

Abstract
Background
The Candida albicans cell wall is the first point of contact with the host, and its outer surface is heavily enriched in mannoproteins modified through the addition of N- and O-mannan. Previous work, using mutants with gross defects in glycosylation, has clearly identified the importance of mannan in the host-pathogen interaction, immune recognition and virulence. Here we report the first analysis of the MNN1 gene family, which contains six members predicted to act as α-1,3 mannosyltransferases in the terminal stages of glycosylation.
Findings
We generated single null mutants in all members of the C. albicans MNN1 gene family, and disruption of MNN14 led to both in vitro and in vivo defects. Null mutants in other members of the family demonstrated no phenotypic defects, suggesting that these members may display functional redundancy. The mnn14Δ null mutant displayed hypersensitivity to agents associated with cell wall and glycosylation defects, suggesting an altered cell wall structure. However, no gross changes in cell wall composition or N-glycosylation were identified in this mutant, although an extension of phosphomannan chain length was apparent. Although the cell wall defects associated with the mnn14Δ mutant were subtle, this mutant displayed a severe attenuation of virulence in a murine infection model.
Conclusion
Mnn14 plays a distinct role from other members of the MNN1 family, demonstrating that specific N-glycan outer chain epitopes are required in the host-pathogen interaction and virulence.
Keywords: Candida albicans, Glycosylation, Mannoproteins, Cell wall, MNN1, Virulence
Findings
Background
Candida albicans is the most common opportunistic fungal pathogen of humans causing superficial infections of the mucosa, and life threatening systemic infections in immunocompromised and severely ill patients [1-3]. The fungal cell wall is a dynamic structure required for maintaining cell shape and providing protection from changes in the extracellular environment. In addition, the cell wall acts as the first point of contact with the host and plays an essential role in the host-fungal interaction. The cell wall is composed of an inner skeletal layer of β-glucans and chitin, decorated with an outer layer enriched in mannoproteins that are heavily post-translationally modified through the addition of N- and O-linked mannans [4-6]. These mannans have been shown to play a vital role in cell wall integrity, adhesion and virulence, and constitute one of the main C. albicans pathogen associated molecular patterns (PAMPs) recognised by the host innate immune system [7-16].
In C. albicans structural studies of O-mannan have shown that it typically consists of one to five α1,2-linked mannose residues attached to serine or threonine, and that these are required for full virulence [11,12]. In addition studies using anti-β-mannan specific antibodies have demonstrated that O-mannan may also contain β1,2-linked mannose residues [13], presumably added by members of the BMT family [14]. This is different to Saccharomyces cerevisiae where O-mannan consists of one to two α1,2-linked mannose residues capped with α1,3-linked mannose residues transferred by members of the Mnn1 family. N-glycosylation is initiated in the endoplasmic reticulum (ER) with the transfer of the N-mannan precursor (Glc3Man9GlcNAc2) to specific asparagine residues in the protein. The precursor is then processed by ER resident glycosidases to form the mature Man8GlcNAc2 core conserved across eukaryotes [10]. In C. albicans as well as other fungi, the core is then extensively modified through outer chain elaboration as the protein passes through the Golgi [7,14,16,17]. Outer chain elaboration is initiated through the addition of a single α1,6-linked mannose residue to the core by Och1 [7], and the subsequent extension of the α1,6-backbone by the sequential action of the mannan polymerase I and II enzyme complexes [17,18]. The α1,6-backbone is then further elaborated with side chains, which in C. albicans consists of α1,2-, α1,3-, and β1,2-linked mannose residues [14,16,19]. In addition both N-mannan and O-mannan structures can be further modified through the addition of phosphomannan, which in C. albicans consists of a chain of β1,2-linked mannose residues attached through a phosphodiester linkage [10,20,21].
Previous work has clearly identified outer chain N-glycosylation as an important factor in the host-fungal interaction and virulence [7,8,10,14-16]. However, most work to date has focused on the early stages of outer chain elaboration where the mutants display gross defects in glycosylation. In S. cerevisiae the MNN1/2 family contains genes involved in the elaboration of O- and N-mannan; in particular the MNN1 sub-family encodes α1,3-mannosyltranferases important for the addition of the terminal mannose residues in O- and N-mannan [22,23]. In C. albicans we have identified six MNN1 family members and here we report the first analysis of this gene family in C. albicans. Null mutants in most of its members displayed no alteration of phenotype, suggesting possible functional redundancy. However, the mnn14Δ mutant, whilst displaying only subtle cell wall changes, was severely attenuated in virulence, demonstrating that specific glycans are important in the host-fungal interaction.
Results and discussion
Analysis and disruption of C. albicans MNN1 family members
Our analysis of assembly 21 of the C. albicans genome identified a gene family of 12 members homologous to the S. cerevisiae MNN1/2 family. Similar to S. cerevisiae this family could be divided into two subfamilies based on homology to either MNN1 or MNN2. Six members of the C. albicans MNN1 family were identified (orf19.4279, orf19.4900, orf19.4270, orf19.6996, orf19.753 and orf19.6313) and designated MNN1, MNN12, MNN13, MNN14, MNN15 and MNN16. The proteins encoded by this family share 19-44% homology between members (Table 1, Figure 1) and a characteristic type II membrane protein topology, with a single N-terminal membrane spanning region (17–19 amino acids) preceded by a short (5–13 amino acid) cytosolic tail. All Mnn1 family members, except Mnn16, contain the conserved DxD motif known to be important for substrate and co-factor binding [24], suggesting that Mnn16 may have altered functionality. The MNN1 family shows varying numbers in the sequenced CUG clade species [25] with C. albicans, Candida tropicalis and Candida parapsilosis encoding 6, 8, and 4 members respectively, compared to the non-pathogenic or infrequent pathogens, Lodderomyces elongisporus, Candida guilliermondii, Candida lusitaniae, and Debaryomyces hansenii which have 5, 4, 3 and 3 members respectively.
Table 1.
