

Abstract
Significance
Both chronic wounds and excessive scar formation after cutaneous injury create a formidable clinical problem resulting in considerable morbidity and healthcare expenditure. The deposition and remodeling of extracellular matrix (ECM) components are critical processes in cutaneous healing. Understanding the role of the ECM in directing progenitor and reparative cell fate and activities during wound repair is required to improve wound-care strategies.
Recent Advances
In addition to providing structural integrity, the ECM is recognized to play critical roles in regulating progenitor and reparative cell behaviors such as migration, differentiation, proliferation, and survival. The ECM dictates these activities through its binding of adhesion receptors as well as its ability to regulate growth factor bioavailability and signaling. More recently, a key role for mechanical control of cell fate through interaction with the ECM has emerged.
Critical Issues
Despite significant advances in understanding the pathophysiology of cutaneous wound repair, problematic wounds remain a significant healthcare challenge. Regenerative medical strategies that either target endogenous stem cells or utilize applications of exogenous stem cell populations have emerged as promising approaches to pathologic wounds. However, the identification of smart biomaterials and matrices may allow for further optimization of such therapies.
Future Directions
An efficient and appropriate healing response in the skin postinjury is regulated by a fine balance of the quantity and quality of ECM proteins. A more complete understanding of ECM regulation of the cell fate and activities during cutaneous wound repair is vital for the development of novel treatment strategies for improvement of cutaneous healing.

Susan W. Volk, VMD, PhD

Ardeshir Bayat, BSc (Hons), MBBS, MRCS, PhD
Scope and Significance
Progenitor cells have been isolated from all layers of the skin and contribute to its repair upon injury. In all phases of wound repair, extracellular matrix (ECM) proteins have been shown to play an integral role in a dynamic relationship between stem/progenitor and other reparative cell populations, growth factors, and matrix modifying-enzymes. This review provides a current understanding of how the ECM plays a critical role in healing of both the epidermal and dermal compartments by directing the progenitor cell fate after cutaneous injury and how this knowledge may be applied to develop improved therapies for chronic wounds and targeted strategies to prevent excessive scar formation.
Translational Relevance
The role of progenitor cells in cutaneous wound healing and tissue regeneration is well established. It is also widely accepted that the ECM plays a key role in tissue repair beyond its ability to provide a structural framework for cells through its ability to modulate cell behavior. Elucidating mechanisms by which ECM controls the behavior of progenitor cells during the course of wound repair will provide novel strategies for the treatment of pathologic healing of the skin.
Clinical Relevance
Chronic nonhealing wounds, such as venous leg, diabetic foot, and pressure ulcers, are a significant source of morbidity for more than 6 million Americans, resulting in an annual healthcare cost exceeding $25 billion.1 Changes in the ECM associated with pathologic states may halt progression of the sequence of events critical for timely wound healing. On the other end of the scale, excessive production of a collagenous matrix can be equally problematic, resulting in cutaneous scarring that causes functional impairment, discomfort, and esthetic disfigurement. Through the manipulation of both ECM components and stem/progenitor cells, regenerative medicine approaches have shown promise for the treatment of both extremes of cutaneous healing.
Background
The skin is the largest organ in the body and acts as the first line of defense against pathogens, toxins, and trauma. It also plays a critical role in fluid homeostasis, and provides sensory functions and thermal regulation. Damage or loss of skin integrity resulting from an injury or disease can lead to significant morbidity and even death. After injury, a complex overlapping process to repair the two layers of skin, the dermis of mesodermal origin and the overlying epidermis of ectodermal origin, is initiated to restore tissue integrity. Analogous to repair processes in many noncutaneous tissues, repair of the skin can be divided into three phases: an initial inflammatory phase, followed by a proliferative/repair phase, and concluding with a remodeling phase, which results in scar formation in postnatal mammals.
In all phases of wound repair, ECM proteins play a key role in directing progenitor and reparative cell fate and activities. Immediately after injury, the ECM orchestrates the recruitment of platelets and directs the inflammatory cell response to initiate the hemostatic and the cellular debridement phases.2 These cells, which migrate into the wound bed on the ECM of the initial hemostatic plug and then into the provisional matrix, respond to individual ECM components and growth factors (which may be bound to this matrix). These cells, in turn, recruit and direct stem/progenitor and reparative cells from both distant sites and locally to mediate the proliferative/repair phase of healing. Particularly, in this rebuilding phase of healing, adult stem cells play a critical role in replenishing cells damaged or lost after injury. In addition to their role post-trauma, adult stem cells participate in maintenance of the skin as well as healing, which occurs as a consequence of disease. Therefore, understanding how the ECM directs stem/progenitor cell fate and activities in the skin has potentially far-reaching implications for a variety of cutaneous pathologies.
Stem cells reside in a specialized microenvironment that has been described as the stem cell niche,3 comprised of a complex milieu of directional inputs from surrounding cells, signaling molecules, matrix components, and their modifiers, as well other environmental factors such as pH and oxygen tension. In both noninjured and injured skin, quantitative and qualitative differences in the ECM components provide instructive cues for stem cells to differentiate or self-renew. Aberrant ECM expression as well as post-translational modification of matrix molecules that occurs secondary to aging, diseases (such as vascular disease and diabetes), pathogens, or steroid therapy can contribute to pathologic cellular wound responses.4
The four general categories of ECM proteins in a healing wound include structural ECM proteins, adhesive glycoproteins, proteoglycans, and matricellular proteins (Table 1). These molecules direct cutaneous stem and reparative cell fate and activities by direct engagement of cells via cell–ECM interactions or indirectly through their modulation of growth factor activities. These matrix components communicate with cells of the repair process through integrins and nonintegrin receptors such as discoidin domain receptors, cell surface proteoglycans, syndecans, and hyaluronan receptors. These interactions direct stem and/or other reparative cells to both sense and respond to biophysical signals and to maintain a balance between the forces originating from the ECM and those that arise from the contractility of the intracellular cytoskeleton. Further, these mechanotransduction processes can combine with growth factor-mediated signaling to determine the stem cell fate.4 Finally, growth factor signaling can be altered through direct interactions of individual domains of ECM proteins with growth factor receptors or by controlling either the bioavailability or activity of the ligands (Table 2).5–15
Table 1.
Major classes of extracellular matrix proteins that participate in cutaneous wound repair
| Class | Examples |
|---|---|
| Structural | Collagens |
| Elastin | |
| Fibrillins | |
| Fibulin | |
| Multidomain adhesive glycoproteins | Fibronectins |
| Vitronectin | |
| Laminins | |
| Matrilins | |
| Glycosaminoglycans and proteoglycans | Hyaluronan |
| Proteoglycans | |
| Decorin | |
| Biglycan | |
| Perlecan | |
| Syndecans | |
| Versican | |
| Matricellular proteins | Thrombospondins |
| Tenascin C and X | |
| CCN (such as CCN2/CTGF) | |
| SPARC/BM-40/Osteonectin | |
| Osteopontin |
Table 2.
