169, and 827; AR shRNA-5; a nonspecific 18-bp spacer; and empty lentivirus controls. Cells were harvested using trypsin-EDTA and plated in 10-cm dishes in medium containing 3 μg/ml puromycin dihydrochloride (Cellgro) for cell selection after lentivirus transduction. After 4 days in culture, cells were incubated with 10 nm DHT for 24 h and extracted using immunoblot lysis buffer.
Acrylamide gels (8 or 10%) containing SDS were calibrated using dual color Precision Plus protein standards (Bio-Rad). Immunoblots were probed using the following antibodies from Santa Cruz Biotechnology, Inc. at 1:200 dilution unless indicated otherwise: p107 C-18 (sc-318) affinity-purified rabbit polyclonal antibody to a carboxyl-terminal human p107 peptide, E2F1 KH95 (sc-251) mouse monoclonal antibody to human E2F1 amino acid residues 342–386, E2F1 C-20 (sc-193) rabbit polyclonal antibody to a carboxyl-terminal epitope, E2F4 C-108 (sc-512) rabbit polyclonal antibody to human E2F4 amino acid residues 108–300, p130 C-20 (sc-317) affinity-purified rabbit polyclonal antibody to a human p130 carboxyl-terminal peptide, and DP-1 K-20 (sc-610) rabbit polyclonal antibody. Additional antibodies included MAGE-A11 rabbit polyclonal antibody against FLAG-tagged human MAGE-A11 expressed in baculovirus (0.2 μg/ml for expressed MAGE-A11 and 10 μg/ml for endogenous MAGE-A11), AR32 rabbit polyclonal antibody to human AR 9–28 amino acid peptide (1 μg/ml) (42), AR52 rabbit polyclonal antibody to human AR 544–558 amino acid peptide (10 μg/ml) (30), anti-FLAG M2 F3165 monoclonal antibody (Sigma, 1:2000 dilution), Rb Ab-1 clone 1F8 mouse monoclonal antibody to human Rb amino acid residue epitope 703–722 (Thermo Scientific, 1:500 dilution), p27Kip1 (p27) purified mouse antibody (BD Transduction Laboratories, 1:50 dilution), and rabbit polyclonal HA antibody ab9110 (Abcam, 1:1000 dilution). In some experiments, blots were stripped by incubating for 23 min using a hybridization rotator at 55 °C in 10 ml of prewarmed 2% SDS, 62.5 mm Tris-HCl (pH 6.7) containing 64 μl of β-mercaptoethanol and reprobing with antibody.
Quantitative Real-time RT-PCR
LAPC-4 cells (2.4 × 106 cells/6-cm dish) plated in growth medium containing 10% fetal calf serum were grown for at least 48 h to ∼50% confluence and transferred to serum-free medium. The next day, cells were placed in medium containing 5% charcoal-stripped serum and incubated with or without DHT. Cells were harvested in 1 ml TRIzol reagent (Invitrogen). RNA was isolated using chloroform extraction and precipitated using isopropanol. First-strand cDNA was prepared using SuperScript II reverse transcriptase (Invitrogen) and oligo(dT) primer. Real-time PCR was performed using an Applied Biosystems StepOnePlus real-time PCR system and p27 forward primer 5′-GTTAACCCGGGACTTGGA-3′ and p27 reverse primer 5′-CACCTCTTGCCACTCGTA-3′ and peptidylprolyl isomerase A housekeeping control forward primer 5′-ATCTTGTCCATGGCAAATGC-3′ and reverse primer 5′-GCCTCCACAATATTCATGCC-3′ (Integrated DNA Technologies). Reactions (20 μl) contained 4 μl of cDNA (40 ng), 10 μl of Sso Advanced SYBR Green Supermix (Bio-Rad), 0.4 μl of ROX reference dye (Invitrogen), 2 μl of 2 μm amplification primers, and 1.6 μl of RNase-free H2O. Real-time PCR amplification was 1 cycle at 95 °C for 20 min followed by 45 cycles at 95 °C for 30 s, 57.5 °C for 30 s, 72 °C for 40 s, and 79 °C for 20 s. p27 and peptidylprolyl isomerase A standard curves were performed on the basis of 10-fold dilutions of cDNA.
Cell Growth Assays
LAPC-4 cells (4 × 105 cells/well) were plated in triplicate in 24-well plates in 0.5 ml of medium containing 10% charcoal-stripped serum without phenol red. The next day and 3 days later, 100 μl of serum-free medium with or without DHT were added. Cells were harvested daily, beginning 24 h after the first addition of DHT, by aspirating the medium and adding 0.2 ml of serum-free medium and 20 μl of cell counting kit 8 reagent (Dojindo Laboratories). After 2.5 h at 37 °C, 0.1 ml was analyzed spectrophotometrically at 485 nm.
Immunostaining
Benign prostate and prostate cancer tissues were obtained from prostatectomy specimens after informed consent and approval by institutional review boards at the University of North Carolina at Chapel Hill. Adjacent formalin-fixed, paraffin-embedded, 8-μm sections of androgen-stimulated benign prostate and androgen-stimulated and castration-recurrent prostate cancer were immunostained for endogenous p107 and MAGE-A11 using affinity-purified human p107 rabbit antibody C-18 (sc-318, 1:100 dilution, Santa Cruz Biotechnology) and affinity-purified human MAGE-A11 rabbit polyclonal anti-peptide MAGE-(94–108) antibody (6 μg/ml). Sections were pretreated with 4% H2O2 in 83% methanol for 30 min at room temperature, blocked using 2% goat serum, incubated overnight with primary antibody at 4 °C, blocked, and incubated for 30 min with biotinylated rabbit secondary antibody at room temperature. This was followed by 30 min with avidin DH-biotinylated horseradish peroxidase H complex using the Vectastain Elite ABC kit (Vector Laboratories). Slides were incubated for 10 min using the Vector Laboratories peroxidase 3,3′-diaminobenzidine substrate kit and counterstained using 0.05% toluidine blue in 30% ethanol (14). Images were captured using a SPOT Insight-4 camera (Diagnostic Instruments) and a Nikon Eclipse E600 microscope.
RESULTS
Regulation of Human AR Transcriptional Activity by MAGE-A11 and p107
MAGE-A11 is expressed at very low levels in normal tissues of the human male reproductive tract and at higher levels in prostate cancer, where it increases AR transcriptional activity (4, 10, 11, 14). MAGE-A11 influences AR levels in the absence and presence of androgen (1). To explore the mechanisms by which MAGE-A11 increases AR signaling in prostate cancer, we investigated the effects on the retinoblastoma family. Initial studies were performed in CV1 cells that do not express MAGE-A11, HeLa cells with lower MAGE-A11 expression, and LAPC-4 prostate cancer cells with higher MAGE-A11 expression (14).
MAGE-A11 and p300 increased androgen-dependent AR transactivation of the prostate-specific antigen enhancer in CV1 cells (Fig. 1A), as reported previously (4, 9). p107 inhibited the increase in AR activity by MAGE-A11 and p300 but did not inhibit AR activity without expressing MAGE-A11 and p300. AR activity was not inhibited by p130, another member of the Rb family, with or without MAGE-A11 and p300 (Fig. 1A). In HeLa cells with low MAGE-A11 expression relative to LAPC-4 cells (Fig. 1B), low levels of p107 slightly stimulated AR activity in the presence of MAGE-A11, but higher levels of p107 were inhibitory with or without MAGE-A11 (C). In contrast, the highest amounts of p107 that inhibited the MAGE-A11-dependent increase in AR activity in CV1 or HeLa cells did not inhibit AR in LAPC-4 cells (Fig. 1F, right).
FIGURE 1.

Inhibition of MAGE-A11-dependent AR transcriptional activity by p107. A, pCMV5 (p5) or pCMV-AR (25 ng) was expressed in CV1 cells with 0.25 μg of PSA-Enh-Luc and 25 ng of empty vector (−), CMV-p107, or pcDNA3-p130 with or without 50 ng of pSG5-MAGE (M) and 50 ng pSG5-HA-p300 (P). Cells were plated in medium containing 5% charcoal-stripped serum and incubated for 24 h the day after transfection with or without 1 nm DHT. B, LAPC-4 (LA, lane 1) and HeLa (HL, lane 2) cell extracts (80 μg of protein/lane) from cells cultured in serum-free medium for 24 h before harvest were prepared in immunoblot lysis buffer. Lane 3 contains 0.1 μg of protein extract from COS1 cells (C) expressing pSG5-MAGE. The immunoblot was probed using 10 μg/ml MAGE-A11 antibody. C, pCMV-AR (25 ng) was expressed in HeLa cells with 0.1 μg of PSA-Enh-Luc and 50 ng of pSG5 (−) or pSG5-MAGE with or without 50 ng of pSG5 (−) or 10, 25, or 50 ng of CMV-p107. Cells were incubated with or without 1 nm DHT. D, pCMV5 (25 ng) or 25 ng of pCMV-AR-(1–660) was expressed in HeLa cells with 0.1 μg of PSA-Enh-Luc with or without 50 ng of pSG5-MAGE and pCMV5 (−), CMV-p107 and pSG5 (−), or pSG5-MAGE and CMV-p107. E, pCMV5 or pCMV-AR-(1–660) (10 ng) was expressed in HeLa cells with 0.1 μg of PSA-Enh-Luc, 50 ng of pSG5 (−) or pSG5-MAGE, and 50 ng of p5 (−) or 10, 25, or 50 ng of CMV-p107. F, pCMV5 (5 ng) (−), 5 ng of pCMV-AR-(1–660), or 25 ng of pCMV-AR was expressed in LAPC-4 cells with 0.1 μg of PSA-Enh-Luc and 50 ng of pCMV5 (−) or 50 ng of CMV-p107. Cells were incubated with or without 1 nm DHT. In A and C–F, luciferase activity is the mean ± S.D. (error bars) and representative of three experiments.
The influence of MAGE-A11 and p107 on AR transcriptional activity was investigated further using a constitutively active AR-(1–660) NH2-terminal and DNA binding fragment that contains NH2-terminal activation function 1 and the FXXLF motif region that interacts with MAGE-A11 but lacks the AR ligand binding domain (1, 31). AR-(1–660) is similar to splice variants reported at low levels in prostate cancer (43, 44). Similar to full-length AR, p107 inhibited AR-(1–660) activity in a dose-dependent manner in HeLa cells with or without MAGE-A11 (Fig. 1, D and E). In contrast, p107 slightly increased AR-(1–660) activity in LAPC-4 cells (Fig. 1F, left).
The ability of p107, but not p130, to influence MAGE-A11 activation of the AR suggests that MAGE-A11 interacts selectively with Rb-related proteins. This possibility was investigated by comparing the immunoprecipitation of p107, Rb, and p130 with FLAG-MAGE or FLAG-MAGE-(112–429), a carboxyl-terminal fragment with the conserved MAGE homology domain (13). FLAG-MAGE was used because endogenous MAGE-A11 levels were low. p107 (Fig. 2A) and, to a lesser extent, Rb (B) immunoprecipitated with FLAG-MAGE and FLAG-MAGE-(112–429). In contrast, p130 did not associate with either form of FLAG-MAGE (Fig. 2C). The specificity of MAGE-A11 interaction with Rb-related proteins was consistent with the ability of p107, but not p130, to modulate the MAGE-A11-dependent increase in AR activity.