Homology of MNN1 gene family products
| Protein |
% Identity (% similarity)a |
|||||
|---|---|---|---|---|---|---|
| Mnn1 | Mnn12 | Mnn13 | Mnn14 | Mnn15 | Mnn16 | |
| Mnn1 |
100% |
- |
- |
- |
- |
- |
| (100%) |
|
|
|
|
|
|
| Mnn12 |
43.8% |
100% |
- |
- |
- |
- |
| (60.7%) |
(100%) |
|
|
|
|
|
| Mnn13 |
42.8% |
37.7% |
100% |
- |
- |
- |
| (62.0%) |
(55.9%) |
(100%) |
|
|
|
|
| Mnn14 |
31.3% |
32.9% |
32.2% |
100% |
- |
- |
| (47.3%) |
(49.34%) |
(49.1%) |
(100%) |
|
|
|
| Mnn15 |
24.3% |
24.8% |
25.9% |
26.1% |
100% |
- |
| (39.5%) |
(39.6%) |
(41.8%) |
(42.8%) |
(100%) |
|
|
| Mnn16 | 21.9% |
19.0% |
21.2% |
24.6% |
21.4% |
100% |
| (35.4%) | (31.8%) | (36.1%) | (39.8%) | (36.5%) | (100%) | |
a Identity and similarity were calculated over the full length of the protein.
Figure 1.

Phylogram of the C. albicans MNN1 gene family. A multiple sequence alignment and phylogram of the six MNN1 orthologues was generated in Clustal Omega (version 1.1.0).
Single null mutants in all MNN1 family members were generated through sequential gene deletion, and the URA3 marker was introduced at the RPS1 locus to avoid problems associated with its ectopic expression. To control against second site mutations two independently constructed null mutants were generated for each member of the family and shown to display comparable phenotypes. The mutants all had growth rates similar to the wild type control strain (doubling time 1.4 h) in SC medium at 30°C, and displayed no morphological defects, such as cellular aggregation, which has previously been seen in mutants displaying glycosylation defects [7-10,16]. In terms of hyphal development all of the MNN1 family mutants underwent morphogenesis in response to 20% serum. However, the mnn14Δ null mutant displayed a defect in response to the weaker hypha-inducing signal of pH and temperature, with cells constitutively forming pseudohyphae in response to Lee’s medium at pH 6.5 and 37°C. In addition, the mnn14Δ null mutant also failed to form filaments on solid spider medium (Figure 2).
Figure 2.
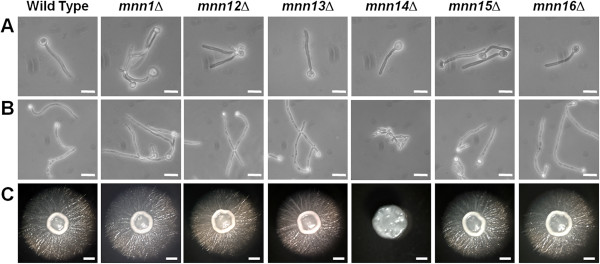
Cell and colony morphology of MNN1 family mutants. Cell morphology after growth at 37°C in 20% serum for 4 h (A) and at 37°C in Lee’s at pH6.5 for 6 h (B). Scale bar, 10 μm. (C) Colony morphology after 6 days growth at 30°C on solid Spider medium. Scale bar, 1 mm.
Glycosylation defects in the MNN1 family mutants
The effect of disruption of the MNN1 family members on N-linked glycosylation was initially assessed through monitoring the electrophoretic mobility of secreted Hex1 (β-N-acetylhexosaminidase) by activity staining following native gel electrophoresis [7]. None of the MNN1 family mutants displayed the increased electrophoretic mobility of Hex1 that is normally associated with a deficiency in N-glycosylation, as demonstrated by och1Δ and pmr1Δ mutants [7,8], indicating no gross N-glycosylation defects in the MNN1 family mutants (Figure 3A). However, the mnn14Δ null mutant instead demonstrated a noticeable decrease in Hex1 mobility following native gel electrophoresis (Figure 3A). This decrease in electrophoretic mobility could be suggestive of an increased level of glycosylation of Hex1 in this mutant. Alternatively, an alteration in the level, or extent of, phosphomannan modification could affect the surface charge of Hex1, which would impact on its mobility in a native gel.
Figure 3.

Glycosylation defects in C. albicans MNN1 family null mutants. The extent of N-glycosylation was determined by activity staining of Hex1 (β-N-acetylhexosaminidase) after protein samples were separated by non-denaturing gel electrophoresis (A). The continuous and dotted lines indicate the normal and decreased electrophoretic mobility of Hex1 respectively. The degree of N-glycosylation modification was also determined by western blot analysis of Hex1-V5-6× His tagged strains (B). Hex1-V5-6× His is apparent in both an unmodified (67 kDa) and a heavily glycosylated (~125 kDa) form. Acid-labile phosphomannan (C) and β-eliminated O-mannan (D) were released from [2-3H] mannose-labelled cells and separated by TLC. Samples are as follows: lane 1, wild type; lane 2, mnn1∆; lane 3, mnn12∆; lane 4, mnn13∆; lane 5, mnn14∆; lane 6, mnn15∆; lane 7, mnn16∆; lane 8, och1∆; lane 9, pmr1∆; lane 10, mnt1∆-mnt2∆.
To screen for more subtle glycosylation defects, and to discount the impact of altered surface charge on electrophoretic mobility, we epitope tagged Hex1 with V5-6xHis in the null mutants and conducted Western blot analysis of Hex1-V5-6xHis following standard denaturing gel electrophoresis. Following Western blot analysis Hex1-V5-6xHis was apparent in both an unmodified (67 kDa) and a heavily glycosylated (~125 kDa) form in soluble protein extracts. In the pmr1Δ mutant, which is known to display glycosylation defects [8], the glycosylated form of Hex1-V5-6xHis demonstrated a clear increase in electrophoretic mobility characteristic of this mutant’s gross defect in glycosylation. However, no change in electrophoretic mobility of the glycosylated form of Hex1-V5-6xHis was observed for any of the MNN1 family mutants (Figure 3B), demonstrating that none of these mutants displayed a gross defect in N-glycosylation. In addition, this would suggest that the decreased electrophoretic mobility of Hex1 seen in the mnn14Δ null mutant following native gel electrophoresis and activity staining is likely to be due to differences in the proteins surface charge and not the overall level of glycosylation.