Select examples of extracellular matrix modulation of growth factor activities
| ECM–Growth Factor Interaction | Example | Consequence |
|---|---|---|
| ECM binding to growth factor | Binding of FGF-2 and VEGF to heparin sulfate10,11 | Binding of growth factor to ECM facilitates/enhances signaling |
| Binding of TGFβ1 to decorin, beta-glycan, biglycan, collagens, fibrillin5,7,12 | Sequestration and inactivation of TGFβ by matrix molecules | |
| Binding of FGF to fibrin and fibrinogen6 | Protection of growth factor from degradation | |
| Activation of growth factors from a latent form | TSP-1 activation of latent TGFβ8 | ECM component directly activates latent growth factor |
| Stiffness of matrix activates TGFβ9,13 | Activation of TGFβ mediated by the stiff ECM through integrin sensing or enhanced stimulation via the PI3/Akt signaling pathway | |
| Matrikine–growth factor receptor interaction | Tenascin-C and laminin bind to EGF receptors14 | Matrikine induced signaling through growth factor receptor binding |
| ECM–integrin interaction-mediated growth factor signaling | Expression of αvβ3 is required for TNFα and VEGF induced angiogenesis15 | ECM modulation of integrin expression/activation modulates growth factor signaling |
ECM, extracellular matrix; FGF, fibroblast growth factor; VEGF, vascular endothelial growth factor; TGFβ, transforming growth factor beta; TSP, thrombospondins; EGF, epidermal growth factor; TNF, tumor necrosis factor.
Discussion of Findings and Relevant Literature
Interactions of the ECM and progenitors via cell surface receptors
Integrin–ECM interactions
Integrins constitute one of the major families, and arguably the best characterized, of cell adhesion receptors capable of mediating the interaction of cells with their surrounding ECM.16 As in other tissues, integrins play an important role in maintaining the cutaneous stem cell niche and also provide pivotal cues for directing differentiation of stem cells.17 Integrins are expressed on the cell membrane as transmembrane heterodimers and act as ECM receptors for proteins such as collagens, laminins (LNs), and fibronectin (FN). As many as 24 distinct integrin heterodimers can be generated through the combination of 18 α- and 8 β-integrin subunits.16 The extracellular domain of unique integrin heterodimers directly links a repertoire of the ECM ligands to the cytoskeleton through intracellular cytoplasmic domains, allowing for bidirectional transduction of biochemical signals and mechanical tension across the plasma membrane. This outside-in and inside-out signaling allows integrins to mediate such key cellular events in wound repair as adhesion, migration, proliferation, apoptosis, and differentiation (Fig. 1). Both quantitative as well as qualitative differences in integrin display by cells within the epidermis and dermis modulate the affinity for specific ECM components that further regulates cell responsiveness to the surrounding matrix.
Figure 1.

Bidirectional signaling by integrins. The heterodimeric integrins composed of an α- and a β- chain. In an inactivated state, the two chains are closely linked together at the cytosolic end, while the extracellular heads are found to be in a bent-down position. When activated from inside by attachment of talin to the β- chain, the two polypeptides move apart, and their heads at the extracellular end form an upright posture. This inside-out signal allows the integrins to interact with the extracellular matrix (ECM; in this case, we have only shown collagen). This conformational change and subsequent ligand binding activate Src, which binds to focal adhesion kinase (FAK) and other kinases. This intracellular signaling cascade initiates transcription of key genes that promote cell survival, proliferation, etc. This phenomenon has been termed the outside-in signaling. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Integrin expression in both injured and uninjured skin has been extensively characterized. The major epidermal integrins are α2β1, α3β1, and α6β4, although other integrin heterodimers formed by pairing with either an αv- or a β1-subunit have been described in the nontraumatized epidermis or when induced during wound repair.17 Within the interfollicular epidermis (IFE), elevated integrin expression by epidermal stem cells compared to adjacent more-differentiated progeny has been suggested to increase adhesion to the underlying ECM components of the basement membrane (BM).18 When epidermal stem cells commit to differentiate terminally, they downregulate integrin expression, and migrate away from the BM toward the skin surface.19 Studies with cultured human epidermal cells have shown that β1-integrin activation suppresses stem cell terminal differentiation,20 although studies in transgenic mice lacking β1-integrin failed to demonstrate an effect on the terminal differentiation program of epidermal stem cells in vivo.21 However, subsequent studies in mice in which epidermal β1 expression is attenuated, rather than deleted, suggest that β1-integrin expression is essential for expansion of epidermal stem cells.22 In addition to controlling epidermal stem cell differentiation by regulating extracellular signal-related kinase/mitogen-activated protein kinase (ERK/MAPK), β1-integrins have also been shown to play a role in controlling asymmetric cell division, which promotes epidermal stratification and differentiation of mammalian skin.23 Overexpression of α2β1-integrins leads to epidermal hyperproliferation as well as enhanced inflammation associated with production of high levels of proinflammatory cytokines.24 Indeed, appropriate and balanced integrin expression in the suprabasal epidermis permits the interaction of epidermal stem cells and a more-differentiated progeny with the ECM to facilitate physiologic repair in the proliferative phase of wound healing. Numerous studies have shown the critical role that multiple integrin subunits and specific heterodimers play in re-epithelialization by the differentiated progeny of epidermal stem cells and have been reviewed elsewhere.17,25
Similarly, the integrin–ECM interactions that direct the restoration of the dermis during wound repair have been the subject of intense investigation. Initial migration of cells on provisional matrix components such as FN and vitronectin is mediated by integrin heterodimers composed of β1-, β3-, and β5-subunits. Integrins also mediate several critical inputs in the differentiation of myofibroblasts from mesenchymal progenitors in granulation tissue. Compared to their unactivated fibroblast counterparts, myofibroblasts possess a contractile phenotype, are characterized by their expression of alpha smooth muscle actin (α-SMA), and are hypersecretory with respect to their production of collagens.26 Thus, myofibroblasts play a role in the normal wound repair process through their ability to augment wound contraction. Notably, however, the overabundance and persistence of myofibroblasts have been implicated in the disorders of scarring and fibrosis. Therefore, the ability to control myofibroblast differentiation and survival has important implications for maximizing a reparative response postinjury with minimal scarring. The induction of myofibroblast differentiation is dependent upon three factors: the interaction of fibroblasts with the extra domain-A (ED-A) splice variant of fibronectin (ED-A FN),27 the sensing of mechanical tension,28 and active transforming growth factor beta (TGFβ) signaling.29 While the engagement of ED-A FN by cells and the sensing of mechanical forces by expressed integrins involve direct interaction with the ECM, the bioactivity of TGFβ1 has also been shown to be indirectly regulated by the ECM through both direct activation of TGFβ1 as well as modulating the intracellular response to TGFβ signaling.30 Many integrins have been implicated in myofibroblast differentiation, including α3β1, α11β1, αvβ3, α4β7, and β1.31–35 In addition, αvβ6 and αvβ5 may also influence this process and the development of subsequent scarring through their role in modulating TGFβ signaling in epithelial and mesenchymal cells, respectively.36,37
Beyond their role in guiding mesenchymal progenitor cell recruitment and differentiation, several integrins expressed on both endothelial cell (EC) and endothelial progenitor cell (EPC) mediate neovascularization of the granulation tissue bed. While α4β1 regulates EPC mobilization from the bone marrow, class switching and expression of α5β1, α6β1, αvβ3, and αvβ5 are critical to EPC homing, invasion, and differentiation.38 The spatiotemporal regulation of both integrin heterodimers allows EPC and ECs to respond to the evolving ECM during the course of angiogenesis.
