

Background: Multiple signaling pathways are disrupted in ovarian cancer, but the role of protein phosphatases is not appreciated.
Results: PTP1B antagonizes signaling by IGF-1R and BRK/PTK6 in ovarian cancer cells.
Conclusion: Decreased expression of PTP1B in ovarian cancer cells promotes migration, invasion, proliferation, and survival.
Significance: PTP1B, which dephosphorylates the insulin receptor and is an important therapeutic target in diabetes, may antagonize IGF-1-induced signaling in specific contexts.
Keywords: Cell Death, Ovarian Cancer, Phosphorylation, Tyrosine-protein Kinase (Tyrosine Kinase), Tyrosine-protein Phosphatase (Tyrosine Phosphatase), BRK, Dephosphorylation, IGF-1R, PTP1B
Abstract
Ovarian cancer, which is the leading cause of death from gynecological malignancies, is a heterogeneous disease known to be associated with disruption of multiple signaling pathways. Nevertheless, little is known regarding the role of protein phosphatases in the signaling events that underlie the disease; such knowledge will be essential to gain a complete understanding of the etiology of the disease and how to treat it. We have demonstrated that protein-tyrosine phosphatase 1B (PTP1B) was underexpressed in a panel of ovarian carcinoma-derived cell lines, compared with immortalized human ovarian surface epithelial cell lines. Stable restoration of PTP1B in those cancer cell lines substantially decreased cell migration and invasion, as well as proliferation and anchorage-independent survival. Mechanistically, the pro-survival IGF-1R signaling pathway was attenuated upon ectopic expression of PTP1B. This was due to dephosphorylation by PTP1B of IGF-1R β-subunit and BRK/PTK6, an SRC-like protein-tyrosine kinase that physically and functionally interacts with the IGF-1R β-subunit. Restoration of PTP1B expression led to enhanced activation of BAD, one of the major pro-death members of the BCL-2 family, which triggered cell death through apoptosis. Conversely, inhibition of PTP1B with a small molecular inhibitor, MSI-1436, increased proliferation and migration of immortalized HOSE cell lines. These data reveal an important role for PTP1B as a negative regulator of BRK and IGF-1Rβ signaling in ovarian cancer cells.
Introduction
Ovarian cancer is the leading cause of death from gynecological malignancies and ranks fifth of all cancer-related deaths in women (1). According to the Ovarian Cancer National Alliance, a woman's risk of developing invasive ovarian cancer during her life is 1 in 72. Despite recent advances in surgery and chemotherapy, major problems remain. In particular, the disease is often diagnosed late, when it is already at an advanced stage with metastasis to other organs, and it is difficult to treat. In contrast, survival for those patients diagnosed at an early stage, when the disease is still limited to the ovaries, improves dramatically. The standard therapy for advanced disease is tumor debulking, followed by platinum- and taxane-based chemotherapies. Unfortunately, the vast majority of these patients will relapse and develop resistance to the chemotherapy. Consequently, there is an urgent need to elucidate the molecular basis for the regulation of signaling pathways underlying ovarian tumorigenesis and cancer progression as a first step toward identifying better strategies for therapeutic intervention, in addition to potential biomarkers for the disease (2).
Despite the fact that most ovarian cancers are of epithelial origin, this is a remarkably heterogeneous disease associated with multiple genetic and epigenetic changes (3). Fifteen tumor suppressors and 16 oncogenes have been implicated in the disease, suggesting disruption of multiple signaling pathways (3). Of the tumor suppressors, mutation or loss of p53 is one of the most frequent changes in high grade tumors, together with DNA repair defects induced by mutations in BRCA1 and BRCA2. A second category of low grade tumors is defined by mutations in KRAS, BRAF, and the PIK3CA genes encoding the catalytic subunit of PI 3-kinase (3). Multiple signaling pathways are disrupted, including PI 3-kinase, driven not only by activating mutations in the kinase and AKT, but also inactivating mutations in PTEN (phosphatase and tensin homolog), overexpression of IL-6 leading to activation of JAK-STAT signaling, up-regulation of lysophosphatidic acid receptors, and constitutive activation of NF-κB (3). More recently, attention has also focused on the protein-tyrosine kinase (PTK)3 MET (4), the Hedgehog signaling pathway (5), mammalian target of rapamycin (6), and GRB7/ERK (7) as potential avenues for therapeutic intervention. Although the complexity of the signaling changes underlying the disease is apparent, this also represents an opportunity for approaches to therapy that involve combinatorial strategies to inhibit multiple targets and pathways simultaneously.
A signaling pathway that represents a major focus of research in cancer in general, including ovarian cancer, is that triggered by insulin-like growth factor-1 (IGF-1). This is important because IGF-1 exerts its effects at the level of the whole organism, as well as more local effects in cells and tissues (8, 9). The receptor for IGF-1 displays a similar subunit composition and organization to that of the insulin receptor. Each includes dimers of an α- and β-subunit pair, in which α is responsible for ligand binding and β is the PTK that is activated in response to ligand. In fact, the similarities are such that there is the potential for signaling from hybrid insulin/IGF-1 receptor dimer pairs (8, 9). High IGF-1 levels in patients are associated with increased risk of various cancers. IGF-1, which is normally produced in the liver, is also generated by tumors to trigger autocrine activation of pro-survival pathways. Hyperactivation of IGF-1 receptor signaling has also been implicated in resistance mechanisms to therapies, including resistance to cisplatin in ovarian cancer (10). Consequently, attention has focused on the potential to target IGF-1 signaling therapeutically. Various strategies have been adopted, including attempts to reduce the levels and activity of IGF-1, small molecule inhibitors of the IGF-1 receptor β-subunit PTK activity, which face the challenge of specificity relative to the insulin receptor, and targeting the IGF-1 receptor with antibodies (8, 9, 11). This latter approach, which has been developed most extensively, has encountered complications due to induction of increased levels of growth hormone and IGF-1 and hyperglycemia (8, 9, 11). This has drawn attention to the importance of identifying predictive biomarkers, to ensure that the trials are conducted on the optimal patient populations. Also, considering the similarities in their receptors, there is a need to identify differences in the activities of insulin and IGF-1 and the role in triggering signaling. For example, unlike insulin, the bioavailability of IGF-1 is regulated by binding proteins (8, 9). Furthermore, an important regulatory component of the insulin and IGF-1 signaling pathways that has not been considered extensively is the protein-tyrosine phosphatases (PTPs).
PTPs are represented by a large and structurally diverse family of receptor-like and cytoplasmic enzymes that play a vital role in reversible tyrosine phosphorylation-dependent signaling in coordination with PTKs (12). Deregulation of the expression and activity of PTPs has been implicated in many major diseases, including metabolic disorders and cancers (13). PTP1B, which was the first PTP to be purified and characterized (14, 15), plays a well established role in attenuating insulin receptor kinase activity and signaling through dephosphorylation of Tyr(P) residues in the activation loop of the β-subunit of the receptor, as well as IRS-1, the adaptor protein and immediate substrate of the insulin receptor (12). Considering the similarities between the insulin and IGF-1 receptors, including in the activation loop, their engagement of IRS-1 as a signaling intermediate, and the fact that the receptors can heterodimerize, we investigated whether PTP1B could down-regulate IGF-1 receptor signaling. In this report, we describe a role for PTP1B in attenuating the pro-survival signaling events induced by IGF-1 in ovarian cancer cells.
