

Abstract
Background
Nucleoside phosphorylases (NPs) have been extensively investigated in human and bacterial systems for their role in metabolic nucleotide salvaging and links to oncogenesis. In plants, NP-like proteins have not been comprehensively studied, likely because there is no evidence of a metabolic function in nucleoside salvage. However, in the forest trees genus Populus a family of NP-like proteins function as an important ecophysiological adaptation for inter- and intra-seasonal nitrogen storage and cycling.
Results
We conducted phylogenetic analyses to determine the distribution and evolution of NP-like proteins in plants. These analyses revealed two major clusters of NP-like proteins in plants. Group I proteins were encoded by genes across a wide range of plant taxa while proteins encoded by Group II genes were dominated by species belonging to the order Malpighiales and included the Populus Bark Storage Protein (BSP) and WIN4-like proteins. Additionally, we evaluated the NP-like genes in Populus by examining the transcript abundance of the 13 NP-like genes found in the Populus genome in various tissues of plants exposed to long-day (LD) and short-day (SD) photoperiods. We found that all 13 of the Populus NP-like genes belonging to either Group I or II are expressed in various tissues in both LD and SD conditions. Tests of natural selection and expression evolution analysis of the Populus genes suggests that divergence in gene expression may have occurred recently during the evolution of Populus, which supports the adaptive maintenance models. Lastly, in silico analysis of cis-regulatory elements in the promoters of the 13 NP-like genes in Populus revealed common regulatory elements known to be involved in light regulation, stress/pathogenesis and phytohormone responses.
Conclusion
In Populus, the evolution of the NP-like protein and gene family has been shaped by duplication events and natural selection. Expression data suggest that previously uncharacterized NP-like proteins may function in nutrient sensing and/or signaling. These proteins are members of Group I NP-like proteins, which are widely distributed in many plant taxa. We conclude that NP-like proteins may function in plants, although this function is undefined.
Keywords: Nucleoside phosphorylases, Vegetative storage proteins, Bark storage proteins, Nitrogen cycling, Populus trichocarpa
Background
Nucleotides, nucleotide precursors and derivatives are essential components for life. They compose nucleic acids, act as signaling molecules, intercellular energy transporters, and can be converted to essential enzymatic cofactors. Nucleotide metabolism is therefore a necessary cellular function [1,2]. In mammalian and bacterial systems, nucleoside phosphorylases (NPs) salvage nucleosides by cleaving the glycosidic bond of (deoxy-) ribonucleosides in the presence of inorganic phosphate (Pi) to yield (deoxy-) ribose-1-phosphate and a nucleobase [1,3]. The free nucleobase can be synthesized into organic molecules or degraded, and the (deoxy-) ribose-1-phosphate can be utilized by the Pentose Phosphate Pathway and glycolysis [3]. The most widely studied NPs are purine nucleoside phosphorylases (PNPs), which are a focus in clinical and cancer research for their role in mutation-related immunodeficiency diseases, prostate cancer, leukemia and periodontal disease [4-6].
The ability to salvage purines is particularly important for N-limited plants in the context of retaining and remobilizing nitrogen (N) [7,8]. The proposed physiological role of purine degradation in plants is that it promotes nitrogen-use efficiency (NUE) through mobilization from source to sink [8]. However, there is no evidence that NPs are involved in purine nucleoside salvage in plants; instead hydrolysis of nucleosides occurs by nucleosidases (EC 3.2.2x) [9-11]. Notably, genes in purine salvage and degradation pathways are induced by wounding, drought, abscissic acid (ABA), dark conditions and dark-induced senescence consistent with a role in NUE [12-14]. Although purine salvage appears to be a component of NUE, the role of NP-like proteins in plants in relation to nucleoside salvaging and NUE is not known.
There is a large body of research on a family of proteins that share sequence similarity with PNPs in Populus (reviewed by [15]). In this genus, these proteins have an eco-physiological role in seasonal and short-term nitrogen storage. One subfamily of these proteins are termed Bark Storage Proteins (BSPs) since they accumulate in bark parenchyma and xylem rays in autumn and decline in abundance when growth resumes in spring [16-18]. The autumn accumulation of BSP and associated gene expression is a phytochrome mediated photoperiod response [19,20]. In addition, BSPs and related Vegetative Storage Proteins (VSPs) genes are also expressed following wounding, high nitrogen and drought stress suggesting a role in short-term storage [21-23]. Seasonal storage facilitated by BSPs is likely an important evolutionary adaptation that facilitates perennial growth in low nutrient forest systems while short-term storage helps conserve nutrients in response to stresses. This seasonal adaptation uncouples N demands for growth from uptake and assimilation and provides a competitive advantage under conditions of low N supply [24,25]. Populus provides a model system to study NP-like proteins and genes not only because of their importance in seasonal and short-term storage but also because Populus possesses more NP-like proteins than any other known plant genera.
The involvement of NP-like proteins in seasonal N cycling could result from the expansion and functional evolution of protein and gene families which provides innovation for adaptation and speciation [26,27]. Gene duplication is a mechanism for such innovation and occurs through whole genome duplications (WGD) or small-scale genome duplications such as tandem duplications (TD) [28]. Following duplication, genes can have many fates: duplicates may amplify or buffer original function [29-31], gain a novel function (neofunctionalization) [32,33], accumulate mutations that subdivide the original function (subfunctionalization) [34,35] or become non-functional (pseudogenization) [32]. Because seasonal N cycling is likely an adaptive trait, we considered the main models of adaptive maintenance and gene expansion which are positive dosage, neofunctionalization, subfunctionalization, diversification of multifunctional genes and the dosage balance model (reviewed by [36]). Positive dosage describes the retention of duplicate genes that increase fitness by buffering or functional redundancy [29,30,37]. Neofunctionalization is the gain of new function in the duplicate genes through neutral mutations followed by positive selection while preserving the parent copy [32,33]. The subfunctionalization model suggests that neutral mutations in duplicate genes weaken or alter the original function so that both copies are maintained to perform the original function [35,36]. Under the diversification of multifunctional genes model, a multifunctional parent gene is uncoupled among gene duplicates [38]. The dosage balance hypothesis is useful for explaining the retention of genes following the type of duplication event: the duplicate maintaining the stoichiometric balance of protein complexes, favoring high retention rates for genes and proteins with many interactions [39,40].
The aims of this research were to investigate the phylogenetic and evolutionary relationships of NP-like proteins in Populus and the plant kingdom to further our understanding of their functional evolution and the extent to which they may be involved in nutrient salvaging and in particular N cycling and NUE. We first constructed the evolutionary relationships among 13 NP-like genes in Populus, examined transcript abundance in four tissue-types under long day (LD) and short day (SD) conditions and compared the structure of promoter regions of these genes to gain insight into gene expression and regulation. We further examined the functional divergence of the genes by testing whether any of the genes are under divergent natural selection and when gene expression diverged. Finally, we conducted phylogenetic analyses of NP-like proteins across the plant kingdom. Our analyses of natural selection and the evolution of gene expression patterns were then considered in light of this broader phylogenetic context to draw conclusions about the model of evolution and possible gene fates of NP-like genes and proteins within Populus.
Results
Protein phylogeny and chromosome location of NP-like gene family in Populus
To better understand the relationship between the NP-like proteins within Populus we constructed an evolutionary tree based on full-length protein sequences retrieved from the Populus trichocarpa genome through Phytozome (Table 1). This tree indicates three subfamilies of clustered proteins with strong support, posterior probabilities of 1.0 and bootstrap support over 99% (Figure 1A). The first subfamily is comprised of BSP A, BSP B and BSP C and is designated as the BSP subfamily (Figure 2). The second cluster of proteins includes WIN4 and WIN4-like proteins and is designated as the WIN4-like subfamily. A third subfamily includes four uncharacterized NP-like proteins that we designated as the NP-like subfamily. PNI 288 clustered within the cluster composed of WIN4 and BSP subfamilies, but based on this analysis the protein is distinct from the WIN4 and BSP subfamilies (Figure 1A). Gene structure of the primary NP-like transcripts reveals common intron-exon boundaries found within each gene subfamily (Figure 2).
Table 1.