FIGURE 2.

MAGE-A11 interacts with p107 and Rb but not p130. pCMV-FLAG (−), pCMV-FLAG-MAGE, or FLAG-MAGE-(112–429) (5 μg) was expressed in COS1 cells with 5 μg of CMV-p107 (A), pCMV-hRb (B), or pcDNA3-p130 (C). Cells were incubated for 24 h in serum-free medium containing 0.1 μg/ml EGF and harvested in immunoprecipitation lysis buffer without deoxycholate or glycerol. Cell extracts (50 μg of protein/lane, right) and immunoprecipitates (IP, left) were probed using p107, Rb, p130, and MAGE-A11 antibodies. Arrows, p107, Rb, p130, FLAG-MAGE, and IgG.
The results suggest that MAGE-A11 interacts preferentially with p107, less with Rb, and does not interact with p130 of the Rb family. The specificity of MAGE-A11 interaction with Rb-like proteins supported the physiological relevance of MAGE-A11 interaction with p107 and modulation of transcriptional activity.
MAGE-A11 Mediates an Interaction between AR and p107
The modulation of AR transcriptional activity by MAGE-A11 and p107 suggests that MAGE-A11 might mediate an interaction between these proteins, which was investigated by immunoprecipitation of MAGE-A11 and p107 with FLAG-AR. MAGE-A11 associated with FLAG-AR with or without p107 (Fig. 3, lanes 10–17), which agreed with previous findings (1). However, an interaction between FLAG-AR and p107 was seen only when MAGE-A11 was expressed (Fig. 3, lanes 6–13). Treatment with EGF, a growth factor that increases phosphorylation in the MAGE homology domain and enhances MAGE-A11 interaction with the AR (13), promoted the MAGE-A11-dependent interaction between the AR and p107 (Fig. 3, lanes 10–13).
FIGURE 3.

MAGE-A11 mediates AR interaction with p107. pCMV-FLAG (4 μg) and 2 μg of CMV-p107 or 4 μg of pCMV-FLAG-AR with or without 2 μg of pCMV5 (p5), 2 μg of CMV-p107, and/or 2 μg of pSG5-MAGE was expressed in COS1 cells. Cells were incubated with or without 10 nm DHT and 0.1 μg/ml EGF for 24 h before harvest. Cell extracts (40 μg of protein/lane) and immunoprecipitates (IP, top) were probed using p107, MAGE-A11, and AR32 antibodies.
p107 has multiple interaction domains for regulatory proteins similar, but not identical, to other retinoblastoma family members (Fig. 4A). The domains of p107 that interact with MAGE-A11 were investigated using FLAG-MAGE and HA-tagged p107 fragments. p107 NH2-terminal fragments (1–180) and (1–385), carboxyl-terminal fragment (385–1068) containing the pocket region, and Δ409–826 with the pocket region deleted each associated with FLAG-MAGE (Fig. 4B, lanes 9–12) similar to full-length p107 (lane 8). FLAG-MAGE interacted with endogenous p107 in HEK293 cells (Fig. 4C) that have relatively high levels of p107 (see Fig. 12A). FLAG-MAGE also associated with endogenous E2F1 and E2F4 transcription factors important in cell cycle control (Fig. 4B, lanes 7–12).
FIGURE 4.

MAGE-A11 interacts with multiple regions of p107, endogenous p107, and endogenous E2Fs. A, schematic of human p107 protein interaction domains. B, pCMV-FLAG or pCMV-FLAG-MAGE (4 μg) was expressed in COS1 cells with 1 μg of full-length CMV-p107-(1–1068), CMV-HA-p107-(1–180), or CMV-HA-p107-(1–385) or 3 μg of CMV-HA-p107-(385–1068) or CMV-HA-p107Δ409–826. Cells were incubated for 24 h in serum-free medium containing 0.1 μg/ml EGF. Immunoprecipitates (IP, top panel) and cell extracts (40 μg of protein/lane, bottom panel) were probed using p107, HA, and MAGE-A11 antibodies. Endogenous E2F4 and E2F1 were detected using E2F4 (sc-512) and E2F1 (sc-251) antibodies. C, pCMV-FLAG or pCMV-FLAG-MAGE (3 μg) was expressed in HEK293 cells. Cells were incubated for 24 h before harvest in serum-free medium containing 0.1 μg/ml EGF and 1 μm MG132 proteasome inhibitor. Immunoprecipitates (top panel) and cell extracts (150 μg, center and bottom panels) were probed for endogenous p107 and FLAG-MAGE.
FIGURE 12.

Transcriptional activation by p107 in LAPC-4 cells. A, HeLa (106), HeLa-AR3A-PSA-ARE4 (2 × 106), CWR-R1 (1.5 × 107), LNCaP (1.2 × 107), LAPC-4 (9 × 106), PWR-1E (3 × 106), RWPE-2 (3 × 106), CV1 (1.5 × 106), COS1 (1.5 × 106), and HEK-293 cells (5 × 106) were plated in 10-cm dishes in serum-containing medium. The next day, the medium was changed to 5% charcoal-stripped serum without phenol red. Cells were treated for 48 h with or without 10 nm DHT. Cell extracts (50 μg of protein/lane) were analyzed by probing the transblot using p107, AR32, AR52, E2F1, DP-1, and MAGE-A11 antibodies. B, pCMV5 (p5, 1 μg) or 1 μg of CMV-p107 was expressed in COS1 cells using Lipofectamine with or without 5 nm nonspecific (NS) siRNAs or p107 siRNA-5, 6, 8, or 9. The transblot of cell extracts (25 μg of protein/lane) was probed using p107 antibody. C, pCMV5 (25 ng) (−) or 25 ng of CMV-E2F1 was expressed in LAPC-4 cells with 0.1 μg of E2F1-Luc and 10 nm nonspecific siRNA or p107 siRNA-5, 6, 8, or 9 using Lipofectamine 2000. D, E2F1-Luc (0.1 μg) was expressed in LAPC-4 cells with 10 nm nonspecific or p107 siRNA-5, 6, 8, or 9 using Lipofectamine 2000. In C and D, luciferase activity is the mean ± S.D. (error bars) representative of three experiments.
The results suggest that MAGE-A11 interacts with multiple regions of p107 to modulate AR transcriptional activity. The association of MAGE-A11 with endogenous p107 and endogenous E2Fs places it in important cell growth regulatory pathways.
MAGE-A11 Stabilizes p107 and Modulates Ubiquitination
The effect of MAGE-A11 on p107 was investigated in stability studies. MAGE-A11 increased p107 levels in cells treated with or without EGF (Fig. 5A) but did not increase p130 (B). A second, more slowly migrating form of p107 was evident with the coexpression of MAGE-A11 (Fig. 5, A and B). Both full-length MAGE-A11 and the (112–429) carboxyl-terminal fragment that interacted with p107 (Fig. 2) associated with the fast- and slow-migrating forms of p107. However, MAGE-A11 fragments 112–307 and 112–298 had no effect on p107 levels (Fig. 5C).
FIGURE 5.

MAGE-A11 interacts with endogenous p107, stabilizes p107, and modulates p107 ubiquitination. A, pCMV5 or CMV-p107 (2 μg) was expressed in COS1 cells in 10-cm dishes with or without 2 μg of pSG5-MAGE. Cells were incubated for 24 h in serum-free medium with or without 0.1 μg/ml EGF. Cell extracts prepared in immunoblot lysis buffer (40 μg of protein/lane) were probed using p107 and MAGE-A11 antibodies. B, CMV-p107 (3 μg), 3 μg of pCMV5 (−), or 3 μg of pcDNA3-p130 was expressed in COS1 cells in 6-cm dishes with or without 1 μg of pSG5-MAGE. Cells were incubated for 24 h in serum-free medium with 0.1 μg/ml EGF. Cell extracts (40 μg of protein/lane) prepared in immunoblot lysis buffer were probed using p107, p130, and MAGE-A11 antibodies. C, pCMV5 (2 μg) alone (−) or 2 μg of CMV-p107 with or without 100 ng of WT pSG5-HA-MAGE-(2–429), 25 ng of pSG5-HA-MAGE-(112–429), 50 ng of pSG5-HA-MAGE-(112–307), or 100 ng of pSG5-HA-MAGE-(112–298) were expressed in COS1 cells in 10-cm dishes. Cells were incubated for 24 h in serum-free medium. Cell extracts (40 μg of protein/lane) prepared in immunoblot buffer were probed on transblots using p107 and HA antibodies. D, CMV-p107 (2 μg) and/or 2 μg of pSG5-MAGE were expressed in COS1 cells in 10-cm dishes. Cells were incubated for 24 h in serum-free medium. The next day, medium was exchanged, and cells were treated with 5 μm cycloheximide. Cells were harvested at 0, 8, 16, and 24 h in immunoblot lysis buffer. Cell extracts (50 μg of protein/lane) were probed using p107 and MAGE-A11 antibodies. E, pCMV-FLAG (6 μg) or 6 μg of pCMV-FLAG-ubiquitin (FLAG-Ub) was expressed in COS1 cells with or without 1 μg of pSG5-MAGE and/or 3 μg of CMV-p107. Cells were incubated for 24 h in serum-free medium containing 0.1 μg/ml EGF and immunoprecipitated using FLAG antibody affinity resin. Transblots of immunoprecipitates (IP, top panels) and cell extracts (40 μg of protein/lane, bottom panels) were probed using p107 and MAGE-A11 antibodies.
The apparent stabilization of p107 by MAGE-A11 was investigated further by inhibiting protein synthesis using cycloheximide. In these experiments, p107 was detected in a time-dependent manner only when MAGE-A11 was expressed (Fig. 5D, lanes 1–8). p107 migrated as two distinct bands whose intensity declined with time, similar to MAGE-A11. There was a time-dependent shift from the faster-migrating to the slower-migrating form of p107. Treatment of cell extracts with λ protein phosphatase did not alter the double-band migration of p107 (data not shown), which indicates that phosphorylation was not responsible. Immunoprecipitation of p107 from cells expressing FLAG-ubiquitin demonstrated that p107 undergoes ubiquitination in association with the stabilizing effects of MAGE-A11 (Fig. 5E). MAGE-A11 appeared to inhibit ubiquitination of p107.
These results suggest that MAGE-A11 stabilizes p107 by inhibiting ubiquitination. The specificity of MAGE-A11 stabilization of p107 provided further evidence that MAGE-A11 interacts preferentially with p107 of the Rb family.