EXPERIMENTAL PROCEDURES
Cell Culture
Ovarian carcinoma-derived cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/ml), and streptomycin (100 μg/ml). Normal HOSE 11-12 and 6-3 control cell lines were grown in 199/MCDB 105 (1:1) medium containing 10% FBS. Ectopic expression of PTP1B was established in ovarian carcinoma-derived cell lines, CAVO-1 and CAOV-4, by retroviral infection following hygromycin selection. Specifically, retrovirus stably expressing wild type and C215S mutant PTP1B was generated in Phoenix-Ampho packaging cells by co-transfecting plasmids, including pWZL-PTP1B WT or C215S, VSVG, and the pCL-Ampho Retrovirus Packaging vector at a ratio of 3:1:1. Twenty four hours after transfection, recombinant retrovirus supernatants were harvested and passed through 0.45-μm filter to remove cell debris. The cleared supernatants were then incubated with CAOV4 and CAOV1 ovarian cancer cells in the presence of Polybrene (8 μg/ml final conc.). Twenty four hours after infection, cells were placed under hygromycin selection (200 μg/ml final concentration), and the effectiveness of infection was further confirmed by immunoblotting with antibody against PTP1B. Cells were maintained at 37 °C in 5% CO2. For suspension culture, cells were seeded in the 12-well plates pre-coated with polyhema (Sigma).
Cell Proliferation Assays
Growth curves were determined by cell counting in the presence of trypan blue exclusion dye (Invitrogen). The BrdU assay (BrdU cell proliferation assay kit, Cell Signaling Technology) followed the manufacturer's protocol.
Wound Healing Assay
To measure cell migration, a confluent monolayer of cells was “wounded” by scraping a 10-μl pipette tip across the monolayer to produce lesions with constant length. Any loose cells were removed by washing three times with PBS. Phase images were taken by Zeiss Axiovert 200 M using AxioVision 4.4 software.
Cell Invasion Assays
Cell invasion was quantitated using BioCoatTM BD MatrigelTM Invasion Chambers, 8.0-μm pore size. Cells (2 × 105) were grown in the insert. After 24 h, those cells retained inside the insert were removed; those that migrated to the other side of the insert were fixed and stained with KARYOMAX Giemsa stain (Invitrogen).
Soft Agar Assay
The bottom layer of soft agar contained 0.7% agar with DMEM and 10% serum. Cells were seeded (2,500 per well) on a top layer that contained 0.35% agar in DMEM and 10% serum. At each time point, an image of each well was taken at ×20 magnification (Zeiss Axiovert 200 M using AxioVision 4.4 software).
Immunoblotting and Immunoprecipitation
Cell extracts were prepared in RIPA lysis buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) and analyzed by immunoblotting. Antibodies used in this study were against the following: PTP1B (FG6) and TCPTP (CF4); PTPN14 (from R&D Systems); β-catenin, poly(ADP-ribose) polymerase, PTP-PEST, phospho-Tyr-1131, Tyr-Y1135, and total IGF-1Rβ, phospho-Srt-112 and total BAD, phospho-Tyr-416, Tyr-527, and total SRC, phospho-Thr-308 and total AKT, phospho- and total ERK (from Cell Signaling); 4G10, phospho-Tyr-342-BRK, phospho-Tyr-612 IRS-1 (from Millipore); IRS-1 (from Invitrogen); phospho-p38 (from Promega); p38 and BRK (from Santa Cruz Biotechnology); and β-tubulin, β-actin, and FLAG (from Sigma). Anti-PTPN23 polyclonal antibody was a gift from J. Maier (University of Milan Medical School).
Statistics
All statistics were performed using a standard Student's t test.
RESULTS
PTP1B Was Down-regulated in Ovarian Carcinoma-derived Cell Lines
We used immunoblotting to compare the expression of PTP1B protein between two HOSE cell lines immortalized by the human papilloma viral oncogenes E6 and E7 (16) and 11 ovarian carcinoma-derived cell lines. We observed that relative to the HOSE control cells, the level of PTP1B was significantly down-regulated in all of the ovarian carcinoma-derived cell lines (Fig. 1). This observation was confirmed in a parallel analysis by iTRAQ quantitative proteomics (data not shown).
FIGURE 1.
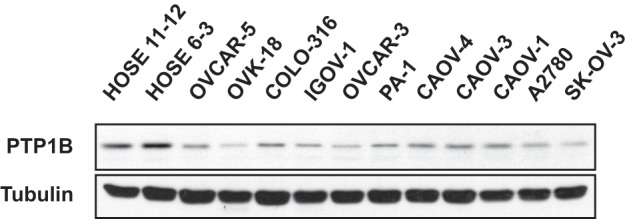
PTP1B was underexpressed in ovarian carcinoma-derived cell lines. Two immortalized human normal ovarian epithelial cell lines (HOSE 11-12 and HOSE 6-3) and 11 ovarian carcinoma-derived cell lines were examined by immunoblotting to demonstrate decreased expression of PTP1B in all of the ovarian carcinoma-derived cell lines compared with normal controls. Tubulin was probed as the loading control.
Effects of Altering PTP1B Expression on Proliferation, Migration, and Invasion of Ovarian Carcinoma-derived Cell Lines
To assess the biological consequence of PTP1B loss in ovarian carcinoma-derived cells, we established stable lines (CAOV-1 w/PTP1B and CAOV-4 w/PTP1B) in which PTP1B protein was ectopically expressed to levels that were comparable with those in the control immortalized HOSE 6-3 ovarian epithelial cells (Fig. 2A). Of note, restoration of PTP1B expression delayed cell proliferation, with similar results observed when proliferation was assessed either by cell counts or BrdU incorporation (Fig. 2B). These effects required the phosphatase activity of PTP1B (Fig. 2B). Expression of PTP1B also decreased the rate of cell migration in wound healing assays (Fig. 2C). Because wound recovery was monitored over a time period that was less than the doubling time of both cell lines (CAOV-1, 0–14 h; CAOV-4, 0–6 h), the effects on cell migration were not due to a decreased rate of proliferation. Furthermore, we also observed that ectopic expression of PTP1B impaired invasion of the ovarian cancer cells (Fig. 2D), highlighting a potential role of PTP1B in restraining ovarian tumor cell function. Consistent with these findings, we observed the opposite effects of treatment of both HOSE cell lines with a specific, small molecule inhibitor of PTP1B, MSI-1436 (17, 18). Cell proliferation, which was monitored by BrdU incorporation (Fig. 2E), and cell motility (Fig. 2F) were increased significantly following treatment with the PTP1B inhibitor.