List and description of NP-like genes within Populus trichocarpa used for quantitative gene expression analyses and qPCR primer information
| Gene symbol | Gene name | Locus name Phytozome v2.2 (POPTR) and v3.0 (Potri) | Primers (5′-3′) | Product size (bp) | Annealing temp (°C) | PCR efficiency |
|---|---|---|---|---|---|---|
|
BSP A |
Bark Storage Protein A |
POPTR_0013s10380 |
F: TGGAGAGAACTTGTTGGGGAC |
81 |
55 |
1.981 |
| Potri.013G100700 |
R: CAGAAAACTTCCTTGGGCG |
|||||
|
BSP B |
Bark Storage Protein B |
POPTR_0013s10370 |
F: ATGTTCTCTCCAAGTGAAGCAC |
130 |
57.6 |
1.981 |
| Potri.013G082800 |
R: CGGGCAGGCATTTATCTG |
|||||
|
BSP C |
Bark Storage Protein C |
POPTR_0013s10350 |
F: TTCGTGGTGTTCCAAGGTG |
85 |
54 |
1.936 |
| Potri.013G101000 |
R: AGGCGTTGTAGGAGGCTAAG |
|||||
|
WIN4 |
Wound Induced 4 |
POPTR_0423s00200 |
F: AGGATTTTCGCCTGCTGG |
64 |
62.6 |
2.032 |
| Potri.013G080500 and/or Potri.013G082800 |
R: AATGAACTTGGCTGCGGC |
|||||
|
PNI 288 |
Poplar nitrogen-regulated cDNA 288 |
POPTR_0019s07690 |
F: TGCCAATAGATTCAATGCCAC |
60 |
55 |
1.976 |
| Potri.019G050200 |
R: GAAGCCAAAGCAACAGCAG |
|||||
|
VSP 87A |
WIN-like VSP 87A |
POPTR_0013s07850 |
F: TGAACGGAGAGAACTTGTTGGC R: AGGATGTGGTGCTGGGAAGC |
105 |
54 |
1.912 |
| Potri.013G082600 | ||||||
|
VSP 425 |
WIN-like VSP 425 |
POPTR_0013s07810 |
F: CAAATGTAGCAGGTGAAGCAAG |
154 |
54 |
1.983 |
| Potri.013G080400 |
R: TCAAACGACTCAGAAGCAGATAC |
|||||
|
VSP XIII |
WIN-like VSP XIII |
POPTR_0013s07800 |
F: TCCAGGATTATCGCCTGCTA |
110 |
55 |
2.040 |
| Potri.013G080300 |
R: AATCCCATCACTCACAAGCC |
|||||
|
VSP 840 |
WIN-like VSP 840 |
POPTR_0013s07840 |
F: CCTCCTACAATGCTTTCCTTGCTG |
91 |
55 |
1.96 |
| Potri.013G082700 |
R: GCAGATACAAAATCCCATCACTCAC |
|||||
|
NP 157 |
NP-like 157 |
POPTR_0006s16610 |
F: GCTGTAGATGCTTCACTTAGGTTC |
65 |
54.4 |
1.986 |
| Potri.T096300 |
R: GCCTTATTCGGTAGTTCCAAC |
|||||
|
NP 860 |
NP-like 860 |
POPTR_0008s02860 |
F: TCAAACGGGTATCCTGTGATTGTC |
96 |
61 |
2.07 |
| Potri.008G028500 |
R: TGCTAAGGGTCCAAATGTCTGG |
|||||
|
NP 870 |
NP-like 870 |
POPTR_0008s02870 |
F: AGGGGATGGAACTGGAGAAGTG |
102 |
60 |
2.01 |
| Potri.008G028600 |
R: CCACGAAAATGTCTGCGGTTG |
|||||
|
NP 880 |
NP-like 880 |
POPTR_0008s02880 |
F: GCTACCAGGATACAACTCTCCATTG |
127 |
55 |
1.99 |
| Potri.008G028700 | R: GCTGAAGAACCCCTAAAGATGTCTC |
Figure 1.

NP-like family in Populus trichocarpa. (A) Phylogenetic relationship of 13 NP-like proteins in Populus. Numbers at branches indicate posterior probabilities and bootstrap percentages based on 1000 replicates, respectively. Branches in red indicate significant evidence for experiencing episodic diversifying selection based on the branch-site REL test implemented in HyPhy. (B) Heat map representing the relative transcript expression of NP-like genes in shoot tips, young leaves, mature leaves and bark after 8 weeks long-day (LD) conditions and after 3, 6, 8 and 12 weeks short-day (SD) conditions. The 12 week short-day treatment was combined with low-temperature for the final 4 weeks of SD (i.e. after 8 weeks SD). Values were rescaled within each gene between 0 and 1 with 1 indicating highest relative expression levels.
Figure 2.

Intron-exon structure for NP-like genes in Populus trichocarpa. A comparison of gene structure was constructed using the primary NP-like transcripts from Phytozome (http://www.phytozome.net) and the gene structure draw server found at the PIECE database (http://wheat.pw.usda.gov/piece/index.php). NP-like gene subfamilies are indicated by brackets.
The chromosome locations of the genes revealed that the Populus NP-like genes comprising the three subfamilies reside on four chromosomes (Figure 3). There are two clusters of NP-like genes on chromosome XIII that includes one cluster of all the BSP subfamily members and a second cluster that includes the WIN4-like subfamily gene members. The NP-like subfamily genes are clustered together on chromosome VIII. Only PNI 288 and NP 157 were found to not be clustered with other members of the NP-like genes with PNI 288 located on chromosome XIX while NP 157 is on chromosome VI.
Figure 3.
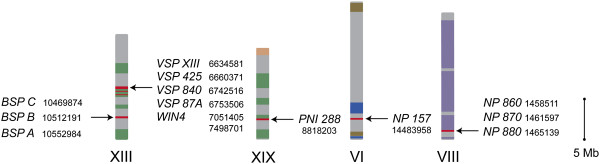
Chromosome location of NP-like genes in Populus trichocarpa. Arrows indicate approximate location of genes. Roman numerals indicate chromosome designation and numbers reflect start site location according to Phytozome (http://www.phytozome.net). Colored blocks indicate known syntenic regions within the Populus trichocarpa genome v2.0 and the link for these coordinates can be found in the Methods.
Expression patterns within the Populus NP-like gene family
Gene expression analysis of the NP-like gene family in P. trichocarpa was assessed in four tissue types (shoot tips, young leaves, mature leaves and bark) of plants treated with LD, SD and SD combined with low-temperatures using qPCR (Table 1 and Figure 1B). Overall, expression of genes within each subfamily was associated with a particular type of tissue and environmental treatment. Expression of all three BSPs increased during SD treatment in bark and after SD combined with LT in shoot tips (Figure 1B). Although expression of all three BSPs was associated with SD, some differences in expression were observed, notably the induction of BSP C in young and mature leaves after 12 weeks SD when the last 4 weeks of SD were combined with low-temperatures. The expression of BSP A and BSP B were very similar and these two genes are more closely related to each other while BSP C is more distant.
In contrast to the BSP subfamily where expression was mostly associated with SD and perennial tissues (shoot tips and bark), expression of the WIN4-like subfamily members was greater only in bark of LD treated plants and during the early stages of SD photoperiod (3 and 6 weeks) treatment for shoot tips and young and mature leaves. For all the WIN-4-like genes, continued exposure to SD resulted in a decline in the steady state abundance of mRNA. This decline in expression of WIN4-like genes in plants treated with SD was most dramatic in bark. These patterns of gene expression show that expression of BSP subfamily members are closely associated with SD in perennial tissues such as shoot tips and bark while members of the WIN4-subfamily are associated with LD in these same perennial tissues. Furthermore, compared to the BSP subfamily the WIN4-subfamily members are also expressed to a greater extent in both young and mature leaves. PNI 288 expression was similar to members of the WIN4-like subfamily and was detected in all tissues with expression declining during SD treatment.
Except for NP 157, expression of the NP-like genes (NP 880, NP 870, NP 860) was observed to occur at lower levels in shoot-tips, young leaves, matures leaves and bark when compared to members of the BSP and WIN4-like subfamilies. Expression of the NP-like subfamily genes also tended to be associated with SD conditions in all tissues. NP 157 was expressed to a greater level than other members of the NP-like subfamily and expression was associated with SD in all tissues. The greatest levels of expression for NP 157 were observed in both young and mature leaves after 12 weeks of SD with the last 4 weeks SD combined with low temperature.
In summary, members of each of the three NP-like subfamilies (BSP, WIN4-like and NP-like) were observed to have similar expression patterns that were associated with each subfamily. Members of the BSP subfamily are expressed in SD and tend to be associated with perennial tissues. Members of the WIN4-like subfamily are found in both perennial and deciduous tissues but are repressed by SD. The NP-like subfamily is found in both perennial and deciduous tissues but only one member, NP 157, appears to show a consistent SD response among the various tissues. Combined with the phylogenetic analysis our analysis of gene expression demonstrates a correlation between phylogenetically defined NP-like subfamilies and gene expression. This relationship is also supported by PCA based on the expression data (Figure 4), which shows that members of the same gene subfamily are closer to one another than to members of the other gene subfamilies. Within this analysis, PCA Axis 1 and Axis 2 explained 44% and 16% of the variance, respectively. To determine the number of non-trivial components produced through the PCA, we compared our observed eigenvalues to the distribution created by conducting the analyses on 10000 randomized matrices of the observed dataset (e.g., λk). Based on this approach, we found that only the first axis had an eigenvalue significantly greater (P < 0.05) than that expected by random.
Figure 4.

Principal component analysis (PCA) of NP-like gene expression in Populus trichocarpa. PCA illustrating the relationships of NP-like genes in Populus trichocarpa based upon the expression data presented in Figure 1B. The percentage of variation explained by each axis is given in parentheses next along each axis.
Natural selection and continuous character evolution
Based on the Branch-site REL method, we found statistically significant evidence for episodic diversifying selection within VSP 425, VSP 840, VSP 87A, BSP A, BSP B and BSP C (Holm-Bonferroni corrected p-value < 0.05) (Figure 1A and Table 2). We also found evidence for diversifying selection at the base of the gene tree corresponding to the initial gene duplication that gave rise to phylogenetic clusters referred to as Group I and Group II NP-like proteins (Figure 1A and Table 2).
Table 2.
Results of tests for a significant phylogenetic signal of the expression data as measured by Pagel's lambda transformation
| Condition | λBark | λShoot tips | λMature leaves | λYoung leaves |
|---|---|---|---|---|
| LD |
0.658 |
0.414 |
0.972** |
0.974** |
| SD 3 |
0.000 |
0.502 |
0.845* |
0.986*** |
| SD 6 |
0.901 |
0.000 |
0.792* |
0.815* |
| SD 8 |
1.000* |
1.000* |
1.000* |
0.000 |
| SD 12 | 0.936* | 0.667 | 1.000* | 1.000* |
A value of 0 represents no signal with a value of 1 signifying complete phylogenetic patterning. P-values denoted as P-value < 0.05, ** P-value < 0.01 and *** P-value < 0.001.
Tests for whether there is a phylogenetic signal, λ, to gene expression patterns were significantly different than the alternative of no signal (i.e., Brownian motion model of character evolution) (Table 3). Additionally, our tests of evolutionary differences between the NP-like proteins within Populus illustrated that expression divergence was concentrated late in the evolutionary process. Specifically, δ was greater than 1 which is indicative of evolutionary changes occurring recently and that a model raising all branches to the power δ was a better fit than a Brownian motion model (Table 3).
Table 3.