Matrikine ligand presentation
In addition to binding to integrins and other adhesion receptors, the subdomains of ECM proteins are capable of binding cell surface receptors belonging to the growth factor, cytokine, chemokine, or ion channel families.39 These ligands, termed matrikines, regulate gene expression, as well as cell differentiation, mitogenesis, apoptosis, and migration. Cryptic matrikines or matricryptins refer to the ligands that are exposed after structural or conformational alterations. Through their ability to direct cellular activities and fate, matrikines have been shown to regulate inflammation, angiogenesis, fibroblast activities, and matrix remodeling within the granulation tissue, while enhancing migration of overlying keratinocytes to cover the wound bed.40–43
During wound healing, tenascin-C (TN-C) and LN-332/LN-5 are expressed at the leading edge of the epithelial tongue.39,42 Epidermal growth factor-like (EGF-L) repeats found within TN-C and similar matrix metalloproteinase 2–released repeats from LN-332 at this epidermal–dermal junction are thought to provide migratory signals to keratinocytes by binding to EGF receptors.39,44 EGF-L repeats have been shown to provide promitotic and migratory signals to fibroblasts as well. Fibroblast proliferation and migration have also been shown to increase in response to binding of fibrillar collagens to the discoidin domain tyrosine kinase receptor 2.45 Although several matrikines have been shown to modulate the activity of the cellular mediators of cutaneous wound repair (Table 3), less is currently known about their specific roles in regulating progenitor/stem cell activity and fate during this process.
Table 3.
Select examples of matrikines involved in wound repair
| Matrikine | Receptor | Target Cell Type(s) | Effect |
|---|---|---|---|
| EGF-L of TN-C and LN-332 | EGFR | Fibroblasts; keratinocytes (?) | Fibroblast migration and proliferation; proposed to promote re-epithelialization through its promigratory properties |
| Leucine-rich domain of decorin | EGFR | Fibroblasts; endothelial cells | Sustained EGFR activation downregulates EGFR function and results in quiescent effect |
| Triple-helical domain of collagens | DDR1/DDR2 | Endothelial cells, epithelial cells (mainly DDR1) and fibroblasts (DDR2) | Regulates migration, proliferation, and MMP production |
| XGXXPG-containing peptides of elastin | Elastin-binding protein | Fibroblasts; endothelial cells | Chemotaxis, proliferation, and protease release |
TN-C, tenascin-C; LN, laminin; DDR, discoidin domain tyrosine kinase receptor; MMP, matrix metalloproteinase.
ECM regulation of growth factor bioavailability and activity
ECM proteins may also modulate growth factor signaling through their ability to bind ligands directly. By doing so, the ECM may increase growth factor signaling by activation of latent forms, presentation of growth factors to their cognizant receptor, release of sequestered growth factors from the matrix, and protection from proteolytic degradation.5,6,30 Sequestration may also serve to quench bioavailability or localize its activity based upon expression of individual ECM components spatially and temporally.7,30 Qualitative and quantitative changes to the ECM throughout the phases of healing can therefore enhance or diminish the effects of individual growth factors on both progenitor and reparative cells. The ability to fine-tune growth factor signaling is particularly advantageous in the process of wound repair when processes such as inflammation, angiogenesis, and fibroplasia must resolve to achieve timely wound healing without excessive scar formation. For example, ECM proteins such as fibrillin, collagens, and small leucine-rich proteoglycans (such as decorin, biglycan, and aspirin) may bind to TGFβ or its associated latent TGFβ-binding protein, resulting in quenching of activity.5,7,12,30,46 The ECM may also be critical for integrin-dependent activation of TGFβ, as occurs with FN, or by direct activation of latent TGFβ by induction of the conformational change such as that occurs with thrombospondin-1 (TSP1).8 Matrix stiffening may also increase activation of latent TGFβ and alter the response of cells to TGFβ stimulation via the phophatidylinositol-3-phosphate/Akt signaling pathway.9 Finally, ECM proteins may confer stability to growth factors and/or their receptors, as occurs with two fibroblast growth factor (FGF) molecules and receptors as a tetrameric complex with either the proteoglycan heparin or heparin sulfate.47
Regulation of epidermal homeostasis and re-epithelialization by the ECM
Epidermal maintenance and repair postinjury require controlled proliferation and differentiation of stem cells. Epidermal stem cells reside and are maintained in the basal layer of the IFE, the sebaceous gland, as well as the permanent portion of the hair follicle (bulge region).48 The basal layer of the IFE is separated from the dermis by the BM, a thin network of proteoglycans, and other ECM proteins, including LNs and collagen IV. Enrichment for epidermal stem cells is possible by their propensity for adhesion to the BM ECM components.18,49
Previously, epidermal stem cells of the IFE were identified as a superior source of epidermal cells for the use in tissue-engineered skin substitutes compared to their more-differentiated progeny by their ability to support epidermal maintenance.50 Therefore, there is a considerable interest in better understanding the epidermal stem cell niche components that would support the ability to isolate and cultivate epidermal stem cells in vitro for tissue-engineering strategies. In addition to the BM ECM components on which epidermal stem cells and their differentiated progeny reside in the uninjured state, the provisional matrix and ECM components of the newly formed granulation tissue in a wound also provide epidermal stem and reparative cells with key contextual cues to mediate cell fate. The importance of fibroblasts and their elaborated matrix in stem cell survival, proliferation, and differentiation is highlighted by the importance of using lethally irradiated 3T3 feeder cells in the cultivation of epidermal stem cells. Recently, Takagi et al. identified six ECM molecules with significantly altered expression in 3T3 cells compared to the cells incapable of supporting epidermal stem cell growth.51 Of those, microfibrillar type VI collagen was uniquely capable of promoting colony formation of epithelial cells in vitro. Moreover, a comparative analysis of ECM protein expression by stem cells in the bulge region of the hair follicle relative to other epidermal cells revealed significant differences in the ECM niche of epidermal stem cells.25 However, further investigation is required to assess the role for individual components in maintaining this niche.