FIGURE 2.

Loss of PTP1B-enhanced proliferation, migration, and invasion of ovarian carcinoma-derived cells. A, immunoblot to demonstrate ectopic expression of PTP1B, both wild type and inactive CS mutant, in ovarian carcinoma-derived cell lines CAOV-1 and CAOV-4 to similar levels as those detected in HOSE 6-3 normal control cells. B, ectopic expression of PTP1B decreased proliferation of ovarian carcinoma-derived cell lines. Proliferation was assessed by cell counting (CAOV-1 cells) and BrdU incorporation (CAOV-4 cells) at the indicated time intervals. Results represent mean ± S.D. from three independent experiments. C, ectopic expression of PTP1B decreased migration of ovarian carcinoma-derived cell lines. Phase images are presented to illustrate the extent of cell migration at the indicated times following introduction of a scratch wound in the cell monolayer. The dotted line represents the boundary of cell migration. D, ectopic expression of PTP1B decreased invasion of ovarian carcinoma-derived cell lines. The data are from a Boyden chamber assay, in which invasion was monitored after 24 h. Results represent mean ± S.D. from three independent experiments. E, inhibition of PTP1B with the small molecule MSI-1436 increased proliferation of both HOSE cell lines. Proliferation was monitored using the BrdU assay 48 h after incubation with MSI-1436 (5 μm). Results represent mean ± S.D. from three independent experiments. F, PTP1B inhibition by MSI-1436 (5 μm) enhanced migration of both HOSE cell lines, as visualized with a wound healing assay.
Effects of PTP1B Expression on Survival of Ovarian Carcinoma-derived Cell Lines
In addition to attenuating proliferation, expression of PTP1B impaired anchorage-independent survival of the ovarian cancer cell lines; cell death was observed as early as day 5 in the soft agar assay (Fig. 3A). We used polyhema-coated plates for suspension culture to mimic anchorage-independent growth, and we observed increased poly(ADP-ribose) polymerase cleavage following expression of PTP1B in CAOV-1 cells (Fig. 3B), consistent with cell death through apoptosis. This change in apoptotic phenotype in nonadherent cultures suggests that PTP1B sensitizes cells to anoikis, apoptosis caused by lack of basement membrane adherence. Consistent with these phenotypic observations, CAOV-1 and CAOV-4 cells ectopically expressing PTP1B displayed decreased activation of the ERK signaling pathway (Fig. 3C). We did not detect AKT activation in parental or engineered cancer cell lines. Interestingly, p38 phosphorylation levels were elevated in those cells with high PTP1B compared with parental controls, concomitant with the decreased phosphorylation of BAD at Ser-112, consistent with the initiation of apoptosis following restoration of PTP1B expression.
FIGURE 3.

Ectopic expression of PTP1B decreased anchorage-independent survival of ovarian carcinoma-derived cell lines through BAD-mediated apoptosis. A, soft agar assay in which cells were monitored at the indicated time intervals to demonstrate that ectopic expression of PTP1B decreased anchorage-independent survival of both CAOV-1 and CAOV-4 ovarian carcinoma-derived cell lines. B, parental and PTP1B-expressing CAOV-1 cells were harvested at indicated time points and lysed, and total and cleaved poly(ADP-ribose) polymerases (PARP) were detected by immunoblotting, to illustrate that ectopic expression of PTP1B triggered cell death through apoptosis in suspension culture. Tubulin was probed as the loading control. C, lysates of parental and PTP1B-expressing CAVO-1 and CAOV-4 cell lines were immunoblotted with the indicated antibodies to illustrate the effects of PTP1B on specific signaling pathway components. Actin was probed as the loading control.
PTP1B Directly Dephosphorylated IGF-1Rβ and Antagonized IGF-1R-mediated Survival Signaling in Ovarian Carcinoma Cell Lines
Given the dramatic effects of restoring PTP1B expression on the cancer cell phenotype, we were encouraged to identify the potential substrate(s) of PTP1B, first by examining the change in global tyrosine phosphorylation profile after ectopic PTP1B expression. Using the anti-Tyr(P) antibody 4G10, we detected a decrease in phosphorylation of a protein of 90–100 kDa, which coincided with the β-subunit of the IGF1 receptor tyrosine kinase (Fig. 4A). Treatment with BMS-536924, a specific inhibitor of IGF-1Rβ (19), resulted in decreased phosphorylation of Tyr-1135 of IGF-1Rβ in both CAOV-1 and CAOV-4 cell lines (Fig. 4B, lane 4 versus 2 and lane 8 versus 6). Furthermore, BMS-536924 delayed proliferation of the ovarian tumor cells, but not the HOSE controls (Fig. 4C).
FIGURE 4.

IGF-1R-induced pro-survival signaling was antagonized by PTP1B; recognition of IGF-1R β-subunit as a substrate by PTP1B. A, anti-phosphotyrosine antibody immunoblot to examine effects of ectopic expression of PTP1B on tyrosine phosphorylation in CAOV-4 cells. The blot was re-probed with antibodies against IGF-1Rβ and loading control actin (left panel). Tyrosine phosphorylation of IGF-1Rβ was examined by immunoprecipitation (IP) and blotting with anti-phosphotyrosine antibody 4G10 (right panel). exp, exposure. B, cells were subjected to serum starvation for 16 h, and then IGF-1Rβ inhibitor BMS-536924 (125 nm) was added 90 min prior to stimulation with recombinant human IGF-1 (100 ng/ml, 30 min). Levels of phospho- and total IGF-1Rβ were examined by immunoblotting (IB), with tubulin used as loading control. C, BrdU assay of cell proliferation performed 72 h after incubation with BMS-536924 to demonstrate that inhibition of IGF-1R signaling decreased proliferation of ovarian carcinoma-derived cell lines but not of normal HOSE cell lines. Results represent mean ± S.D. from three independent experiments. n.s., not significant. D, cells were stimulated with hIGF-1 (100 ng/ml) for the indicated times, lysed, and immunoblotted with the designated antibodies to illustrate that ectopic expression of PTP1B inhibited IGF-1-induced activation of IGF-1R signaling pathway. E, 293T cells were transiently transfected with the indicated constructs, lysed, and immunoblotted with the indicated antibodies. In addition, PTP1B was immunoprecipitated, and the interactions between the wild type and substrate-trapping mutant forms of the phosphatase were assessed by immunoblotting. Actin was probed as the loading control. F, 293T cells were transiently transfected with the indicated constructs, lysed, and immunoblotted as indicated to illustrate dephosphorylation of the IGF-1R by PTP1B but not by the other PTPs that were tested. Actin was probed as the loading control.
