

Background: Phosphorylation of the connexin43 C-terminal (Cx43CT) domain regulates gap junction intercellular communication (GJIC).
Results: Phosphorylation alters the α-helical propensity of the Cx43CT.
Conclusion: Altering the conformational preference of the Cx43CT presents a novel mechanism for regulation of GJIC.
Significance: Cx43CT residues susceptible to structural alterations are prime targets for chemical modulators of GJIC.
Keywords: Circular Dichroism (CD), Gap Junctions, Nuclear Magnetic Resonance, Phosphorylation, Protein Dynamics
Abstract
Phosphorylation of the connexin43 C-terminal (Cx43CT) domain regulates gap junction intercellular communication. However, an understanding of the mechanisms by which phosphorylation exerts its effects is lacking. Here, we test the hypothesis that phosphorylation regulates Cx43 gap junction intercellular communication by mediating structural changes in the C-terminal domain. Circular dichroism and nuclear magnetic resonance were used to characterize the effects of phosphorylation on the secondary structure and backbone dynamics of soluble and membrane-tethered Cx43CT domains. Cx43CT phospho-mimetic isoforms, which have Asp substitutions at specific Ser/Tyr sites, revealed phosphorylation alters the α-helical content of the Cx43CT domain only when attached to the membrane. The changes in secondary structure are due to variations in the conformational preference and backbone flexibility of residues adjacent and distal to the site(s) of modification. In addition to the known direct effects of phosphorylation on molecular partner interactions, the data presented here suggest phosphorylation may also indirectly regulate binding affinity by altering the conformational preference of the Cx43CT domain.
Introduction
Gap junctions enable the direct cytoplasmic exchange of ions and low molecular mass metabolites between adjacent cells (1). They provide a pathway for propagating and/or amplifying the signal transduction cascades triggered by cytokines, growth factors, and other cell signaling molecules involved in growth and development. Dysfunctional gap junction intercellular communication (GJIC)5 has been implicated in many human diseases (2, 3). Gap junction channels are formed by the apposition of two connexons from adjacent cells. Each connexon consists of six connexin proteins, which are named based on their molecular mass (e.g. 43-kDa isoform, connexin43 (Cx43)). Connexins share a common topology of four membrane-spanning domains with two extracellular loops, one cytoplasmic loop (CL), and cytoplasmic N- and C-terminal domains. Although the 21 connexin isoforms share significant sequence homology, they are the most divergent in the CL and C-terminal domains.
Early structural studies of Cx43, which used electron crystallography, and the more recent x-ray crystal structure of Cx26 have provided a significant amount of information about channel architecture (4, 5). However, neither technique was able to address the structure of the CL or CT because of the dynamic nature of these domains. These same characteristics that interfere with crystallographic techniques make NMR and CD ideal tools for studying these domains (6). For example, NMR was used to solve the structure of the soluble Cx43CT (Ser-255–Ile-382) domain, which was predominantly disordered with two dynamic α-helical regions (Ala-315–Thr-326; Asp-340–Ala-348 (7)). Analysis of eukaryotic genomes estimates 41% of membrane proteins have intrinsically disordered regions with more than 30 consecutive residues preferentially localized to the cytoplasmic side (8). Intrinsically disordered proteins (IDPs) fall into the following four groups: entropic chains, molecular recognition, molecular assembly, and protein modification (9). The major functions of these, including molecular partner binding, flexibility, and phosphorylation, all apply to the Cx43CT.
The degree of disorder among IDPs, like Cx43, spans the entire protein folding pathway from highly extended structures to more compact molten globule structures with varying degrees of residual secondary structural elements (10). The rapid inter-conversion of an IDP between heterogeneous conformations confers several unique kinetic and thermodynamic advantages (11–13). The large hydrodynamic radius of a disordered region can increase the capture radius of the protein (14, 15). “Fly casting” contributes to the high kon rates often associated with IDPs (16). The ability to adopt multiple conformations enables the molecular promiscuity typical of many IDPs (17). From a thermodynamic perspective, the entropic cost of coupled folding and binding results in high specificity, low affinity interactions, which are advantageous in cellular signaling and regulatory response (12, 13). Additionally, many IDPs have a conformational preference consistent with the bound state (14). Altering the conformational preference of an IDP could modulate the favorability of a protein partner interaction (18). One cellular event regulating protein function that causes significant structural rearrangement and is disproportionately high for IDPs is phosphorylation (9, 19).
The Cx43CT is differentially phosphorylated at a dozen or more residues at various times throughout the Cx43 life cycle (20–24). Many kinases have been identified to phosphorylate Cx43 with the predominant effect being a decrease in GJIC. For example, following an ischemic event, the resulting intracellular acidification stimulates changes in the Cx43CT phosphorylation, which leads to closure of gap junction channels (25). Unfortunately, an understanding of the mechanism(s) by which Cx43 phosphorylation alters channel communication is lacking. This is best exemplified by the functional opposition of Cx43 phosphorylation at Ser-365 (PKA-mediated) and Ser-368 (PKC). Although the sites are in close proximity, phosphorylation of Ser-368 results in closure of Cx43 gap junction channels, whereas phosphorylation at Ser-365 increases GJIC and acts as a gatekeeper by blocking Ser-368 phosphorylation-induced channel closure (26). Evidence has emerged that a phosphate can directly affect binding of the Cx43CT domain with molecular partners by blocking the binding site (e.g. ZO-1 (27) and tubulin (28)) or by being directly involved in the interaction (e.g. Nedd4 (29, 30)). Alternatively, phosphorylation could affect indirectly interactions between IDPs and protein partners by altering the conformational preference of the disordered region (18, 31), a mechanism that has not been previously investigated for Cx43.
In this study, CD and NMR were used to characterize global and local effects of phosphorylation on the secondary structure and backbone dynamics of soluble and membrane-tethered Cx43CT domains. Asp substitutions were used to mimic phosphorylation by kinases that play an important role in the regulation of Cx43 (Table 1) (for review, see Ref. 32). The results indicate phosphorylation can alter the α-helical propensity of the Cx43CT at residues proximal and distal to the site(s) of modification. The changes in secondary structure are due to variations in the conformational preference and backbone flexibility of residues adjacent and distal to the site(s) of modification. Changes in the conformational preference of an IDP is one mechanism by which protein partner interactions can be regulated and it represents a novel mechanism for regulation of GJIC.
TABLE 1.
Cx43CT phosphorylation
mp means mediated phosphorylation.
| Kinase | Phosphorylation site | GJIC |
|---|---|---|
| Src | Tyr-247, Tyr-265 | Decrease |
| MAPKa | Ser-255, Ser-279, Ser-282 | Decrease |
| PKC | Ser-368, Ser-372 | Decrease |
| PKA-mp | Ser-364, Ser-365, Ser-369, Ser-373 | Increase |
| CK1 | Ser-325, Ser-328, Ser-330 | Increase |
| cdc2 | Ser-255, Ser-262 | Decrease |
a TM4-Cx43CT MAPK isoform Asp-substituted only at S282D.
EXPERIMENTAL PROCEDURES
Protein Purification
The expression and purification of the soluble Cx43CT (Val-236–Ile-382) and Cx43CT domain when attached to the 4th transmembrane domain (TM4-Cx43CT; Asp-197–Ile-382) in detergent micelles have been previously described (33, 34). Phospho-mimetic isoforms were created by substituting sites of Ser/Tyr phosphorylation (Table 1) with Asp using site-directed mutagenesis (QuikChange Lightning Kit, Agilent Technologies). Purifications of the phospho-mimetic isoforms were done using the same protocols as the soluble and membrane-tethered wild-type (WT) proteins. All plasmid sequences were verified at the University of Nebraska Medical Center DNA Sequencing Core Facility. Protein purity was assessed using SDS-PAGE.