While it is clear that specific ECM components may direct epidermal stem cell fate decisions such as survival, growth, and activation of terminal differentiation programs, it is also becoming apparent that the micropatterning of matrix molecules in the extracellular environment also plays a role in directing such activities.52 In addition, studies examining the effect of substrates of increasing stiffness reveal that a low elastic modulus (0.5 kPa) led to differentiation of epidermal stem cells secondary to decreased activation of ERK/MAPK signaling.53 Further, ECM components may also indirectly influence epidermal stem cell and more-differentiated reparative cell fate through their effects on growth factor and/or matrix metalloproteinase activities.
While not much is known about the role of specific ECM components play in driving epithelial stem cell differentiation during re-epithelialization, their importance is highlighted by their role in directing keratinocyte proliferation and migration. The role of the ECM in modulating the keratinocyte activity and fate has been the subject of several reviews.2,54 With injury beneath the BM, epithelial cells migrate as a sheet from the edge of the damaged BM (or from epidermal appendages in partial-thickness injury). To do so, basal keratinocytes must dissolve hemi-desmosomes and disengage from the BM ECM components such as LNs and type IV collagen. As they begin to migrate over the newly forming neodermis, epidermal cells express new integrin heterodimers and relocate integrins from the lateral to the basal membrane surface to interact with FN, vitronectin, TN-C, and type I and III collagens of the exposed wound bed.55,56
In addition to the ECM components of the exposed wound bed, type IV collagen and LNs of the BM have also been shown to modulate keratinocyte migration and stratification of the neoepidermis. LNs belong to a large family of conserved membrane-bound glycoproteins that are major constituents of the BM separating epidermal cells from the underlying dermis, as well as that of blood vessels.55 The isoform LN-332/LN-5 is a major component of the BM and can either promote or inhibit migration of keratinocytes during wound healing in in-vitro and in-vivo studies, depending on its proteolytic processing.57 These findings highlight the importance of understanding not only the role of specific ECM molecules on the cells during wound repair but also the effects of post-translational modification and proteolytic processing of native matrices that occur during the course of wound repair. As re-epithelialization progresses, keratinocytes are able to reform the BM to re-establish the normal barrier between the epidermis and dermis. Reformation of the BM, with reappearance of its components such as LN-111/LN-1, is thought to provide keratinocytes with the proper signals to inhibit keratinocyte migration and remodel the neoepidermis.54
The role of the ECM in granulation tissue formation
The source of mesenchymal progenitors that populate the developing granulation tissue and possess the capacity for differentiation to myofibroblasts during cutaneous wound healing may include resident fibroblasts, smooth muscle cells, pericytes, fibrocytes, mesenchymal stem cells (MSCs), and epithelial or ECs undergoing epithelial- or endothelial-to-mesenchymal transition (Fig. 2).31,58 Myofibroblasts both respond to the ECM as well as play a critical role in the elaboration and remodeling of the ECM in physiologic and pathologic wound healing in a process referred to as dynamic reciprocity.2 Given the role of the myofibroblast in physiologic wound repair and its role as the key effector cell in pathologic scar formation and fibrosis, understanding how the ECM regulates myofibroblast differentiation and fate is key to optimize wound healing.
Figure 2.
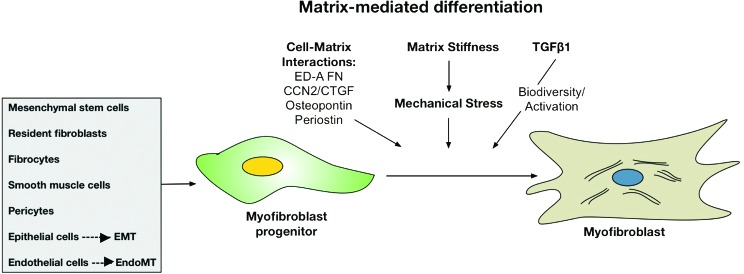
ECM regulation of myofibroblast differentiation. Several cell types may serve as myofibroblast progenitors that are directed to transform into fully differentiated myofibroblasts by several factors. The ECM has been shown to directly promote this process by cell–ECM interactions, mediating tissue stiffness, and regulating the bioavailability and activity of the profibrogenic growth factor TGFβ1. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
In addition to ED-A FN, several ECM components regulate myofibroblast differentiation (Fig. 2). Production of endogenous hyaluronan (HA) is essential for TGFβ1-dependent myofibroblast activation and persistence;59 however, exogenously applied HA attenuates myofibroblast differentiation. Further control of this process may also occur earlier in the differentiation process, as low-molecular-weight HA has been shown to inhibit differentiation of fibrocytes derived from peripheral blood monocytes, whereas high-molecular-weight HA promotes their differentiation.60 Our data support a role for type III collagen (Col3) in the early healing phase to limit myofibroblast differentiation.61 Diminished Col3 promotes myofibroblast differentiation, which then paradoxically leads to increased collagen production and scar formation at later stages of wound repair. Several matricellular proteins, including connective tissue growth factor,62 periostin,63 osteopontin,64 and TN-C,65 have also been shown to regulate myofibroblast recruitment and/or differentiation. Finally, the post-translational modification of the ECM components provides additional regulation of the cellular response to the ECM, which can drive fibrotic response to injury. Cross-linking of ECM proteins through the activities of lysyl oxidases and transglutaminases, or via glycation, increases tissue stiffness and may render the resulting ECM resistant to degradation and remodeling.31 Fibroblasts and myofibroblasts sense and modulate stiffness within the ECM through the development of focal adhesions. In fact, mechanical stiffness has been shown to drive myofibroblast activation in a TGFβ-independent manner, thus promoting and amplifying progressive fibrosis.32
Beyond their role in guiding mesenchymal progenitor cell recruitment and differentiation during wound repair, several ECM components direct EC and EPC neovascularization of the wound bed. EPCs and ECs respond to an evolving ECM during the course of angiogenesis. In addition to the provisional matrix components (fibrin, FN, vitronectin, and von Willebrand factor) that mediate adhesion and migration of ECs via its αvβ3-integrin receptor, other ECM components (particularly LN) that are released by proteolytic degradation of the EC BM can enhance the proliferation of ECs.66 Members of the thrombospondin family of matricellular proteins have also been identified as potent regulators of angiogenesis, primarily through their ability to bind CD36 on ECs and induce apoptosis in both liganded and surrounding ECs.67 While TSP-1 is expressed early in wound healing, TSP-2 is expressed at a later stage. 67 Absence of TSP-1 delays wound healing due to its effect on macrophage recruitment and TGFβ signaling, whereas its overexpression limits healing through the effects on angiogenesis and granulation tissue formation.67 In contrast, TSP-2 deficiency is associated with accelerated wound healing with exaggerated neovascularization and irregular collagen fibrillogenesis. Beyond its role in directly interacting with ECs, TSP-1 may also exert its antiangiogenic effects by direct binding and clearance of vascular endothelial growth factor through receptor-mediated endocytosis. While TSP-1 inhibits angiogenesis through direct protein binding, FGF interaction with heparin-like moieties in the ECM and subsequent mobilization by heparinase enhance the angiogenic response during healing.68
The use of the ECM in advanced wound therapies
Tissue engineering and regenerative medicine have shown significant promise in treatment of problematic wounds. Initial proof-of-principle studies investigating the use of exogenously applied adult stem cells for the treatment of chronic wounds provided support for their vulnerary effects.69,70 MSCs have been shown to improve wound healing through their ability to directly contribute cells and ECM components to the repair process, as well as their ability to direct other cells participating in the repair process through their production of paracrine mediators. Although initial attempts showed improvement in healing with injection of cells alone, it is clear that delivery strategies that recapitulate complex stem cell niches will be required to maximize their potential. Given the ability of the ECM to influence many key aspects of that niche such as signaling events, regulation of growth factor bioavailability, and mechanosensation, the identification of specific ECM components that most accurately recreate a functional stem cell niche for incorporation into therapeutic biomaterials remains a focus of intense investigation.