Consistent with the hypothesis that IGF-1Rβ is a direct substrate of PTP1B, we observed attenuated IGF-1-induced activation of IGF-1Rβ in both CAOV-1 and CAOV-4 cells expressing PTP1B, coincident with decreased phosphorylation of BAD at Ser-112 (Fig. 4D). We used the substrate-trapping strategy, which was developed in the laboratory (20), to investigate further whether or not PTP1B can dephosphorylate IGF-1Rβ. PTP1B, or its trapping mutant D181A, was either expressed alone or co-expressed with IGF-1Rβ in HEK 293T cells. We observed increased phosphorylation of IGF-1Rβ on Tyr-1131 and Tyr-1135 of the activation loop (Fig. 4E, lane 5 versus 4) in response to hIGF-1 stimulation. PTP1B D181A (Fig. 4E, lanes 8 and 9) but not the wild type enzyme (Fig. 4E, lanes 6 and 7) formed a complex with IGF-1Rβ, and the extent of complex formation was enhanced by hIGF-1 stimulation, coincident with increased tyrosine phosphorylation of IGF-1Rβ (Fig. 4E, lane 9 versus 8). Furthermore, wild type PTP1B efficiently dephosphorylated IGF-1Rβ at Tyr-1131 and Tyr-1135 (Fig. 4E, lane 7 versus 5). In addition, none of the other PTPs that were tested (PTPN12, -N14, and -N23) displayed activity toward IGF-1Rβ (Fig. 4F), highlighting the importance of PTP1B for these effects.
To examine specificity in these effects of PTP1B, we tested the consequences of overexpressing the phosphatase in OVK-18 cells, an ovarian cancer cell line that did not express IGF-1Rβ (Fig. 5A). In these cells, PTP1B expression did not affect proliferation (Fig. 5B) or migration (Fig. 5C). Furthermore, the effects on cell signaling were distinct from those seen in IGF-1Rβ-positive ovarian cancer cells. In particular, overexpression of PTP1B led to enhanced phosphorylation of ERK, with no detectable effect on the phosphorylation of p38 or AKT (Fig. 5D).
FIGURE 5.

PTP1B did not antagonize proliferation in the IGF-1R-negative ovarian carcinoma-derived cell line OVK-18. A, immunoblot to demonstrate that neither phospho- nor total IGF-1R was detected, in the presence or absence of hIGF-1 (100 ng/ml), in the OVK-18 cell line. CAOV-4 cells were included as a positive control, and tubulin was used as the loading control. B, ectopic expression of PTP1B had no apparent effect on proliferation of OVK-18 cells. Results represent mean ± S.D. from three independent experiments. n.s., not significant. C, phase images of a scratch/wound-healing assay to illustrate that ectopic expression of PTP1B did not affect migration of OVK-18 cells. The dotted line represents the boundary of cell migration. D, lysates of parental and PTP1B-expressing OVK-18 cell line were immunoblotted with the indicated antibodies to assess signaling output in these cells.
BRK/PTK6 Physically Interacted with and Activated IGF-1Rβ
Considering the abnormally high levels of BRK/PTK6 in ovarian tumors (21), and the fact that it has been implicated in IGF-1R-induced anchorage-independent survival (22), we investigated whether there was an interaction between the two. First, we observed that co-expression of an inactive mutant form of IGF-1Rβ, in which there was a mutation in the ATP-binding site, with either BRK or the constitutively active BRK Y447F mutant, led to trans-phosphorylation of IGF-1Rβ, illustrating that IGF-1Rβ is a potential substrate of BRK (Fig. 6A). Furthermore, in OVK-18 cells, which lack the IGF-1 receptor, there was no activation of BRK in response to hIGF-1 (Fig. 6B). Finally, we expressed FLAG-tagged wild type or constitutively active mutant Y447F BRK in HEK 293T cells, either alone or co-expressed with IGF-1Rβ, to examine their potential interaction (Fig. 6C). As expected, both wild type and constitutively active Y447F BRK were active, with the latter displaying higher activity, and able to phosphorylate both endogenous (Fig. 6C, lanes 3 and 4) and ectopically expressed IGF-1Rβ (Fig. 6C, lanes 5 and 6). By examining anti-FLAG antibody immunoprecipitates, to pull down ectopically expressed BRK, we detected a complex of IGF-1Rβ with both wild type and constitutively active Y447F BRK (Fig. 6C).
FIGURE 6.
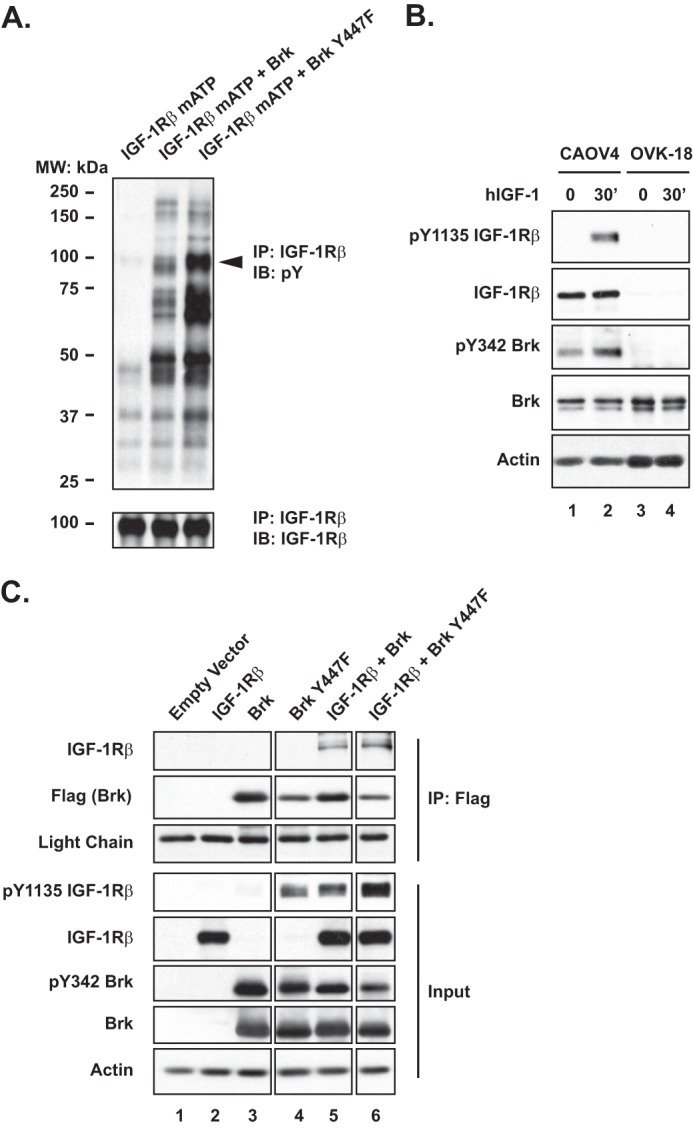
Regulation of IGF-1 signaling by BRK. A, 293 cells were transiently transfected with a mutant IGF-1R that was defective in the ATP-binding site, in the presence or absence of the indicated BRK expression plasmids. The IGF-1R was immunoprecipitated (IP) from cell lysates and blotted with an anti-phosphotyrosine antibody, to examine the extent of tyrosine phosphorylation, or with an antibody to IGF-1R, to determine loading. B, immunoblot (IB) to demonstrate that, unlike in CAOV-4 cells, BRK was not phosphorylated in response to hIGF-1 stimulation (100 ng/ml) in OVK-18 cells. Actin was probed as the loading control. C, 293T cells were transiently transfected with indicated constructs, and FLAG-tagged BRK was immunoprecipitated and tested by immunoblotting for association with IGF-1R. Immunoglobin light chain and actin were probed as loading control for immunoprecipitation and input, respectively.