Results of the tests for episodic diversifying selection among the 13 NP-like genes assayed within Populus trichocarpa
| Branch | ω- | Pr[ω = ω-] | ωN | Pr[ω = ωN] | ω+ | Pr[ω = ω+] | LRT | p-value | Corrected p-value | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| VSP425 |
0.911 |
1.0 |
0.888 |
1.0 |
0.009 |
10000.0 |
0.102 |
120.182 |
0.0 |
0.0 |
|
| VSP840 |
1.078 |
0.607 |
0.968 |
1.0 |
0.004 |
72.780 |
0.028 |
12.596 |
0.0 |
0.004 |
|
| VSP87A |
10.0 |
0.0 |
0.748 |
0.0 |
0.071 |
10000.0 |
0.180 |
8.721 |
0.002 |
0.033 |
|
| Node3 |
1.855 |
0.0 |
0.653 |
0.0 |
0.052 |
21.384 |
0.296 |
8.193 |
0.002 |
0.042 |
|
| BSPA |
0.394 |
0.279 |
0.996 |
0.281 |
0.0 |
10000.0 |
0.004 |
8.079 |
0.002 |
0.043 |
|
| BSPC |
0.899 |
0.0 |
0.673 |
0.0 |
0.236 |
12.791 |
0.091 |
8.013 |
0.002 |
0.042 |
|
| Node2 |
0.324 |
0.202 |
0.934 |
1.0 |
0.002 |
24.413 |
0.064 |
5.244 |
0.011 |
0.187 |
|
| BSPB |
0.595 |
0.059 |
0.959 |
0.197 |
0.001 |
21.161 |
0.040 |
3.666 |
0.028 |
0.444 |
|
| NP880 |
0.155 |
0.094 |
0.959 |
0.841 |
0.0 |
593.764 |
0.041 |
2.912 |
0.044 |
0.660 |
|
| NP157 |
0.545 |
0.0 |
0.499 |
0.857 |
0.0 |
2.940 |
0.501 |
0.754 |
0.193 |
1.0 |
|
| NP860 |
0.174 |
0.108 |
0.855 |
0.730 |
0.120 |
684.793 |
0.026 |
0.587 |
0.222 |
1.0 |
|
| Node12 |
0.446 |
0.302 |
0.881 |
0.650 |
0.0 |
3.380 |
0.119 |
0.277 |
0.299 |
1.0 |
|
| Node9 |
10.0 |
0.0 |
0.240 |
0.0 |
0.111 |
10000.0 |
0.649 |
0.245 |
0.310 |
1.0 |
|
| Node14 |
0.375 |
0.150 |
0.832 |
0.993 |
0.0 |
2.068 |
0.168 |
0.186 |
0.333 |
1.0 |
|
| Node5 |
0.163 |
0.0 |
0.404 |
0.321 |
0.554 |
3333.11z |
0.042 |
0.172 |
0.339 |
1.0 |
|
| PNI288 |
0.540 |
0.0 |
0.428 |
0.999 |
0.0 |
1.192 |
0.572 |
0.108 |
0.371 |
1.0 |
|
| VSPXIII |
0.973 |
0.986 |
0.0 |
0.984 |
0.0 |
1.040 |
1.0 |
0.001 |
0.486 |
1.0 |
|
| NP870 |
0.301 |
0.165 |
0.867 |
1.0 |
0.060 |
3.224 |
0.073 |
-0.380 |
0.500 |
1.0 |
|
| Node7 |
0.0 |
0.0 |
0.885 |
0.996 |
0.0 |
2.106 |
0.115 |
-0.001 |
0.500 |
1.0 |
|
| Node1 |
10.0 |
0.093 |
0.971 |
0.894 |
0.028 |
8.630 |
0.001 |
-0.001 |
0.500 |
1.0 |
|
| Node21 |
10.0 |
0.095 |
0.009 |
0.897 |
0.010 |
8.819 |
0.980 |
-0.001 |
0.500 |
1.0 |
|
| Node17 |
10.0 |
0.054 |
0.370 |
0.492 |
0.232 |
0.293 |
0.398 |
0.0 |
1.0 |
1.0 |
|
| WIN4 | 0.545 | 0.714 | 0.0 | 0.722 | 1.0 | 1.578 | 0.0 | 0.0 | 1.0 | 1.0 |
Branch = The name of the branch (see tree plot on the main analysis page for the location of automatically named internal branches).
= The ω ratio inferred under the MG94xREV model that permits lineage-to-lineage but no site- to-site ω variation.
ω- = The maximum likelihood estimate (MLE) of the first rate class with ω ≤ 1.
Pr[ω = ω-] = The MLE of the proportion of sites evolving at ω-.
ωN = The MLE of the second rate class with ω ≤ 1.
Pr[ω = ωN] = The MLE of the proportion of sites evolving at ωN.
ω+ = The MLE of the rate class with unconstrained ω.
Pr[ω = ω+] = The MLE of the proportion of sites evolving at ω + .
LRT = Likelihood ratio test statistic for ω + = 1 (null) versus ω + unrestricted (alternative).
p-value = The uncorrected p-value for the LRT test.
Corrected p-value = The p-value corrected for multiple testing using the Holm-Bonferroni method.
In silico promoter sequence analyses of the NP-like gene family in Populus
We identified overrepresented motifs present in the 2 kb regions upstream of Populus NP-like genes by performing in silico analyses using two prediction programs, MEME and MotifClick [41,42]. We identified motifs that correspond to known cis-regulatory elements (CREs) from the transcription factor (TF) database consolidator program PlantPAN [43]. Of the 200 motifs identified by MEME and MotifClick, 23 motifs correspond to known CREs (Figure 5, Table 4, Additional file 1: Table S6). These regions have been shown to bind TFs, which possibly regulate transcription in a range of tissues and in response to phytohormones, stress and light (Table 4). CREs were found in the Populus NP-like gene promoters that may contribute to tissue-specific expression (AG, AGL3, Athb-1, -5, -9, CDC5, MYB.ph3, O2 and SQUA), phytohormone responses (ARR10, ERELEE4, RAV1 and RYREPEATVFLEB4), stress responses (RAV1, UPRE2AT and UPRMOTIFIAT) and light responses (CIACADIANLELHC and PIF3). Both motif prediction programs identified motifs corresponding to the known CREs AG, AGL3, Athb-9, O2 and RAV1 (Table 4).
Figure 5.

Cis-regulatory element distribution in the promoter regions of NP-like genes in Populus trichocarpa. CREs identified by two methods of motif prediction are represented as colored bars and correspond to the CRE name.
Table 4.
Known cis-regulatory elements (CREs) in the promoter regions of NP-like genes within Populus trichocarpa identified by the motif prediction programs MEME and MotifClick (MC), CRE database and the transcription factors and/or functions
| CRE name | Prediction Program | CRE database | Transcription factor/function |
|---|---|---|---|
| AG |
MC |
TRANSFAC |
AGAMOUS, expressed in flowers |
| MEME | |||
| AGL3 |
MEME |
TRANSFAC |
AGAMOUS-like (AGL) 3, expressed in vegetative and floral above ground tissue |
| MC | |||
| ANT |
MEME |
TRANSFAC |
ANT (a member of AP2/EREBP TFs) |
| ARR10 |
MC |
JASPER |
ARR10, involved in 2 component regulation and possibly cytokinin signaling |
| Athb-1 |
MC |
TRANSFAC |
AtHB-1, involved in cell differentiation in leaves, expressed in leaves |
| Athb-5 |
MC |
TRANSFAC |
AtHB-5, expressed in vegetative tissues, preferentially in leaf tissues. Function in mature vegetative tissues. |
| Athb-9 |
MC |
TRANSFAC |
AtHB-9, possibly involved in dorsiventral patterning of lateral organs (leaves). |
| MEME | |||
| bZIP910 |
MC |
TRANSFAC |
BZIP transcript factor from snapdragon. |
| C8GCARGAT |
MC |
PLACE |
AGL15, possibly involved in gibberellin metabolic signaling. |
| CARGCW8GAT |
MC |
PLACE |
AGL15, possibly involved in gibberellin metabolic signaling. |
| CDC5 |
MEME |
TRANSFAC |
AtCDC5, required for function of shoot apical meristem. Silencing accelerates cell death in leaves. Possibly involved in cell cycle regulation. |
| CIACADIANLELHC |
MC |
PLACE |
Region necessary for circadian expression of light harvesting complex genes |
| ERELEE4 |
MC |
PLACE |
Ethylene responsive element, senescence-related expression |
| GMHDLGMVSPB |
MC |
PLACE |
GmHdl56/GmHd157, found in the promoter vegetative storage protein conferring function vacuolar glycoprotein acid phosphatase in soybean |
| HMG-IY |
MEME |
JASPER |
Binding regions for proteins similar to histone H1/H5 family |
| MYB.ph3 |
MC |
JASPER |
MYB.ph3, petal epidermis-specific, possibly GA regulated and may bind chromatin |
| TRANSFAC | |||
| O2 |
MC |
TRANSFAC |
Opaque-2, found in Maize endosperm |
| MEME | |||
| PIF3 |
MEME |
TRANSFAC |
PIF3, present in many light-regulated promoters, including PhyB |
| RAV1 |
MC |
TRANSFAC |
RAV1, may be negative regulator of plant development, down regulated by brassinosteroids. Touch-, drought-, salt- cold-, bacteria-induced. Positively regulates leaf senescence. Ethylene mediated signaling. |
| MEME | |||
| RYREPEATVFLEB4 |
MEME |
PLACE |
FUS3/TRAB1, ABA and auxin responsive, found in seed proteins |
| SQUA |
MC |
JASPER |
SQUA, required for inflorescence development |
| UPRE2AT |
MEME |
PLACE |
Found in promoters of many genes associated with ER stress |
| UPRMOTIFIAT | MEME | PLACE |
The results of the principal component analysis based on the number of predicted motifs within each gene were generally consistent with the phylogenetic relationships. Along PCA Axis 1, which explains 28% of the variance in the data, motifs in the promoter regions of VSP XIII and WIN4 genes are primarily separated from the BSP and NP genes (Figure 6). PCA Axis 2, which explains 13% of the variance in the data, primarily separates the motifs in the promoter regions of the BSP genes from the other NP-like genes. However, along axis two the motifs in the promoter regions of BSP A and BSP B are found close together compared to motifs of the promoter of BSP C which is isolated at the negative end of the axis. Based on the λk approach described above to assessing the number of non-trivial components, we found that axes 1 and 2 had eigenvalues significantly greater (P < 0.05) than those created under the randomizations.