Phosphomannan, attached to O- and N- mannan through a phosphodiester linkage, can affect the native charge of proteins. The overall level of phosphomannan incorporation into the cell wall can be estimated through the use of the phthalocyanine dye, Alcian Blue, which binds to phosphorylated polysaccharides [20,26]. None of the MNN1 family null mutants displayed altered Alcian Blue binding (Table 2), indicating that each displays a similar level of phosphomannan at the cell surface. This would also suggest that the decreased native gel mobility of Hex1 from mnn14Δ cells was not due to a decrease in the overall level of phosphomannan present. The surface charge associated with phosphomannan is also dependent on the chain length of β1,2-mannose residues present [26,27]. Therefore, we also directly analysed phosphomannan structure in the MNN1 family null mutants by TLC. The wild type strain, and the majority of the mutants, displayed the expected profile of one to five β1,2-mannose residues. However, the mnn14Δ null mutant clearly displayed an altered profile with a preponderance for longer chains and an increased chain length of one to eight residues (Figure 3C). Extension of the phosphomannan chain has previously been identified as playing a major role in the modulation of cell surface charge, with longer phosphomannan chains resulting in the loss of charge demonstrated by an increase in hydrophobicity of the cell wall [26,27]. Therefore the increase in the degree of phosphomannan extension seen in the mnn14Δ null mutant could reduce the surface charge associated with Hex1 which would then explain its decreased mobility under native conditions even though its overall level of modification is not grossly alerted.
Table 2.
Alcian blue binding
| Strain genotype | Alcian blue binding ± SD (μg bound/OD600 1.0) |
|---|---|
| Wild type |
183.1 ± 8.7 |
|
mnn1Δ |
148.2 ± 21.6 |
|
mnn12Δ |
143.3 ± 37.4 |
|
mnn13Δ |
175.3 ± 7.4 |
|
mnn14Δ |
181.9 ± 3.9 |
|
mnn15Δ |
184.1 ± 2.6 |
|
mnn16Δ |
191.1 ± 6.7 |
| och1Δ | 25.2 ± 2.2 |
The structure of O-linked mannans was also assessed by TLC and no significant differences were observed in any of the MNN1 family mutants (Figure 3D). This is consistent with the previous finding that Mnt1 and Mnt2 are the principal enzymes involved in the extension of O-glycans, and that C. albicans lacks α1,3-linked residues in its O-mannan [11].
Cell wall associated defects
To examine cell wall integrity we screened the MNN1 family null mutants for sensitivity to a range of cell wall perturbing agents. The mnn14Δ null mutant was clearly hypersensitive to SDS, tunicamycin, and hygromycin B which is characteristic of glycosylation mutants (Figure 4). However, no increase in sensitivity was seen to the cell wall perturbing agent Calcofluor White, suggesting that the mnn14Δ null mutant does not have a defect in the general robustness of the cell wall. No other family members displayed altered sensitivity to any of the compounds tested. Changes in cell wall structure were also assessed through HPLC sugar composition analysis, but no gross alteration in the relative proportions of the three main cell wall polysaccharides (glucan, mannan and chitin) was identified for any of the MNN1 family null mutants (data not shown).
Figure 4.
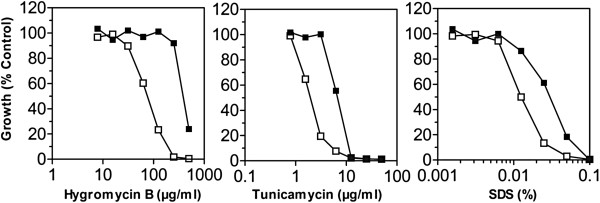
Sensitivity of C. albicans mnn14Δ null mutant to cell wall perturbing agents. The sensitivities of the wild type (closed squares) and Camnn14Δ null mutant (open squares) to cell wall perturbing agents were determined by a broth microdilution method. The agents to which the Camnn14Δ null mutant displayed hypersensitivity are shown (hygromycin B, tunicamycin, and SDS).
Defects in the initiation of O-glycosylation or treatment with tunicamycin, which inhibits N-glycosylation, have previously been shown to impair biofilm formation [28,29]. We therefore screened the MNN1 family mutants for defects in biofilm formation, using a 96 well microplate model and XTT reduction assay to measure metabolic activity of the cells within the biofilm. As predicted the och1∆ mutant [7], which has a gross defect in N-linked glycosylation, displayed a severe biofilm defect demonstrating a 90% reduction in biofilm formation at 48 h, therefore confirming the importance of N-mannan in biofilm formation. In addition the mnt1Δ-mnt2Δ double mutant [11], which lacks the 4 terminal mannose residues in O-mannan, also displayed a clear biofilm defect (80% reduction). This indicates that the extension of O-mannan, in addition to the initiation of its synthesis, is also important for biofilm formation. However, none of the MNN1 family single mutants displayed a significant defect in biofilm formation (Figure 5). This lack of a phenotypic defect may be due to functional redundancy in the MNN1 family. Alternatively, it may suggest that only gross N-glycosylation defects impact upon biofilm formation rather than its being dependent on specific epitopes. Indeed, the pmr1Δ null mutant, which displays both O- and N-linked glycosylation defects, but to a lesser extent than the individual defects seen in the och1Δ or mnt1Δ-mnt2Δ mutants, only displays a subtle biofilm defect (40% reduction at 48 h). Therefore, biofilm formation is dependent both on correct O-glycosylation and the overall level of N-glycosylation, presumably through their impact on cellular interactions.