BRK Was a Direct Substrate of PTP1B
BRK is a nonreceptor tyrosine kinase distantly related to the SRC family, members of which have been identified previously as substrates of PTP1B (23). Interestingly, we noted sequence similarity between the BRK autophosphorylation motif and substrate motifs reported for PTP1B (24). This suggests that unlike SRC, for which PTP1B dephosphorylates the inhibitory C-terminal tyrosyl residue leading to kinase activation (25), BRK may be a direct substrate of PTP1B in which dephosphorylation inhibits kinase function. To address this point, we tested the effects of overexpressing PTP1B in CAOV-4 cells on BRK phosphorylation and pro-survival signaling (Fig. 7A). The cells were serum-starved for 16 h and then stimulated with hIGF-1. We were able to detect the phosphorylated, active form of BRK (pY342 BRK) in parental CAOV-4 cells after 30 min of stimulation; however, in contrast, activation of BRK was not detected in cells overexpressing PTP1B. Phosphorylation of BAD was also markedly attenuated in the cells overexpressing PTP1B.
FIGURE 7.

PTP1B was a negative regulator of BRK. A, parental and PTP1B-expressing ovarian carcinoma-derived cell line CAOV-4 was stimulated with hIGF-1 (100 ng/ml) for the times indicated, lysed, and immunoblotted with the indicated antibodies. B, 293T cells were transiently transfected with BRK, PTP1B, and TCPTP expression plasmids, as indicated. In the left panel, PTP1B was immunoprecipitated (IP), and its association with BRK was examined by immunoblotting. The phosphorylation status of BRK was determined by immunoblotting with phosphospecific antibodies to Tyr-342. Actin was probed as the loading control. C, CAOV-4 cells were serum-starved (16 h) prior to treatment with the PTP1B inhibitor MSI-1436 (4 h) at the indicated concentrations, lysed, and then immunoblotted with the indicated antibodies to examine the signaling status of the cells. Phospho- and total IRS-1 were also probed to confirm PTP1B inhibition.
To test whether PTP1B dephosphorylated BRK directly, we applied the strategy involving substrate trapping mutant forms of the phosphatase. Although we detected some co-precipitation of BRK with wild type PTP1B (Fig. 7B, lane 5), the extent of the association was greater with substrate trapping mutant PTP1B D181A (Fig. 7B, lane 6). Consistent with the trapping data, wild type PTP1B dephosphorylated BRK at Tyr-342. The extent of dephosphorylation was much less pronounced following expression of TCPTP, the closest relative of PTP1B (Fig. 7B, right panel). Furthermore, dephosphorylation was not observed with any of the other PTPs (PTPN12, -N14, and -N23) that were tested (data not shown), confirming the specificity of PTP1B in this context.
We also tested the effects of a small molecular inhibitor of PTP1B, MSI-1436 (17, 18), in CAOV-4 cells to determine whether inhibition of the phosphatase had the opposite effect to that observed in the PTP1B overexpression setting. Tyrosine phosphorylation of IRS1, a known substrate of PTP1B (23), increased dramatically following treatment with 5 μm MSI-1436 (Fig. 7C), indicating the effectiveness of the inhibitor at this dosage. Compared with the untreated control, the active form of BRK was detected in the presence of 5 μm MSI-1436 (Fig. 7C). Inhibitor treatment also resulted in substantially enhanced phosphorylation of ERK and BAD (Fig. 7C), illustrating that attenuation of PTP1B function led to enhanced signaling in these cells.
DISCUSSION
In this study, we have demonstrated that PTP1B is down-regulated at the protein level in a panel of ovarian cancer cell lines, relative to normal control cells. This is consistent with the report from Lu et al. (26), in which they observed by microarray analysis that PTP1B mRNA levels were significantly lower in serous, endometrioid, and mucinous subtypes of human ovarian cancer samples than in normal epithelial controls. When we overexpressed PTP1B in the cancer cell lines, to the levels detected in normal epithelial cells, it resulted in impaired cell invasion and migration, delayed proliferation, and increased cell death through apoptosis. Conversely, we observed that treatment of the normal ovarian surface epithelial cells with a small molecule inhibitor of PTP1B (17, 18) led to enhanced proliferation and motility. These data are consistent with a role for PTP1B in attenuating signals that are important for the tumor phenotype.
Considerable attention has focused on the role of IGF-1 receptors in promoting tumorigenesis (8, 9). In this study, we have demonstrated that PTP1B targeted the β-subunit of the IGF-1 receptor as a substrate in the ovarian cancer cells and that there was no inhibitory effect of overexpressing PTP1B in ovarian cancer cells that do not express IGF-1R. A potential effect of PTP1B as an antagonist of IGF-1 signaling was first suggested by O'Connor and co-workers (27, 28), who tested the effects of the phosphatase on IGF-1R β-subunit in yeast and COS cell systems and examined IGF-1-mediated motility and protection from serum withdrawal-induced apoptosis in PTP1B-depleted mouse embryo fibroblasts. More recently, bioluminescence resonance energy transfer was used to monitor an interaction between IGF-1R β-subunit and a substrate trapping mutant form of PTP1B in 293 cell co-transfection studies (29). Our data now reveal that the suppressive effects of PTP1B on migration, invasion, proliferation, and survival of the panel of ovarian cancer cells are mediated, at least in part, through dephosphorylation of the IGF-1R β-subunit. Furthermore, our data highlight an important role for another PTK, BRK, as a target of PTP1B in these suppressive effects on ovarian cancer cell signaling.
BRK/PTK6 is a member of a family of PTKs that bear structural similarity to SRC (30, 31). Although both feature SH2, SH3, and PTK domains, there are differences in the regulatory roles of these domains between BRK and SRC (30, 31). BRK activity is regulated by autophosphorylation, with the SH2 and SH3 domains serving in an autoinhibitory capacity (32). Changes in subcellular localization, between nucleus, cytosol, and membrane, are also of regulatory significance (33). Levels of BRK are low in normal tissues, where its expression is restricted to differentiating epithelial cells (34, 35); however, its expression is up-regulated in a variety of tumors, particularly breast and ovary, primarily at a transcriptional or post-transcriptional level, rather than by mutation (36). In fact, BRK is highly expressed in >80% of breast cancers (31, 37, 38) and has been suggested as a potential therapeutic target (39, 40).