MAGE-A11 Activates E2F Transcriptional Activity
The association of endogenous E2F1 and E2F4 with FLAG-MAGE (Fig. 4B) suggests that MAGE-A11 interacts with additional cell cycle regulatory proteins influenced by phosphorylation (45). Immunoprecipitation studies of endogenous E2F1 in COS1 and HeLa cells showed that FLAG-MAGE associated with a faster-migrating form of endogenous E2F1 (Fig. 6A, lanes 4 and 6). MAGE-A11 interaction with hypophosphorylated E2F1 was confirmed by the shift to the faster-migrating form after treatment with λ protein phosphatase (Fig. 6B). The results show that MAGE-A11 interacts with hypophosphorylated E2F1 and not with hyperphosphorylated E2F1.
FIGURE 6.
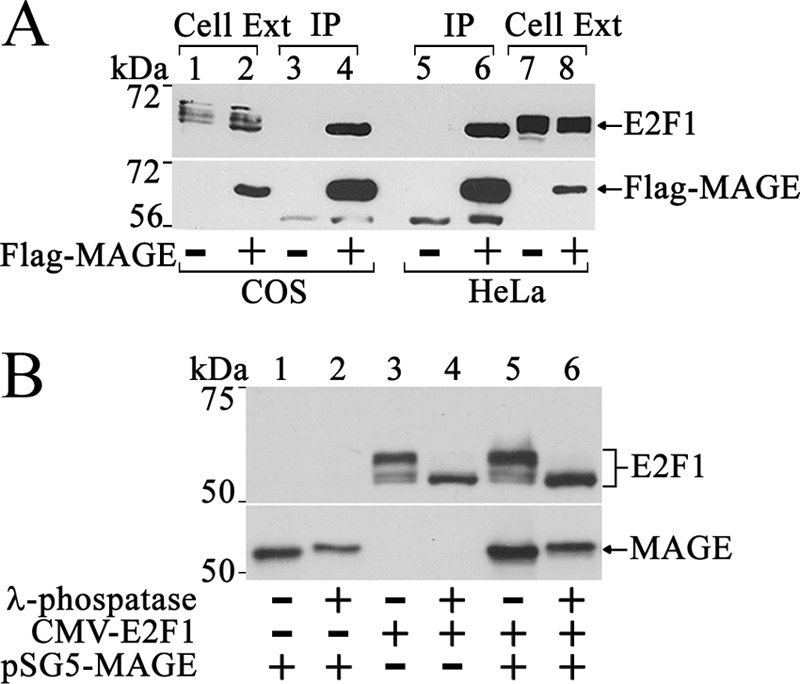
MAGE-A11 interacts with hypophosphorylated E2F1. A, pCMV-FLAG (3 μg) (−) or 3 μg of pCMV-FLAG-MAGE was expressed in COS1 cells (1.8 × 106 cells/10-cm dish, left) using DEAE-dextran and 2 μg of pCMV-FLAG (−) or pCMV-FLAG-MAGE in HeLa cells (7.5 × 105 cells/10-cm dish, right) using FuGENE 6 (Roche Applied Science). Cell extracts (40 μg of protein/lane) and immunoprecipitates (IP) were probed using MAGE-A11 antibody and E2F1 (sc-251) antibody for endogenous E2F1. B, pSG5-MAGE (2 μg) was expressed in COS1 cells with or without 2 μg of CMV-E2F1. Cells were incubated with 0.1 μg/ml EGF and 1 μm MG132 proteasome inhibitor for 24 h before harvest in immunoprecipitation lysis buffer without NaF, deoxycholate, sodium vanadate, or EDTA. Cell extracts (30 μg of protein/lane) were treated with or without λ protein phosphatase as described under “Experimental Procedures.” Transblots were probed using E2F1 (sc-193) and MAGE-A11 antibodies.
A MAGE-A11-dependent release of active E2Fs from p107 or Rb would be expected to increase E2F transcriptional activity. This possibility was tested by expressing MAGE-A11 with E2F1-Luc, an E2F1-responsive luciferase reporter gene that contains the −728 nucleotide E2F1 promoter region (38). Increased expression of MAGE-A11 caused a dose-dependent increase in E2F1-Luc transactivation (Fig. 7A). The MAGE-A11-dependent increase in E2F1-Luc transactivation was similar to that seen with E1A (Fig. 7B), an early adenoviral protein that interacts with Rb-related proteins and releases transcriptionally active E2Fs (29). Multiple inactivating mutations in the E2F1 response element region of E2F1-Luc in E2F1-Luc(-E2F) that disrupted activation by E2F1 (38) eliminated activation by MAGE-A11 or E1A (Fig. 7B, right).
FIGURE 7.

MAGE-A11 activates endogenous E2F1. A, pSG5 (100 ng) (−) or 2, 10, 25, or 100 ng of pSG5-MAGE was expressed in HeLa cells with 0.1 μg of E2F1-Luc. B, pSG5, pSG5-MAGE, or CMX-E1A (10, 25, 50, or 100 ng) was expressed in HeLa cells with 0.1 μg of E2F1-Luc or 0.1 μg of E2F1-Luc(-E2F) with inactivating mutations in E2F1 response elements. C, pSG5 (10 ng) (−) or 10 ng of pSG5-MAGE was expressed in HeLa cells with or without 100 ng of pSG5 (−) or pSG5-HA-p300 and 0.1 μg of E2F1-Luc (left) or 0.1 μg of E2F4-Luc (right). D, pSG5 (50 ng) (−) or 10 ng of pSG5-MAGE WT or I188A,F189A mutant was expressed in HeLa cells with 0.1 μg of E2F1-Luc with or without 50 ng of pSG5 (−) or pSG5-HA-p300. E, pSG5 (0.1 μg) (−) or 0.1 μg of pSG5-MAGE WT or 111-429, T360A, K240A,K245A, S174A, I188A,F189A, L358A,L359A, or Y368A,L369A mutant was expressed in HeLa cells with 0.1 μg of E2F1-Luc. Luciferase activity is the mean ± S.D. (error bars) representative of three experiments.
The specificity of MAGE-A11 activation of the two major classes of E2Fs was tested using E2F1-Luc and E2F4-Luc, where the latter contains a 3-kb E2F4 promoter region that is activated by E2F4 (38). Studies were performed with or without p300 on the basis of the synergistic actions of MAGE-A11 and p300 (4). MAGE-A11 increased transactivation of E2F1-Luc and E2F4-Luc to a similar extent, but the synergy between MAGE-A11 and p300 was greater for E2F1-Luc (Fig. 7C).
Several previously characterized MAGE-A11 mutations disrupt amino acid residues critical for steroid receptor coregulator activity (2–4, 13). Some of these site-specific mutants were tested for their effects on E2F1 transactivation. The synergistic effect of MAGE-A11 and p300 on E2F1-Luc transactivation was diminished with MAGE-I188A,F189A, in which the p300 interaction site was disrupted (Fig. 7D). This finding suggests that the synergy between MAGE-A11 and p300 in E2F transactivation depends on MAGE-A11 interaction with p300. The NH2-terminal deletion mutant MAGE-(111–429), Chk1 kinase phosphorylation site mutant T360A, monoubiquitination site mutant K240A,K245A, and hydrophobic F-box mutants L358A, L359A and V368A,L369A, which express at levels similar to the wild type (4, 10, 13), each inhibited MAGE-A11 activation of E2F1-Luc (Fig. 7E).
These results suggest that the interaction of MAGE-A11 with p107 releases transcriptionally active E2Fs, similar to adenovirus oncoprotein E1A. MAGE-A11 interaction with hypophosphorylated E2F1 is consistent with the release of active hyperphosphorylated E2F1. Amino acid residues important for steroid receptor coregulatory activity were required for MAGE-A11 to increase E2F transcriptional activity.
Transcriptional Regulation by MAGE-A11 and p107
The influence of MAGE-A11 on transcriptional activator or repressor activity of p107 was explored further by determining the effects of MAGE-A11 and p107 on E2F1-Luc transactivation. Synergy between MAGE-A11 and p300 and between E1A and p300 in E2F1-Luc transactivation was inhibited by p107 in HeLa cells (Fig. 8, A and B). p107Δ409–826 (DE), in which the pocket region was deleted, inhibited MAGE-A11 activation of E2F1-Luc less than full-length p107, and p130 did not inhibit E2F1-Luc transactivation by MAGE-A11 (Fig. 8C).
FIGURE 8.
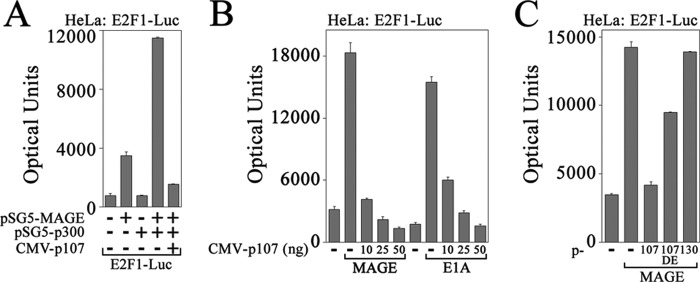
p107 inhibits MAGE-A11 activation of E2F1 in HeLa cells. A, pSG5 (25 ng) (−) or 25 ng of pSG5-MAGE was expressed in HeLa cells with 0.1 μg of E2F1-Luc with or without 50 ng of pSG5 (−) or pSG5-HA-p300 and 10 ng of pCMV5 (−) or CMV-p107. B, pSG5 (50 ng) alone (−), 25 ng of pSG5-MAGE (left), or 25 ng of CMX-E1A (right) was expressed in HeLa cells with 0.1 μg of E2F1-Luc with 50 ng of pCMV5 (−) or 10, 25, or 50 ng of CMV-p107. C, pCMV5 (100 ng) (−) or 100 ng of pSG5-MAGE was expressed in HeLa cells with 0.1 μg of E2F1-Luc and 10 ng of pCMV5 (−), CMV-p107, CMV-p107Δ409–826 (DE) or pcDNA3-p130. Luciferase activity is the mean ± S.D. (error bars) representative of three experiments.
In LAPC-4 cells, E2F1 or E1A activated E2F1-Luc to a greater extent than MAGE-A11 (Fig. 9A), possibly because of higher endogenous MAGE-A11 levels in LAPC-4 cells. When E2F1 expression was inhibited using siRNA (Fig. 9B), transactivation of E2F1-Luc by expressed or endogenous E2F1 was inhibited (C and D). However, increasing p107 did not inhibit, and slightly increased, E2F1-Luc transactivation in LAPC-4 cells (Fig. 9E).
FIGURE 9.