Figure 6.

Principal component analysis (PCA) of NP-like cis-regulatory element motifs in Populus trichocarpa. PCA based upon the abundance of cis-regulatory element motifs within promoter regions of NP-like genes in Populus trichocarpa. The percentage of variation explained by each axis is given in parentheses next along each axis.
Phylogenetic distribution in the plant kingdom
To investigate the diversity of plant NP-like proteins, we retrieved 142 complete and non-redundant amino acid sequences across the plant kingdom (Additional file 2: Table S5) with NP sequence homology based on BLASTP search. Phylogenetic analyses based on alignments of the predicted NP-like region for 38 genera revealed 2 general groups of NP-like proteins (Figure 7). Group I and II have strong support, with posterior probabilities of 1.0 as well as bootstrap support of 95%. Group I includes NP-like proteins from all represented genera having complete genome sequences with subclasses that generally cluster according to Rosids and monocot lineages [44,45]. Group I also includes Populus proteins that are not known to be involved in storage (NP-like subfamily members). Phylogenetic analyses that include a bacterial outgroup can be found in Additional file 3: Figure S1. The addition of an outgroup does not change the topology of the plant NP groups.
Figure 7.

Phylogenetic analyses of NP-like proteins in the plant kingdom. Phylogenetic relationships were constructed using Bayesian and maximum-likelihood methods. Numbers at branches indicate posterior probabilities and bootstrap percentages based on 1000 replicates, respectively. Numbers in parentheses correspond to Phytozome or NCBI sequence identifiers, which can be found in the Additional file 7: Table S4. The five predominant taxonomic families are indicated by the highlighted colors.
Group II is mainly composed of proteins from Populus, Manihot esculenta and Ricinus communis. There is a smaller cluster of proteins from monocot genera and two citrus proteins. The Populus proteins in Group II include the BSPs, WIN4-like proteins and PNI 288 (Figure 7).
Discussion
Protein phylogeny and chromosomal distribution of NP-like gene family in Populus
Previous studies identified three BSP genes, three WIN4-like genes and PNI 288[15]. In this study we examined the Populus genome sequence and identified an additional 6 genes to expand this gene family to 13 members. Based on our phylogenetic analyses we propose this gene family be designated the NP-like gene family with three subfamilies that include the BSP subfamily (three gene members), the WIN4-like subfamily (five gene members) and the NP-like subfamily (four gene members). In addition, PNI 288 also is a member of the NP-like gene family but does not cluster with any of the three subfamilies.
The protein phylogeny of the evolutionary relationships among NP-like proteins across the plant kingdom and the chromosomal distribution indicates that the NP-like gene family in Populus expanded through WGD and TD events. This is consistent with previous studies investigating the evolutionary history of the Populus genome [46-48]. A WGD event occurred in Populus approximately 65 mya (millions years ago) or earlier, followed by genome wide reorganization that resulted in, among other changes, paralogous sets of chromosomes of which XIII and XIX are a pair (where PNI 288 and the BSP/WIN4-like subfamilies reside, respectively) [46-48]. Chromosomes VI and VIII (where NP 157 and the NP-like subfamily reside, respectively) are not a paralogous set [46]. The members of each gene subfamily are located within a 100 Kb region and are likely the result of TD (Figure 2). This proximity of the genes to each other is generally a good indication of TD [49,50]. The type of duplication event (i.e. TD or WGD) has implications on gene function: genes families in Populus that have expanded through TD are enriched for functions involving defense responses, apoptosis and protein kinases [49].
In silico promoter analyses of the NP-like gene family in Populus
Promoter analyses are an important component of investigating the functional evolution of genes particularly in cases where genes are created by TD. This is because TD mainly occurs through unequal recombination, which can result in subfunctionalization if regulatory regions are not also duplicated [51]. These analyses also provide information regarding possible regulation and tissue-specific expression—crucial for future investigations to determine function.
Despite the potential for informative conclusions to be gained from in silico promoter analyses, motif discovery is difficult from a bioinformatics perspective [52]. Mapping CREs in promoter regions using only databases of known CREs results in a high rate of false positives and does not prove that these regions are TF binding sites. To increase confidence, we utilized 2 motif prediction programs (MEME and MotifClick) to identify over-represented 8 bp motifs in the 2 kb promoter regions of NP-like genes in Populus. Known CREs were identified from the resulting motifs and mapped (Table 4, Figure 5). Both programs predicted motifs that correspond to CREs associated with gene activation by the TFs AG, AGL3, Athb-9, O2 and RAV1. These CREs are involved in a wide range of developmental and metabolic processes. AG has been shown to bind the AGAMOUS TF that functions in temporal development of floral stem cells [53] while the related TF AGL3 is likely involved in development of above-ground organs and binds to regions identified as the AGL3 CRE [54]. Athb-9 appears to regulate patterning of leaves while O2 or Opaque-2 is a TF responsible for expression of a prominent storage protein in Zea mays and contribute to endosperm metabolism [55,56]. Lastly, RAV1 is a TF that promotes and possibly regulates leaf senescence [57]. Overall, we identified CREs that have been shown to be involved in light, stress and defense and hormone responses. The presence of these potential CRE in the promoters of the Poplar NP-like genes provides possible clues related to the regulation of these genes. The potential regulation of NP-like genes in Populus by hormone signaling pathways would be particularly interesting to pursue.
The principal component analysis of predicted promoter motifs is consistent with the similarity tree expression data (Figure 1A) and phylogenetic analyses (Figure 6). This is not unexpected since gene expression is regulated by TFs and their interaction with CREs in promoter regions. The distances between BSP A, BSP B and BSP C, which represent differences in the motifs in their promoter regions, could result from unequal recombination combined with diversifying selection (Figure 6, 1A) [51]. This may also explain the distances between the motifs in the promoter regions of WIN4-like genes (Figure 6, 1A).
Expression divergence and natural selection analyses
Our evaluation of selection pressure indicates diversifying selection at the initial duplication event and more recent divergent selection of BSP and WIN4-like subfamily members. This succession of duplication events is consistent with phylogenetic studies in plants (e.g. [58-60]). Populus specific duplication events have also been evaluated [46,48,60]. In Populus there are reports that correlate tandemly duplicated genes to the expansion of plant defense and stress gene families, consistent with the gene dosage model [49,50,61]. Strong patterns between opposing functional gene groups retained following either WGD or TD events have been observed in P. trichocarpa[49,58]. This fits well with observations of BSPs and VSPs accumulating following wounding and drought stress [22,23].
Our results show that BSP and WIN4-like subfamilies gene expression patterns evolved relatively recently and members of these subfamilies are also under selection pressure; this suggests they are undergoing subfunctionalization or diversification of multifunctional genes. This is supported by the expression data showing differential co-expression among the BSP and WIN4-like gene subfamilies (Figure 1B). Perhaps the data reflect amplification, buffering or “near” subfunctionalization, all of which can facilitate functional redundancy that enables organisms to respond to a greater range of cellular, environmental and/or genetic perturbations [62,63]. This positive dosage model is particularly applicable to genes involved in stress and environmental responses [29,30,37]. Such a strategy would functionally promote optimum nutrient cycling and storage pathways [24]. The amplification model describes the potential functional evolution and fixation of genes from unstable (i.e. tandem) duplications [31]. The high degree of nucleic acid similarity between BSP genes (BSP A vs. BSP B: 98.2%; BSPA + B vs. BSP C ~94%) also supports the amplification model since sequence divergence is loosely correlated with expression divergence [49].
We qualify our predictions of the functional gene fates, acknowledging that classification is restricted to the current gene duplication models, which are limited by a poor understanding of the role of population size, selection pressure and fixation preceding and following duplication [26,64,65]. Research is trending toward developing a more general model of adaptive maintenance duplication [64-66]. Without such information it is particularly difficult to distinguish the subfunctionalization models and the diversification of a multifunctional genes [66]. Another complexity involves multiple types of functionalization occurring over the course of evolution [67].
NP-like gene family expansion order within Populus trichocarpa
From the results presented here, we propose a hypothetical origin of NP-like genes within Populus trichocarpa, depicted in Figure 8. The NP-like parent protein was retained following an ancient WGD event after which the NP-like parent protein and the duplicate became the progenitors of Group I and II NP-like proteins, respectively. These genes could have been retained if they had a function involvement in signaling networks or pathways, the dosage balance model. The progenitor of Group II NP-like proteins may have undergone a functionalization process that conferred fitness or became advantageous following a change (i.e. environmental) followed by stabilizing selection pressure. Another WGD event occurred, specific to the Populus lineage and was followed by tandem duplications where upon the amplification of this function would result in immediate stabilizing selection pressure, the positive dosage model.
Figure 8.
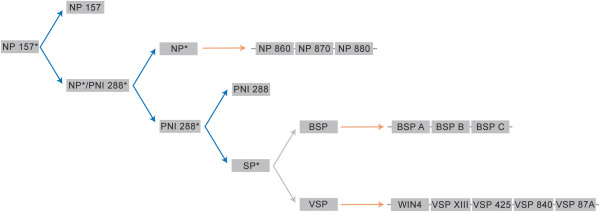
Hypothetical origin of the NP-like gene family in Populus trichocarpa. Blue arrows represent whole genome duplication events and orange arrows represent tandem duplication events. Asterisks denote ancestral genes with unknown sequence similarity to present-day genes.