A wide variety of biological functions are regulated by BRK (30, 31). In normal cells, it has been implicated in the control of differentiation and apoptosis; in contrast, in cancer cells BRK has been reported to enhance mitogenic signals and cell proliferation, protect breast cancer cells from anchorage-independent autophagic cell death, and promote migration and invasion (31, 41). BRK has been implicated in regulating EGF signaling. It sensitizes mammary epithelial cells to EGF (42) and enhances EGF receptor (EGFR) signaling (43, 44), and its suppression impairs the signaling response to EGF (45). BRK mediates these effects, at least in part, by inhibiting receptor down-regulation, both through phosphorylation of EGFR, which disrupts ubiquitination of the receptor by CBL (46), and by phosphorylation of the BRK-binding protein ARAP1 (47). BRK also affects the function of other EGFR family members. There have been reports of co-amplification of chromosome 20q13.3, encoding the BRK gene, and 17q21-22, encoding the gene for HER2 (40, 48), and complex formation between the BRK and HER2 proteins, which promotes HER2 signaling in breast cancer (31). The effects of BRK are not restricted to the EGFR family; for example, it is also activated downstream of MET, which is associated with increased invasiveness of breast cancer cell lines (49). Nevertheless, despite all of this progress, very little is known about the impact of protein phosphatases on the function of BRK.
BRK/PTK6 was also identified in a high throughput siRNA screen that was designed to discover components responsible for IGF-1R-induced anchorage-independent survival of mammary epithelial cells (22). In addition, down-regulation of BRK was shown to induce apoptosis of breast and ovarian cancer cells deprived of matrix attachment (22), which is similar to the effects we observed following overexpression of PTP1B. In fact, our data highlight not only a physical and functional interaction between IGF-1R β-subunit and BRK, as reported in Ref. 22, but also illustrate that a major aspect of the inhibitory effects of PTP1B on ovarian cancer cell signaling was exerted through its dephosphorylation of both IGF-1R and BRK. Furthermore, our data emphasize the exquisite specificity of PTPs and the importance of context in determining their signaling function. There is now a considerable body of evidence to establish that members of the PTP family not only have the expected capacity to antagonize tyrosine phosphorylation-dependent signaling but also may function positively to promote signaling. The classic example is CD45, which dephosphorylates the C-terminal inhibitory site of tyrosine phosphorylation in SRC family PTKs to promote antigen receptor signaling (12). Furthermore, it is now also clear that some individual PTPs may function either negatively or positively depending on context, with PTP1B serving as an excellent case in point (12, 23). PTP1B is well established as an antagonist of signaling by insulin (by dephosphorylation of insulin receptor β-subunit and IRS-1) and leptin (by dephosphorylation of JAK PTKs) and is an established therapeutic target for diabetes and obesity (12, 23, 50). In the context of cancer, PTP1B is known to play a suppressive role in inhibiting the action of several growth factor receptor PTKs (23). In addition, loss of PTP1B in p53-null mice increases susceptibility to B cell lymphomas (51). However, despite such growth inhibitory functions, PTP1B-null mice do not show an increase in spontaneous tumors, nor do they show increased IGF-1 receptor signaling (50). In contrast, PTP1B is highly expressed in some tumors, such as breast, and plays a positive role in signaling by the HER2 oncoprotein-tyrosine kinase. Although the mechanism is unclear and may involve dephosphorylation and activation of SRC (52) or activation of RAS-MAPK signaling via dephosphorylation of p62DOK (53), this raises the possibility that PTP1B may also be a therapeutic target for treatment of HER2-positive cancer (54). Nevertheless, consistent with the importance of context and specificity in PTP function, signaling through the EGF receptor, which is from the same family of PTKs as HER2, is not augmented by PTP1B; in fact, PTP1B is recognized for its inhibitory effects on EGFR signaling (23).
Overall, our data suggest that in ovarian cancer cells PTP1B has the potential to attenuate IGF-1R- and BRK-dependent signaling and that the observed decreased expression of the phosphatase relative to control normal cells contributes to promoting the signaling pathways associated with cancer cell proliferation, survival, migration, and invasion. In particular, the suppression of PTP1B may play an important role in the ability of tumor cells in ascites to bypass suspension-induced apoptosis, or anoikis, to spread to distant organs. The mechanisms underlying the overt differences in function of PTP1B as a regulator of signaling in the context of normal versus cancer cells remain to be defined, but these observations not only provide important insights into the regulation of IGF-1R signaling in cancer cells but also draw our attention to the importance of assessing directly whether there are any deleterious effects of inhibiting PTP1B in any clinical trials designed to test the effects of inhibitors of the phosphatase in diabetes, obesity, or cancer.
Acknowledgments
We thank Senthil Muthuswamy (Ontario Cancer Institute), Todd Miller (State University of New York at Stony Brook), Steven Rosenzweig (Medical University of South Carolina), and Olle Larsson (Karolinska Institutet) for the gift of plasmids and all Tonks laboratory members for fruitful discussions during the course of this work. PTP1B inhibitor MSI-1436 was provided by Ohr Pharmaceuticals and Genaera Corp.
This work was supported, in whole or in part, by National Institutes of Health Grants CA53840 and GM55989 (to N. K. T.). This work was also supported by a grant from the STARR Foundation, Department of Defense Congressional funds (to R. L. and N. K. T.), Cold Spring Harbor Laboratory Cancer Centre Support Grant CA45508, and from The Gladowsky Breast Cancer Foundation, Hansen Foundation, Irving Hansen Foundation, Islip Breast Cancer Coalition, Fannie Rippel Foundation, Robertson Research Fund, and the Masthead Cove Yacht Club Carol Marcincuk Fund.
This article was selected as a Paper of the Week.
- PTK
- protein-tyrosine kinase
- PTP
- protein-tyrosine phosphatase
- SH
- Src homology
- HOSE
- human ovarian surface epithelial
- hIGF
- human IGF
- EGFR
- EGF receptor.