The use of acellular and cell-based tissue-engineered dermal substitutes has become increasingly routine.71 Dermal matrices and other biomimetic scaffolds for delivery of adult stem cells have been shown to augment MSC regenerative potential and enhance wound healing through their presumptive ability to recreate the aspects of a regenerative niche.72–74 Despite their ability to improve the efficiency and quality of cutaneous wound healing, existing products have yet to be able to promote repair to that which approximates uninjured skin. The evolution of superior biomaterials will depend upon a greater understanding of the role of individual components play in the regenerative niches of uninjured and injured skin, particularly the ECM constituents. Further, the development of successful biomimetic and bioresponsive substrates will incorporate the knowledge of not only how cells interact with these substrates but also how these cells will then remodel and deposit their own ECM in response.
Summary
Complications of inadequate wound closure or excessive scar formation result in significant morbidity and healthcare expenditure. Scar formation after cutaneous injury in the postnatal organism is thought to be a consequence of events facilitating rapid closure to re-establish the protective barrier of the skin. The ability of the mid-gestational fetuses to heal cutaneous wounds in a scarless manner with restitution of epidermal adnexal structures75 suggests that an improved understanding of the progenitor cell niche, including contribution by ECM components, will be critical to improving healing in the postnatal patient. Dynamic interactions of the ECM components with each other, with progenitor and other reparative cells, as well as growth factors and matrix modifiers are all essential for wound repair. Therefore, understanding the role of individual ECM proteins in directing cutaneous healing is essential for harnessing the potential of regenerative medicine and tissue-engineering strategies to improve or prevent pathologic healing.
Take-Home Messages.
Temporospatial expression of individual ECM components during cutaneous wound healing directs stem and reparative cell activities in all phases of repair.
During wound healing, the ECM plays critical roles in regulating stem cell behaviors such as migration, differentiation, proliferation, and survival by binding cellular adhesion receptors such as integrins, regulating the bioavailability and activity of growth factors, and direct binding to growth factor receptors.
Class switching of integrins by cells involved in wound repair allows cells to respond to injury by interacting with different ECM ligands in the course of healing.
Post-translational modification of ECM components by lysyl oxidases, transglutaminases, and glycation modulates tissue stiffness and therefore provides additional control of cell fate during wound healing.
Mechanosensitivity and responsiveness of stem cells play a key role in the regulation of cell fate. The force balance is transmitted across the mechanical continuum of ECM–integrin–cytoskeleton and can further modulate other signaling events mediated by integrins and other cell surface receptors.
Evolution of smart biomaterials in wound-healing therapeutics will continue through elucidation of mechanisms by which the ECM directs tissue repair.
Abbreviations and Acronyms
- α-SMA
alpha smooth muscle actin
- BM
basement membrane
- Col3
type III collagen
- CTGF
connective tissue growth factor
- EC
endothelial cell
- ECM
extracellular matrix
- ED-A FN
ED-A splice variant of fibronectin
- EGF-L
epidermal growth factor–like
- EPC
endothelial progenitor
- ERK
extracellular signal–related kinase
- FGF
fibroblast growth factor
- FN
fibronectin
- HA
hyaluronan
- IFE
interfollicular epidermis
- LN
laminin
- MAPK
mitogen-activated protein kinase
- MSC
mesenchymal stem cells
- PBMC
peripheral blood monocytes
- TGFβ
transforming growth factor beta
- TSP
thrombospondins
- VEGF
vascular endothelial growth factor
Acknowledgments and Funding Sources
The authors wish to apologize to colleagues whose work could not be cited due to formatting constraints.
Author Disclosure and Ghostwriting
No competing financial interests exist for the authors. The content of this article was expressly written by the authors listed. No ghostwriters were used to write this article.