Figure 5.

Biofilm formation of MNN1 family mutants. Biofilm formation was assessed in 96 well plates using a tetrazolium salt (XTT) reduction assay following growth in SC medium for 24 h (closed bars) and 48 h (open bars).
Host-pathogen interaction and virulence
As cell wall mannan has been shown to play an important role in the stimulation of the host response [15] we tested the ability of the null mutants to induce cytokine production by human PBMCs. All the MNN1 family mutants were as potent at stimulating TNFα and IL-6 production as wild type C. albicans (data not shown). Virulence of the single mutants was tested in a Galleria mellonella larvae model of infection. In this model all larvae infected with the wild type strain succumbed to infection by day 2, whereas 75% of larvae infected with the pmr1Δ mutant, which displays a gross defect in glycosylation, survived to the end of the experiment. However, none of the MNN1 family mutants displayed a virulence defect, demonstrating that none of the individual family members are required for virulence in this model. Because the mnn14Δ mutant displayed an altered phenotype in vitro we tested its virulence in a murine model of disseminated infection; in addition the mnn1Δ and mnn12Δ mutants were also tested. The mnn1Δ and mnn12Δ null mutants were unaltered in virulence. However, the mnn14Δ null mutant was clearly attenuated in virulence (log-rank test; p < 0.001) with all mice surviving to the end of the experiment at 28 days compared to a median survival time of 9 days for the wild type control. Mice infected with the mnn14Δ null mutant also displayed a clear reduction in tissue burdens at 28 days compared to that seen post-mortem following infection with the wild type strain, with a >2 log reduction in kidney burdens (log10 CFU/g 4.0 ± 1.3 cf. 6.5 ± 0.4) and a 1 log reduction in the brain (log10 CFU/g 3.3 ± 0.9 cf. 4.3 ± 0.4). Hence the mnn14Δ null mutant was significantly attenuated in virulence in the murine model of systemic candidiasis.
Conclusion
The C. albicans cell wall is the immediate point of contact between the invading fungus and the host, and previous work has clearly identified both O- and N- mannan structures as important in the host-pathogen interaction and virulence. In this study, we present the first analysis of the C. albicans MNN1 gene family, which is predicted to encode capping enzymes involved in the terminal stages of glycosylation. We generated single null mutants in the six members of the C. albicans MNN1 gene family. The majority of the mutants generated, with the exception of the mnn14Δ null mutant, demonstrated no discernible change in phenotype potentially due to functional redundancy as seen with some other C. albicans mannosyltransferase gene families [9,11,16]. Disruption of MNN14 however led to both in vitro and in vivo defects. The mnn14Δ null mutant displayed subtle morphogenesis defects and hypersensitivity to agents associated with cell wall and glycosylation defects, suggesting altered cell wall structure or permeability. However, no gross changes in cell wall composition or N-glycan extension were identified, although an extension of phosphomannan chains was apparent. This extension of phosphomannan in the mnn14Δ null mutant could be a compensatory mechanism for its absence, through an increase in the expression, or activity, of other mannosyltransferases. Alternatively Mnn14 could potentially act as a capping enzyme to terminate extension similar to the proposed role of MNN1 in S. cerevisiae[30]. Although the mnn14Δ null mutant only displayed subtle cell wall defects this mutant did display a severe defect in virulence in a murine infection model, apparent both in terms of overall survival and associated tissue burdens. This virulence defect was not associated with differences in the induction of inflammatory responses, as no defect was seen in the TNFα and IL-6 host cytokine production in vitro, which suggests that the virulence defect may not be due to an alteration of the host response. Overall, therefore, the MNN1 family appears to display redundancy, with the exception of MNN14, which may therefore play a distinct role either in the synthesis of specific epitopes or in the modification of a discrete protein(s) required for virulence and the host-pathogen interaction.
Methods
Strains, media and culture conditions
C. albicans strains were grown in yeast and hyphal forms as described previously [7]. To induce β-N-acetylhexosaminidase expression strains were grown in SC-GlcNAc (0.67% yeast nitrogen base, 0.079% complete supplement mixture, 25 mM N-acetylglucosamine). Biofilm formation in the mutants was assessed using a tetrazolium salt (XTT) reduction assay following growth in SC medium for 24 and 48 h [31]. Null mutants in MNN1 (orf19.4279) and MNN12 (orf19.4900) were generated by the “Ura-blaster” protocol in strain CAI-4 [32]. Briefly, the 5′ and 3′ regions of homology were amplified by PCR (MNN1 5′ primer pair MNN1-5H/5S, 3′ primer pair MNN1-3A/3B; MNN12 5′ primer pair MNN12-5H/5S, 3′ primer pair MNN12-3A/3B) and cloned into the HindIII/SphI and Asp718/BanII sites of the disruption vector pMB-7 respectively. The ura-blaster cassettes were then released by digestion with HindIII and BanII and genes disrupted in CAI-4 by sequential gene replacement and recycling of the URA3 marker by selection on SD medium plus 5-fluoroorotic acid (1 mg/ml) and uridine (50 μg/ml). Single null mutants in the other family members, MNN13 (orf19.4270), MNN14 (orf19.6996) MNN15 (orf19.753) and MNN16 (orf19.6313), were generated through PCR mediated disruption [33]. Primers (MNNxx-KO-F/-R) were designed to contain 70 bp homology to the gene of interest, and disruption cassettes amplified from pSN40 (C. maltosa LEU2) and pSN52 (C. dubliniensis HIS1). Amplified cassettes were then sequentially transformed into strain SN78 to generate null mutants. The URA3 marker was integrated at the RPS1 locus in all mutants by transformation with the StuI-digested CIp10 vector [34], to avoid problems associated with its expression [35]. To control against second site mutations two independently constructed null mutants were generated and screened for each member of the family. Strain genotypes and the oligonucleotides used in their construction are listed in Tables 3 and 4. The wild type strain used for phenotypic studies throughout was NGY152 [35] derived from CAI-4 with the CIp10 vector integrated at the RPS1 locus.