Circular Dichroism (CD)
CD experiments were performed using a Jasco J-815 spectrophotometer fitted with a Peltier temperature control system. The soluble Cx43CT (100 μm) spectra were recorded at 7 °C in 1× phosphate-buffered saline (PBS) and 1 mm DTT at pH 5.8 or pH 7.5. Spectra for the TM4-Cx43CT (100 μm) were recorded at 42 °C in 20 mm MES buffer, 50 mm NaCl, 1 mm EDTA, 1 mm DTT, 8% (w/v) 1-palmitoyl-2-hydroxy-sn-glycero-3-[phospho-rac-(1-glycerol)] (LPPG; Avanti) at pH 5.8 or 7.5. Additional CD spectra were collected for the TM4-Cx43CT isoforms in the presence of 10% (v/v) 2,2,2-trifluoroethanol (TFE). For each sample, five scans (wavelength range, 190–300 nm; response time, 1 s; scan rate, 50 nm/min; bandwidth, 1.0 nm) were collected using a 0.01-cm quartz cell and processed using Spectra Analysis (Jasco). Protein concentrations for the soluble Cx43CT constructs were determined using a NanoDrop 1000 UV-visible spectrophotometer (Thermo Scientific). The concentration of the TM4-Cx43CT samples was ascertained using the high tension voltage to optical density conversion at 280 nm in spectra analysis. CD data were deconvoluted with Dichroweb (35–38).
Nuclear Magnetic Resonance (NMR)
NMR data were acquired using a 600 MHz Varian INOVA NMR spectrometer fitted with a cryo-probe (Agilent Technologies). All NMR data for the soluble Cx43CT (200 μm) and TM4-Cx43CT (800 μm) were collected at 7 and 42 °C, respectively. Backbone assignments of the soluble Cx43CT were obtained using the following three-dimensional experiments: HNCACB, HN(CO)CACB, HNCO, HN(CA)CO, and HNCACO. NMR data for the TM4-Cx43CT constructs were obtained in the presence of 10% TFE. Gradient-enhanced 15N HSQC experiments used to observe backbone amide resonances were acquired with 1024 complex points in the direct dimension and 256 complex points in the indirect dimension. 15N NOESY-HSQC data were collected with a mixing time of 50 ms. The backbone 15N R1 (inversion recovery) and R2 (Carr-Purcell-Meinboom-Gill) NMR relaxation measurements were collected with 1024 and 128 complex points in 1H and 15N, respectively, with 32 scans per R1 point and a recycle delay of 1 s. The R1 longitudinal relaxation delays were 10, 20, 40, 80, 140, 200, 300, 400, 600, 800, 1200, 2000, and 4000 ms. The R2 relaxation delays were 10, 30, 50, 70, 110, 150, 190, and 230 ms, with a 1-ms delay between 15N pulses. The 1H-15N NOE data were acquired with and without 1H saturation. All NMR spectra were processed using NMRPipe (39) and analyzed using NMRView (40). Relaxation data were processed by rate analysis in NMRView.
RESULTS
Rationale for Asp-substituted Cx43CT Isoforms
Phosphorylation of connexins has been implicated in the regulation of GJIC. However, several problems have contributed to the difficulty of elucidating the mechanism(s) by which a single kinase affects GJIC as follows: the transient nature of a particular Cx43CT phosphorylation state, the ability of many kinases to phosphorylate more than one Cx43CT residue, the ability of various kinases to phosphorylate the Cx43CT at the same time, and the inability to precisely control which residue(s) are phosphorylated (of the 27 Ser/Tyr residues in the Cx43CT, all but 8 have been observed to be phosphorylated (41, 42)). Strategies employed to overcome these difficulties include the use of phosphorylation-specific Cx43CT antibodies (43) and short Cx43CT phosphopeptides (27, 28). Also well appreciated is the use of Asp (or Glu) substitutions as a mimetic for phosphorylation. Asp substitutions allow for the precise control of the site(s) modified and the production of enough protein for biophysical studies. Although the size and charge of an Asp(−1) is not identical to a phosphate (−2 at pH 7.5 and >−1 at pH 5.8), in vivo and in vitro studies have confirmed Asp (or Glu) substitutions can elicit a response similar to a phosphate in Cx43 gap junction channels (26, 44, 45). Data were collected at either physiological (pH 7.5) or acidic (pH 5.8) conditions. Intracellular acidification, which is a major consequence of tissue ischemia during a myocardial infarction, leads to closure and degradation of gap junction channels and can be a substrate for malignant ventricular arrhythmias (46).
Electrophoretic Mobility of Asp-substituted Soluble Cx43CT Isoforms
Numerous studies have shown that differentially phosphorylated Cx43 results in multiple electrophoretic isoforms as follows: a fast migrating isoform, which includes the nonphosphorylated isoform (P0), and multiple slower migrating isoforms (P1 and P2) (20, 47, 48). Here, Asp substitutions were cloned into the soluble version of Cx43CT (Val-236–Ile-382) and each phospho-mimetic isoform was analyzed by SDS-PAGE (Fig. 1). The data indicate that the wild-type (WT) Cx43CT (nonphosphorylated) and the Src, PKC, and cdc2 phospho-mimetic isoforms migrate at P0, MAPK at P1, and CK1 at P2 (Fig. 1A, top and middle). These results are consistent with the known phosphorylation state(s) that compose the P0, P1, and P2 isoforms. Thus, Asp-substituted Cx43CT isoforms mimic characteristics of endogenously phosphorylated Cx43. PKA-mediated phosphorylation at Ser-365 is typically present in the P1 isoform; however, the soluble Cx43CT S365D phospho-mimetic isoform migrated at P0 (Fig. 1A, top). In addition to Ser-365, PKA can also phosphorylate Cx43 at Ser-364, Ser-369, and Ser-373. Therefore, the migration pattern for each PKA-mediated phosphorylation site alone and together was analyzed (Fig. 1A, bottom). The data reveal a shift in migration from P0 to P1 is dependent upon PKA-mediated phosphorylation at multiple Cx43CT sites. The effect of single-site phosphorylation by each of the kinases was also tested, and none caused a shift in the migration pattern (results are summarized in Fig. 1B).
FIGURE 1.

Electrophoretic mobility of Cx43. SDS-PAGE analysis of WT and phospho-mimetic isoforms of the Cx43CT indicating shifts in electrophoretic mobility. Substitutions are listed along with the kinase responsible for the phosphorylation of Cx43. The soluble Cx43CT is shown in A along with a summary of the soluble CT results in B. The membrane-tethered TM4-Cx43CT is shown in C. P0, P1, and P2 SDS-PAGE mobility shifts are typical of full-length, endogenously phosphorylated Cx43.
Phosphorylation-dependent shifts in electrophoretic mobility are not phenomena unique to connexins. Protein phosphorylation occurring on residues adjacent to a Pro has been reported to induce SDS-resistant changes in structure that alter electrophoretic mobility (49). The altered mobility is likely due to structural perturbations that interfere with the formation of an “ideal” random coil in the presence of SDS. Additionally, the negative charges on the phosphate group can interfere with protein-SDS interactions, thereby disrupting the charge-to-mass ratio and altering the electrophoretic mobility of the protein. However, this mechanism requires a number of phosphates to be close together. For the Cx43CT domain, both mechanisms likely contribute to the electrophoretic mobility of the phospho-isoforms. Next, the possibility that phosphorylation alters the secondary structure of the soluble Cx43CT domain was investigated by evaluating the residue-specific effects of phosphorylation using NMR.