Activation of E2F1 in LAPC-4 cells. A, pCMV5 (p5, 50 ng) or 10, 25, 50, or 100 ng of CMV-E2F1, pSG5-MAGE, or CMX-E1A was expressed in LAPC-4 cells with 0.1 μg of E2F1-Luc. B, pCMV5 (1 μg) or 1 μg of CMV-E2F1 was expressed in COS1 cells in 6-well plates using Lipofectamine 2000 with or without 10 nm nonspecific siRNA (NS) or E2F1 siRNA-5, -6, -7, or -8. Cell extracts (10 μg of protein/lane) prepared in immunoblot lysis buffer were probed on the transblot using E2F1 (sc-251) antibody. C, pCMV5 (25 ng) (−) or 25 ng of CMV-E2F1 was expressed in LAPC-4 cells using Lipofectamine 2000 with 0.1 μg of E2F1-Luc and 1 nm nonspecific or E2F1 siRNA. D, pSG5 (150 ng) was expressed in LAPC-4 cells with 0.1 μg of E2F1-Luc and 1 nm E2F1 siRNA or two different nonspecific siRNAs. E, pCMV5 (50 ng) (−) alone or 50 ng of CMV-E2F1 was expressed in LAPC-4 cells with 0.1 μg of E2F1-Luc and 50 ng of pCMV5 (−) or 10, 25, or 50 ng of CMV-p107. In A and C–E, luciferase activity is the mean ± S.D. (error bars) representative of three experiments.
These results suggest that transcriptional repression by p107 in HeLa cells that have low levels of MAGE-A11 is lost in LAPC-4 cells with higher MAGE-A11. The cellular environment of LAPC-4 cells that includes higher MAGE-A11 contributes to the transcriptional activator activity of p107.
Regulation of an endogenous E2F1-dependent Gene
p27 is a cyclin-dependent kinase inhibitor and tumor suppressor that inhibits the G1-to-S phase transition of the cell cycle (46). p27 is transcriptionally up-regulated by E2F1 (47, 48), but its levels are often low in advanced prostate cancer (49, 50). In agreement with this finding, endogenous p27 protein was almost undetectable in LAPC-4 cells, but its levels increased slightly in a dose-dependent manner in response to DHT (Fig. 10A, left). The DHT-dependent increase in p27 protein was more evident in cells treated with MG132, a proteasome inhibitor (Fig. 10A, right), and there was a transient increase in p27 mRNA in response to DHT (B).
FIGURE 10.

AR and MAGE-A11 regulation of p27 expression. A, LAPC-4 cells grown for 48 h in 5% charcoal-stripped serum medium were incubated for 24 h in serum-free medium with increasing concentrations of DHT without (lanes 1–4) or with 1 μm MG132 added 24 and 1 h before harvest (lanes 5–8). Cells were extracted in immunoblot lysis buffer, and extracts (80 μg of protein/lane) were probed on the transblot using purified mouse p27 antibody. B, LAPC-4 cells placed in serum-free medium for 24 h were transferred to 10% charcoal-stripped serum medium with or without 10 nm DHT for increasing times. RNA was extracted and analyzed by quantitative real-time RT-PCR as described under “Experimental Procedures.” C, LAPC-4 cell growth was assayed with or without increasing DHT concentrations as described under “Experimental Procedures.” Data are the mean ± S.E. of triplicate measurements. D, immunoblot analysis of LAPC-4 cell extracts after transduction of cells without lentivirus (NV) or with lentivirus for nonspecific 18-bp control; MAGE-A11 shRNA-827, 947, and 169; nonspecific empty vector control; and AR5 shRNA. Transblots of cell extracts (40 or 80 μg of protein/lane) were probed using AR32, MAGE-A11, and p27 (BD Biosciences, 1:200) antibodies.
The apparent rapid degradation of p27 mRNA and/or protein in LAPC-4 cells was consistent with the faster LAPC-4 cell growth in the presence of 1 or 10 nm DHT (Fig. 10C) and the increase in p27 protein with AR and MAGE-A11 lentivirus shRNA knockdown (D). The increase in p27 with AR or MAGE-A11 knockdown may reflect, in part, the dependence of LACP-4 cell growth on AR and MAGE-A11 (10) because loss of AR signaling can arrest androgen-dependent prostate cancer cells in G0/G1 phase of the cell cycle (51). The results suggest that AR and MAGE-A11 are involved in the regulation of endogenous p27.
MAGE-A11 Links E2F1 and p107 for Transcriptional Activation
Controversy regarding p107 association with E2F1 in transcriptional activation or repression (52–54) led us to determine the effects of MAGE-A11 on E2F1 interaction with p107. E2F1 was stabilized by MAGE-A11 (Fig. 11A), similar to the effect of MAGE-A11 on p107 (Fig. 5A). E2F1 was not detected without expression of MAGE-A11 (Fig. 11A, lanes 4 and 5) but was readily detected in an EGF-dependent manner when MAGE-A11 was expressed (Fig. 11A, lanes 6 and 7).
FIGURE 11.
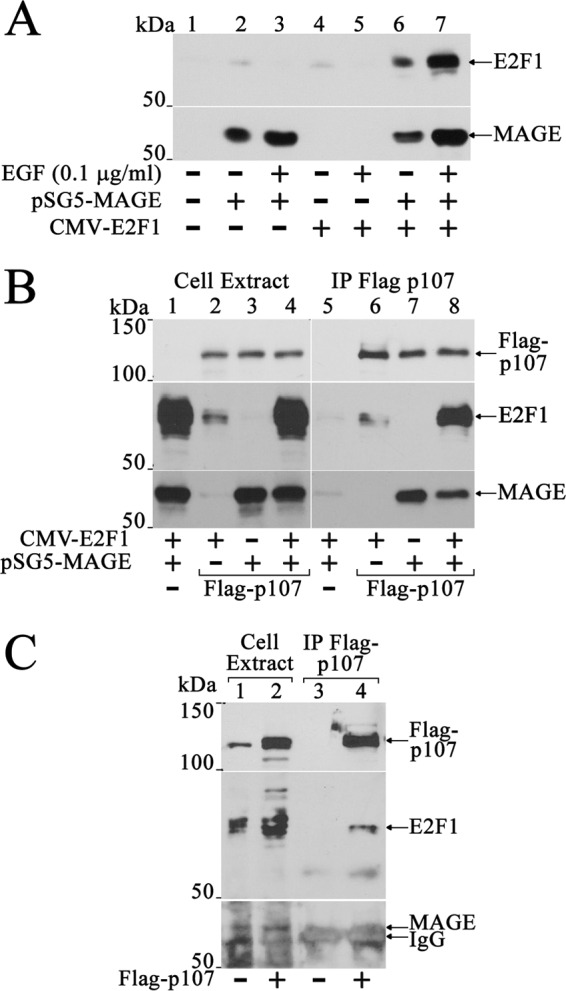
MAGE-A11 stabilizes E2F1 and mediates an interaction between E2F1 and p107. A, pCMV5 (1 μg) (lane 1), 1 μg of pSG5-MAGE (lanes 2 and 3), 0.5 μg of CMV-E2F1 (lanes 4 and 5), or 1 μg of pSG5-MAGE and 0.5 μg of CMV-E2F1 together (lanes 6 and 7) was expressed in COS1 cells. Cells were incubated for 24 h before harvest in serum-free medium with or without 0.1 μg/ml EGF. Cell extracts (40 μg of protein/lane) were probed on the transblot using E2F1 (sc-193) and MAGE-A11 antibodies. B, pCMV-FLAG (4 μg) (−) or 4 μg of pCMV-FLAG-p107 was expressed in COS1 cells with or without 3 μg of pSG5-MAGE or 4 μg of CMV-E2F1 alone or together. Cells were incubated for 24 h in serum-free medium with 0.1 μg/ml EGF and immunoprecipitated using FLAG affinity resin. Transblots of cell extracts (40 μg of protein/lane, left) or immunoprecipitates (IP, right) were probed using p107, E2F1 (sc-193), and MAGE-A11 antibodies. C, pCMV-FLAG, or pCMV-FLAG-p107 (4 μg/dish) was expressed in three 10-cm dishes plated at 7 × 106 LAPC-4 cells/dish using 8 μl of X-tremeGENE in 160 μl of medium added/dish. The next day, cells were transferred to serum-free medium with 0.1 μg/ml EGF and incubated for 24 h. The cell extract (40 μg of protein/lane) prepared in immunoprecipitation lysis buffer and immunoprecipitates were probed overnight at 4 °C on transblots using p107 (sc-318, 1:200 dilution) and MAGE-A11 antibodies (15 μg/ml). The blot was stripped and reprobed using E2F1 (sc-193) antibody (1:100 dilution).
Stabilization of E2F1 and p107 by MAGE-A11 raised the possibility that MAGE-A11 links these regulatory proteins, as seen for AR and p107. Immunoprecipitation studies showed that E2F1 strongly associated with FLAG-p107 in the presence of MAGE-A11 (Fig. 11B). Endogenous E2F1 also associated with FLAG-p107 in LAPC-4 cells, although endogenous MAGE-A11 was too low for detection in the immunoprecipitate (Fig. 11C).
To obtain further evidence for p107 transcriptional activator activity in prostate cancer, the effect of p107 knockdown was assessed in LAPC-4 cells. p107 was detected in LAPC-4 cell extracts at levels similar to CWR-R1 and LNCaP prostate cancer cells and was not significantly different from levels in benign human prostate epithelial PWR-1E and RWPE-2 cells or CV1 or COS1 cells but greater than HeLa or HeLa cells expressing the AR (Fig. 12A). HEK-293 cells had the highest endogenous p107 (Fig. 12A, lane 18). AR was detected in prostate cancer and HeLa cells stably expressing AR (Fig. 12A, lanes 3–10). E2F1 was detected in all cell types examined. However, the mobility of E2F1 differed between benign and cancer cells that suggested differences in phosphorylation. E2F1 migrated as two major bands in prostate cancer cells (Fig. 12A, lanes 5–10) but as the single faster-migrating hypophosphorylated form in PWR-1E and RWPE-2 benign prostate cells (lanes 11–14). Our earlier results (Fig. 6A) showed that MAGE-A11 interacts with the faster-migrating hypophosphorylated form of E2F1. The E2F1 dimer partner DP-1 was detected in all cell types. MAGE-A11 was detected in LAPC-4 cells that express endogenous AR (Fig. 12A, lanes 9 and 10) and in HeLa cells that stably express AR (lanes 3 and 4), which suggests that an increase in AR is associated with an increase in endogenous MAGE-A11.
When p107 levels were lowered using siRNA (Fig. 12B), activation of E2F1-Luc by expressed or endogenous E2F1 was inhibited in LAPC-4 cells (Fig. 12, C and D). p107 siRNA-5, 6, and 9 decreased p107 levels and inhibited E2F1-Luc transactivation, whereas nonspecific siRNA or p107 siRNA-8 did not alter p107 levels or inhibit E2F1-Luc transactivation.
Immunostaining MAGE-A11 and p107 in representative samples of benign prostate and prostate cancer showed that MAGE-A11 and p107 are expressed weakly in stromal and epithelial cells of benign prostate tissue (Fig. 13, A–D). MAGE-A11 and p107 immunostaining increased in androgen-stimulated prostate cancer (Fig. 13, E and F) and was most intense in castration-recurrent prostate cancer (G and H). These results support the hypothesis that MAGE-A11 facilitates the transcriptional activator activity of p107 during prostate cancer progression.
FIGURE 13.