About the Authors
Susan W. Volk is an Assistant Professor of Small-Animal Surgery at the University of Pennsylvania School of Veterinary Medicine. She completed the Veterinary Medical Scientist Training Program (VMSTP; VMD-PhD), followed by a small-animal internship and surgical residency at the University of Pennsylvania. She became a diplomate of the American College of Veterinary Surgery in 2003 and joined the faculty of the School of Veterinary Medicine at the University of Pennsylvania in 2007. The research interests of her laboratory focus on defining the regulatory cues important to direct progenitor cell homing to the sites of injury, as well as in vitro and in vivo signals important to direct their differentiation into appropriate target cell types for the use in tissue-engineering strategies in human and veterinary medicine. Her current work focuses on defining the cellular and molecular mechanisms by which mesenchymal progenitor cells improve cutaneous wound repair and elucidating the role of the ECM in modulation of reparative and progenitor cell activities during tissue repair and regeneration. This work has been funded by both NIH-sponsored and private funding mechanisms. Dr. Syed Amir Iqbal graduated with an MSc in Biotechnology and did his PhD in Biomolecular Sciences from the University of Manchester. Since his PhD in 2008, he has worked as a postdoctoral fellow in the laboratory of Dr. Ardeshir Bayat in the Department of Plastic and Reconstructive Surgery Research at Manchester Institute of Biotechnology, University of Manchester. His research has been focused on exploring the role of stem cells in tissue fibrosis. His work led to the identification of the populations of MSCs and HSCs in keloid tissue and fibrocytes in keloid tissue. Recently, he identified for the first time the MSCs and fibrocytes in Dupuytren's disease tissue. Ardeshir Bayat, BSc (Hons), MBBS, MRCS, PhD, is a clinician as well as a scientist in plastic and reconstructive surgery. He is currently a principal investigator in plastic and reconstructive surgery research at the University of Manchester. The focus of his research has been in wound repair, fibrosis, and tissue regeneration. Currently, his group (www.plasticsurgeryresearch.org) is comprised of research nurses, post-docs, experimental officers, and PhDs. He has made in excess of 350 presentations as of yet and has had personal invitations to talk at more than 60 events in the United Kingdom and worldwide. He has published extensively in peer-reviewed journals (in excess of 170 articles to date). In addition, he is currently on the editorial boards of several journals, and is a consultant to industry and advises companies such as Johnson & Johnson, Sanofi, Allergan, GSK, Astra Zeneca, and others. His mission is to develop research programs that combine high-quality basic research with strong clinical interactions to make a major impact on human health (with emphasis on skin) through translation into better outcomes for patients suffering from abnormal cutaneous scarring and tissue fibrosis. His research thrives upon the multidisciplinary and interactive research environment that currently exists, which allows translation of basic science research to major therapeutic developments that will have a direct impact and specific utilization in clinical practice.
References
- 1.Sen CK. Gordillo GM. Roy S. Kirsner R. Lambert L. Hunt TK. Gottrup F. Gurtner GC. Longaker MT. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 2009;17:763. doi: 10.1111/j.1524-475X.2009.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schultz GS. Davidson JM. Kirsner RS. Bornstein P. Herman IM. Dynamic reciprocity in the wound microenvironment. Wound Repair Regen. 2011;19:134. doi: 10.1111/j.1524-475X.2011.00673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuchs E. Tumbar T. Guasch G. Socializing with the neighbors: stem cells and their niche. Cell. 2004;116:769. doi: 10.1016/s0092-8674(04)00255-7. [DOI] [PubMed] [Google Scholar]
- 4.Kurtz A. Oh SJ. Age related changes of the extracellular matrix and stem cell maintenance. Prev Med. 2012;54:S50. doi: 10.1016/j.ypmed.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Y. Oganesian A. Keene DR. Sandell LJ. Type IIA procollagen containing the cysteine-rich amino propeptide is deposited in the extracellular matrix of prechondrogenic tissue and binds to TGF-beta1 and BMP2. J Cell Biol. 1999;144:1069. doi: 10.1083/jcb.144.5.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahni A. Baker CA. Sporn LA. Francis CW. Fibrinogen and fibrin protect fibroblast growth factor-2 from proteolytic degradation. Thromb Haemost. 2000;83:736. [PubMed] [Google Scholar]
- 7.Yamaguchi Y. Mann DM. Rouslahti E. Negative regulation of transforming growth factor beta by the proteoglycan decorin. Nature. 1990;346:281. doi: 10.1038/346281a0. [DOI] [PubMed] [Google Scholar]
- 8.Murphy-Ullrich JE. Poczatek M. Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11:59. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]
- 9.Leight JL. Wozniak MA. Chen S. Lynch ML. Chen CS. Matrix rigidity regulates a switch between TGF-β 1 induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell. 2012;23:677. doi: 10.1091/mbc.E11-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rapraeger AC. Kruftka A. Olwin BB. Requirement of heparin sulfate for bFGF-mediated fibroblast growth and myoblast differenitation. Science. 1991;252:1705. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- 11.Ortega N. L'Faqihi FE. Plouet J. Control of vascular endothelial growth factor angiogenic activity by the extracellular matrix. Biol Cell. 1998;90:381. doi: 10.1111/j.1768-322x.1998.tb01047.x. [DOI] [PubMed] [Google Scholar]
- 12.Chaudry SS. Cain SA. Morgan A. Dallas SL. Shuttleworth CA. Kielty CM. Fibrillin-1 regulates the bioavailability of TGFβ 1. J Cell Biochem. 2007;176:355. doi: 10.1083/jcb.200608167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wipff PJ. Rifkin DB. Meister JJ. Hinz B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J Cell Biol. 2007;179:1311. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran KT. Lamb P. Deng JS. Matrikines and matricryptins: implications for cutaneous cancers and skin repair. J Dermatol Sci. 2005;40:11. doi: 10.1016/j.jdermsci.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Brooks PC. Clark RA. Cheresh DA. Requirement of vascular integrin αvβ3 for angiogenesis. Science. 1994;264:569. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 16.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 17.Margadant C. Charafeddine RA. Sonnenberg A. Unique and redundant functions of integrins in the epidermis. FASEB J. 2010;24:4133. doi: 10.1096/fj.09-151449. [DOI] [PubMed] [Google Scholar]
- 18.Jones PH. Watt FM. Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell. 1993;73:713. doi: 10.1016/0092-8674(93)90251-k. [DOI] [PubMed] [Google Scholar]