Secondary Structural Studies Using the Soluble Cx43CT Domain
The structure of the soluble Cx43CT (Ser-255–Ile-382) was determined by NMR (45). In this study, a longer construct (Val-236–Ile-382) is used, which incorporates the other Src phosphorylation site at Tyr-247. All WT Cx43CT backbone 1H, 15N, and 13C resonances (147 residues; 15 Pro) were assigned to identify specific residues and/or regions that are affected by phosphorylation. Fig. 2A is the assigned WT Cx43CT 15N HSQC spectrum. When the WT Cx43CT 15N HSQC spectrum is compared with spectra from all the phospho-mimetic isoforms, several residues shifted. However, these shifts are small, of similar line width, and localized next to the site of substitution (e.g. CK1 isoform; Fig. 2B). The small chemical shift differences could be the result of altered secondary structure or due to changes in the chemical environment of residues neighboring the Asp substitutions. Therefore, the CD spectrum of the WT Cx43CT was compared with each of the phospho-mimetic isoforms to determine whether the differences detected by NMR were the result of a change in secondary structure.
FIGURE 2.

15N HSQC spectra of the soluble Cx43CT (Val-236–Ile-382). A, peak assignments for the backbone amides are indicated with numbering corresponding to the full-length Cx43 protein. B, 15N HSQC spectrum of wild-type Cx43CT (black) overlaid with the CK1 phospho-mimetic isoform spectrum (red). Residues affected by the amino acid substitutions are numbered in the spectra.
CD spectra of proteins have distinct MRE values for random coil, β-sheet, and α-helical structures (50). The CD spectrum of the soluble WT Cx43CT at pH 5.8 has a peak minimum near 198 nm and a small minimum at 222 nm (Fig. 3A), characteristic of an IDP with a small amount of α-helical content. This observation is consistent with previous CD data and the NMR solution structure of the shorter Cx43CT (Ser-255–382) domain (7, 51). Surprisingly, the CD spectra for all of the phospho-mimetic isoforms are nearly identical to the WT Cx43CT at pH 5.8 (Fig. 3A). The phospho-mimetic isoforms are also compared at pH 7.5 (Fig. 3B) to determine whether a phosphorylation-induced structural change was contingent upon pH (46). The CD data indicate phosphorylation does not alter the secondary structure of the soluble Cx43CT. This suggests the differences in NMR chemical shifts are solely due to changes in the amino acid sequence. The possibility exists that the lack of a phosphorylation-induced structural change is due to the increased flexibility of the soluble Cx43CT domain.
FIGURE 3.
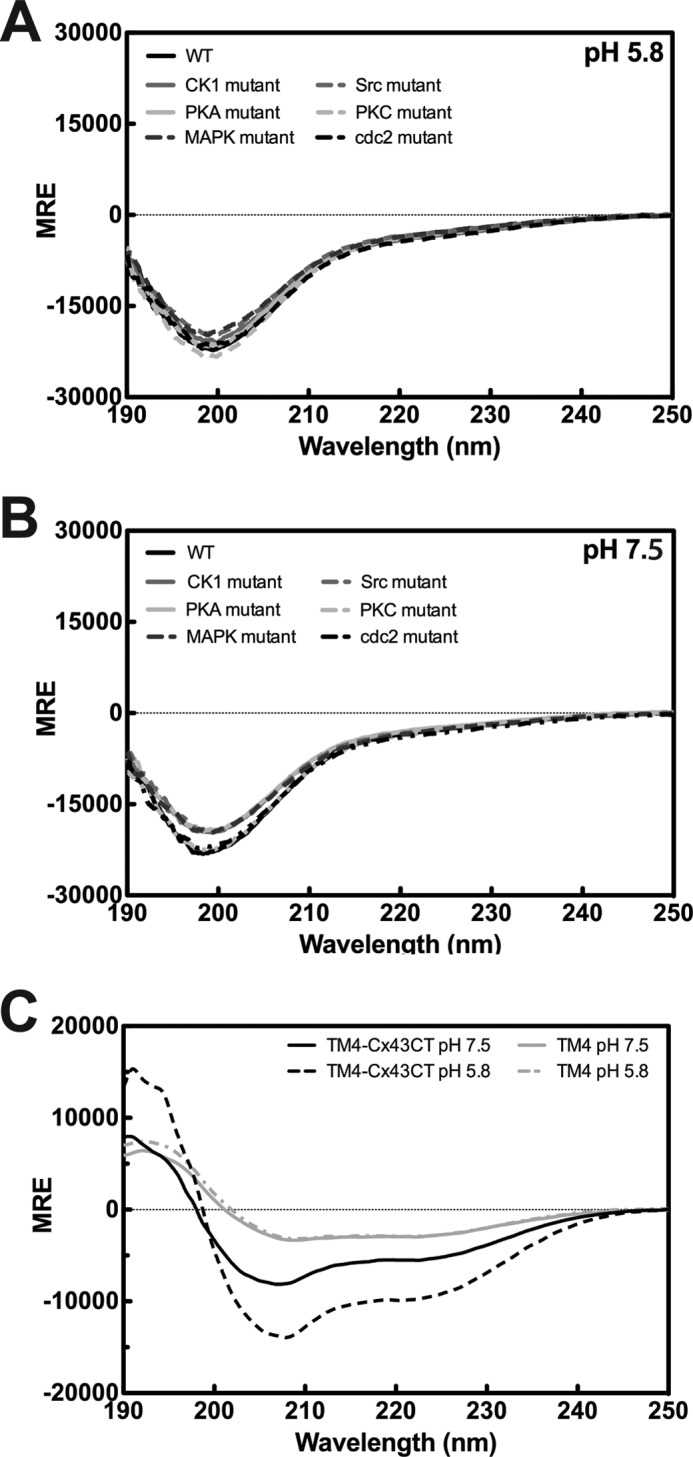
Secondary structure the Cx43CT. The CD spectra for the soluble Cx43CT and the phospho-mimetic isoforms at pH 5.8 (A) and pH 7.5 (B) (WT, black line; isoform name for kinase responsible for the endogenous phosphorylation, grayscale). C, CD spectra of the Cx43CT attached to the 4th transmembrane domain (TM4-Cx43CT; black lines) and the TM4 (Asp-197–Val-240; gray lines), which consists of the fourth transmembrane domain and lacks the C-terminal domain. Spectra were collected at pH 7.5 (solid lines) and pH 5.8 (dotted lines). Units are in MRE.
Asp-substituted Membrane-tethered Cx43CT Isoforms
Our previous study determined that the Cx43CT, when attached to the 4th transmembrane domain (TM4-Cx43CT), has more native-like characteristics than the soluble Cx43CT (34). Therefore, the TM4-Cx43CT construct was used to investigate the effects of phosphorylation on the secondary structure of the Cx43CT. SDS-PAGE analysis of the soluble Cx43CT indicated multiple site modifications are needed to mimic the shifts seen with endogenously phosphorylated Cx43 (Fig. 1B). Therefore, multiple Asp substitutions were cloned into the TM4-Cx43CT to mimic complete phosphorylation by the kinases listed in Table 1, except MAPK. For the MAPK isoform (S255D,S279D,S282D), various attempts (e.g. changes in growth and expression conditions; various cell lines), including a double substitution (S279D,S282D), were made to express the phospho-mimetic isoform without success. Two MAPK single substitutions were expressed and purified, TM4-Cx43CTS279D and TM4-Cx43CTS282D. The TM4-Cx43CTS282D was chosen for characterization given that phosphorylation at Ser-282 is sufficient to induce Cx43 channel closure (52). The membrane-tethered phospho-isoforms exhibited SDS-PAGE mobility shifts consistent with the soluble Cx43CT phospho-isoforms Cx43 (Fig. 1C). Notably, the different electrophoretic mobility between MAPK S282D (Fig. 1C, TM4-Cx43CT) and S255D,S279D,S282D (Fig. 1A, soluble Cx43CT) is likely caused by the number of negative charges needed to cause a shift in the gel.