Table 3.
C. albicans strains
| Strain | Parent strain | Genotype | Reference |
|---|---|---|---|
| CAI-4 |
- |
ura3Δ::imm434/ura3Δ::imm434 |
[32] |
| SN78 |
- |
ura3Δ::imm434/ura3Δ::imm434, leu2Δ/leu2Δ, his1Δ/his1Δ |
[33] |
| NGY152 |
CAI-4 |
As CAI-4 but RPS1/rps1Δ::CIp10 |
[35] |
| SBC106 |
CAI-4 |
As CAI-4 but MNN1/mnn1Δ::hisG-URA3-hisG |
This study |
| SBC107 |
SBC106 |
As CAI-4 but MNN1/mnn1Δ::hisG |
This study |
| SBC108 |
SBC107 |
As CAI-4 but mnn1Δ::hisG/mnn1Δ::hisG-URA3-hisG |
This study |
| SBC109 |
CAI-4 |
As CAI-4 but MNN12/mnn12Δ::hisG-URA3-hisG |
This study |
| SBC110 |
SBC109 |
As CAI-4 but MNN12/mnn12Δ::hisG |
This study |
| SBC111 |
SBC110 |
As CAI-4 but mnn12Δ::hisG/mnn12Δ::hisG-URA3-hisG |
This study |
| SBC112 |
SBC108 |
As CAI-4 but mnn1Δ::hisG/mnn1Δ::hisG |
This study |
| SBC113 |
SBC111 |
As CAI-4 but mnn12Δ::hisG/mnn12Δ::hisG |
This study |
| SBC114 |
SBC112 |
As CAI-4 but mnn1Δ::hisG/mnn1Δ::hisG, RPS1/rps1Δ::CIp10 |
This study |
| SBC115 |
SBC113 |
As CAI-4 but mnn12Δ::hisG/mnn12Δ::hisG, RPS1/rps1Δ::CIp10 |
This study |
| SBC122 |
SN78 |
As SN78 but MNN13/mnn13Δ::CmLeu2 |
This study |
| SBC123 |
SN78 |
As SN78 but MNN14/mnn14Δ::CmLeu2 |
This study |
| SBC124 |
SN78 |
As SN78 but MNN15/mnn15Δ::CmLeu2 |
This study |
| SBC135 |
SN78 |
As SN78 but MNN16/mnn16Δ::CmLeu2 |
This study |
| SBC126 |
SBC122 |
As SN78 but mnn13Δ::CmLeu2/mnn13Δ::CdHIS1 |
This study |
| SBC127 |
SBC123 |
As SN78 but mnn14Δ::CmLeu2/mnn14Δ::CdHIS1 |
This study |
| SBC128 |
SBC124 |
As SN78 but mnn15Δ::CmLeu2/mnn15Δ::CdHIS1 |
This study |
| SBC129 |
SBC135 |
As SN78 but mnn16Δ::CmLeu2/mnn16Δ::CdHIS1 |
This study |
| SBC130 |
SBC126 |
As SN78 but mnn13Δ::CmLeu2/mnn13Δ::CdHIS1, RPS1/rps1Δ::CIp10 |
This study |
| SBC131 |
SBC127 |
As SN78 but mnn14Δ::CmLeu2/mnn14Δ::CdHIS1, RPS1/rps1Δ::CIp10 |
This study |
| SBC132 |
SBC128 |
As SN78 but mnn15Δ::CmLeu2/mnn15Δ::CdHIS1, RPS1/rps1Δ::CIp10 |
This study |
| SBC133 |
SBC129 |
As SN78 but mnn16Δ::CmLeu2/mnn16Δ::CdHIS1, RPS1/rps1Δ::CIp10 |
This study |
| SBC163 |
SBC130 |
As SN78 but mnn13Δ::CmLeu2/mnn13Δ::CdHIS1, RPS1/rps1Δ::CIp10, HEX1/HEX1-V5-6xHis-NAT1 |
This study |
| SBC164 |
SBC131 |
As SN78 but mnn14Δ::CmLeu2/mnn14Δ::CdHIS1, RPS1/rps1Δ::CIp10, HEX1/HEX1-V5-6xHis-NAT1 |
This study |
| SBC165 |
SBC132 |
As SN78 but mnn15Δ::CmLeu2/mnn15Δ::CdHIS1, RPS1/rps1Δ::CIp10, HEX1/HEX1-V5-6xHis-NAT1 |
This study |
| SBC166 |
SBC133 |
As SN78 but mnn16Δ::CmLeu2/mnn16Δ::CdHIS1, RPS1/rps1Δ::CIp10, HEX1/HEX1-V5-6xHis-NAT1 |
This study |
| SBC161 |
SBC114 |
As CAI-4 but mnn1Δ::hisG/mnn1Δ::hisG, RPS1/rps1Δ::CIp10, HEX1/HEX1-V5-6xHis-NAT1 |
This study |
| SBC162 |
SBC115 |
As CAI-4 but mnn12Δ::hisG/mnn12Δ::hisG, RPS1/rps1Δ::CIp10, HEX1/HEX1-V5-6xHis-NAT1 |
This study |
| SBC158 |
NGY152 |
As NGY152 but HEX1/HEX1-V5-6xHis-NAT1 |
[36] |
| SBC159 |
- |
pmr1Δ::hisG/pmr1Δ::hisG HEX1/HEX1-V5-6xHis-URA3 |
[36] |
| NGY355 |
- |
As CAI-4 but pmr1Δ::hisG/pmr1Δ::hisG, RPS1/rps1Δ::CIp10 |
[8] |
| NGY357 |
|
As CAI-4 but och1Δ::hisG/och1Δ::hisG, RPS1/rps1Δ::CIp10 |
[7] |
| NGY337 | As CAI-4 but mnt1-mnt2Δ::hisG/mnt1-mnt2Δ::hisG, RPS1/rps1Δ ::CIp10 |
[11] |
Table 4.