- 19.Watt FM. Kubler MD. Hotchin NA. Nicholson LJ. Adams JC. Regulation of keratinocyte terminal differentiation by integrin-extracellular matrix interactions. J Cell Sci. 1993;106:175. doi: 10.1242/jcs.106.1.175. [DOI] [PubMed] [Google Scholar]
- 20.Evans RD. Perkins VC. Henry A. Stephens PE. Robinson MK. Watt FM. A tumor-associated beta 1 integrin mutation that abrogates epithelial differentiation control. J Cell Biol. 2003;160:589. doi: 10.1083/jcb.200209016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raghavan S. Bauer C. Mundschau G. Li Q. Fuchs E. Conditional ablation of β1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J Cell Biol. 2000;150:1149. doi: 10.1083/jcb.150.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piwko-Czuchra A. Koegel H. Meyer H. Bauer M. Werner S. Brakebusch C. Fässler R. β1 integrin-mediated adhesion signalling is essential for epidermal progenitor cell expansion. PLoS One. 2009;4:e5488. doi: 10.1371/journal.pone.0005488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lechler T. Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437:205. doi: 10.1038/nature03922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teige I. Backlund A. Svensson L. Kvist PH. Peterson TK. Kemp K. Induced keratinocyte hyperproliferation in α2β1 integrin transgenic mice results in systemic immune cell activation. Int Immunopharmacol. 2010;10:107. doi: 10.1016/j.intimp.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Watt FM. Fujiwara H. Cell-extracellular matrix interations in normal and diseased skin. Cold Spring Harb Perspect Biol. 2011;3:1. doi: 10.1101/cshperspect.a005124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 27.Serini G. Gabbiani G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res. 1999;250:273. doi: 10.1006/excr.1999.4543. [DOI] [PubMed] [Google Scholar]
- 28.Hinz B. Mastrangelo D. Iselin CE. Chaponnier C. Gabbiani G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am J Pathol. 2001;159:1009. doi: 10.1016/S0002-9440(10)61776-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desmouliere A. Geinoz A. Gabbiani F. Gabbiani G. Transforming growth factor-β1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horiguchi M. Ota M. Rifkin DB. Matrix control of transforming growth factor-β function. J Biochem. 2012;152:321. doi: 10.1093/jb/mvs089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klinberg F. Hinz B. White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol. 2013;229:298. doi: 10.1002/path.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu F. Mih JD. Shea BS. Kho AT. Sharif AS. Tager AM. Tschumperlin DJ. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190:693. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohan M. Muro AF. White WS. Berkman N. EDA-containing cellular fibronectin induces fibroblast differentiation through binding to α4β7 integrin receptor and MAPK/Erk 1/2-dependent signaling. FASEB J. 2010;24:4503. doi: 10.1096/fj.10-154435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horan GS. Wood S. Ona V. Li DJ. Lakashev ME. Weinreb PH. Simon KJ. Hahm K. Allaire NE. Rinaldi NJ. Goyai J. Feghall-Bostwick CA. Matteson EL. O'Hara C. Lafyatis R. Davis GS. Huang X. Sheppard D. Violette SM. Partial inhibition of integrin αvβ6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. 2008;177:56. doi: 10.1164/rccm.200706-805OC. [DOI] [PubMed] [Google Scholar]
- 35.Carracedo S. Lu N. Popova SN. Jonsson R. Eckes B. Gullberg D. The fibroblast integrin α11β1 is induced in a mechanosensitive manner involving activin A and regulates myofibroblast differntiation. J Biol Chem. 2010;285:10434. doi: 10.1074/jbc.M109.078766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim KK. Wei Y. Szekeres C. Kugler MC. Wolters PJ. Hill ML. Frank JA. Brumwell AN. Wheeler SE. Kreidberg JA. Chapman HA. Epithelial cell α3β1 integrin links β-catenin and Smad signaling to promote myofibroblast formaiton and pulmonary fibrosis. J Clin Invest. 2009;119:213. doi: 10.1172/JCI36940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Y. Hagood JS. Lu B. Merryman WD. Murphy-Ullrich JE. Thy-1-integrin avb5 interactions inhibit lung fibroblast contraction-induced latent transforming growth factor-β1 activation and myofibroblast differentiation. J Biol Chem. 2010;285:22382. doi: 10.1074/jbc.M110.126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caiado F. Dias S. Endothelial progenitor cells and integrins: adhesive needs. Fibrogenesis Tissue Repair. 2012;5:1. doi: 10.1186/1755-1536-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swindle CS. Tran KT. Johnson TD. Banerjee P. Mayes AM. Griffith L. Wells A. Epidermal growth factor (EGF)-like repeats of human tenascin-C as ligands for EGF receptor. J Cell Biol. 2001;154:459. doi: 10.1083/jcb.200103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bellon G. Martiny L. Robinet A. Matrix metalloproteinases and matrikines in angiogenesis. Crit Rev Oncol Hematol. 2004;49:203. doi: 10.1016/j.critrevonc.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 41.Adair-Kirk TL. Senior RM. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol. 2008;40:1101. doi: 10.1016/j.biocel.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tran KT. Griffith L. Wells A. Extracellular matrix signaling through growth factor receptors during wound healing. Wound Repair Regen. 2004;12:262. doi: 10.1111/j.1067-1927.2004.012302.x. [DOI] [PubMed] [Google Scholar]
- 43.Schultz GS. Wysocki A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009;17:153. doi: 10.1111/j.1524-475X.2009.00466.x. [DOI] [PubMed] [Google Scholar]
- 44.Giannelli G. Falk-Marzillier J. Schiraldi O. Stetler-Stevenson WG. Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277:225. doi: 10.1126/science.277.5323.225. [DOI] [PubMed] [Google Scholar]
- 45.Olaso E. Labrador JP. Wang L. Ikeda K. Eng FJ. Klein R. Lovett DH. Lin HC. Friedman SL. Discoidin domain receptor 2 regulates fibroblast proliferation and migration through the extracellular matrix in association with transcriptional activation of matrix metalloproteinase-2. J Biol Chem. 2002;277:3606. doi: 10.1074/jbc.M107571200. [DOI] [PubMed] [Google Scholar]
- 46.Hildebrand A. Romaris M. Rasmussen LM. Heinegård D. Twardzik DR. Border WA. Ruoslahti E. Interaction of the small intestinal proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem J. 2004;302(pt 2):527. doi: 10.1042/bj3020527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schlessinger J. Plotnikov AN. Ibrahimi OA. Eliseenkova AV. Yeh BK. Yayon A. Linhardt RJ. Mohammadi M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6:743. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 48.Blanpain C. Fuchs E. Epidermal stem cells of the skin. Annu Rev Cell Dev Biol. 2006;22:339. doi: 10.1146/annurev.cellbio.22.010305.104357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J. Miao C. Guo W. Jia L. Zhou J. Ma B. Peng S. Liu S. Cao Y. Duan E. Enrichment of putative human epidermal stem cells based on cell size and collagen type IV adhesiveness. Cell Res. 2008;18:360. doi: 10.1038/cr.2007.103. [DOI] [PubMed] [Google Scholar]