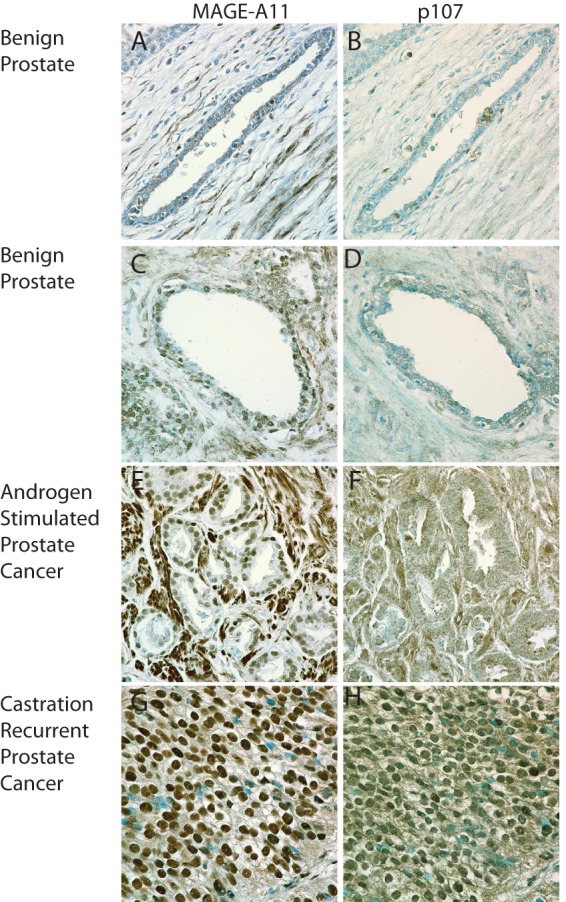
MAGE-A11 and p107 immunostaining in benign prostate and prostate cancer. Adjacent formalin-fixed, paraffin-embedded sections of benign prostate-14C (A and B) and 4740 (C and D), androgen-stimulated prostate cancer MS99-10751-198 (E and F), and castration-recurrent prostate cancer MS97-11832-2 (G and H) were immunostained using affinity-purified MAGE-(94–108) rabbit polyclonal anti-peptide antibody (6 μg/ml) (left) and affinity-purified p107 C-18 (sc-318) rabbit polyclonal antibodies (1:100) (right) as described under “Experimental Procedures.” Brown positive reaction product is shown against toluidine blue staining. The original magnification is ×60.
DISCUSSION
Primate-specific Transcriptional Coregulator MAGE-A11
We have shown that MAGE-A11 interacts selectively with p107 of the Rb family, increases E2F1 transcriptional activity and contributes to the transcriptional activator function of p107. Our findings are consistent with the established role of p107 in cell cycle regulation (24), the cell cycle-dependent expression of MAGE-A11 (4), the cell growth promoting effects of MAGE-A11 (10), and the regulation of MAGE-A11 by a cell cycle-dependent kinase (4, 13).
The ability of MAGE-A11 to link p107 to AR and E2F1 and enhance prostate cancer cell growth suggests that MAGE-A11 is a transcriptional amplifier in primates important in cell cycle control. siRNA knockdown of p107 in prostate cancer cells showed that p107 is a transcriptional activator that has lost its ability to repress AR and E2F1 transcriptional activity. MAGE-A11 appears to act in a molecular hub for transcription regulation in primates by linking p107 to E2F1 and AR. The increase in MAGE-A11 (10, 11, 14) and p107 in prostate cancer suggests that MAGE-A11 is a proto-oncogene with properties similar to the adenoviral oncoprotein E1A.
Regulation of p107 by MAGE-A11
The retinoblastoma family members Rb, p107, and p130 regulate the cell cycle and have tumor suppressor activity through the modulation of E2F transcription factors (55–57). Rb is a tumor suppressor, whereas p107 and p130 are involved in cell cycle regulation (24). Hypophosphorylated Rb-related proteins interact with E2Fs and corepressors to actively repress E2F-dependent gene transcription and cause cell cycle arrest in G0/G1 (58–61). Mitogen-induced hyperphosphorylation of Rb or p107 by cyclin-dependent kinases releases active E2Fs to up-regulate genes involved in cell cycle control (18, 62). Rb is hypophosphorylated in G0/G1 and phosphorylated during the G1-S transition (63). p107 levels vary during the cell cycle, are low in G0, and accumulate during re-entry into S phase (64, 65). p107 associates with E2Fs and becomes phosphorylated by cyclin-dependent kinases during S phase (24, 66, 67).
We have shown that MAGE-A11 interacts preferentially with p107 and less with Rb and did not interact with p130. MAGE-A11 interaction with p107 resulted in stabilization and time-dependent ubiquitination of p107. MAGE-A11 may be part of a ubiquitin ligase complex as suggested for another member of the MAGE family (68). MAGE-A11 itself undergoes monoubiquitination on lysine residues in the MAGE homology domain required to stimulate transcriptional activity of human AR (13), human progesterone receptor-B (2) and E2F1. The increase in MAGE-A11 and its ability to stabilize p107 may contribute to the increase in p107 in prostate cancer and its function as a transcriptional activator.
Rb has been implicated in cross-talk with the AR NH2-terminal region (69, 70). Overexpression of Rb increased AR transcriptional activity that was lost in Rb-deficient cells (71). However, whether Rb has direct effects on AR or indirect effects through coregulators remains unclear. MAGE-A11 interacts with the AR NH2-terminal FXXLF motif region and could mediate the effects of Rb. Our studies suggest that p107 and possibly Rb are modulated by the AR coregulator MAGE-A11.
Regulation of E2F Transcriptional Activity by MAGE-A11
E2F transcription factors are DNA binding proteins that recognize the consensus sequence G/CTTTG/C in promoter regions of genes that regulate cell cycle entry and exit (72, 73). E2F binding to DNA is modulated by phosphorylation. Activator or repressor activity of E2Fs is influenced by interactions with transcription factors and coregulators (45, 74). E2Fs are negatively regulated by the Rb family (75, 76). Mitogens activate cyclin-dependent kinases to phosphorylate Rb-like proteins, release E2Fs, and induce E2F-regulated genes that control cell exit from G1 and entry into S phase (20). Upon release in response to mitogen-induced phosphorylation or viral transformation, E2Fs form heterodimeric complexes with the dimer partner DP-1 or DP-2 (77).
Most E2F-responsive genes regulate DNA synthesis and cell cycle progression. E2F1, E2F2, and E2F3 are thought to complex primarily with Rb. When activated, they induce entry into S phase or, depending on their levels, induce apoptosis (78, 79). E2F1 up-regulates its own expression during the cell cycle (38, 80). E2F4 and E2F5 have repressive activity and are involved in cell differentiation. E2F4 associates with p107 and E2F5 with p130 (81–83). E2F6, E2F7 and E2F8 are transcriptional repressors that do not interact with Rb-related proteins (73). E2F1, E2F2, and E2F3 are predominantly nuclear, whereas E2F4 and E2F5 are cytoplasmic and nuclear (53). Although p107 is considered a transcriptional repressor that associates primarily with E2F4 (84–86) and not with E2F1 (81, 87), recent studies in addition to our own suggest that p107 can have activator or repressor activity, with an interaction between p107 and E2F1 modulated during the cell cycle (52).
MAGE-A11 increased the interaction between p107 and E2F1, which was reported to be weaker than p107 interaction with E2F4 (88), and enhanced the transcriptional activity of E2F1. E2F1 is regulated by phosphorylation (89–91). We noted that a major hyperphosphorylated form of E2F1 in cancer cells was not evident in benign prostate cells. MAGE-A11 forms a stable complex with hypophosphorylated E2F1. The faster migration of E2F1 after treatment with λ protein phosphatase suggests that activation of E2F1 by phosphorylation is associated with release from MAGE-A11. Similar to E1A, MAGE-A11 appears to sequester hypophosphorylated E2F1 and promote the release of activated phosphorylated E2F1 from p107.
MAGE-A11 interaction with E2F transcription factors resembles, in some respects, other members of the MAGE gene family. Necdin, necdin-like protein 1 (MAGE-L2), and necdin-like protein 2 (MAGE-G1) are neuron-specific cell growth suppressors whose absence is associated with Prader-Willi syndrome and autistic disorders (92). Necdin and MAGE-G1 bind E2F1 and repress E2F1-dependent gene transcription (93, 94). We found that MAGE-A11 stabilized E2F1 and p107 and mediated an interaction of p107 with AR and E2F1 that caused transcriptional repression or activation, depending on MAGE-A11 levels.
MAGE-A11 activation of E2F1 may promote normal cycling cells to enter S phase of the cell cycle. A threshold model was proposed where genes that regulate cell proliferation or apoptosis are induced by different levels of E2F that act as positive or negative regulators of cell growth (53). Positive and negative cooperation in gene regulation by AR and E2F1 were reported in prostate cancer cells (95–97). Relative levels of MAGE-A11 and p107 influence AR and E2F1 transcriptional activation or repression.
MAGE-A11 and Viral Oncogene E1A
Human adenoviral early region protein E1A transforms cells indirectly by activating E2Fs through competitive interaction with Rb-related proteins (27, 28, 98–100). E1A interaction with Rb-related proteins releases active E2F transcription factors that activate target genes and cause cell cycle progression (29). E1A inactivates Rb in G0/G1 of the cell cycle, causing exit from G1 and induction of DNA synthesis in S phase in association with uncontrolled cell cycle progression and immortalization (27, 101–104). The effect of oncoprotein E1A overrides the inhibitory effects of p16 on cell cycle progression (105).
MAGE-A11 shares a remarkable similarity with E1A. MAGE-A11 and E1A form strong complexes with p300 (4, 106, 107) and interact with E2F transcription factors (27) and Rb-related proteins to activate or repress transcription (101, 108). Like E1A, MAGE-A11 relieves the constraint by p107 on E2F1 promotion of cell cycle progression. MAGE-A11 interaction with p107 caused a dose-dependent increase in E2F activity similar to E1A. MAGE-A11 shares a sequence similarity with E1A and E2F1 motifs that bind the pocket region of Rb-related proteins. MAGE-A11 amino acid sequence 410DPYSYPDLYE419 is similar to E1A 39EPPTLHELYD48, which binds Rb-related protein pocket regions, and E2F1 417EGEGIRDLFD426 in the transactivation domain.
However, although the primate-specific MAGE-A11 gene is expressed at very low levels in normal cells and at higher levels in prostate cancer, E1A is a viral oncogene. Neither MAGE-A11 nor E1A bind DNA directly (109). E1A interacts indirectly with promoters of a large number of cell cycle regulatory genes by stimulating E2F DNA binding through increased E2F phosphorylation (45). E1A interaction with p300 and p107 follows a temporal sequence during cell replication (108), which also may occur with MAGE-A11. Synergy between MAGE-A11 and p300 in E2F-dependent gene transcription depends on E2F response element DNA and MAGE-A11 interaction with p300. MAGE-A11 also amplifies AR transactivation of androgen-dependent genes (3).