Oligonucleotides used for strain construction
| Primer | Sequence (5′-3′) a |
|---|---|
| MNN1-5H |
AAGCTTtctacagtggctgttaataatg |
| MNN1-5S |
GCATGCgaaaactcatacaattcactaag |
| MNN1-3A |
GGTACCtgcttactcaagtgttggtg |
| MNN1-3B |
GAGCTCgttagtggcttgtatcaaagg |
| MNN12-5H |
AAGCTTatcataacacgttcgttcagc |
| MNN12-5S |
GCATGCtgacagatcatcgtcttcatc |
| MNN12-3A |
GGTACCatgccattattccaccaatgg |
| MNN12-3B |
GAGCTCgttgtgactgtggctccac |
| MNN13-KO-F |
gttgactttgaagagccactgtatactttccctaagacggtgctatttgaaaacaacaacaatttggatgatttattatGCTCGGATCCACTAGTAACG |
| MNN13-KO-R |
ttaatattttccaacccatacatctcctaagtaattgaaatatgcaatttcttcaggggtatactcaaccaataaaccaCCAGTGTGATGGATATCTGC |
| MNN14-KO-F |
atgcattttatttggcaatcacaagccaattctggattaatactaaaaaatgaattatcactgacatcaagtatcccccGCTCGGATCCACTAGTAACG |
| MNN14-KO-R |
ctataaataattatatcccctcatccatatctccccaagctttttaaaatgttctatttcctctggtgaataattaattCCAGTGTGATGGATATCTGC |
| MNN15-KO-F |
cgtataagggaacttttacatgatagtgaaaaatttgatctcgagagtctaaagcaagctagcttggagataagaaaagGCTCGGATCCACTAGTAACG |
| MNN15-KO-R |
ctttatccgaaccagttccaagtttctcagtttctaatttaacttttggcttcattttggatccaccagtgtgccatatCCAGTGTGATGGATATCTGC |
| MNN16-KO-F |
gaagttccatcttaaaagatatgtcatagttacttcaatactactatcttttttcttattattcagacgacaatttcttGCTCGGATCCACTAGTAACG |
| MNN16-KO-R | ttaatcttgtaaccaaatttcaattaatttcttatattttttttgtaaactttgttcaacattaatagataatcctctaCCAGTGTGATGGATATCTGC |
a Gene-specific sequences are lowercase.
Cell wall and glycosylation analysis
Null mutants were screened for sensitivity to cell wall stressing agents by the microdilution method as previously reported [7]. Briefly, standardised inocula (A600 = 0.01) of strains in YEPD medium were incubated with cell wall stressing agents, across a range of doubling dilutions, for 16 h at 30°C and the A600 was determined. The agents tested (maximum concentrations in parentheses) included Calcofluor White (500 μg/ml), Congo Red (500 μg/ml), SDS (0.1%), hygromycin B (500 μg/ml), NaCl (2 M) and tunicamycin (50 μg/ml). Phosphomannan content and N-glycosylation status were determined by Alcian Blue binding assays and β-N-acetylhexosaminidase (Hex1) native PAGE zymograms as reported elsewhere [7]. In addition the N-glycosylation status was assessed by Western blot analysis of mutant strains carrying the V5-6xHis tagged Hex1 protein as a glycosylation reporter as described previously [36]. Cell wall carbohydrate composition was determined by acid hydrolysis of the cell wall carbohydrate polymers and quantification of constituent monosaccharides following high performance anion exchange chromatography [37]. Finally, for the analysis of phosphomannan and O-linked mannan, strains were labelled with D-[2-3H] mannose, mannans released through mild acid treatment and β-elimination respectively and separated through thin layer chromatography (TLC) [7].
Virulence assays
Isolation of human peripheral blood mononuclear cells (PMBCs) and cytokine stimulation assays were carried out as described previously [15]. For the wax moth larvae infection model groups of 8 larvae were infected with 3×105 CFU of C. albicans through the last left proleg and survival monitored over 7 days at 37°C. To test virulence in a murine infection model groups of 6 female BALB/c mice were intravenously challenged with 3.6×104 CFU/g (body weight) C. albicans and monitored over 28 days. Animals showing signs of illness or disease were humanely terminated and recorded as dying the following day, and those surviving the course of the experiment were terminated on day 28. The kidneys and brain were aseptically removed post-mortem, homogenised, and tissue burdens determined by viable counting. Experiments were conducted under the terms of a UK Home Office licence and were approved by the University of Aberdeen Ethical Review Committee.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SB conducted the experimental work and drafted the manuscript. RAH and JC assisted with the experimental work. MGN carried out the immunoassays. DMM conducted the murine infection model. SB, AJPB, FCO and NARG conceived the study, participated in its design and co-ordination. All authors read and approved the final manuscript.
Contributor Information
Steven Bates, Email: s.bates@ex.ac.uk.
Rebecca A Hall, Email: r.a.hall@bham.ac.uk.
Jill Cheetham, Email: j.cheetham@ex.ac.uk.
Mihai G Netea, Email: m.netea@aig.umcn.nl.
Donna M MacCallum, Email: d.m.maccallum@abdn.ac.uk.
Alistair JP Brown, Email: al.brown@abdn.ac.uk.
Frank C Odds, Email: f.odds@abdn.ac.uk.
Neil AR Gow, Email: n.gow@abdn.ac.uk.
Acknowledgements
This work was supported by a Wellcome Trust Programme grant (080088) to NG, FO and AB and by a FP7-2007-2013 grant agreement (HEALTH-F2- 2010-260338–ALLFUN) and BBSRC SABR (CRISP) award. MGN was supported by a Vici grant of the Netherlands Organization for Scientific Research. JC was supported by a UK Biotechnology and Biological Sciences Research Council project grant (BB/F009232/1) to SB.