Secondary Structural Studies Using the Membrane-tethered Cx43CT Domain
In Fig. 4, the CD spectrum of the WT TM4-Cx43CT is compared with the phospho-mimetic isoforms at pH 5.8 and 7.5. MRE at 222 nm was used as a gauge for changes in α-helical content (50). The data indicate phosphorylation can alter both the α-helical content of the TM4-Cx43CT as well as the structural responsiveness to acidification (Table 2). The CK1 and MAPK isoforms (Fig. 4, A and B) are more α-helical than the WT TM4-Cx43CT at pH 7.5 and exhibit an increase in α-helical content upon acidification. Conversely, the α-helical content of the Src isoform is similar to the WT TM4-Cx43CT at pH 7.5 (Fig. 4C) but is unaffected by a decrease in pH. Likewise, the PKA-mediated and PKC isoforms show little to no change in secondary structure upon acidification (Fig. 4, D and E), but the α-helical content is similar to the WT TM4-Cx43CT at pH 5.8. Finally, the α-helical content of the cdc2 isoform at both pH values (Fig. 4F) exceeds WT TM4-Cx43CT at pH 5.8 (similar to the CK1 and MAPK isoforms), but it undergoes little to no change upon acidification (similar to the Src isoform). Including the observation that pH does not affect the secondary structure of the TM4 domain (residues Asp-197–Val-240; Fig. 3C), the data suggest phosphorylation alters the secondary structure of the membrane-tethered Cx43CT domain.
FIGURE 4.

Global effect of phosphorylation on the secondary structure of the TM4-Cx43CT. CD spectra of the TM4-Cx43CT phospho-mimetic isoforms (gray lines) compared with wild type (WT; black lines) at pH 7.5 (solid lines) and pH 5.8 (dotted lines) are given in A–F. Isoforms are named based on kinase responsible for the endogenous phosphorylation. Units are in MRE. Changes in α-helical content are determined by evaluation of the MRE at 222 nm (vertical solid line).
TABLE 2.
Effects of pH and phosphorylation on TM4-Cx43CT secondary structure
Helical content was determined using DichroWeb Provencher and Glöckner method analysis using the SP175 reference. The = , ↑, and ↓ indicate α-helical content relative to WT. mp means mediated phosphorylation.
| pH 7.5 |
pH 5.8 |
pH-mediated change in α-helical content | |||
|---|---|---|---|---|---|
| MREλ⎭ = 222 nm | α-Helical content | MREλ⎭ = 222 nm | α-Helical content | ||
| WT | −8221.3 | 29.6% | −13453.2 | 45.6% | Yes |
| Src | −8692.6 | 29.5% = | −8968.2 | 31.0% ↓ | No |
| cdc2 | −18447 | 51.4% ↑ | −16894.3 | 50.7% ↑ | No |
| CK1 | −11286.8 | 42.9% ↑ | −20873.9 | 57.3% ↑ | Yes |
| MAPK | −14747.7 | 44.6% ↑ | −25592.6 | 64.7% ↑ | Yes |
| PKA-mp | −11557.1 | 42.9% ↑ | −12418.9 | 41.5% ↓ | No |
| PKC | −10786.2 | 39.5% ↑ | −12298.3 | 39.3% ↓ | No |
To complement the global perspective provided by the CD data, NMR was used to investigate phosphorylation-mediated changes in secondary structure on a residue-by-residue basis. Previously, we determined the NMR backbone assignment of the WT TM4-Cx43CT at pH 5.8 in the presence of 10% (v/v) TFE (53). TFE was used to stabilize innate α-helical content and improve NMR spectra (33). Importantly, 10% TFE did not induce α-helical structure in the WT TM4-Cx43CT (34) or the phospho-mimetic isoforms as assayed by CD (data not shown). Therefore, NMR 15N HSQC spectra of all TM4-Cx43CT phospho-mimetic isoforms were collected and superimposed onto the TM4-Cx43CT WT spectrum (Fig. 5, A–F). All affected Cx43CT residues from each phospho-mimetic isoform were mapped onto the Cx43CT sequence (Fig. 5G; Table 3). Many of the peaks that were altered in the phospho-mimetic isoforms either disappeared or were unable to be identified due to such large changes in chemical shift. Unlike the soluble Cx43CT, chemical shift changes in the TM4-Cx43CT phospho-mimetic isoforms were localized not only to regions adjacent to the site of amino acid substitution but propagated throughout the entire Cx43CT domain.
FIGURE 5.

Residue-specific effects of phosphorylation on the TM4-Cx43CT. The 15N HSQC spectra of the TM4-Cx43CT (black) overlaid with 15N HSQC spectra of each of the phospho-mimetic isoforms (red; A–F). Phospho-mimetic isoform chemical shifts that were different from WT are mapped to the Cx43CT sequences given in G. Blue letters, residues with minor changes in chemical shift (peaks are still slightly overlapped); red letters, residues with large changes in chemical shift or peaks that disappeared; green, underlined* letters, sites of Asp substitution; black lines indicate the seven regions previously predicted to be α-helical. Phospho-mimetic isoforms are named based on kinase responsible for the endogenous phosphorylation.
TABLE 3.
Residues with significant changes in chemical shift
mp means mediated phosphorylation.
| Totala | Used in relaxation analysisb | |
|---|---|---|
| Src | 21 | 2 (Ala-276, Ser-279) |
| cdc2 | 15 | 2 (Tyr-286, Asp-340) |
| CK1 | 23 | 4 (Ile-327, Asn-329, Asp-340, Ile-358) |
| MAPK | 29 | 7 (Met-281, Tyr-286, Lys-346, Ser-368, Ser-369, Ala-371, Ser-372) |
| PKA-mp | 44 | 8 (Ser-279, Gly-291, Glu-307, Asn-312, Tyr-313, Ala-344, Ala-349, Ala-371) |
| PKC | 14 | 4 (Ser-306, Ala-357, Ile-358, Ala-371) |
a Residues with large chemical shifts or that disappeared (red peaks in Fig. 5G) are indicated.
b Residues were identified based on WT assignment (residue number in parentheses).
The 15N HSQC spectra at pH 5.8 of the CK1, MAPK, and cdc2 isoforms each show upfield peak shifts (Fig. 5, A–C, arrows). Upfield shifts were expected because these isoforms have more α-helical content (as determined by CD) than the WT TM4-Cx43CT. Many of the CK1, MAPK, and cdc2 isoform peaks are weak or have increased line widths (Fig. 5, A–C, circles). The increase in line width suggests these residues are in exchange between a disordered and ordered structure. The Src isoform has several peaks that shift downfield and/or increase line widths compared with the WT TM4-Cx43CT at pH 5.8 (Fig. 5D, arrows). These observations are consistent with the CD data; Src phosphorylation destabilizes α-helical structure to favor a disordered conformation. The 15N HSQC for the PKC isoform is similar to the WT TM4-Cx43CT (Fig. 5E) with only a few chemical shift differences near the sites of Asp substitution. This observation is also consistent with the CD data. Conversely, the 15N HSQC spectrum of the PKA-mediated isoform is different (Fig. 5F), even though the CD data indicate the α-helical content is similar to both the WT TM4-Cx43CT and PKC isoforms. Numerous 15N HSQC peaks of the PKA-mediated isoform shift and/or change in line width. Additionally, there is an increase in the total number of resonance peaks, which is likely due to the presence of more than one stable conformation. This would explain the double band observed for the PKA-mediated isoform in the SDS-PAGE (Fig. 1C).