Our selection and phylogenetic analyses suggest NP 157 is the likely progenitor gene. Further, the expression patterns of NP 157 were distinctly different from those of the BSP and WIN4-like gene subfamilies. It seems unlikely that resources would be directed toward transcription of high levels of NP 157 transcripts, and presumably the translation of proteins, of a nonfunctional or pseudogene in senescing leaves. Two explanations are that NP 157 is scavenging nucleosides prior to senescence or has retained regulatory elements similar to those of other genes involved in purine salvage and degradation. Increased expression of other genes involved in purine salvage and degradation has been observed under dark-induced senescence conditions [14]. Additionally, knocking-down the enzyme considered to be the key bottleneck in purine degradation, xanthine dehydrogenase, resulted in reduced growth, early senescence and infertility [12,68]. Yet another explanation could be that NP 157 acts as transient storage protein or is otherwise involved in nutrient signaling. At the very least, NP 157 as well as BSP and WIN4-like genes may be responding to changes in nutrient signals induced by SD photoperiod and/or the onset of senescence. This seems plausible when compared to NP expression in other systems. In the bacteria Bacillus subtilis, PNP transcription is induced by nucleosides in growth media and repressed by glucose, pointing to modulation of PNP by carbon and energy availability [69,70]. In humans, PNPs are up-regulated in diseased and cancerous tissues where metabolic shifts occur [4-6]. While more research is needed to conclusively define the function of NP 157 we posit that NP-like proteins may still be involved in nutrient signaling in plants.
Phylogenetic distribution across the plant kingdom
Our data suggest that NP-like proteins were conserved in all higher plants examined with complete genomes (Figure 7). Interestingly, NP-like proteins were not identified in Physcomitrella and green algae genomes. These taxa could have lost NP-like genes. In bacteria, there are multiple phyla that do not have PNPs or related uridine phosphorylases and it is hypothesized that this may have occurred due to different resource requirements among phyla in bacteria [71]. The non-vascular plant Selaginella moellendorffii has two NP-like representatives that cluster at the base of Group I NP-like proteins suggesting that these proteins are the most ancestral (Figure 7). The inclusion of a bacterial outgroup supports the hypothesis that NP-like proteins are monophyletic and evolved from NPs (Additional file 3: Figure S1).
The phylogeny of NP-like proteins across the plant kingdom reveals that the P. trichocarpa subfamily designations are consistent with those found in the tree of the 13 NP-like proteins in Populus (Figures 1 and 5). The BSP and WIN4-like subfamilies and PNI 288 cluster within Group II and the NP-like subfamily belong to the Group I. If NP-like proteins functioned as storage proteins in all plants, we would expect that NP-like proteins from other woody perennial genera would cluster near Populus storage proteins yet Prunus, Picea and Malus NP-like proteins do not cluster near Populus storage proteins. Instead, proteins from these genera cluster in Group I near proteins from the NP-like subfamily members in Populus. This suggests that NP-like proteins are not storage proteins throughout the plant kingdom.
In addition to the conclusions based on our phylogenetic analyses, the finding that NP-like proteins are not general storage proteins is consistent with previous reports on seasonal storage proteins in other woody plant genera. For example, Prunus persica and Picea sitchensis are both woody perennials. In the bark of Prunus persica, the predominant storage proteins are a 60 kDa dehydrin, a 19 kDa allergen-related protein and a 16 kDa protein with no known homology [72]. In Picea glauca x engelmannii complex, the predominant storage proteins were 20 and 27 kDa, loosely suggesting that 32 kDa BSPs are not the dominant storage proteins in Picea[73].
Our results point to an expansion of the NP-like gene family in a common ancestor of the order Malpighiales of which Populus, Manihot, Ricinus and Linum are representatives [44,74]. While proteins from Populus, Manihot and Ricinus often clustered together in both Groups, Linum, has only three NP-like proteins that cluster in Group I. We propose that Linum lost Group II NP-like proteins. The recent assembly of the Linum genome revealed that Linum has experienced a recent WGD followed by the loss of one or more chromosomes [75]. The many proteins found in Manihot and Ricinus that cluster near the Populus storage proteins indicates a storage function in these genera.
Conclusions
Our investigation into the functional significance and evolutionary history of NP-like proteins and genes within Populus and across the plant kingdom illustrates the importance of both microevolutionary (e.g., natural selection) and macroevolutionary (e.g., genome duplication and tandem duplications) forces in shaping patterns of diversity within this protein family. Of particular note is that we found evidence that hitherto uncharacterized NP-like genes might serve a functional role within Populus based on expression data; the functional significance of such proteins is unknown. Further, analyses of promoter regions that showed the presence of motifs associated with light responses, phytohormone responses, stress and tissue-specificity that also support a functional role for NP-like proteins. We also found significant evidence for episodic diversifying selection acting on the NP-like proteins within Populus and that changes in gene expression levels had occurred relatively recently in the evolution of this gene family. Consistent with the other findings, our inferred phylogeny implicates both historical genome duplication events and more recent taxon specific independent duplication events as mechanism that gave rise to the extant diversity of NP-like proteins within Populus. In conclusion, our results are the first to examine NP-like proteins in plants and we present the possibility that they may have functional significance within plants.
Methods
Plant material
Populus trichocarpa (Torr. and Gray) genotype ‘Nisqually-1’ cuttings were grown in 6 L pots in controlled environmental chambers at 18°C with a PAR range of 310–470 μmol m–2 s–1. Plants were fertilized one week after transplanting with 5 g of controlled release fertilizer (18-3-3) (Nutricote, Florikan, Sarasota, FL, USA). Tissue samples were collected after 8 weeks under LD conditions (16 h light/8 h dark) and after 3, 6, 8 and 12 weeks under SD conditions (8 h light/16 h dark). The temperature was lowered to 10°C day/4°C night for the last 4 weeks of SD (i.e. from 8 to 12 weeks SD). Shoot tips or apical buds, young leaves (leaf plastochron index (LPI) 3), mature leaves (LPI 8) and bark (between LPI 8–9) were harvested and frozen in liquid N2 and then stored at -80°C until RNA extraction. One replicate was composed of pooled tissues from three plants. Two replicates were used for gene expression analysis.
Identification and phylogenetic analysis of NP-like genes in the Populus genome
To identify NP-like proteins in the Populus genome a BLASTP search against the Populus trichocarpa genome v2.2 (http://www.phytozome.net) was performed using default parameters (expect (E) threshold = -1, BLOSUM62 comparison matrix) with the P. trichocarpa BSP A (locus name: POPTR_0013s10380) as the query sequence. NP-like protein sequences meeting these criteria were then retrieved from the Phytozome database and used for phylogenetic analyses.
QPCR primer design and validation
Primers for the NP-like gene family and reference genes were designed using MacVector v10 (MacVector Inc., Cary, NC, USA). All primers were synthesized by Invitrogen (Carlsbad, CA, USA). Optimum annealing temperatures were determined by a temperature gradient and amplification efficiencies calculated from a five-point calibration curve of ten-fold serial dilutions. To confirm a single amplification product, melt curves were performed for all qPCR reactions.
RNA extraction, cDNA synthesis, qPCR detection and statistical analyses
Total RNA was purified using RNeasy plant mini kits and the automated QIAcube (Qiagen, Valencia, CA, USA). Tissues were ground in liquid nitrogen and added to RLT extraction buffer containing 1% polyvinylpyrrolidone, and 1% beta-mercaptoethanol. Samples were vortexed and 0.4 volumes of 5 M potassium acetate, pH 6.5 was added to the sample, mixed and incubated on ice for 15 min. Samples were then centrifuged at 15,000 g for 15 min at 4°C. The supernatant was transferred to the QIAcube for RNA extraction with on-column DNase I digestion (Qiagen, Valencia, CA, USA). Microfluidic analyses using the ExperionTM automated electrophoresis system and RNA StdSens Chips were used to determined RNA quality and quantity (Bio-Rad, Hercules, CA, USA). First strand cDNA was synthesized in triplicate reactions from 1 μg of total RNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific Inc., Rockford, IL, USA) and oligo dT primers. cDNA was pooled and used as template for qPCR detection. Amplification reactions were performed with the iQ5 Real-Time Detection System (Bio-Rad, Hercules, CA, USA) using Maxima SYBR green qPCR master mix (Thermo Scientific Inc., Rockford, IL, USA). Amplification reactions consisted of 10 min at 95°C followed by 40 cycles of 15 sec at 95°C and 1 min at the optimum annealing temperature. Stable reference genes were determined for each tissue type by geNormPLUS in qbasePLUS version 3 (http://www.qbaseplus.com). (Additional file 4: Table S1) and relative expression analyses were performed with qbasePLUS (Additional file 5: Table S2). Relative expression data are scaled to the maximum relative expression units of each tissue.
We also performed a principal component analysis based on differences in expression patterns within each tissue type across the treatments for each of the 13 genes. The analysis was performed using the ADE4 package [76] within R [77].
In silico promoter sequence analyses of the NP-like gene family in Populus
Promoter sequences 2 kb upstream from the transcription start site (ATG codons) of each Populus NP-like genes were retrieved from the Populus trichocarpa genome v2.2 in Phytozome (http://www.phytozome.net/). In silico analyses of these regions were performed using the prediction programs MEME (http://meme.nbcr.net) and MotifClick (http://motifclick.uncc.edu/) to determine overrepresented 8 bp motifs [41,42]. MEME uses an expected-maximization strategy to detect overrepresented motifs whereas MotifClick employs a graph-based algorithm that determines motifs by identifying complete subgraphs also known as cliques [41,42]. MEME was performed with the following settings: any number of repetitions for a single motif distributed among the sequences, the minimum and maximum motif width of 8 bp and the maximum number of motifs to find was 100. Settings for MotifClick analyses were performed with motif length set to 8 bp, 100 maximum motif number, more degenerate sites and sum of square distance between the binding site and background sequence run at 0.06 and 0.1. The output from runs at SSD 0.06 and 0.1 were 93% similar, with output from SSD 0.06 being chosen for further comparison (Additional file 6: Table S3). The resulting 8 bp motifs were compared against the known CRE sequences in these regions determined by PlantPAN (Plant Promoter Analysis Navigator) [43]. PlantPAN collected known TF binding motifs from PLACE, TRANSFAC, AGRIS and Jaspar [43] (see Additional file 7: Table S4 for output from PlantPAN). Motif lengths of 8 bp were chosen for three reasons: TFs commonly bind to DNA regions that are between 8–10 and 16–20 bp in length, the length is suitable for prediction programs and cross-referencing with output of PlantPAN [52]. Smaller regions can result in an increased number of false-positives [52].