MAGE-A11 in Prostate Cancer
Interactions between Rb-related proteins and E2Fs are interrupted frequently in cancer, which result in E2F transactivation of cell cycle regulatory genes. Almost all cancers functionally inactivate Rb by gene mutation or deletion, dysregulation by cyclin-dependent kinases, or sequestration of Rb-related proteins by oncogenic proteins such as E1A. In agreement with Rb mutations in late-stage cancer, loss of Rb function is associated with advanced prostate cancer and increased AR expression (70). E2F1 and E2F3 increase during prostate cancer progression (110, 111).
Here we provide evidence that MAGE-A11 is a proto-oncogene that disrupts the tumor suppressor function of Rb-related proteins. MAGE-A11 increases during prostate cancer progression, enhances AR signaling, and promotes prostate cancer growth (10, 11, 14). The increase in MAGE-A11 in prostate and epithelial ovarian cancer results from promoter hypomethylation, and is associated with early cancer recurrence and poor survival (112). The cell growth-promoting effects of MAGE-A11 appear to be mediated through AR signaling and Rb-related proteins that increase E2F1 transactivation.
Prostate cancer is associated with greater AR sensitivity to increased levels of coregulators, such as MAGE-A11 (11, 14, 113, 114). The ability of AR and MAGE-A11 to regulate p27, an important physiological brake on the cell cycle, has implications for prostate cancer. In LNCaP prostate cancer cells, a biphasic androgen dose response influences p27 protein levels where higher DHT increases p27 protein and arrests cell growth (118, 119). Androgen-dependent changes in proteasomal degradation have been reported to influence p27 levels (116, 117). We found that higher DHT concentrations stimulated LAPC-4 cell growth in association with rapid degradation of p27. The increase in p27 protein in LAPC-4 cells after knockdown of AR or MAGE-A11 could reflect the loss of androgen-dependent proteasomal degradation of p27 (115, 120) and the dependence of prostate cancer cells on androgen-stimulated growth.
The biphasic LNCaP cell growth response to androgen has been linked previously to the Rb and E2F pathways (119). Increased LNCaP cell growth in response to 0.1 nm R1881 was attributed to increased hypophosphorylated Rb. With 10 nm R1881, increased p27 protein inhibited cyclin-dependent kinase phosphorylation of Rb (119). An increase in E2F1 at low androgen levels was inhibited at higher androgen levels. Our findings suggest that MAGE-A11 contributes to androgen and E2F1 regulation of p27.
Increased AR expression may be a molecular basis for prostate cancer progression (113). However, MAGE-A11 mRNA increases during prostate cancer progression in an inverse relationship with AR mRNA (14). A subset of castration-recurrent prostate cancer specimens had 1000-fold higher MAGE-A11 mRNA, whereas AR mRNA was undetectable. p107 was also increased in castration-recurrent prostate cancer. These findings suggest that MAGE-A11 not only functions synergistically with AR and could compensate for lower levels of AR but also contributes to the activator function of p107. The increase in primate-specific expression of MAGE-A11 in prostate cancer facilitates AR transactivation and the inappropriate activation of E2F1 through selective interaction with Rb-related protein p107.
Acknowledgments
We thank Andrew T. Hnat for assistance and Frank S. French for reviewing the manuscript.
This work was supported, in whole or part, by National Institutes of Health Grant HD16910, by United States Public Health Service NICHD, National Institutes of Health Eunice Kennedy Shriver Grant HD067721, and by Cooperative Agreement NICHD, National Institutes of Health Eunice Kennedy Shriver Grant U54-HD35041 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research. This work was also supported by National Cancer Institute Grant P01-CA77739.
- MAGE-A11
- melanoma antigen-A11
- AR
- androgen receptor
- Rb
- retinoblastoma
- E1A
- early region 1A
- PSA
- prostate-specific antigen
- DHT
- dihydrotestosterone
- Luc
- luciferase.
REFERENCES
- 1. Bai S., He B., Wilson E. M. (2005) Melanoma antigen gene protein MAGE-11 regulates androgen receptor function by modulating the interdomain interaction. Mol. Cell Biol. 25, 1238–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Su S., Blackwelder A. J., Grossman G., Minges J. T., Yuan L., Young S. L., Wilson E. M. (2012) Primate-specific melanoma antigen-A11 regulates isoform-specific human progesterone receptor-B transactivation. J. Biol. Chem. 287, 34809–34824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Askew E. B., Bai S., Hnat A. T., Minges J. T., Wilson E. M. (2009) Melanoma antigen gene protein-A11 (MAGE-11) F-box links the androgen receptor NH2-terminal transactivation domain to p160 coactivators. J. Biol. Chem. 284, 34793–34808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Askew E. B., Bai S., Blackwelder A. J., Wilson E. M. (2010) Transcriptional synergy between melanoma antigen gene protein-A11 (MAGE-11) and p300 in androgen receptor signaling. J. Biol. Chem. 285, 21824–21836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. He B., Kemppainen J. A., Wilson E. M. (2000) FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J. Biol. Chem. 275, 22986–22994 [DOI] [PubMed] [Google Scholar]
- 6. He B., Lee L. W., Minges J. T., Wilson E. M. (2002) Dependence of selective gene activation on the androgen receptor NH2- and carboxyl-terminal interaction. J. Biol. Chem. 277, 25631–25639 [DOI] [PubMed] [Google Scholar]
- 7. He B., Bowen N. T., Minges J. T., Wilson E. M. (2001) Androgen-induced NH2- and carboxyl-terminal interaction inhibits p160 coactivator recruitment by activation function 2. J. Biol. Chem. 276, 42293–42301 [DOI] [PubMed] [Google Scholar]
- 8. He B., Gampe R. T., Jr., Kole A. J., Hnat A. T., Stanley T. B., An G., Stewart E. L., Kalman R. I., Minges J. T., Wilson E. M. (2004) Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol. Cell 16, 425–438 [DOI] [PubMed] [Google Scholar]
- 9. Liu Q., Su S., Blackwelder A. J., Minges J. T., Wilson E. M. (2011) Gain in transcriptional activity by primate-specific coevolution of melanoma antigen-A11 and its interaction site in the androgen receptor. J. Biol. Chem. 286, 29951–29963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Minges J. T., Su S., Grossman G., Blackwelder A. J., Pop E. A., Mohler J. L., Wilson E. M. (2013) Melanoma antigen-A11 (MAGE-A11) enhances transcriptional activity by linking androgen receptor dimers. J. Biol. Chem. 288, 1939–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wilson E. M. (2010) Androgen receptor molecular biology and potential targets in prostate cancer. Therap. Adv. Urol. 2, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bai S., Grossman G., Yuan L., Lessey B. A., French F. S., Young S. L., Wilson E. M. (2008) Hormone control and expression of androgen receptor coregulator MAGE-11 in human endometrium during the window of receptivity to embryo implantation. Mol. Hum. Reprod. 14, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bai S., Wilson E. M. (2008) Epidermal growth factor-dependent phosphorylation and ubiquitinylation of MAGE-11 regulates its interaction with the androgen receptor. Mol. Cell Biol. 28, 1947–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karpf A. R., Bai S., James S. R., Mohler J. L., Wilson E. M. (2009) Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol. Cancer Res. 7, 523–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burkhart D. L., Sage J. (2008) Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 8, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morris E. J., Dyson N. J. (2001) Retinoblastoma protein partners. Adv. Cancer Res. 82, 1–54 [DOI] [PubMed] [Google Scholar]
- 17. Genovese C., Trani D., Caputi M., Claudio P. P. (2006) Cell cycle control and beyond. Emerging roles for the retinoblastoma gene family. Oncogene 25, 5201–5209 [DOI] [PubMed] [Google Scholar]
- 18. Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 [DOI] [PubMed] [Google Scholar]
- 19. Cobrinik D. (2005) Pocket proteins and cell cycle control. Oncogene 24, 2796–2809 [DOI] [PubMed] [Google Scholar]
- 20. Blais A., Dynlacht B. D. (2007) E2F-associated chromatin modifiers and cell cycle control. Curr. Opin. Cell Biol. 19, 658–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer. The next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 22. Beroukhim R., Mermel C. H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J. S., Dobson J., Urashima M., Mc Henry K. T., Pinchback R. M., Ligon A. H., Cho Y. J., Haery L., Greulich H., Reich M., Winckler W., Lawrence M. S., Weir B. A., Tanaka K. E., Chiang D. Y., Bass A. J., Loo A., Hoffman C., Prensner J., Liefeld T., Gao Q., Yecies D., Signoretti S., Maher E., Kaye F. J., Sasaki H., Tepper J. E., Fletcher J. A., Tabernero J., Baselga J., Tsao M. S., Demichelis F., Rubin M. A., Janne P. A., Daly M. J., Nucera C., Levine R. L., Ebert B. L., Gabriel S., Rustgi A. K., Antonescu C. R., Ladanyi M., Letai A., Garraway L. A. (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kondo T., Higashi H., Nishizawa H., Ishikawa S., Ashizawa S., Yamada M., Makita Z., Koike T., Hatakeyama M. (2001) Involvement of pRB-related p107 protein in the inhibition of S phase progression in response to genotoxic stress. J. Biol. Chem. 276, 17559–17567 [DOI] [PubMed] [Google Scholar]
- 24. Wirt S. E., Sage J. (2010) p107 in the public eye. An Rb understudy and more. Cell Div. 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bellacchio E., Paggi M. G. (2013) Understanding the targeting of the RB family proteins by viral oncoproteins to defeat their oncogenic machinery. J. Cell Physiol. 228, 285–291 [DOI] [PubMed] [Google Scholar]
- 26. Felsani A., Mileo A. M., Paggi M. G. (2006) Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene 25, 5277–5285 [DOI] [PubMed] [Google Scholar]
- 27. Fattaey A. R., Harlow E., Helin K. (1993) Independent regions of adenovirus E1A are required for binding to and dissociation of E2F-protein complexes. Mol. Cell Biol. 13, 7267–7277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ikeda M. A., Nevins J. R. (1993) Identification of distinct roles for separate E1A domains in disruption of E2F complexes. Mol. Cell Biol. 13, 7029–7035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu X., Marmorstein R. (2007) Structure of the retinoblastoma protein bound to adenovirus E1A reveals the molecular basis for viral oncoprotein inactivation of a tumor suppressor. Genes Dev. 21, 2711–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lubahn D. B., Joseph D. R., Sar M., Tan J., Higgs H. N., Larson R. E., French F. S., Wilson E. M. (1988) The human androgen receptor. Complementary DNA cloning, sequence analysis and gene expression in prostate. Mol. Endocrinol. 2, 1265–1275 [DOI] [PubMed] [Google Scholar]