The phosphorylation-mediated effects could be the result of stabilizing an existing dynamic α-helix or disordered residues now transitioning between α-helical and random coiled structures (i.e. changing the conformational preference). Therefore, 15N NOESY-HSQC experiments were performed to determine which of these two possibilities account for the changes in Cx43CT secondary structure. Chemical shift analysis of the WT TM4-Cx43CT predicted seven α-helical regions along the C-terminal domain (53). However, many of the expected NOEs were not present in the 15N NOESY-HSQC spectrum indicating the α-helical regions are flexible and in exchange between a structured and unstructured conformation (Fig. 6, left panel). The CK1 isoform was selected as a representative phospho-mimetic isoform to study because the α-helical content of the CK1 isoform is greater than WT and increases upon acidification. Additionally, unlike the MAPK isoform, all of the CK1 phosphorylation sites are Asp-substituted. The CK1 isoform 15N NOESY-HSQC spectrum was collected using the identical NMR parameters, protein concentrations, and solution conditions as WT. Upon visualization of the CK1 isoform data, NOE peaks had broad line widths and weak signal intensity, when compared with the WT TM4-Cx43CT (Fig. 6, middle panel). A ×3 magnification of the CK1 spectrum was required to obtain the same signal intensity as the WT spectrum (Fig. 6, right panel). If the change in CT phosphorylation state had stabilized α-helical structure, then additional peaks would have been detected in the CK1 isoform 15N NOESY-HSQC. Even though the CD data indicate an increase in the overall α-helical content, the NOESY data suggest the TM4-Cx43CT CK1 isoform is in intermediate exchange. This observation is consistent with phosphorylation altering the fluctuation of Cx43CT residues between random coiled and α-helical structures.
FIGURE 6.

Effects of phosphorylation on TM4-Cx43CT inter-residue NOEs. 15N NOESY-HSQC spectra, WT TM4-Cx43CT (left panel) and CK1 isoform (middle) shown at level = 1×; the right panel is the spectrum of the CK1 isoform at level = 3×.
NMR Relaxation Studies of the Membrane-tethered Cx43CT Domain
All proteins undergo time-dependent molecular motions that affect NMR relaxation rates. Molecular motions, such as protein folding, side chain rotation, molecular tumbling, and bond vibrations occur over a broad time scale (1–10−15 s (54)). The relaxation parameters R1, R2, and 1H-15N NOE describe site-specific chemical exchange processes (e.g. a nucleus exchanging between environments) occurring on the picosecond to nanosecond time scale (12, 55). For a disordered protein, the folding energy landscape is relatively flat compared with a structured protein (i.e. no energetically favored conformation) resulting in greater protein picosecond to nanosecond dynamics. One of the difficulties of working with an IDP is low NMR peak dispersion, which often results in peak overlap and limits the number of peaks suitable for measurement. NMR relaxation data were collected for WT TM4-Cx43CT and each of the phospho-mimetic isoforms to determine the effect(s) of phosphorylation on the backbone dynamics of the Cx43CT domain. Relaxation measurements were obtained for 80 of the 125 assigned WT TM4-Cx43CT non-proline peaks. For the phospho-mimetic isoforms, fewer peaks were used due to additional overlap, disappearance, and/or ambiguous identification (Table 3). In total, R1 and R2 relaxation measurements were collected for 67 peaks of the MAPK and Src isoforms, 65 for the cdc2 isoform, 54 for the PKC isoform, 53 for the CK1 isoform, and 51 for the PKA-mediated isoform. Of note, 15N HSQC assignment of the phospho-mimetic isoforms was determined by comparing the chemical shifts of the each of the isoforms to the previously determined WT chemical shifts (53). Unfortunately, the most interesting phospho-mimetic isoform peaks, which are those that have significant changes in chemical shift, could not be matched to a corresponding WT peak (e.g. CK1 isoform, only 4 of 23 peaks with significant changes could be unambiguously identified). Table 3 provides the phospho-mimetic isoform peaks of interest that were suitable for relaxation analysis.
The WT TM4-Cx43CT R1 measurements show a relatively flat profile with a steep decline at the C terminus, characteristic of a disordered protein (Fig. 7A) (51). The mean R1 rate of 1.46 s−1 (minimum value, 0.81 s−1; maximum value, 1.70 s−1) and R2 rate of 5.25 s−1 (minimum value, 1.08 s−1; maximum value, 12.37 s−1; Fig. 7B) also indicate fast nanosecond to picosecond time scale dynamics consistent with the TM4-Cx43CT being an IDP. R1 and R2 values of structured protein are ∼2 and 4 times greater than the corresponding values of a disordered protein of similar size, respectively (56, 57). 1H-15N NOE data are also consistent with fast dynamics (Fig. 7C, mean = −0.15).
FIGURE 7.

TM4-Cx43CT backbone dynamics. 15N relaxation parameters R1 (A), R2 (B), and 15N heteronuclear NOE (C) were measured at 600 MHz. Bars indicate the curve-fit root mean square deviation for each point. The residue specific R2/R1 and R2R1 are given in D and E, respectively. Peaks with chemical shift overlap were excluded from the analysis. Mean values (solid line) and standard deviations (dotted) are indicated.
Several residues along the Cx43CT have R1 and R2 values >1 standard deviation (S.D.) above the mean (R1, Ala-311, Ala-315–Gln-317, Ala-367, and Ala-371; R2, Tyr-286, Gly-291, Asn-295, Tyr-301 Ala-357, and Ile-358), suggesting they are less flexible. These residues overlap with α-helical regions predicted from the chemical shifts of the WT TM4-Cx43CT domain (53), which encompasses the two α-helical regions of the soluble Cx43CT (7). However, the R1 and R2 values of the α-helical regions are still within the range of an IDP and are not suggestive of rigid secondary structure. R1 and R2 values ≥1 S.D. below the mean were observed from the largest stretch of consecutive disordered residues (Ala-323–Asp-339) from the WT TM4-Cx43CT predicted structure, indicating increased flexibility. Another observation was that many of the residues differ between the R1 and R2 data. One possible explanation is the contribution of microsecond to millisecond chemical exchange to R2 (58).
A common approach used in analysis of NMR R1 and R2 relaxation data is to consider the R2/R1 ratio and the R2R1 product (59, 60). If the sequence-specific rates of protein motion are isotropic, as is typical for a well folded protein, then the R2/R1 ratio for each residue will be similar. R2/R1 values that differ from the mean indicate anisotropic motions on the picosecond to nanosecond time scale. One caveat to this analysis is that R2 relaxation rates often have a large contribution by exchange processes on the microsecond to millisecond time scale (59). The R2R1 product mitigates the contribution of anisotropic motions on the picosecond to nanosecond time scale such that deviations >1 S.D. above the mean are indicative of chemical exchange on the microsecond to millisecond time scale (60). Residues within the regions Ser-282–Tyr-286, Arg-293–Cys-298, and Leu-354–Cys-358 have R2/R1 ratios >1 S.D. above the mean indicating less picosecond to nanosecond motion (Fig. 7D). However, the WT TM4-Cx43CT R2/R1 ratio profile is nearly identical to the R2R1 product profile (Fig. 7E). This suggests deviations >1 S.D. above the mean are due to chemical exchange on the microsecond to millisecond time scale rather than anisotropic motion.
Also examined were the relaxation profiles of the phospho-mimetic isoforms. The R2/R1 ratios for each of the phospho-mimetic isoforms (data not shown) are nearly identical to their respective R2R1 product profiles (Fig. 8), suggesting values >1 S.D. above the mean are also the result of chemical exchange processes. With the exception of the PKA isoform, all of the phospho-mimetic isoforms have two major areas with R2R1 values >1 S.D. above the mean. The areas overlap with WT regions Ser-282–Tyr-286 and Leu-354–Cys-358 but encompass slightly different residues. As an example, two regions of the CK1 isoform have increased R2R1 that include more residues (Fig. 8A, Ala–276–Thr-290 and Ser-357-Ser-362). The CK1 isoform also has residues >1 S.D. below the mean (Lys-346–Glu-352) indicating increased picosecond to nanosecond motion in this region. The PKA-mediated isoform R2R1 profile (Fig. 8F), which is the least similar to the WT profile, indicates four regions with R2R1 values greater than the mean (Gly-285–Asp-292, Asn-294–Arg-299, Ser-314–Gln-317, and Ala-344–Gly-350).