- 50.Dunnwald M. Monanek-Chalkley A. Alexandrunas D. Fishbaugh J. Bickenbauch JR. Isolating a pure population of epidermal stem cells for use in tissue engineering. Exp Dermatol. 2001;10:45. doi: 10.1034/j.1600-0625.2001.100106.x. [DOI] [PubMed] [Google Scholar]
- 51.Takagi R. Yamato M. Kushida A. Nishida K. Okano T. Profiling of extracellular matrix and cadherin family gene expression in mouse feeder layer cells: type VI collagen is a candidate molecule inducing colony formation of epithelial cells. Tissue Eng Part A. 2012;18:2539. doi: 10.1089/ten.TEA.2011.0428. [DOI] [PubMed] [Google Scholar]
- 52.Connelly JT. Gautrot JE. Trappmann B. Tan DW. Donati G. Huck WT. Watt FM. Actin and serum response factor transduce physical cues from the microenvironment to regulate epidermal stem cell fate decisions. Nat Cell Biol. 2010;12:711. doi: 10.1038/ncb2074. [DOI] [PubMed] [Google Scholar]
- 53.Trappmann B. Gautrot JE. Connelly JT. Strange DG. Li Y. Oyen ML. Cohen Stuart MA. Boehm H. Li B. Vogel V. Spatz JP. Watt FM. Huck WT. Extracellular-matrix tethering regulates stem-cell fate. Nat Mater. 2012;11:642. doi: 10.1038/nmat3339. [DOI] [PubMed] [Google Scholar]
- 54.O'Toole EA. Extracellular matrix and keratinocyte migration. Clin Exp Dermatol. 2001;26:525. doi: 10.1046/j.1365-2230.2001.00891.x. [DOI] [PubMed] [Google Scholar]
- 55.Larjava H. Salo T. Happasalmi K. Kramer RH. Heino J. Expression of integrins and basement membrane components by wound keratinocytes. J Clin Invest. 1993;92:1425. doi: 10.1172/JCI116719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clark RA. Ashcroft GS. Spencer MJ. Larjava H. Fergusen MW. Re-epithelialization of normal human excisional wound is associated with a switch from αvβ5 to αvβ6 integrins. Br J Dermatol. 1996;135:46. [PubMed] [Google Scholar]
- 57.Kariya Y. Sato H. Katou N. Kariya Y. Miyazaki K. Polymerized laminin-332 matrix supports rapid and tight adhesion of keratinocytes, suppressing cell migration. PLoS One. 2012;7:e35546. doi: 10.1371/journal.pone.0035546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iqbal SA. Sidgwick GP. Bayat A. Identification of fibrocytes from mesenchymal stem cells in keloid tissue: a potential source of abnormal fibroblasts in keloid scarring. Arch Dermatol Res. 2012;304:665. doi: 10.1007/s00403-012-1225-5. [DOI] [PubMed] [Google Scholar]
- 59.Webber J. Jenkins RH. Meran S. Phillips A. Steadman R. Modulation of TGFβ 1-dependent myofibroblast differentiation by hyaluronan. Am J Pathol. 2009;175:148. doi: 10.2353/ajpath.2009.080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maharjan AS. Pilling D. Gomer RH. High and low molecular weight hyaluronic acid differntially regulate human fibrocyte differentiation. PLoS One. 2011;6:e26078. doi: 10.1371/journal.pone.0026078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Volk SW. Wang Y. Mauldin EA. Liechty KW. Adams SL. Diminished type III collagen promotes myofibroblast differentiation and increases scar deposition in cutaneous wound healing. Cells Tissues Organs. 2011;194:25. doi: 10.1159/000322399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu S-W. Leask A. Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008;19:133. doi: 10.1016/j.cytogfr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 63.Elliott CG. Wang J. Guo X. Xu S-W. Eastwood M. Guan J. Leask A. Conway SJ. Hamilton DW. Periostin modulates myofibroblast differentiation during full-thickness cutaneous wound repair. J Cell Sci. 2011;125:121. doi: 10.1242/jcs.087841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lenga Y. Koh A. Perera AS. McMulloch CA. Sodek J. Zohar R. Osteopontin expression is required for myofibroblast differentiation. Circ Res. 2008;102:319. doi: 10.1161/CIRCRESAHA.107.160408. [DOI] [PubMed] [Google Scholar]
- 65.Tamaoki M. Imanaka-Yoshida K. Yokoyama K. Nishioka T. Inada H. Hiroe M. Sakakura T. Yoshida T. Tenascin-C regulares recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol. 2005;167:71. doi: 10.1016/S0002-9440(10)62954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Form DM. Pratt BM. Madri JA. Endothelial cell proliferation during angiogenesis. In vitro modulation by basement membrane components. Lab Invest. 1986;55:521. [PubMed] [Google Scholar]
- 67.Bornstein P. Thrombospondins function as regulators of angiogenesis. J Cell Commun Signal. 2009;3:189. doi: 10.1007/s12079-009-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zcharia E. Zilka R. Yaar A. Yacoby-Zeevi O. Zetser A. Metzger S. Sarid R. Naggi A. Casu B. Ilan N. Vlodavsky I. Abramovitch R. Heparinase accelerates wound angiogenesis and wound healing in mouse and rat models. FASEB J. 2005;19:211. doi: 10.1096/fj.04-1970com. [DOI] [PubMed] [Google Scholar]
- 69.Volk SW. Radu A. Zhang L. Liechty KW. Stromal progenitor cell therapy corrects the wound healing defect in the ischemic rabbit ear model of chronic wound repair. Wound Repair Regen. 2007;15:736. doi: 10.1111/j.1524-475X.2007.00277.x. [DOI] [PubMed] [Google Scholar]
- 70.Badillo AT. Redden RA. Zhang L. Doolin EJ. Liechty KW. Treatment of diabetic wounds with fetal murine mesenchymal stromal cells enhances wound closure. Cell Tissue Res. 2007;329:301. doi: 10.1007/s00441-007-0417-3. [DOI] [PubMed] [Google Scholar]
- 71.Hodgkinson T. Bayat A. Dermal substitute-assisted healing: enhancing stem cell therapy with novel biomaterial design. Arch Dermatol Res. 2011;303:301. doi: 10.1007/s00403-011-1131-2. [DOI] [PubMed] [Google Scholar]
- 72.Rustad KC. Wong VW. Sorkin M. Glotzbach JP. Major MR. Rajadas J. Longaker MT. Gurtner GC. Enhancement of mesenchymal stem cell angiogenic capacity and stemness by a biomimetic hydrogel scaffold. Biomaterials. 2012;33:80. doi: 10.1016/j.biomaterials.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou Y. Yan Z. Zhang H. Lu W. Liu S. Huang X. Luo H. Jin Y. Expansion and delivery of adipose-derived mesenchymal stem cells on three microcarriers for soft tissue regeneration. Tissue Eng Part A. 2011;17:2981. doi: 10.1089/ten.tea.2010.0707. [DOI] [PubMed] [Google Scholar]
- 74.Altman AM. Matthias N. Yan Y. Song Y-H. Bai X. Chiu ES. Slakey DP. Alt EU. Dermal matrix as a carrier for in vivo delivery of human adipose-derived stem cells. Biomaterials. 2008;29:1431. doi: 10.1016/j.biomaterials.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 75.Adzick NS. Lorenz HP. Cells, matrix, growth factors, and the surgeon: the biology of scarless fetal wound repair. Ann Surg. 1994;220:10. doi: 10.1097/00000658-199407000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]