References
- Leroy O, Gangneux JP, Montravers P, Mira JP, Gouin F, Sollet JP, Carlet J, Reynes J, Rosenheim M, Regnier B, Lortholary O. Epidemiology, management, and risk factors for death of invasive Candida infections in critical care: a multicenter, prospective, observational study in France (2005–2006) Crit Care Med. 2009;37:1612–1618. doi: 10.1097/CCM.0b013e31819efac0. [DOI] [PubMed] [Google Scholar]
- Perlroth J, Choi B, Spellberg B. Nosocomial fungal infections: epidemiology, diagnosis, and treatment. Med Mycol. 2007;45:321–346. doi: 10.1080/13693780701218689. [DOI] [PubMed] [Google Scholar]
- Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol. 2010;36:1–53. doi: 10.3109/10408410903241444. [DOI] [PubMed] [Google Scholar]
- Klis FM, de Groot P, Hellingwerf K. Molecular organization of the cell wall of Candida albicans. Med Mycol. 2001;39(Suppl 1):1–8. [PubMed] [Google Scholar]
- Mora-Montes HM, Ponce-Noyola P, Villagomez-Castro JC, Gow NA, Flores-Carreon A, Lopez-Romero E. Protein glycosylation in Candida. Future Microbiol. 2009;4:1167–1183. doi: 10.2217/fmb.09.88. [DOI] [PubMed] [Google Scholar]
- Ruiz-Herrera J, Elorza MV, Valentin E, Sentandreu R. Molecular organization of the cell wall of Candida albicans and its relation to pathogenicity. FEMS Yeast Res. 2006;6:14–29. doi: 10.1111/j.1567-1364.2005.00017.x. [DOI] [PubMed] [Google Scholar]
- Bates S, Hughes HB, Munro CA, Thomas WP, MacCallum DM, Bertram G, Atrih A, Ferguson MA, Brown AJ, Odds FC, Gow NA. Outer chain N-glycans are required for cell wall integrity and virulence of Candida albicans. J Biol Chem. 2006;281:90–98. doi: 10.1074/jbc.M510360200. [DOI] [PubMed] [Google Scholar]
- Bates S, MacCallum DM, Bertram G, Munro CA, Hughes HB, Buurman ET, Brown AJ, Odds FC, Gow NA. Candida albicans Pmr1p, a secretory pathway P-type Ca2+/Mn2+-ATPase, is required for glycosylation and virulence. J Biol Chem. 2005;280:23408–23415. doi: 10.1074/jbc.M502162200. [DOI] [PubMed] [Google Scholar]
- Mora-Montes HM, Bates S, Netea MG, Castillo L, Brand A, Buurman ET, Diaz-Jimenez DF, Jan Kullberg B, Brown AJ, Odds FC, Gow NA. A multifunctional mannosyltransferase family in Candida albicans determines cell wall mannan structure and host-fungus interactions. J Biol Chem. 2010;285:12087–12095. doi: 10.1074/jbc.M109.081513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora-Montes HM, Bates S, Netea MG, Diaz-Jimenez DF, Lopez-Romero E, Zinker S, Ponce-Noyola P, Kullberg BJ, Brown AJ, Odds FC. et al. Endoplasmic reticulum alpha-glycosidases of Candida albicans are required for N glycosylation, cell wall integrity, and normal host-fungus interaction. Eukaryot Cell. 2007;6:2184–2193. doi: 10.1128/EC.00350-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro CA, Bates S, Buurman ET, Hughes HB, Maccallum DM, Bertram G, Atrih A, Ferguson MA, Bain JM, Brand A. et al. Mnt1p and Mnt2p of Candida albicans are partially redundant alpha-1,2-mannosyltransferases that participate in O-linked mannosylation and are required for adhesion and virulence. J Biol Chem. 2005;280:1051–1060. doi: 10.1074/jbc.M411413200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouabhia M, Schaller M, Corbucci C, Vecchiarelli A, Prill SK, Giasson L, Ernst JF. Virulence of the fungal pathogen Candida albicans requires the five isoforms of protein mannosyltransferases. Infect Immun. 2005;73:4571–4580. doi: 10.1128/IAI.73.8.4571-4580.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradin C, Slomianny MC, Mille C, Masset A, Robert R, Sendid B, Ernst JF, Michalski JC, Poulain D. β-1,2 Oligomannose adhesin epitopes are widely distributed over the different families of Candida albicans cell wall mannoproteins and are associated through both N- and O-glycosylation processes. Infect Immun. 2008;76:4509–4517. doi: 10.1128/IAI.00368-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mille C, Bobrowicz P, Trinel PA, Li H, Maes E, Guerardel Y, Fradin C, Martinez-Esparza M, Davidson RC, Janbon G. et al. Identification of a new family of genes involved in beta-1,2-mannosylation of glycans in Pichia pastoris and Candida albicans. J Biol Chem. 2008;283:9724–9736. doi: 10.1074/jbc.M708825200. [DOI] [PubMed] [Google Scholar]
- Netea MG, Gow NA, Munro CA, Bates S, Collins C, Ferwerda G, Hobson RP, Bertram G, Hughes HB, Jansen T. et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and toll-like receptors. J Clin Invest. 2006;116:1642–1650. doi: 10.1172/JCI27114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RA, Bates S, Lenardon MD, MacCallum DM, Wagener J, Lowman DW, Kruppa MD, Williams DL, Odds FC, Brown AJP. et al. The Mnn2 mannosyltransferase family modulates mannoprotein fibril length, immune recognition and virulence of Candida albicans. PLoS Pathog. 2013;9:e1003276. doi: 10.1371/journal.ppat.1003276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean N. Asparagine-linked glycosylation in the yeast Golgi. Biochim Biophys Acta. 1999;1426:309–322. doi: 10.1016/S0304-4165(98)00132-9. [DOI] [PubMed] [Google Scholar]