FIGURE 8.

Effects of phosphorylation on TM4-Cx43CT R2R1 backbone dynamics. 15N relaxation parameters (R1 and R2) for the phospho-mimetic isoforms were measured at 600 MHz. The R2R1 values are given for each of the isoforms in A–F. Residues with chemical shift overlap or ambiguous assignments (including * sites of substitution for the cdc2 and Src isoforms) were excluded from the analysis. Vertical lines indicate sites of Cx43CT phosphorylation; mean values (solid line) and standard deviations (horizontal dotted lines) are provided.
Finally, the 1H-15N NOE values for all phospho-mimetic isoforms were consistent with fast amide N–H bond vector dynamics (data not shown). Similar to the R1 and R2 data, there are slight differences in the phospho-mimetic isoform NOE profiles (e.g. CK1 isoform, residue Phe-337 shows slower dynamics than WT). Altogether, the NMR data suggest that even for the phospho-mimetic isoforms determined by CD to have the greatest α-helical content (e.g. MAPK, CK1, cdc2), the increased α-helical content is not manifesting in rigid structure. Rather more random coiled residues have now transitioned to fluctuating between random coiled and α-helical structure.
DISCUSSION
Utility of the TM4-Cx43CT Construct for Investigating Phosphorylation-induced Changes in Structure
The soluble Cx43CT has proven to be a useful model for studying the structure-function mechanisms regulating gap junctions. However, several results indicate the soluble Cx43CT may not be the best model system. For example, the electron crystallography structure of the CT-truncated Cx43 mutant (terminates at Thr-263) suggested that region Ser-255–Thr-263 adopts an α-helical structure (4, 61). NMR data for the soluble CT identified that peaks were weak from this region, suggesting these residues are in exchange between unstructured and α-helical conformations (62). Additionally, not all of the expected NOEs were observed in the two α-helical regions of the soluble CT structure. The differences can be attributed to the constraints imposed when attached to the membrane. Therefore, expression, purification, and NMR solution conditions were optimized for a more native-like construct, the TM4-Cx43CT solubilized in detergent micelles (34, 63). CD data indicated the TM4-Cx43CT has more α-helical content than can be attributed solely to the addition of the individual TM4 and C-terminal domains. The TM4-Cx43CT also undergoes a pronounced increase in α-helical content upon solvent acidification (i.e. ischemic conditions), unlike the soluble Cx43CT (Fig. 3C). The structural responsiveness to changes in pH and phosphorylation indicates the TM4-Cx43CT is a better model than the soluble Cx43CT for investigating structure-based mechanisms of Cx43 regulation.
Previously, we reported the backbone assignments and predicted secondary structure of the TM4-Cx43CT (53). The total amount of α-helical structure observed from CD data of the TM4-Cx43CT construct (46%) is consistent with the combined content from the seven predicted α-helical regions of the C-terminal domain and α-helical TM4 domain (34). However, not all of the NOEs were apparent in these α-helical regions suggesting they are flexible and in exchange between random coiled and α-helical conformations. Here, comparison of the TM4-Cx43CT CD spectra with a construct containing only the TM4 portion (Asp-197–Val-240) confirmed pH-dependent structural changes occurred only in the C-terminal domain; the α-helical content of the TM4 is unaffected by changes in pH. Therefore, we expect phosphorylation-mediated changes in secondary structure to also only occur in the C-terminal domain.
Phosphorylation Alters the Secondary Structure and Dynamics of the Cx43CT
One difficulty in understanding how Ser phosphorylation regulates protein function is the inability to predict whether phosphorylation will act to stabilize or destabilize secondary structure (31). For a disordered protein, the large negative charge of a phosphate could alter backbone hydration facilitating formation of intramolecular hydrogen bonds that stabilize secondary structure. The phosphate could also provide electrostatic (or steric) interactions that affect the formation of secondary structures. For the soluble Cx43CT domain, the effect on secondary structure correlated with the location of the phosphorylation. The PKA-mediated and PKC isoforms, which are modified at the C terminus of the Cx43CT, are both more α-helical than the WT TM4-Cx43CT at pH 7.5 but show little to no change in secondary structure upon acidification. The CK1 and MAPK isoforms, which have phosphorylation sites centrally located, have greater α-helical content than WT at pH 7.5 and become more α-helical upon acidification. Conversely, cdc2, which is closer to the N terminus, has more α-helical content at both pH 5.8 and 7.5 than WT but undergoes little to no change in α-helical content upon acidification. Although the use of an Asp substitution to mimic Tyr phosphorylation is not optimal, Asp was chosen over Glu for the Src isoform to maintain consistency with the other phospho-mimetic constructs. The Src isoform is less α-helical than the WT at pH 5.8, as would be expected if a true phosphate were present; Tyr phosphorylation, unlike Ser phosphorylation, generally disrupts α-helical structure (64). Furthermore, the Asp-substituted Tyr residues in the Src isoform elicited an opposite effect on α-helical content compared with Asp-substituted Ser residues in a similar location (i.e. cdc2 isoform).
The 15N HSQC spectra of the phospho-mimetic isoforms, except the PKC isoform, show the effects of phosphorylation to propagate along the C-terminal domain. The line widths of the peaks in the 15N HSQC spectra suggest residues are in intermediate exchange between more than one conformation. Collectively, the CD and 15N HSQC NMR data indicate phosphorylation alters the conformational preference of the C-terminal domain between random coiled and α-helical structure. Conformational exchange is consistent with the WT TM4-Cx43CT and CK1 isoform 15N NOESY-HSQC data. Most of the expected NOEs were not present for residues in the α-helical regions, leaving a limited number of NMR peaks suitable for relaxation analysis. The NMR relaxation data indicate the α-helical structure is dynamic and in exchange with a disordered conformation. Although slight differences were observed between WT and the phospho-mimetic isoforms, the most interesting peaks to analyze would be those with significant changes (Table 3; Fig. 5G, red residues) as these residues would have the greatest variation in flexibility and/or secondary structure. NMR is similar to CD as both are averages of the ensemble of protein conformations in solution. The increased α-helical content in some of the TM4-Cx43CT phospho-mimetic isoforms is not due to stabilization of rigid secondary structure; phosphorylation shifts the conformational preference of random coiled residues to form dynamic α-helical structure.
An NMR study using a nonphosphorylated and phosphorylated Cx43CT peptide (Lys-234–Asp-259) determined Src phosphorylation at Tyr-247 regulates the interaction between Cx43 and tubulin (28). The nonphosphorylated peptide adopted an α-helical conformation upon interaction with tubulin. However, phosphorylation at Tyr-247 had a negative impact on tubulin recognition. In the α-helical conformation, Tyr-247 would be on the hydrophobic face of the peptide involved in the tubulin interaction. Although the phosphate at Tyr-247 may act to directly inhibit tubulin binding, the data presented here suggest the phosphate may also inhibit this interaction by altering the conformational preference of these CT residues to a disordered state. This would destabilize the hydrophobic face necessary for the interaction. Changing the conformational preference of an IDP can fine-tune the thermodynamic favorability of protein partner interactions (14, 18). Backbone dynamics on the picosecond to nanosecond time scale have been implicated in determining ligand specificity and affinity and have been suggested as a regulatory mechanism for IDPs in general (17, 54).