- 31. Simental J. A., Sar M., Lane M. V., French F. S., Wilson E. M. (1991) Transcriptional activation and nuclear targeting signals of the human androgen receptor. J. Biol. Chem. 266, 510–518 [PubMed] [Google Scholar]
- 32. Li Y., Nichols M. A., Shay J. W., Xiong Y. (1994) Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb. Cancer Res. 54, 6078–6082 [PubMed] [Google Scholar]
- 33. Claudio P. P., Howard C. M., Baldi A., De Luca A., Fu Y., Condorelli G., Sun Y., Colburn N., Calabretta B., Giordano A. (1994) p130/pRb2 has growth suppressive properties similar to yet distinctive from those of retinoblastoma family members pRb and p107. Cancer Res. 54, 5556–5560 [PubMed] [Google Scholar]
- 34. Hsia E. Y., Kalashnikova E. V., Revenko A. S., Zou J. X., Borowsky A. D., Chen H. W. (2010) Deregulated E2F and the AAA+ coregulator ANCCA drive proto-oncogene ACTR/AIB1 overexpression in breast cancer. Mol. Cancer Res. 8, 183–193 [DOI] [PubMed] [Google Scholar]
- 35. Zhu L., van den Heuvel S., Helin K., Fattaey A., Ewen M., Livingston D., Dyson N., Harlow E. (1993) Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes Dev. 7, 1111–1125 [DOI] [PubMed] [Google Scholar]
- 36. Chen C. R., Kang Y., Siegel P. M., Massagué J. (2002) E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell 110, 19–32 [DOI] [PubMed] [Google Scholar]
- 37. Huang W., Shostak Y., Tarr P., Sawyers C., Carey M. (1999) Cooperative assembly of androgen receptor into a nucleoprotein complex that regulates the prostate-specific antigen enhancer. J. Biol. Chem. 274, 25756–25768 [DOI] [PubMed] [Google Scholar]
- 38. Johnson D. G., Ohtani K., Nevins J. R. (1994) Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 8, 1514–1525 [DOI] [PubMed] [Google Scholar]
- 39. Cherian M. T., Wilson E. M., Shapiro D. J. (2012) A competitive inhibitor that reduces recruitment of androgen receptor to androgen-responsive genes. J. Biol. Chem. 287, 23368–23380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Askew E. B., Gampe R. T., Jr., Stanley T. B., Faggart J. L., Wilson E. M. (2007) Modulation of androgen receptor activation function 2 by testosterone and dihydrotestosterone. J. Biol. Chem. 282, 25801–25816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He B., Minges J. T., Lee L. W., Wilson E. M. (2002) The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J. Biol. Chem. 277, 10226–10235 [DOI] [PubMed] [Google Scholar]
- 42. Quarmby V. E., Kemppainen J. A., Sar M., Lubahn D. B., French F. S., Wilson E. M. (1990) Expression of recombinant androgen receptor in cultured mammalian cells. Mol. Endocrinol. 4, 1399–1407 [DOI] [PubMed] [Google Scholar]
- 43. Dehm S. M., Schmidt L. J., Heemers H. V., Vessella R. L., Tindall D. J. (2008) Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 68, 5469–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun S., Sprenger C. C., Vessella R. L., Haugk K., Soriano K., Mostaghel E. A., Page S. T., Coleman I. M., Nguyen H. M., Sun H., Nelson P. S., Plymate S. R. (2010) Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest. 120, 2715–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bagchi S., Raychaudhuri P., Nevins J. R. (1989) Phosphorylation-dependent activation of the adenovirus-inducible E2F transcription factor in a cell-free system. Proc. Natl. Acad. Sci. U.S.A. 86, 4352–4356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Polyak K., Lee M. H., Erdjument-Bromage H., Koff A., Roberts J. M., Tempst P., Massagué J. (1994) Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 78, 59–66 [DOI] [PubMed] [Google Scholar]
- 47. Wang C., Hou X., Mohapatra S., Ma Y., Cress W. D., Pledger W. J., Chen J. (2005) Activation of p27Kip1 expression by E2F1. A negative feedback mechanism. J. Biol. Chem. 280, 12339–12343 [DOI] [PubMed] [Google Scholar]
- 48. Hodul P. J., Dong Y., Husain K., Pimiento J. M., Chen J., Zhang A., Francois R., Pledger W. J., Coppola D., Sebti S. M., Chen D. T., Malafa M. P. (2013) Vitamin E δ-tocotrienol induces p27(Kip1)-dependent cell-cycle arrest in pancreatic cancer cells via an E2F-1-dependent mechanism. PLoS ONE 8, e52526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tsihlias J., Kapusta L. R., DeBoer G., Morava-Protzner I., Zbieranowski I., Bhattacharya N., Catzavelos G. C., Klotz L. H., Slingerland J. M. (1998) Loss of cyclin-dependent kinase inhibitor p27Kip1 is a novel prognostic factor in localized human prostate adenocarcinoma. Cancer Res. 58, 542–548 [PubMed] [Google Scholar]
- 50. Yang R. M., Naitoh J., Murphy M., Wang H. J., Phillipson J., deKernion J. B., Loda M., Reiter R. E. (1998) Low p27 expression predicts poor disease-free survival in patients with prostate cancer. J. Urol. 159, 941–945 [PubMed] [Google Scholar]
- 51. Knudsen K. E., Arden K. C., Cavenee W. K. (1998) Multiple G1 regulatory elements control the androgen-dependent proliferation of prostatic carcinoma cells. J. Biol. Chem. 273, 20213–20222 [DOI] [PubMed] [Google Scholar]
- 52. Calbó J., Parreño M., Sotillo E., Yong T., Mazo A., Garriga J., Grana X. (2002) G1 cyclin/cyclin-dependent kinase-coordinated phosphorylation of endogenous pocket proteins differentially regulates their interactions with E2F4 and E2F1 and gene expression. J. Biol. Chem. 277, 50263–50274 [DOI] [PubMed] [Google Scholar]
- 53. Trimarchi J. M., Lees J. A. (2002) Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 3, 11–20 [DOI] [PubMed] [Google Scholar]
- 54. Lees J. A., Saito M., Vidal M., Valentine M., Look T., Harlow E., Dyson N., Helin K. (1993) The retinoblastoma protein binds to a family of E2F transcription factors. Mol. Cell Biol. 13, 7813–7825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chellappan S. P., Hiebert S., Mudryj M., Horowitz J. M., Nevins J. R. (1991) The E2F transcription factor is a cellular target for the RB protein. Cell 65, 1053–1061 [DOI] [PubMed] [Google Scholar]
- 56. Cao L., Faha B., Dembski M., Tsai L. H., Harlow E., Dyson N. (1992) Independent binding of the retinoblastoma protein and p107 to the transcription factor E2F. Nature 355, 176–179 [DOI] [PubMed] [Google Scholar]
- 57. Cobrinik D., Whyte P., Peeper D. S., Jacks T., Weinberg R. A. (1993) Cell cycle-specific association of E2F with the p130 E1A-binding protein. Genes Dev. 7, 2392–2404 [DOI] [PubMed] [Google Scholar]
- 58. Zhang H. S., Gavin M., Dahiya A., Postigo A. A., Ma D., Luo R. X., Harbour J. W., Dean D. C. (2000) Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 101, 79–89 [DOI] [PubMed] [Google Scholar]
- 59. Rayman J. B., Takahashi Y., Indjeian V. B., Dannenberg J. H., Catchpole S., Watson R. J., te Riele H., Dynlacht B. D. (2002) E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 16, 933–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Strobeck M. W., Knudsen K. E., Fribourg A. F., DeCristofaro M. F., Weissman B. E., Imbalzano A. N., Knudsen E. S. (2000) BRG-1 is required for RB-mediated cell cycle arrest. Proc. Natl. Acad. Sci. U.S.A. 97, 7748–7753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. David G., Grandinetti K. B., Finnerty P. M., Simpson N., Chu G. C., Depinho R. A. (2008) Specific requirement of the chromatin modifier mSin3B in cell cycle exit and cellular differentiation. Proc. Natl. Acad. Sci. U.S.A. 105, 4168–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weinberg R. A. (1995) The retinoblastoma protein and cell cycle control. Cell 81, 323–330 [DOI] [PubMed] [Google Scholar]
- 63. Hume A. J., Kalejta R. F. (2009) Regulation of the retinoblastoma proteins by the human herpesviruses. Cell Div. 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Graña X., Garriga J., Mayol X. (1998) Role of the retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene 17, 3365–3383 [DOI] [PubMed] [Google Scholar]
- 65. Nevins J. R. (1998) Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 9, 585–593 [PubMed] [Google Scholar]
- 66. Xiao Z. X., Ginsberg D., Ewen M., Livingston D. M. (1996) Regulation of the retinoblastoma protein-related protein p107 by G1 cyclin-associated kinases. Proc. Natl. Acad. Sci. U.S.A. 93, 4633–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Beijersbergen R. L., Carlée L., Kerkhoven R. M., Bernards R. (1995) Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes Dev. 9, 1340–1353 [DOI] [PubMed] [Google Scholar]
- 68. Doyle J. M., Gao J., Wang J., Yang M., Potts P. R. (2010) MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol. Cell 39, 963–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lu J., Danielsen M. (1998) Differential regulation of androgen and glucocorticoid receptors by retinoblastoma protein. J. Biol. Chem. 273, 31528–31533 [DOI] [PubMed] [Google Scholar]
- 70. Sharma A., Yeow W. S., Ertel A., Coleman I., Clegg N., Thangavel C., Morrissey C., Zhang X., Comstock C. E., Witkiewicz A. K., Gomella L., Knudsen E. S., Nelson P. S., Knudsen K. E. (2010) The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Invest. 120, 4478–4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yeh S., Miyamoto H., Nishimura K., Kang H., Ludlow J., Hsiao P., Wang C., Su C., Chang C. (1998) Retinoblastoma, a tumor suppressor, is a coactivator for the androgen receptor in human prostate cancer DU145 cells. Biochem. Biophys. Res. Commun. 248, 361–367 [DOI] [PubMed] [Google Scholar]
- 72. Xu X., Bieda M., Jin V. X., Rabinovich A., Oberley M. J., Green R., Farnham P. J. (2007) A comprehensive ChIP-chip analysis of E2F1, E2F4, and E2F6 in normal and tumor cells reveals interchangeable roles of E2F family members. Genome Res. 17, 1550–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Attwooll C., Lazzerini Denchi E., Helin K. (2004) The E2F family. Specific functions and overlapping interests. EMBO J. 23, 4709–4716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Freedman J. A., Chang J. T., Jakoi L., Nevins J. R. (2009) A combinatorial mechanism for determining the specificity of E2F activation and repression. Oncogene 28, 2873–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hiebert S. W., Chellappan S. P., Horowitz J. M., Nevins J. R. (1992) The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes Dev. 6, 177–185 [DOI] [PubMed] [Google Scholar]