- Southard SB, Specht CA, Mishra C, Chen-Weiner J, Robbins PW. Molecular analysis of the Candida albicans homolog of Saccharomyces cerevisiae MNN9, required for glycosylation of cell wall mannoproteins. J Bacteriol. 1999;181:7439–7448. doi: 10.1128/jb.181.24.7439-7448.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Shibata N, Osaka T, Miyagawa Y, Ohkubo Y, Suzuki S. Structural study of cell wall mannan of a Candida albicans (serotype A) strain. Phytochemistry. 1992;31:1147–1153. doi: 10.1016/0031-9422(92)80250-I. [DOI] [PubMed] [Google Scholar]
- Hobson RP, Munro CA, Bates S, MacCallum DM, Cutler JE, Heinsbroek SE, Brown GD, Odds FC, Gow NA. Loss of cell wall mannosylphosphate in Candida albicans does not influence macrophage recognition. J Biol Chem. 2004;279:39628–39635. doi: 10.1074/jbc.M405003200. [DOI] [PubMed] [Google Scholar]
- Trinel PA, Lepage G, Jouault T, Strecker G, Poulain D. Definitive chemical evidence for the constitutive ability of Candida albicans serotype A strains to synthesize beta-1,2 linked oligomannosides containing up to 14 mannose residues. FEBS Lett. 1997;416:203–206. doi: 10.1016/S0014-5793(97)01205-2. [DOI] [PubMed] [Google Scholar]
- Romero PA, Lussier M, Veronneau S, Sdicu AM, Herscovics A, Bussey H. Mnt2p and Mnt3p of Saccharomyces cerevisiae are members of the Mnn1p family of alpha-1,3-mannosyltransferases responsible for adding the terminal mannose residues of O-linked oligosaccharides. Glycobiology. 1999;9:1045–1051. doi: 10.1093/glycob/9.10.1045. [DOI] [PubMed] [Google Scholar]
- Yip CL, Welch SK, Klebl F, Gilbert T, Seidel P, Grant FJ, O’Hara PJ, MacKay VL. Cloning and analysis of the Saccharomyces cerevisiae MNN9 and MNN1 genes required for complex glycosylation of secreted proteins. Proc Natl Acad Sci USA. 1994;91:2723–2727. doi: 10.1073/pnas.91.7.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins CA, Munro S. Activity of the yeast MNN1 alpha-1,3-mannosyltransferase requires a motif conserved in many other families of glycosyltransferases. Proc Natl Acad Sci USA. 1998;95:7945–7950. doi: 10.1073/pnas.95.14.7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler G, Rasmussen MD, Lin MF, Santos MA, Sakthikumar S, Munro CA, Rheinbay E, Grabherr M, Forche A, Reedy JL. et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009;459:657–662. doi: 10.1038/nature08064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuoka J, Hazen KC. Differences in the acid-labile component of Candida albicans mannan from hydrophobic and hydrophilic yeast cells. Glycobiology. 1999;9:1281–1286. doi: 10.1093/glycob/9.11.1281. [DOI] [PubMed] [Google Scholar]
- Masuoka J, Hazen KC. Cell wall mannan and cell surface hydrophobicity in Candida albicans serotype A and B strains. Infect Immun. 2004;72:6230–6236. doi: 10.1128/IAI.72.11.6230-6236.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltroche-Llacsahuanga H, Goyard S, D’Enfert C, Prill SK, Ernst JF. Protein O-mannosyltransferase isoforms regulate biofilm formation in Candida albicans. Antimicrob Agents Chemother. 2006;50:3488–3491. doi: 10.1128/AAC.00606-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce CG, Thomas DP, Lopez-Ribot JL. Effect of tunicamycin on Candida albicans biofilm formation and maintenance. J Antimicrob Chemother. 2009;63:473–479. doi: 10.1093/jac/dkn515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier M, Sdicu AM, Bussey H. The KTR and MNN1 mannosyltransferase families of Saccharomyces cerevisiae. Biochim Biophys Acta. 1999;1426:323–334. doi: 10.1016/S0304-4165(98)00133-0. [DOI] [PubMed] [Google Scholar]
- Ramage G, Vande Walle K, Wickes BL, Lopez-Ribot JL. Standardized method for in vitro antifungal susceptibility testing of Candida albicans biofilms. Antimicrob Agents Chemother. 2001;45:2475–2479. doi: 10.1128/AAC.45.9.2475-2479.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonzi WA, Irwin MY. Isogenic strain construction and gene mapping in Candida albicans. Genetics. 1993;134:717–728. doi: 10.1093/genetics/134.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble SM, Johnson AD. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot Cell. 2005;4:298–309. doi: 10.1128/EC.4.2.298-309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murad AM, Lee PR, Broadbent ID, Barelle CJ, Brown AJ. CIp10, an efficient and convenient integrating vector for Candida albicans. Yeast. 2000;16:325–327. doi: 10.1002/1097-0061(20000315)16:4<325::AID-YEA538>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Brand A, MacCallum DM, Brown AJ, Gow NA, Odds FC. Ectopic expression of URA3 can influence the virulence phenotypes and proteome of Candida albicans but can be overcome by targeted reintegration of URA3 at the RPS10 locus. Eukaryot Cell. 2004;3:900–909. doi: 10.1128/EC.3.4.900-909.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne SW, Cheetham J, Lloyd D, Aves S, Bates S. Cassettes for PCR-mediated gene tagging in Candida albicans utilizing nourseothricin resistance. Yeast. 2011;28:833–841. doi: 10.1002/yea.1910. [DOI] [PubMed] [Google Scholar]
- Munro CA, Whitton RK, Hughes HB, Rella M, Selvaggini S, Gow NA. CHS8-a fourth chitin synthase gene of Candida albicans contributes to in vitro chitin synthase activity, but is dispensable for growth. Fungal Genet Biol. 2003;40:146–158. doi: 10.1016/S1087-1845(03)00083-5. [DOI] [PubMed] [Google Scholar]