How Does Phosphorylation Regulate GJIC?
Several hypotheses, which are not mutually exclusive, could explain how phosphorylation regulates GJIC at the plasma membrane as follows: 1) the negative charge of the phosphate could affect the permeability of ions through the pore via electrostatic attraction or repulsion; 2) the phosphate could stabilize favorable electrostatic interactions between the Cx43CT and the positively charged residues of the Cx43CL leading to blockage of the pore, and 3) phosphorylation of the CT could directly alter the binding affinities of proteins involved in Cx43 regulation (e.g. Nedd4 (30), 14-3-3 (65), and ZO-1 (27)). Moreover, the results presented here indicate phosphorylation mediates changes in the secondary structure and flexibility of the Cx43CT domain, providing support for additional mechanisms by which phosphorylation may regulate GJIC. Additionally, CT structural changes induced by phosphorylation could 4) alter channel permeability by indirectly modulating the orientation of the transmembrane α-helices thereby influencing pore size and/or 5) indirectly alter the binding affinity of molecular partners involved in regulating GJIC by altering the conformational preference of the Cx43CT domain.
Conclusion
This study shows the first direct evidence of phosphorylation-mediated structural changes of Cx43. Additionally these structural changes are not due to stabilization of rigid helical structures but rather are due to a change in the conformational preference of the Cx43CT. This is a regulatory mechanism previously demonstrated for other intrinsically disordered proteins but is a novel mechanism for Cx43 regulation. Importantly, qualitative analysis of both CD and NMR relaxation was needed to identify this novel mechanism. Future studies will work toward more detailed analysis of each of the phospho-mimetic isoforms in terms of how changes in the conformational preference mediate specific protein partner interactions involved in regulating Cx43 gap junctional communication.
Acknowledgments
We thank Ed Ezell for assistance with NMR and Jeff Lovelace for computer expertise.
This work was supported, in whole or in part, by National Institutes of Health Grant GM07263 from USPHS.
- GJIC
- gap junction intercellular communication
- Cx43
- connexin43
- Cx43CT
- connexin43 C-terminal domain
- CL
- cytoplasmic loop
- IDP
- intrinsically disordered protein
- TFE
- 2,2,2-trifluoroethanol
- MRE
- mean residue ellipticity.
REFERENCES
- 1. Kumar N. M., Gilula N. B. (1996) The gap junction communication channel. Cell 84, 381–388 [DOI] [PubMed] [Google Scholar]
- 2. Laird D. W. (2008) Closing the gap on autosomal dominant connexin-26 and connexin-43 mutants linked to human disease. J. Biol. Chem. 283, 2997–3001 [DOI] [PubMed] [Google Scholar]
- 3. Dobrowolski R., Willecke K. (2009) Connexin-caused genetic diseases and corresponding mouse models. Antioxid. Redox Signal. 11, 283–295 [DOI] [PubMed] [Google Scholar]
- 4. Unger V. M., Kumar N. M., Gilula N. B., Yeager M. (1999) Three-dimensional structure of a recombinant gap junction membrane channel. Science 283, 1176–1180 [DOI] [PubMed] [Google Scholar]
- 5. Maeda S., Nakagawa S., Suga M., Yamashita E., Oshima A., Fujiyoshi Y., Tsukihara T. (2009) Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature 458, 597–602 [DOI] [PubMed] [Google Scholar]
- 6. Dyson H. J., Wright P. E. (2004) Unfolded proteins and protein folding studied by NMR. Chem. Rev. 104, 3607–3622 [DOI] [PubMed] [Google Scholar]
- 7. Sorgen P. L., Duffy H. S., Sahoo P., Coombs W., Delmar M., Spray D. C. (2004) Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J. Biol. Chem. 279, 54695–54701 [DOI] [PubMed] [Google Scholar]
- 8. Xue B., Li L., Meroueh S. O., Uversky V. N., Dunker A. K. (2009) Analysis of structured and intrinsically disordered regions of transmembrane proteins. Mol. BioSyst. 5, 1688–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dunker A. K., Brown C. J., Lawson J. D., Iakoucheva L. M., Obradović Z. (2002) Intrinsic disorder and protein function. Biochemistry 41, 6573–6582 [DOI] [PubMed] [Google Scholar]
- 10. Dyson H. J., Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 11. Uversky V. N. (2011) Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 43, 1090–1103 [DOI] [PubMed] [Google Scholar]
- 12. Mittag T., Kay L. E., Forman-Kay J. D. (2010) Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 23, 105–116 [DOI] [PubMed] [Google Scholar]
- 13. Mészáros B., Simon I., Dosztányi Z. (2011) The expanding view of protein-protein interactions: complexes involving intrinsically disordered proteins. Physical Biology 8, 035003 [DOI] [PubMed] [Google Scholar]
- 14. Pancsa R., Fuxreiter M. (2012) Interactions via intrinsically disordered regions: what kind of motifs? IUBMB Life 64, 513–520 [DOI] [PubMed] [Google Scholar]
- 15. Shoemaker B. A., Portman J. J., Wolynes P. G. (2000) Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl Acad. Sci. U.S.A. 97, 8868–8873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang Y., Liu Z. (2009) Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the “fly-casting” mechanism. J. Mol. Biol. 393, 1143–1159 [DOI] [PubMed] [Google Scholar]
- 17. Chen J. (2012) Towards the physical basis of how intrinsic disorder mediates protein function. Arch. Biochem. Biophys. 524, 123–131 [DOI] [PubMed] [Google Scholar]
- 18. Wright P. E., Dyson H. J. (2009) Linking folding and binding. Curr. Opin. Struct. Biol. 19, 31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iakoucheva L. M., Radivojac P., Brown C. J., O'Connor T. R., Sikes J. G., Obradovic Z., Dunker A. K. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Solan J. L., Lampe P. D. (2007) Key connexin 43 phosphorylation events regulate the gap junction life cycle. J. Membr. Biol. 217, 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moreno A. P., Lau A. F. (2007) Gap junction channel gating modulated through protein phosphorylation. Prog. Biophys. Mol. Biol. 94, 107–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delmar M., Coombs W., Sorgen P., Duffy H. S., Taffet S. M. (2004) Structural bases for the chemical regulation of connexin43 channels. Cardiovasc. Res. 62, 268–275 [DOI] [PubMed] [Google Scholar]
- 23. Solan J. L., Lampe P. D. (2005) Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim. Biophys. Acta 1711, 154–163 [DOI] [PubMed] [Google Scholar]
- 24. Laird D. W. (2005) Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim. Biophys. Acta 1711, 172–182 [DOI] [PubMed] [Google Scholar]
- 25. Severs N. J. (2007) The carboxyl-terminal domain of connexin43: from molecular regulation of the gap junction channel to supramolecular organization of the intercalated disk. Circ. Res. 101, 1213–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Solan J. L., Marquez-Rosado L., Sorgen P. L., Thornton P. J., Gafken P. R., Lampe P. D. (2007) Phosphorylation at Ser-365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J. Cell Biol. 179, 1301–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen J., Pan L., Wei Z., Zhao Y., Zhang M. (2008) Domain-swapped dimerization of ZO-1 PDZ2 generates specific and regulatory connexin43-binding sites. EMBO J. 27, 2113–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saidi Brikci-Nigassa A., Clement M. J., Ha-Duong T., Adjadj E., Ziani L., Pastre D., Curmi P. A., Savarin P. (2012) Phosphorylation controls the interaction of the connexin43 C-terminal domain with tubulin and microtubules. Biochemistry 51, 4331–4342 [DOI] [PubMed] [Google Scholar]
- 29. Chung T. H., Wang S. M., Chang Y. C., Chen Y. L., Wu J. C. (2007) 18β-Glycyrrhetinic acid promotes src interaction with connexin43 in rat cardiomyocytes. J. Cell. Biochem. 100, 653–664 [DOI] [PubMed] [Google Scholar]