REFERENCES
- 1. Siegel R., Ward E., Brawley O., Jemal A. (2011) Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin. 61, 212–236 [DOI] [PubMed] [Google Scholar]
- 2. Kulasingam V., Pavlou M. P., Diamandis E. P. (2010) Integrating high throughput technologies in the quest for effective biomarkers for ovarian cancer. Nat. Rev. Cancer 10, 371–378 [DOI] [PubMed] [Google Scholar]
- 3. Bast R. C., Jr., Hennessy B., Mills G. B. (2009) The biology of ovarian cancer: new opportunities for translation. Nat. Rev. Cancer 9, 415–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mhawech-Fauceglia P., Afkhami M., Pejovic T. (2012) MET/HGF signaling pathway in ovarian carcinoma: Clinical implications and future direction. Pathol. Res. Int. 2012, 960327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Szkandera J., Kiesslich T., Haybaeck J., Gerger A., Pichler M. (2013) Hedgehog signaling pathway in ovarian cancer. Int. J. Mol. Sci. 14, 1179–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mabuchi S., Hisamatsu T., Kimura T. (2011) Targeting mTOR signaling pathway in ovarian cancer. Curr. Med. Chem. 18, 2960–2968 [DOI] [PubMed] [Google Scholar]
- 7. Chan D. W., Hui W. W., Cai P. C., Liu M. X., Yung M. M., Mak C. S., Leung T. H., Chan K. K., Ngan H. Y. (2012) Targeting GRB7/ERK/FOXM1 signaling pathway impairs aggressiveness of ovarian cancer cells. PLoS One 7, e52578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pollak M. (2008) Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 8, 915–928 [DOI] [PubMed] [Google Scholar]
- 9. Pollak M. (2012) The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat. Rev. Cancer 12, 159–169 [DOI] [PubMed] [Google Scholar]
- 10. Eckstein N., Servan K., Hildebrandt B., Pölitz A., von Jonquières G., Wolf-Kümmeth S., Napierski I., Hamacher A., Kassack M. U., Budczies J., Beier M., Dietel M., Royer-Pokora B., Denkert C., Royer H. D. (2009) Hyperactivation of the insulin-like growth factor receptor I signaling pathway is an essential event for cisplatin resistance of ovarian cancer cells. Cancer Res. 69, 2996–3003 [DOI] [PubMed] [Google Scholar]
- 11. Bruchim I., Werner H. (2013) Targeting IGF-1 signaling pathways in gynecologic malignancies. Expert Opin. Ther. Targets 17, 307–320 [DOI] [PubMed] [Google Scholar]
- 12. Tonks N. K. (2006) Protein-tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846 [DOI] [PubMed] [Google Scholar]
- 13. Julien S. G., Dubé N., Hardy S., Tremblay M. L. (2011) Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 11, 35–49 [DOI] [PubMed] [Google Scholar]
- 14. Tonks N. K., Diltz C. D., Fischer E. H. (1988) Characterization of the major protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 263, 6731–6737 [PubMed] [Google Scholar]
- 15. Tonks N. K., Diltz C. D., Fischer E. H. (1988) Purification of the major protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 263, 6722–6730 [PubMed] [Google Scholar]
- 16. Tsao S. W., Mok S. C., Fey E. G., Fletcher J. A., Wan T. S., Chew E. C., Muto M. G., Knapp R. C., Berkowitz R. S. (1995) Characterization of human ovarian surface epithelial cells immortalized by human papilloma viral oncogenes (HPV-E6E7 ORFs). Exp. Cell Res. 218, 499–507 [DOI] [PubMed] [Google Scholar]
- 17. Lantz K. A., Hart S. G., Planey S. L., Roitman M. F., Ruiz-White I. A., Wolfe H. R., McLane M. P. (2010) Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity 18, 1516–1523 [DOI] [PubMed] [Google Scholar]
- 18. Krishnan N., Fu C., Pappin D. J., Tonks N. K. (2011) H2S-induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signal. 4, ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wittman M., Carboni J., Attar R., Balasubramanian B., Balimane P., Brassil P., Beaulieu F., Chang C., Clarke W., Dell J., Eummer J., Frennesson D., Gottardis M., Greer A., Hansel S., Hurlburt W., Jacobson B., Krishnananthan S., Lee F. Y., Li A., Lin T. A., Liu P., Ouellet C., Sang X., Saulnier M. G., Stoffan K., Sun Y., Velaparthi U., Wong H., Yang Z., Zimmermann K., Zoeckler M., Vyas D. (2005) Discovery of a (1H-benzoimidazol-2-yl)-1H-pyridin-2-one (BMS-536924) inhibitor of insulin-like growth factor I receptor kinase with in vivo antitumor activity. J. Med. Chem. 48, 5639–5643 [DOI] [PubMed] [Google Scholar]
- 20. Flint A. J., Tiganis T., Barford D., Tonks N. K. (1997) Development of “substrate-trapping” mutants to identify physiological substrates of protein-tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 94, 1680–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmandt R. E., Bennett M., Clifford S., Thornton A., Jiang F., Broaddus R. R., Sun C. C., Lu K. H., Sood A. K., Gershenson D. M. (2006) The BRK tyrosine kinase is expressed in high grade serous carcinoma of the ovary. Cancer Biol. Ther. 5, 1136–1141 [DOI] [PubMed] [Google Scholar]
- 22. Irie H. Y., Shrestha Y., Selfors L. M., Frye F., Iida N., Wang Z., Zou L., Yao J., Lu Y., Epstein C. B., Natesan S., Richardson A. L., Polyak K., Mills G. B., Hahn W. C., Brugge J. S. (2010) PTK6 regulates IGF-1-induced anchorage-independent survival. PLoS One 5, e11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yip S. C., Saha S., Chernoff J. (2010) PTP1B: a double agent in metabolism and oncogenesis. Trends Biochem. Sci. 35, 442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Z. Y. (2002) Protein-tyrosine phosphatases: structure and function, substrate specificity, and inhibitor development. Annu. Rev. Pharmacol. Toxicol. 42, 209–234 [DOI] [PubMed] [Google Scholar]
- 25. Bjorge J. D., Pang A., Fujita D. J. (2000) Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J. Biol. Chem. 275, 41439–41446 [DOI] [PubMed] [Google Scholar]
- 26. Lu K. H., Patterson A. P., Wang L., Marquez R. T., Atkinson E. N., Baggerly K. A., Ramoth L. R., Rosen D. G., Liu J., Hellstrom I., Smith D., Hartmann L., Fishman D., Berchuck A., Schmandt R., Whitaker R., Gershenson D. M., Mills G. B., Bast R. C., Jr. (2004) Selection of potential markers for epithelial ovarian cancer with gene expression arrays and recursive descent partition analysis. Clin. Cancer Res. 10, 3291–3300 [DOI] [PubMed] [Google Scholar]
- 27. Buckley D. A., Cheng A., Kiely P. A., Tremblay M. L., O'Connor R. (2002) Regulation of insulin-like growth factor type I (IGF-I) receptor kinase activity by protein-tyrosine phosphatase 1B (PTP-1B) and enhanced IGF-I-mediated suppression of apoptosis and motility in PTP-1B-deficient fibroblasts. Mol. Cell. Biol. 22, 1998–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Buckley D. A., Loughran G., Murphy G., Fennelly C., O'Connor R. (2002) Identification of an IGF-1R kinase regulatory phosphatase using the fission yeast Schizosaccharomyces pombe and a GFP-tagged IGF-1R in mammalian cells. Mol. Pathol. 55, 46–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blanquart C., Boute N., Lacasa D., Issad T. (2005) Monitoring the activation state of the insulin-like growth factor-1 receptor and its interaction with protein-tyrosine phosphatase 1B using bioluminescence resonance energy transfer. Mol. Pharmacol. 68, 885–894 [DOI] [PubMed] [Google Scholar]