We conducted a principal components analysis to elucidate the relationships among the proteins based on the abundance of motifs predicted by both programs. The input for the analysis, which were conducted using the ADE-4 package [76] within R [77], was a matrix of motif counts per gene. To identify non-trivial components produced by PCA, we performed an analysis method [78] of 10000 randomized matrices of the dataset in R [77].
Protein sequence identification and motif prediction of NP-like region for phylogeny
For constructing the evolutionary relationships among NP-like proteins across the plant kingdom, we retrieved amino acid sequences from the Phytozome database (http://www.phytozome.net) using a BLASTP search with default parameters and the P. trichocarpa BSP A (locus name: POPTR_0013s10380) protein sequence for the query. For species absent in Phytozome, a BLASTP search with default parameters was performed at NCBI using the “non-redundant protein sequence” database (http://www.ncbi.nlm.nih.gov/). Sequences for Malus were retrieved from the Rosaceae Database (http://www.rosaceae.org/) and sequences for Picea abies were retrieved from ConGenIE (http://congenie.org/). For all searches, only hits below an E-value of 10-4 were used. Sequences were manually inspected for annotation errors and duplicate sequences were removed. A total of 142 sequences were used in the analysis and 3 bacterial sequences that serve as an outgroup (Additional file 2: Table S5). To confirm that all plant sequences could be classified as NPs, a batch CD-search of the Conserved Domain Database (CDD) using all 142 plant sequences was performed with default setting and the E-value set to 0.01 with all sequences identified as members of the PNP_UDP_1 superfamily (Pfam:01048) [79]. The conserved region was identified with the Gapped Local Alignment of Motifs (GLAM2) program, version 4.8.1, for all sequences with default parameters and 8,000 iterations (http://meme.nbcr.net/meme/cgi-bin/glam2.cgi) [80]. Predicted motifs were cross-referenced with GLAM2SCAN using the NCBI non-redundant protein database with closest matching motifs in proteins within PNP_UDP_1 superfamily (Pfam:01048). To validate GLAM2 predicted motifs, 57 non-plant (bacterial) sequences from the NP family (COG0775, superfamily cl00303) were retrieved from CDD (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) and motifs were predicted for this set of sequences by GLAM2. We then constructed alignments of the predicted motifs from each sequence set (plant and non-plant) with MUSCLE set to default settings to validate the NP-like region in plants [81] (Additional file 8: Figure S2). The validated NP-like motif in plants was used to remove regions outside the NP-like motif sequence and the resulting sequences were used for phylogenetic tree construction.
The coordinates of syntenic regions within the Populus trichocarpa genome v2.2 represented in Figure 2 can be found at http://ftp.jgi-psf.org/pub/compgen/phytozome/v5.0/ Ptrichocarpa/miscelaneous/synteny/allPoplarSegments.seg.
Phylogenetic analyses and protein family evolution
Phylogenetic hypotheses were constructed for each alignment using both a Bayesian (MrBayes v3.0; [82]) and maximum-likelihood based method (Genetic Algorithm for Rapid Likelihood Inference; GARLI; [83]). We used the WAG + I + G amino acid substitution model, determined by ProtTest [84], for all analyses (including the phylogeny of NP-like proteins in Populus with full length amino acid sequences). We present the best topology based on 1000 replicate GARLI analyses on the observed dataset and assessed the statistical support for topological relationships from 1000 bootstrap replicates. All GARLI analyses were performed using the computing resources associated with the LATTICE project [85]. For the Bayesian analyses, we used the default settings (two concurrently running independent analyses of four chains, three of which were heated) but set the amino acid model to mixed. We used a threshold of 0.01 for the standard deviation of the split frequencies as a measure of sufficient convergence and mixing. Analyses of the smaller 13 protein Poplulus dataset and the larger 142 protein dataset were run for 106 and 107 generations, respectively. In both analyses, 25% of the generations served as burn-in. Multiple sequence alignment and phylogenetic tree are available from TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S14494; [86]). Phylogenetic analyses that include a bacterial ourgroup can be found in Additional file 3: Figure S1.
Tests of natural selection and gene expression evolution
We tested for recombination in the 13-gene Populus dataset, which can adversely affect analyses of natural selection, using the Genetic Algorithm for Recombination Detection (GARD) method [87] implemented in the package HyPhy [88]. The results from GARD did not identify any statistically significant evidence for recombination breakpoints. Thus, we used the Branch-site REL (random effects likelihood) method [87] implemented in the package HyPhy to determine whether any of the gene duplicates within the 13 gene Populus dataset showed evidence for being under diversifying selection. We chose this method of detecting selection since our primary interest was to determine which gene duplicates, rather than sites, are under selection. This does not make assumptions as to what branches may be under selection, which can increase the incidence of false positives [87]. In all analyses, the codon sequences were used.
We performed two different tests to assess the evolutionary history of gene expression patterns within each of the four tissue types. First, we estimated the parameter λ, under which 0 represents a complete lack of phylogenetic signal and 1 indicates a strong phylogenetic signal (i.e., similarity due to shared ancestry). Second, we tested whether the evolution of gene expression levels has increased, decreased, or been constant over time by estimating the parameter δ; δ < 1 indicates that the evolution of differences in gene expression patterns occurred early in gene divergence and δ > 1 signifies that evolutionary differences occurred relatively recently. To assess the statistical fit of λ and δ models, we performed a likelihood ratio test (LRT) with respect to the results under a Brownian motion model of character evolution. All analyses were conducted using the GEIGER package [89] within R [77].
Competing interests
The authors declare that there are no competing financial interests.
Authors’ contributions
EP devised experimental setup, performed all work for expression evaluation and promoter analyses and wrote the manuscript. EP and JP conducted analyses. GC offered directional support. JP and GC participated in manuscript editing. All authors have approved the manuscript.
Supplementary Material
Known cis-regulatory elements that correspond to matching predicted motifs. Includes cis-regulatory element (CRE) name, sequence, CRE database and additional functional information.
Phylogenetic tree identifiers. Identifiers from Figure 7 corresponding to the Phytozome locus or NCBI sequence identifier.
Phylogenetic analyses of NP-like proteins in the plant kingdom with bacterial outgroup. Phylogenetic relationships were constructed using Bayesian and maximum-likelihood methods. Numbers at branches indicate posterior probabilities and bootstrap percentages based on 1000 replicates, respectively. Numbers in parentheses correspond to Phytozome or NCBI sequence identifiers, which can be found in the Additional file 7: Table S4. The five predominant taxonomic families are indicated by the highlighted colors.
QPCR reference gene information. Includes qPCR reference gene symbol, gene name, Phytozome locus name, primer sequences, size of amplification product, annealing temperature, PCR efficiency and the tissue type that the reference gene was used for normalization calculations.
Un-scaled relative expression of NP-like gene family in Populus.
Promoter motifs predicted by MEME and MotifClick. Eight bp promoter motifs predicted by MEME (left) and MotifClick (right).
Output from PlantPAN. Includes the cis-regulatory element sequence, NP-like gene following the promoter region, regulatory element name, location within the promoter region, database element was retrieved from and functional category.
Alignment of motifs predicted by MEME for bacterial and plant nucleoside phosphorylase (NP) proteins. Bacterial amino acid sequences were retrieved from the Conserved Domain Database (CDD, http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) from the NP family (COG0775) and plant amino sequences were retrieved from Phytozome (http://www.phytozome.net).
Contributor Information
Emily A Pettengill, Email: emilyannpettengill@gmail.com.
James B Pettengill, Email: fixtgear@gmail.com.
Gary D Coleman, Email: gcoleman@umd.edu.
Acknowledgements
We are grateful to Cécile Parmentier-Line (University of Maryland, College Park MD) for assisting with RNA extractions and assessing the RNA quality and quantity. Funding for this project comes from the NSF grant IOS 0922650 (GC).