- 76. Schwarz J. K., Devoto S. H., Smith E. J., Chellappan S. P., Jakoi L., Nevins J. R. (1993) Interactions of the p107 and Rb proteins with E2F during the cell proliferation response. EMBO J. 12, 1013–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bandara L. R., Lam E. W., Sørensen T. S., Zamanian M., Girling R., La Thangue N. B. (1994) DP-1. A cell cycle-regulated and phosphorylated component of transcription factor DRTF1/E2F which is functionally important for recognition by pRb and the adenovirus E4 orf 6/7 protein. EMBO J. 13, 3104–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. DeGregori J., Leone G., Miron A., Jakoi L., Nevins J. R. (1997) Distinct roles for E2F proteins in cell growth control and apoptosis. Proc. Natl. Acad. Sci. U.S.A. 94, 7245–7250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hallstrom T. C., Nevins J. R. (2009) Balancing the decision of cell proliferation and cell fate. Cell Cycle 8, 532–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Slansky J. E., Li Y., Kaelin W. G., Farnham P. J. (1993) A protein synthesis-dependent increase in E2F1 mRNA correlates with growth regulation of the dihydrofolate reductase promoter. Mol. Cell Biol. 13, 1610–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dyson N., Dembski M., Fattaey A., Ngwu C., Ewen M., Helin K. (1993) Analysis of p107-associated proteins. p107 associates with a form of E2F that differs from pRB-associated E2F-1. J. Virol. 67, 7641–7647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Beijersbergen R. L., Kerkhoven R. M., Zhu L., Carlée L., Voorhoeve P. M., Bernards R. (1994) E2F-4, a new member of the E2F gene family, has oncogenic activity and associates with p107 in vivo. Genes Dev. 8, 2680–2690 [DOI] [PubMed] [Google Scholar]
- 83. Takahashi Y., Rayman J. B., Dynlacht B. D. (2000) Analysis of promoter binding by the E2F and pRB families in vivo. Distinct E2F proteins mediate activation and repression. Genes Dev. 14, 804–816 [PMC free article] [PubMed] [Google Scholar]
- 84. Verona R., Moberg K., Estes S., Starz M., Vernon J. P., Lees J. A. (1997) E2F activity is regulated by cell cycle-dependent changes in subcellular localization. Mol. Cell Biol. 17, 7268–7282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Puri P. L., Cimino L., Fulco M., Zimmerman C., La Thangue N. B., Giordano A., Graessmann A., Levrero M. (1998) Regulation of E2F4 mitogenic activity during terminal differentiation by its heterodimerization partners for nuclear translocation. Cancer Res. 58, 1325–1331 [PubMed] [Google Scholar]
- 86. Moberg K., Starz M. A., Lees J. A. (1996) E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol. Cell Biol. 16, 1436–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ginsberg D., Vairo G., Chittenden T., Xiao Z. X., Xu G., Wydner K. L., DeCaprio J. A., Lawrence J. B., Livingston D. M. (1994) E2F-4, a new member of the E2F transcription factor family, interacts with p107. Genes Dev. 8, 2665–2679 [DOI] [PubMed] [Google Scholar]
- 88. Rubin S. M., Gall A. L., Zheng N., Pavletich N. P. (2005) Structure of the Rb C-terminal domain bound to E2F1-DP1. A mechanism for phosphorylation-induced E2F release. Cell 123, 1093–1106 [DOI] [PubMed] [Google Scholar]
- 89. Lin W. C., Lin F. T., Nevins J. R. (2001) Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 15, 1833–1844 [PMC free article] [PubMed] [Google Scholar]
- 90. Stevens C., Smith L., La Thangue N. B. (2003) Chk2 activates E2F-1 in response to DNA damage. Nat. Cell Biol. 5, 401–409 [DOI] [PubMed] [Google Scholar]
- 91. Yang X. H., Sladek T. L. (1997) Novel phosphorylated forms of E2F-1 transcription factor bind to the retinoblastoma protein in cells overexpressing an E2F-1 cDNA. Biochem. Biophys. Res. Commun. 232, 336–339 [DOI] [PubMed] [Google Scholar]
- 92. Jay P., Rougeulle C., Massacrier A., Moncla A., Mattei M. G., Malzac P., Roëckel N., Taviaux S., Lefranc J. L., Cau P., Berta P., Lalande M., Muscatelli F. (1997) The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nat. Genet. 17, 357–361 [DOI] [PubMed] [Google Scholar]
- 93. Taniura H., Taniguchi N., Hara M., Yoshikawa K. (1998) Necdin, a postmitotic neuron-specific growth suppressor, interacts with viral transforming proteins and cellular transcription factor E2F1. J. Biol. Chem. 273, 720–728 [DOI] [PubMed] [Google Scholar]
- 94. Kuwako K., Taniura H., Yoshikawa K. (2004) Necdin-related MAGE proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. J. Biol. Chem. 279, 1703–1712 [DOI] [PubMed] [Google Scholar]
- 95. Altintas D. M., Shukla M. S., Goutte-Gattat D., Angelov D., Rouault J. P., Dimitrov S., Samarut J. (2012) Direct cooperation between androgen receptor and E2F1 reveals a common regulation mechanism for androgen-responsive genes in prostate cells. Mol. Endocrinol. 26, 1531–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Li Y., Zhang D. Y., Ren Q., Ye F., Zhao X., Daniels G., Wu X., Dynlacht B., Lee P. (2012) Regulation of a novel androgen receptor target gene, the cyclin B1 gene, through androgen-dependent E2F family member switching. Mol. Cell Biol. 32, 2454–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Mallik I., Davila M., Tapia T., Schanen B., Chakrabarti R. (2008) Androgen regulates Cdc6 transcription through interactions between androgen receptor and E2F transcription factor in prostate cancer cells. Biochim. Biophys. Acta 1783, 1737–7144 [DOI] [PubMed] [Google Scholar]
- 98. Harlow E., Whyte P., Franza B. R., Jr., Schley C. (1986) Association of adenovirus early-region 1A proteins with cellular polypeptides. Mol. Cell Biol. 6, 1579–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dyson N., Buchkovich K., Whyte P., Harlow E. (1989) The cellular 107K protein that binds to adenovirus E1A also associates with the large T antigens of SV40 and JC virus. Cell 58, 249–255 [DOI] [PubMed] [Google Scholar]
- 100. Ewen M. E., Xing Y. G., Lawrence J. B., Livingston D. M. (1991) Molecular cloning, chromosomal mapping, and expression of the cDNA for p107, a retinoblastoma gene product-related protein. Cell 66, 1155–1164 [DOI] [PubMed] [Google Scholar]
- 101. Arany Z., Newsome D., Oldread E., Livingston D. M., Eckner R. (1995) A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature 374, 81–84 [DOI] [PubMed] [Google Scholar]
- 102. Rasti M., Grand R. J., Mymryk J. S., Gallimore P. H., Turnell A. S. (2005) Recruitment of CBP/p300, TATA-binding protein, and S8 to distinct regions at the N terminus of adenovirus E1A. J. Virol. 79, 5594–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Whyte P., Williamson N. M., Harlow E. (1989) Cellular targets for transformation by the adenovirus E1A proteins. Cell 56, 67–75 [DOI] [PubMed] [Google Scholar]
- 104. Ben-Israel H., Kleinberger T. (2002) Adenovirus and cell cycle control. Front. Biosci. 7, 1369–1395 [DOI] [PubMed] [Google Scholar]
- 105. Alevizopoulos K., Sanchez B., Amati B. (2000) Conserved region 2 of adenovirus E1A has a function distinct from pRb binding required to prevent cell cycle arrest by p16INK4a or p27Kip1. Oncogene 19, 2067–2074 [DOI] [PubMed] [Google Scholar]
- 106. Eckner R., Ewen M. E., Newsome D., Gerdes M., DeCaprio J. A., Lawrence J. B., Livingston D. M. (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 8, 869–884 [DOI] [PubMed] [Google Scholar]
- 107. Fera D., Marmorstein R. (2012) Different regions of the HPV-E7 and Ad-E1A viral oncoproteins bind competitively but through distinct mechanisms to the CH1 transactivation domain of p300. Biochemistry 51, 9524–9534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ferrari R., Pellegrini M., Horwitz G. A., Xie W., Berk A. J., Kurdistani S. K. (2008) Epigenetic reprogramming by adenovirus e1a. Science 321, 1086–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ferguson B., Krippl B., Andrisani O., Jones N., Westphal H., Rosenberg M. (1985) E1A 13S and 12S mRNA products made in Escherichia coli both function as nucleus-localized transcription activators but do not directly bind DNA. Mol. Cell Biol. 5, 2653–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Davis J. N., Wojno K. J., Daignault S., Hofer M. D., Kuefer R., Rubin M. A., Day M. L. (2006) Elevated E2F1 inhibits transcription of the androgen receptor in metastatic hormone-resistant prostate cancer. Cancer Res. 66, 11897–11906 [DOI] [PubMed] [Google Scholar]
- 111. Foster C. S., Falconer A., Dodson A. R., Norman A. R., Dennis N., Fletcher A., Southgate C., Dowe A., Dearnaley D., Jhavar S., Eeles R., Feber A., Cooper C. S. (2004) Transcription factor E2F3 overexpressed in prostate cancer independently predicts clinical outcome. Oncogene 23, 5871–5879 [DOI] [PubMed] [Google Scholar]
- 112. James S. R., Cedeno C., Sharma A., Zhang W., Mohler J. L., Odunsi K., Wilson E. M., Karpf A. R. (2013) DNA methylation and nucleosome occupancy regulate the cancer germline antigen gene MAGEA11. Epigenetics 8, 849–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Chen C. D., Welsbie D. S., Tran C., Baek S. H., Chen R., Vessella R., Rosenfeld M. G., Sawyers C. L. (2004) Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 10, 33–39 [DOI] [PubMed] [Google Scholar]
- 114. Gregory C. W., Johnson R. T., Jr., Mohler J. L., French F. S., Wilson E. M. (2001) Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 61, 2892–2898 [PubMed] [Google Scholar]
- 115. Fang Z., Zhang T., Dizeyi N., Chen S., Wang H., Swanson K. D., Cai C., Balk S. P., Yuan X. (2012) Androgen receptor enhances p27 degradation in prostate cancer cells through rapid and selective TORC2 activation. J. Biol. Chem. 287, 2090–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Besson A., Dowdy S. F., Roberts J. M. (2008) CDK inhibitors. Cell cycle regulators and beyond. Dev. Cell 14, 159–169 [DOI] [PubMed] [Google Scholar]
- 117. Chu I. M., Hengst L., Slingerland J. M. (2008) The Cdk inhibitor p27 in human cancer. Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 8, 253–267 [DOI] [PubMed] [Google Scholar]
- 118. Tsihlias J., Zhang W., Bhattacharya N., Flanagan M., Klotz L., Slingerland J. (2000) Involvement of p27Kip1 in G1 arrest by high dose 5 α-dihydrotestosterone in LNCaP human prostate cancer cells. Oncogene 19, 670–679 [DOI] [PubMed] [Google Scholar]
- 119. Hofman K., Swinnen J. V., Verhoeven G., Heyns W. (2001) E2F activity is biphasically regulated by androgens in LNCaP cells. Biochem. Biophys. Res. Commun. 283, 97–101 [DOI] [PubMed] [Google Scholar]
- 120. Yuan X., Li T., Wang H., Zhang T., Barua M., Borgesi R. A., Bubley G. J., Lu M. L., Balk S. P. (2006) Androgen receptor remains critical for cell-cycle progression in androgen-independent CWR22 prostate cancer cells. Am. J. Pathol. 169, 682–696 [DOI] [PMC free article] [PubMed] [Google Scholar]