- 30. Brauer P. M., Tyner A. L. (2010) Building a better understanding of the intracellular tyrosine kinase PTK6-BRK by BRK. Biochim. Biophys. Acta 1806, 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ostrander J. H., Daniel A. R., Lange C. A. (2010) Brk/PTK6 signaling in normal and cancer cell models. Curr. Opin. Pharmacol. 10, 662–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qiu H., Miller W. T. (2002) Regulation of the nonreceptor tyrosine kinase Brk by autophosphorylation and by autoinhibition. J. Biol. Chem. 277, 34634–34641 [DOI] [PubMed] [Google Scholar]
- 33. Derry J. J., Prins G. S., Ray V., Tyner A. L. (2003) Altered localization and activity of the intracellular tyrosine kinase BRK/Sik in prostate tumor cells. Oncogene 22, 4212–4220 [DOI] [PubMed] [Google Scholar]
- 34. Haegebarth A., Bie W., Yang R., Crawford S. E., Vasioukhin V., Fuchs E., Tyner A. L. (2006) Protein-tyrosine kinase 6 negatively regulates growth and promotes enterocyte differentiation in the small intestine. Mol. Cell. Biol. 26, 4949–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tupper J., Crompton M. R., Harvey A. J. (2011) Breast tumor kinase (Brk/PTK6) plays a role in the differentiation of primary keratinocytes. Arch. Dermatol. Res. 303, 293–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mitchell P. J., Barker K. T., Shipley J., Crompton M. R. (1997) Characterisation and chromosome mapping of the human nonreceptor tyrosine kinase gene, brk. Oncogene 15, 1497–1502 [DOI] [PubMed] [Google Scholar]
- 37. Mitchell P. J., Barker K. T., Martindale J. E., Kamalati T., Lowe P. N., Page M. J., Gusterson B. A., Crompton M. R. (1994) Cloning and characterisation of cDNAs encoding a novel non-receptor tyrosine kinase, brk, expressed in human breast tumours. Oncogene 9, 2383–2390 [PubMed] [Google Scholar]
- 38. Barker K. T., Jackson L. E., Crompton M. R. (1997) BRK tyrosine kinase expression in a high proportion of human breast carcinomas. Oncogene 15, 799–805 [DOI] [PubMed] [Google Scholar]
- 39. Harvey A. J., Crompton M. R. (2003) Use of RNA interference to validate Brk as a novel therapeutic target in breast cancer: Brk promotes breast carcinoma cell proliferation. Oncogene 22, 5006–5010 [DOI] [PubMed] [Google Scholar]
- 40. Xiang B., Chatti K., Qiu H., Lakshmi B., Krasnitz A., Hicks J., Yu M., Miller W. T., Muthuswamy S. K. (2008) Brk is coamplified with ErbB2 to promote proliferation in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 105, 12463–12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harvey A. J., Pennington C. J., Porter S., Burmi R. S., Edwards D. R., Court W., Eccles S. A., Crompton M. R. (2009) Brk protects breast cancer cells from autophagic cell death induced by loss of anchorage. Am. J. Pathol. 175, 1226–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kamalati T., Jolin H. E., Mitchell P. J., Barker K. T., Jackson L. E., Dean C. J., Page M. J., Gusterson B. A., Crompton M. R. (1996) Brk, a breast tumor-derived non-receptor protein-tyrosine kinase, sensitizes mammary epithelial cells to epidermal growth factor. J. Biol. Chem. 271, 30956–30963 [DOI] [PubMed] [Google Scholar]
- 43. Kamalati T., Jolin H. E., Fry M. J., Crompton M. R. (2000) Expression of the BRK tyrosine kinase in mammary epithelial cells enhances the coupling of EGF signalling to PI 3-kinase and Akt, via erbB3 phosphorylation. Oncogene 19, 5471–5476 [DOI] [PubMed] [Google Scholar]
- 44. Ostrander J. H., Daniel A. R., Lofgren K., Kleer C. G., Lange C. A. (2007) Breast tumor kinase (protein-tyrosine kinase 6) regulates heregulin-induced activation of ERK5 and p38 MAP kinases in breast cancer cells. Cancer Res. 67, 4199–4209 [DOI] [PubMed] [Google Scholar]
- 45. Ludyga N., Anastasov N., Gonzalez-Vasconcellos I., Ram M., Höfler H., Aubele M. (2011) Impact of protein-tyrosine kinase 6 (PTK6) on human epidermal growth factor receptor (HER) signalling in breast cancer. Mol. BioSyst. 7, 1603–1612 [DOI] [PubMed] [Google Scholar]
- 46. Li X., Lu Y., Liang K., Hsu J. M., Albarracin C., Mills G. B., Hung M. C., Fan Z. (2012) Brk/PTK6 sustains activated EGFR signaling through inhibiting EGFR degradation and transactivating EGFR. Oncogene 31, 4372–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang S. A., Lee E. S., Yoon H. Y., Randazzo P. A., Lee S. T. (2010) PTK6 inhibits down-regulation of EGF receptor through phosphorylation of ARAP1. J. Biol. Chem. 285, 26013–26021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Born M., Quintanilla-Fend L., Braselmann H., Reich U., Richter M., Hutzler P., Aubele M. (2005) Simultaneous overexpression of the Her2/neu and PTK6 tyrosine kinases in archival invasive ductal breast carcinomas. J. Pathol. 205, 592–596 [DOI] [PubMed] [Google Scholar]
- 49. Locatelli A., Lofgren K. A., Daniel A. R., Castro N. E., Lange C. A. (2012) Mechanisms of HGF/Met signaling to Brk and Sam68 in breast cancer progression. Horm. Cancer 3, 14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bourdeau A., Dubé N., Tremblay M. L. (2005) Cytoplasmic protein-tyrosine phosphatases, regulation and function: the roles of PTP1B and TC-PTP. Curr. Opin. Cell Biol. 17, 203–209 [DOI] [PubMed] [Google Scholar]
- 51. Dubé N., Bourdeau A., Heinonen K. M., Cheng A., Loy A. L., Tremblay M. L. (2005) Genetic ablation of protein-tyrosine phosphatase 1B accelerates lymphomagenesis of p53-null mice through the regulation of B-cell development. Cancer Res. 65, 10088–10095 [DOI] [PubMed] [Google Scholar]
- 52. Arias-Romero L. E., Saha S., Villamar-Cruz O., Yip S. C., Ethier S. P., Zhang Z. Y., Chernoff J. (2009) Activation of Src by protein-tyrosine phosphatase 1B Is required for ErbB2 transformation of human breast epithelial cells. Cancer Res. 69, 4582–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Julien S. G., Dubé N., Read M., Penney J., Paquet M., Han Y., Kennedy B. P., Muller W. J., Tremblay M. L. (2007) Protein-tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 39, 338–346 [DOI] [PubMed] [Google Scholar]
- 54. Tonks N. K., Muthuswamy S. K. (2007) A brake becomes an accelerator: PTP1B–a new therapeutic target for breast cancer. Cancer Cell 11, 214–216 [DOI] [PubMed] [Google Scholar]