References
- Lewkowicz E, Iribarren A. Nucleoside phosphorylases. Curr Organic Chem. 2006;10:1197–1215. doi: 10.2174/138527206777697995. [DOI] [Google Scholar]
- Zrenner R, Riegler H, Marquard CR, Lange PR, Geserick C, Bartosz CE, Chen CT, Slocum RD. A functional analysis of the pyrimidine catabolic pathway in Arabidopsis. New Phytologist. 2009;183:117–132. doi: 10.1111/j.1469-8137.2009.02843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugmire MJ, Ealick SE. Structural analyses reveal two distinct families of nucleoside phosphorylases. Biochem J. 2002;361:1–25. doi: 10.1042/0264-6021:3610001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravandi F, Gandhi V. Novel purine nucleoside analogues for T-cell-lineage acute lymphoblastic leukaemia and lymphoma. Expert Opin Investig Drugs. 2006;15:1601–1613. doi: 10.1517/13543784.15.12.1601. [DOI] [PubMed] [Google Scholar]
- Batista EL, Deves C, Ayub L, da Silva RG, Filho LCC, Basso LA, Santos DS. Purine nucleoside phosphorylase activity and expression are upregulated in sites affected by periodontal disease. J Periodont Res. 2010;45:664–671. doi: 10.1111/j.1600-0765.2010.01282.x. [DOI] [PubMed] [Google Scholar]
- Kojima S, Chiyomaru T, Kawakami K, Yoshino H, Enokida H, Nohata N, Fuse M, Ichikawa T, Naya Y, Nakagawa M, Seki N. Tumour suppressors miR-1 and miR-133a target the oncogenic function of purine nucleoside phosphorylase (PNP) in prostate cancer. Br J Cancer. 2011;106:405–413. doi: 10.1038/bjc.2011.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PMC, Atkins CA. Purine biosynthesis. Big in cell division, even bigger in nitrogen assimilation. Plant Physiol. 2002;128:793–802. doi: 10.1104/pp.010912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner AK, Witte C-P. The biochemistry of nitrogen mobilization: purine ring catabolism. Trends Plant Sci. 2011;16:381–387. doi: 10.1016/j.tplants.2011.03.012. [DOI] [PubMed] [Google Scholar]
- Guranowski A. Purine catabolism in plants : purification and some properties of inosine nucleosidase from yellow lupin (lupinus luteus L.) seeds. Plant Physiol. 1982;70:344–349. doi: 10.1104/pp.70.2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katahira R, Ashihara H. Profiles of purine biosynthesis, salvage and degradation in disks of potato (Solanum tuberosum L.) tubers. Planta. 2006;225:115–126. doi: 10.1007/s00425-006-0334-9. [DOI] [PubMed] [Google Scholar]
- Jung B, Hoffmann C, Möhlmann T. Arabidopsis nucleoside hydrolases involved in intracellular and extracellular degradation of purines. Plant J. 2011;65:703–711. doi: 10.1111/j.1365-313X.2010.04455.x. [DOI] [PubMed] [Google Scholar]
- Hesberg C, Hänsch R, Mendel RR, Bittner F. Tandem orientation of duplicated xanthine dehydrogenase genes from Arabidopsis thaliana: differential gene expression and enzyme activities. J Biol Chem. 2004;279:13547–13554. doi: 10.1074/jbc.M312929200. [DOI] [PubMed] [Google Scholar]
- Katahira R, Ashihara H. Role of adenosine salvage in wound-induced adenylate biosynthesis in potato tuber slices. Plant Physiol et Biochemi. 2006;44:551–555. doi: 10.1016/j.plaphy.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Brychkova G, Alikulov Z, Fluhr R, Sagi M. A critical role for ureides in dark and senescence-induced purine remobilization is unmasked in the Atxdh1 Arabidopsis mutant. Plant J. 2008;54:496–509. doi: 10.1111/j.1365-313X.2008.03440.x. [DOI] [PubMed] [Google Scholar]
- Cooke JEK, Weih M. Nitrogen storage and seasonal nitrogen cycling in Populus: bridging molecular physiology and ecophysiology. New Phytologist. 2005;167:19–30. doi: 10.1111/j.1469-8137.2005.01451.x. [DOI] [PubMed] [Google Scholar]
- Wetzel S, Demmers C, Greenwood JS. Seasonally fluctuating bark proteins Are a potential form of nitrogen storage in 3 temperate hardwoods. Planta. 1989;178:275–281. doi: 10.1007/BF00391854. [DOI] [PubMed] [Google Scholar]
- Langheinrich U, Tischner R. Vegetative storage proteins in poplar : induction and characterization of a 32- and a 36-kilodalton polypeptide. Plant Physiol. 1991;97:1017–1025. doi: 10.1104/pp.97.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter JJ, Vancleve B. Biochemical, immunochemical, and ultrastructural studies of protein storage in poplar (Populus X Canadensis Robusta) wood. Planta. 1991;183:92–100. doi: 10.1007/BF00197572. [DOI] [PubMed] [Google Scholar]
- Coleman GD, Chen TH, Ernst SG, Fuchigami L. Photoperiod control of poplar bark storage protein accumulation. Plant Physiol. 1991;96:686–692. doi: 10.1104/pp.96.3.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Coleman GD. Phytochrome-mediated photoperiod perception, shoot growth, glutamine, calcium, and protein phosphorylation influence the activity of the poplar bark storage protein gene promoter (bspA) Plant Physiol. 2001;126:342–351. doi: 10.1104/pp.126.1.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman GD, Banados MP, Chen T. Poplar bark storage protein and a related wound-induced gene are differentially induced by nitrogen. Plant Physiol. 1994;106:211–215. doi: 10.1104/pp.106.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence SD, Cooke JEK, Greenwood JS, Korhnak TE, Davis JM. Vegetative storage protein expression during terminal bud formation in poplar. Can J For Res. 2001;31:1098–1103. doi: 10.1139/x01-028. [DOI] [Google Scholar]
- Plomion C, Lalanne C, Claverol S, Meddour H, Kohler A, Bogeat-Triboulot M-B, Barre A, Le Provost G, Dumazet H, Jacob D, Bastien C, Dreyer E, de Daruvar A, Guehl J-M, Schmitter J-M, Martin F, Bonneu M. Mapping the proteome of poplar and application to the discovery of drought-stress responsive proteins. Proteomics. 2006;6:6509–6527. doi: 10.1002/pmic.200600362. [DOI] [PubMed] [Google Scholar]
- Millard P, Grelet GA. Nitrogen storage and remobilization by trees: ecophysiological relevance in a changing world. Tree Physiol. 2010;30:1083–1095. doi: 10.1093/treephys/tpq042. [DOI] [PubMed] [Google Scholar]
- Rennenberg H, Schmidt S. Perennial lifestyle–an adaptation to nutrient limitation? Tree Physiol. 2010;30:1047–1049. doi: 10.1093/treephys/tpq076. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Sémon M, Wolfe KH. Consequences of genome duplication. Curr Opin Genet Dev. 2007;17:505–512. doi: 10.1016/j.gde.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Lynch M. The evolution of genetic networks by non-adaptive processes. Nat Rev Genet. 2007;8:803–813. doi: 10.1038/nrg2192. [DOI] [PubMed] [Google Scholar]
- Chapman BA, Bowers JE, Feltus FA, Paterson AH. Buffering of crucial functions by paleologous duplicated genes may contribute cyclicality to angiosperm genome duplication. Proc Natl Acad Sci USA. 2006;103:2730–2735. doi: 10.1073/pnas.0507782103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino RP, Innan H. Selection for more of the same product as a force to enhance concerted evolution of duplicated genes. Trends Genet. 2006;22:642–644. doi: 10.1016/j.tig.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Pettersson ME, Sun S, Andersson DI, Berg OG. Evolution of new gene functions: simulation and analysis of the amplification model. Genetica. 2008;135:309–324. doi: 10.1007/s10709-008-9289-z. [DOI] [PubMed] [Google Scholar]
- Ohno S. Evolution by Gene and Genome Duplication. New York: Springer; 1970. [Google Scholar]
- Teshima KM, Innan H. Neofunctionalization of duplicated genes under the pressure of gene conversion. Genetics. 2008;178:1385–1398. doi: 10.1534/genetics.107.082933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Force A. The probability of duplicate gene preservation by subfunctionalization. Genetics. 2000;154:459–473. doi: 10.1093/genetics/154.1.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innan H, Kondrashov F. The evolution of gene duplications: classifying and distinguishing between models. Nat Rev Genet. 2010;11:97–108. doi: 10.1038/nrg2689. [DOI] [PubMed] [Google Scholar]
- Veitia RA. Gene dosage balance: deletions, duplications and dominance. Trends Genet. 2005;21:33–35. doi: 10.1016/j.tig.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Marais Des DL, Rausher MD. Escape from adaptive conflict after duplication in an anthocyanin pathway gene. Nature. 2008;454:762–765. doi: 10.1038/nature07092. [DOI] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA. The gene balance hypothesis: from classical genetics to modern genomics. The Plant Cell Online. 2007;19:395–402. doi: 10.1105/tpc.106.049338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edger PP, Pires JC. Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 2009;17:699–717. doi: 10.1007/s10577-009-9055-9. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Zhang S, Li S, Niu M, Pham PT, Su Z. MotifClick: prediction of cis-regulatory binding sites via merging cliques. BMC Bioinformatics. 2011;12:238. doi: 10.1186/1471-2105-12-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W-C, Lee T-Y, Huang H-D, Huang H-Y, Pan R-L. PlantPAN: Plant promoter analysis navigator, for identifying combinatorial cis-regulatory elements with distance constraint in plant gene groups. BMC Genomics. 2008;9:561. doi: 10.1186/1471-2164-9-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Moore MJ, Soltis PS, Bell CD, Brockington SF, Alexandre R, Davis CC, Latvis M, Manchester SR, Soltis DE. Rosid radiation and the rapid rise of angiosperm-dominated forests. Proc Nat Acad Sci. 2009;106:3853–3858. doi: 10.1073/pnas.0813376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EK, Cibrian-Jaramillo A, Kolokotronis S-O, Katari MS, Stamatakis A, Ott M, Chiu JC, Little DP, Stevenson DW, McCombie WR, Martienssen RA, Coruzzi G, DeSalle R. A functional phylogenomic view of the seed plants. PLoS Genet. 2011;7:e1002411. doi: 10.1371/journal.pgen.1002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen GL, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q. et al. The genome of black cottonwood, populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- Sterck L, Rombauts S, Jansson S, Sterky F, Rouzé P, Van de Peer Y. EST data suggest that poplar is an ancient polyploid. New Phytologist. 2005;167:165–170. doi: 10.1111/j.1469-8137.2005.01378.x. [DOI] [PubMed] [Google Scholar]
- Berlin S, Lagercrantz U, Arnold von S, Öst T, Rönnberg-Wästljung A. High-density linkage mapping and evolution of paralogs and orthologs in Salix and Populus. BMC Genomics. 2010;11:129. doi: 10.1186/1471-2164-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers-Melnick E, Mane SP, Dharmawardhana P, Slavov GT, Crasta OR, Strauss SH, Brunner AM, DiFazio SP. Contrasting patterns of evolution following whole genome versus tandem duplication events in Populus. Genome Res. 2012;22:95–105. doi: 10.1101/gr.125146.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehti-Shiu MD, Zou C, Hanada K, Shiu SH. Evolutionary history and stress regulation of plant receptor-like kinase/pelle genes. Plant Physiol. 2009;150:12–26. doi: 10.1104/pp.108.134353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M. Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res. 2006;16:805–814. doi: 10.1101/gr.3681406. [DOI] [PubMed] [Google Scholar]