- 30. Leykauf K., Salek M., Bomke J., Frech M., Lehmann W. D., Dürst M., Alonso A. (2006) Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J. Cell Sci. 119, 3634–3642 [DOI] [PubMed] [Google Scholar]
- 31. Groban E. S., Narayanan A., Jacobson M. P. (2006) Conformational changes in protein loops and helices induced by post-translational phosphorylation. PLoS Comput. Biol. 2, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Solan J. L., Lampe P. D. (2009) Connexin43 phosphorylation: structural changes and biological effects. Biochem. J. 419, 261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duffy H. S., Sorgen P. L., Girvin M. E., O'Donnell P., Coombs W., Taffet S. M., Delmar M., Spray D. C. (2002) pH-dependent intramolecular binding and structure involving Cx43 cytoplasmic domains. J. Biol. Chem. 277, 36706–36714 [DOI] [PubMed] [Google Scholar]
- 34. Grosely R., Kieken F., Sorgen P. L. (2010) Optimizing the solution conditions to solve the structure of the connexin43 carboxyl terminus attached to the 4(th) transmembrane domain in detergent micelles. Cell Commun. Adhes. 17, 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Whitmore L., Woollett B., Miles A. J., Janes R. W., Wallace B. A. (2010) The protein circular dichroism data bank, a Web-based site for access to circular dichroism spectroscopic data. Structure 18, 1267–1269 [DOI] [PubMed] [Google Scholar]
- 36. Whitmore L., Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 37. Lees J. G., Miles A. J., Wien F., Wallace B. A. (2006) A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics 22, 1955–1962 [DOI] [PubMed] [Google Scholar]
- 38. Provencher S. W., Glöckner J. (1981) Estimation of globular protein secondary structure from circular dichroism. Biochemistry 20, 33–37 [DOI] [PubMed] [Google Scholar]
- 39. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 40. Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 [DOI] [PubMed] [Google Scholar]
- 41. Lampe P. D., Lau A. F. (2000) Regulation of gap junctions by phosphorylation of connexins. Arch. Biochem. Biophys. 384, 205–215 [DOI] [PubMed] [Google Scholar]
- 42. Huang R. Y., Laing J. G., Kanter E. M., Berthoud V. M., Bao M., Rohrs H. W., Townsend R. R., Yamada K. A. (2011) Identification of CaMKII phosphorylation sites in connexin43 by high resolution mass spectrometry. J. Proteome Res. 10, 1098–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sosinsky G. E., Solan J. L., Gaietta G. M., Ngan L., Lee G. J., Mackey M. R., Lampe P. D. (2007) The C terminus of connexin43 adopts different conformations in the Golgi and gap junction as detected with structure-specific antibodies. Biochem. J. 408, 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Remo B. F., Qu J., Volpicelli F. M., Giovannone S., Shin D., Lader J., Liu F. Y., Zhang J., Lent D. S., Morley G. E., Fishman G. I. (2011) Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ. Res. 108, 1459–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Léger J., Kempf M., Lee G., Brandt R. (1997) Conversion of serine to aspartate imitates phosphorylation-induced changes in the structure and function of microtubule-associated protein Tau. J. Biol. Chem. 272, 8441–8446 [DOI] [PubMed] [Google Scholar]
- 46. Lau A. F. (2005) c-Src: bridging the gap between phosphorylation- and acidification-induced gap junction channel closure. Sci. STKE 2005, pe33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Crow D. S., Beyer E. C., Paul D. L., Kobe S. S., Lau A. F. (1990) Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol. Cell. Biol. 10, 1754–1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Musil L. S., Cunningham B. A., Edelman G. M., Goodenough D. A. (1990) Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J. Cell Biol. 111, 2077–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith C. L., Debouck C., Rosenberg M., Culp J. S. (1989) Phosphorylation of serine residue 89 of human adenovirus E1A proteins is responsible for their characteristic electrophoretic mobility shifts, and its mutation affects biological function. J. Virol. 63, 1569–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bouvier D., Spagnol G., Chenavas S., Kieken F., Vitrac H., Brownell S., Kellezi A., Forge V., Sorgen P. L. (2009) Characterization of the structure and intermolecular interactions between the connexin40 and connexin43 carboxyl-terminal and cytoplasmic loop domains. J. Biol. Chem. 284, 34257–34271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Warn-Cramer B. J., Cottrell G. T., Burt J. M., Lau A. F. (1998) Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 273, 9188–9196 [DOI] [PubMed] [Google Scholar]
- 53. Grosely R., Kieken F., Sorgen P. L. (2012) 1H, 13C, and 15N backbone resonance assignments of the connexin43 carboxyl-terminal domain attached to the 4th transmembrane domain in detergent micelles. Biomol. NMR, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kleckner I. R., Foster M. P. (2011) An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta 1814, 942–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Choi U. B., Xiao S., Wollmuth L. P., Bowen M. E. (2011) Effect of Src kinase phosphorylation on disordered C-terminal domain of N-methyl-d-aspartic acid (NMDA) receptor subunit GluN2B protein. J. Biol. Chem. 286, 29904–29912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang X., Perugini M. A., Yao S., Adda C. G., Murphy V. J., Low A., Anders R. F., Norton R. S. (2008) Solution conformation, backbone dynamics, and lipid interactions of the intrinsically unstructured malaria surface protein MSP2. J. Mol. Biol. 379, 105–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wikström A., Berglund H., Hambraeus C., van den Berg S., Härd T. (1999) Conformational dynamics and molecular recognition: backbone dynamics of the estrogen receptor DNA-binding domain. J. Mol. Biol. 289, 963–979 [DOI] [PubMed] [Google Scholar]
- 58. Csizmok V., Felli I. C., Tompa P., Banci L., Bertini I. (2008) Structural and dynamic characterization of intrinsically disordered human securin by NMR spectroscopy. J. Am. Chem. Soc. 130, 16873–16879 [DOI] [PubMed] [Google Scholar]
- 59. Kay L. E., Torchia D. A., Bax A. (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28, 8972–8979 [DOI] [PubMed] [Google Scholar]
- 60. Kneller J. M., Lu M., Bracken C. (2002) An effective method for the discrimination of motional anisotropy and chemical exchange. J. Am. Chem. Soc. 124, 1852–1853 [DOI] [PubMed] [Google Scholar]
- 61. Cheng A., Schweissinger D., Dawood F., Kumar N., Yeager M. (2003) Projection structure of full-length connexin 43 by electron cryo-crystallography. Cell Commun. Adhes. 10, 187–191 [DOI] [PubMed] [Google Scholar]
- 62. Sorgen P. L., Duffy H. S., Cahill S. M., Coombs W., Spray D. C., Delmar M., Girvin M. E. (2002) Sequence-specific resonance assignment of the carboxyl-terminal domain of connexin43. J. Biomol. NMR 23, 245–246 [DOI] [PubMed] [Google Scholar]
- 63. Kellezi A., Grosely R., Kieken F., Borgstahl G. E., Sorgen P. L. (2008) Purification and reconstitution of the connexin43 carboxyl terminus attached to the 4th transmembrane domain in detergent micelles. Protein Expr. Purif. 59, 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Muratore K. E., Cole P. A. (2007) A lock on phosphotyrosine signaling. ACS Chem. Biol. 2, 454–456 [DOI] [PubMed] [Google Scholar]
- 65. Park D. J., Freitas T. A., Wallick C. J., Guyette C. V., Warn-Cramer B. J. (2006) Molecular dynamics and in vitro analysis of connexin43: A new 14-3-3 mode-1 interacting protein. Protein Sci. 15, 2344–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]