- Zambelli F, Pesole G, Pavesi G. Motif discovery and transcription factor binding sites before and after the next-generation sequencing era. Briefings Bioinformatics. 2012;14:225–237. doi: 10.1093/bib/bbs016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kim YJ, Muller R, Yumul RE, Liu C, Pan Y, Cao X, Goodrich J, Chen X. AGAMOUS terminates floral stem cell maintenance in Arabidopsis by directly repressing WUSCHEL through recruitment of polycomb group proteins. The Plant Cell Online. 2011;23:3654–3670. doi: 10.1105/tpc.111.091538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Tudor M, Weiss CA, Hu Y, Ma H. The Arabidopsis MADS-box gene AGL3 is widely expressed and encodes a sequence-specific DNA-binding protein. Plant Mol Biol. 1995;28:549–567. doi: 10.1007/BF00020401. [DOI] [PubMed] [Google Scholar]
- McConnell JR, Emery J, Eshed Y, Bao N, Bowman J, Barton MK. Role of PHABULOSA and PHAVOLUTA in determining radial patterning in shoots. Nature. 2001;411:709–713. doi: 10.1038/35079635. [DOI] [PubMed] [Google Scholar]
- Zhang N, Qiao Z, Liang Z, Mei B, Xu Z, Song R. Zea Mays taxilin protein negatively regulates opaque-2 transcriptional activity by causing a change in its Sub-cellular distribution. PLoS ONE. 2012;7:e43822. doi: 10.1371/journal.pone.0043822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo HR, Kim JH, Kim J, Kim J, Lee U, Song IJ, Kim JH, Lee HY, Nam HG, Lim PO. The RAV1 transcription factor positively regulates leaf senescence in Arabidopsis. J Exp Bot. 2010;61:3947–3957. doi: 10.1093/jxb/erq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero-Paulet L, Fares MA. Evolutionary dynamics and functional specialization of plant paralogs formed by whole and small-scale genome duplications. Mol Biol Evol. 2012;29:3541–3551. doi: 10.1093/molbev/mss162. [DOI] [PubMed] [Google Scholar]
- Paterson AH, Bowers JE, Chapman BA. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc Natl Acad Sci USA. 2004;101:9903–9908. doi: 10.1073/pnas.0307901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Leebens-Mack J, Ayyampalayam S, Bowers JE, McKain MR, McNeal J, Rolf M, Ruzicka DR, Wafula E, Wickett NJ, Wu X, Zhang Y, Wang J, Zhang Y, Carpenter EJ, Deyholos MK, Kutchan TM, Chanderbali AS, Soltis PS, Stevenson DW, McCombie R, Pires JC, Wong GK-S, Soltis DE, de Pamphilis CW. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 2012;13:R3. doi: 10.1186/gb-2012-13-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler A, Rinaldi C, Duplessis S, Baucher M, Geelen D, Duchaussoy F, Meyers BC, Boerjan W, Martin F. Genome-wide identification of NBS resistance genes in Populus trichocarpa. Plant Mol Biol. 2008;66:619–636. doi: 10.1007/s11103-008-9293-9. [DOI] [PubMed] [Google Scholar]
- DeLuna A, Vetsigian K, Shoresh N, Hegreness M, Colón-González M, Chao S, Kishony R. Exposing the fitness contribution of duplicated genes. Nat Genet. 2008;40:676–681. doi: 10.1038/ng.123. [DOI] [PubMed] [Google Scholar]
- Qian W, Liao B-Y, Chang AY-F, Zhang J. Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genetics. 2010;26:425–430. doi: 10.1016/j.tig.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad A, Teufel AI, Grahnen JA, Liberles DA. Toward a general model for the evolutionary dynamics of gene duplicates. Genome Biol Evol. 2011;3:1197–1209. doi: 10.1093/gbe/evr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proulx SR. The rate of multi-step evolution in Moran and Wright-Fisher populations. Theor Popul Biol. 2011;80:197–207. doi: 10.1016/j.tpb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Proulx SR. Multiple routes to subfunctionalization and gene duplicate specialization. Genetics. 2012;190:737–751. doi: 10.1534/genetics.111.135590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Zhang J. Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics. 2005;169:1157–1164. doi: 10.1534/genetics.104.037051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa A, Sakamoto S, Takahashi M, Morikawa H, Sakamoto A. The RNAi-mediated silencing of xanthine dehydrogenase impairs growth and fertility and accelerates leaf senescence in transgenic Arabidopsis plants. Plant Cell Physiol. 2007;48:1484–1495. doi: 10.1093/pcp/pcm119. [DOI] [PubMed] [Google Scholar]
- Schuch R, Garibian A, Saxild HH, Piggot PJ, Nygaard P. Nucleosides as a carbon source in Bacillus subtilis: characterization of the drm-pupG operon. Microbiology (Reading, Engl) 1999;145:2957–2966. doi: 10.1099/00221287-145-10-2957. [DOI] [PubMed] [Google Scholar]
- Tozzi MG, Camici M, Mascia L, Sgarrella F, Ipata PL. Pentose phosphates in nucleoside interconversion and catabolism. FEBS J. 2006;273:1089–1101. doi: 10.1111/j.1742-4658.2006.05155.x. [DOI] [PubMed] [Google Scholar]
- Konrad A, Piškur J, Liberles DA. The evolution of catalytic residues and enzyme mechanism within the bacterial nucleoside phosphorylase superfamily 1. Gene. 2012;510:154–161. doi: 10.1016/j.gene.2012.08.046. [DOI] [PubMed] [Google Scholar]
- Wisniewski M, Bassett C, Arora R. Distribution and partial characterization of seasonally expressed proteins in different aged shoots and roots of “Loring” peach (Prunus persica) Tree Physiol. 2004;24:339–345. doi: 10.1093/treephys/24.3.339. [DOI] [PubMed] [Google Scholar]
- Binnie SC, Grossnickle SC, Roberts DR. Fall acclimation patterns of interior spruce seedlings and their relationship to changes in vegetative storage proteins. Tree Physiol. 1994;14:1107–1120. doi: 10.1093/treephys/14.10.1107. [DOI] [PubMed] [Google Scholar]
- Xi Z, Ruhfel BR, Schaefer H. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. In Proc Natl Acad Sci USA. 2012;109:17519–17524. doi: 10.1073/pnas.1205818109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Hobson N, Galindo L, Zhu S, Shi D, McDill J, Yang L, Hawkins S, Neutelings G, Datla R, Lambert G, Galbraith DW, Grassa CJ, Geraldes A, Cronk QC, Cullis C, Dash PK, Kumar PA, Cloutier S, Sharpe A, Wong GK-S, Wang J, Deyholos MK. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012;72:461–473. doi: 10.1111/j.1365-313X.2012.05093.x. [DOI] [PubMed] [Google Scholar]
- Thioulouse J, Chessel D, Dolédec S, Olivier JM. ADE-4: a multivariate analysis and graphical display software. Stat Comput. 1997;7:75–83. doi: 10.1023/A:1018513530268. [DOI] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation; 2008. [Google Scholar]
- Peres-Neto PR, Jackson DA, Somers KM. Giving meaningful interpretation to ordination axes: assessing loading significance in principal component analysis. Ecology. 2003;84(9):2347–2363. doi: 10.1890/00-0634. [DOI] [Google Scholar]
- Marchler-Bauer A, Anderson JB, DeWeese-Scott C, Fedorova ND, Geer LY, He S, Hurwitz DI, Jackson JD, Jacobs AR, Lanczycki CJ, Liebert CA, Liu C, Madej T, Marchler GH, Mazumder R, Nikolskaya AN, Panchenko AR, Rao BS, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Vasudevan S, Wang Y, Yamashita RA, Yin JJ, Bryant SH. CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res. 2003;31:383–387. doi: 10.1093/nar/gkg087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Saunders NFW, Kobe B, Bailey TL. Discovering sequence motifs with arbitrary insertions and deletions. PLoS Comput Biol. 2008;4:e1000071. doi: 10.1371/journal.pcbi.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Zwickl D. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence data sets under the maximum likelihood criterion. PhD thesis. : The University of Texas at Austin; 2006. [Google Scholar]
- Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- Bazinet AL, Cummings MP. In: Distributed & Grid Computing–Science Made Transparent for Everyone Principles, Applications and Supporting Communities. Weber MHW, editor. Marburg: Tectum Publishing House; 2008. The Lattice Project: a Grid research and production environment combining multiple Grid computing models; pp. 2–13. [Google Scholar]
- Sanderson MJ, Donoghue MJ, Piel WH, Eriksson T. TreeBASE: a prototype database of phylogenetic analyses and an interactive tool for browsing the phylogeny of life. Am J Botany. 1994;81:183. [Google Scholar]
- Kosakovsky Pond SL, Murrell B, Fourment M, Frost SDW, Delport W, Scheffler K. A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol. 2011;28:3033–3043. doi: 10.1093/molbev/msr125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Harmon LJ, Weir JT, Brock CD, Glor RE, Challenger W. GEIGER: investigating evolutionary radiations. Bioinformatics. 2007;24:129–131. doi: 10.1093/bioinformatics/btm538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Known cis-regulatory elements that correspond to matching predicted motifs. Includes cis-regulatory element (CRE) name, sequence, CRE database and additional functional information.
Phylogenetic tree identifiers. Identifiers from Figure 7 corresponding to the Phytozome locus or NCBI sequence identifier.
Phylogenetic analyses of NP-like proteins in the plant kingdom with bacterial outgroup. Phylogenetic relationships were constructed using Bayesian and maximum-likelihood methods. Numbers at branches indicate posterior probabilities and bootstrap percentages based on 1000 replicates, respectively. Numbers in parentheses correspond to Phytozome or NCBI sequence identifiers, which can be found in the Additional file 7: Table S4. The five predominant taxonomic families are indicated by the highlighted colors.
QPCR reference gene information. Includes qPCR reference gene symbol, gene name, Phytozome locus name, primer sequences, size of amplification product, annealing temperature, PCR efficiency and the tissue type that the reference gene was used for normalization calculations.
Un-scaled relative expression of NP-like gene family in Populus.
Promoter motifs predicted by MEME and MotifClick. Eight bp promoter motifs predicted by MEME (left) and MotifClick (right).
Output from PlantPAN. Includes the cis-regulatory element sequence, NP-like gene following the promoter region, regulatory element name, location within the promoter region, database element was retrieved from and functional category.
Alignment of motifs predicted by MEME for bacterial and plant nucleoside phosphorylase (NP) proteins. Bacterial amino acid sequences were retrieved from the Conserved Domain Database (CDD, http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) from the NP family (COG0775) and plant amino sequences were retrieved from Phytozome (http://www.phytozome.net).