

Abstract

Metalation-electrophilic fluorination of TMS- and TIPS-protected 1,3-benzothiazol-2-yl (BT) propargyl sulfones gave corresponding BT fluoropropargyl sulfones, Julia-Kocienski reagents for the synthesis of fluoro enynes. Both reagents reacted with aldehydes under mild DBU, or LHMDS-mediated conditions, giving high yields of conjugated fluoro enynes with E- stereoselectivity. In comparison to DBU-mediated reactions, stereoselectivity was higher in low-temperature LHMDS-mediated reactions. Two ketones were shown to react as well, using LHMDS as base. In situ removal of the TMS group gave terminal conjugated 2-fluoro 1,3-enynes. Synthetic utility of the fluoro enynes was demonstrated by conversion to internal alkynes and to stereoisomeric fluoro dienes via Sonogashira and Heck couplings.
INTRODUCTION
Conjugated enynes are valuable synthetic intermediates,1 where chemistry leading to double and triple bond functionalization can be further exploited. Also, the 1,3-enyne structural motif is in a broad range of compounds, from natural products to pharmaceuticals. Some examples are shown in Figure 1.
FIGURE 1.

Examples of natural products and a pharmaceutical with an enyne motif.
Dihydromatricaria acid is secreted by soldier beetles (Cantharidae) for defense,2 (Z)-13-hexadecen-11-ynyl acetate is a sex pheromone of the processionary moth Thaumetopoea pityocampa,3 (+)-brasilenyne and (+)-cis-dihydrorhodophytin are antifeedants from a sea hare,4 (2Z,8S,9Z)-heptadeca-2,9-dien-4,6-diyn-1,8-diol was isolated from Bupleurum salicifolium and demonstrates substantial antibiotic activity,5 whereas Terbinafine (Lamisil) is a synthetic antifungal agent.6
Several methods have been published for the synthesis of conjugated enynes, and in recent years, these have been focused to a large extent on metal-catalyzed approaches.1c,7 Synthesis of fluorinated 1,3-enynes, on the other hand, has been much less explored, despite the widespread interest and importance of the fluoroorganics.8 The few modular approaches reported involve metal-catalyzed synthesis of 1-fluoro,9a–c 2-fluoro,9d–h di9a,9i–k and trifluoro9a,9j 1,3-enynes, as well as the Horner-Wadsworth-Emmons approach to 2-fluoro-1,3-enynes (Figure 2).10 Among natural products, synthesis of a (E)-13-fluoro-13-hexadecen-11-ynyl acetate, a fluorinated analog of a sex pheromone, has been reported.11 Here, the fluoro enyne analog was prepared from the corresponding enyne by dehydration of a fluorohydrin intermediate.11a,b Change in physical, chemical, and biological properties upon introduction of a fluorine atom is well recognized.12 For example, in the case of the pheromone cited earlier, introduction of fluorine induced antipheromone activity.11c
FIGURE 2.

Modular approaches to fluoro enynes.
The versatility of Julia-Kocienski13–16 heteroaryl sulfones for modular assembly of fluoroorganics has been demonstrated by us17 and others.18 Recently, we have developed a bifunctionalizable fluorinated building block, 1,3-benzothiazol-2-yl fluoropropargyl sulfone, and demonstrated its application in the synthesis of fluorovinyl triazoles.17g To the best of our knowledge, the use of Julia-Kocienski methodology for the synthesis of conjugated fluoro enynes has remained unknown. Herein, we explore the utility of this reagent for modular synthesis of conjugated fluoro enynes, and further conversion of fluoro enynes to stereoisomeric fluoro dienes.
RESULTS AND DISCUSSION
Synthesis of 1,3-benzothiazol-2-yl (BT) fluoropropargyl sulfone17g commenced from TMS-protected propargyl bromide and the sodium salt of BT-thiol, to give sulfide 1 (Scheme 1). Oxidation of 1 by m-CPBA to propargyl BT-sulfone 2, followed by metalation-fluorination under heterogeneous conditions gave fluoropropargyl BT-sulfone 3.17g Since our prior communication, we noticed that yields of 3 could vary from batch to batch. In the currently reported improved procedure, to obtain consistent yields of 3 we have found that it is critical to immediately remove any residual fluoride salts from the crude reaction mixture. Therefore, immediately following workup, the reaction mixture was suspended in toluene and filtered through a short silica gel plug, prior to chromatographic purification.
SCHEME 1.

Synthesis of TMS-protected 1,3-benzothiazol-2-yl fluoropropargyl sulfone 3
A sample of fluoropropargyl sulfone 3 was crystallized from hexanes and analyzed by X-ray diffraction (Figure 3). A slight bend of the triple bond from the linearity was observed. The Si-C1-C2 and C1-C2-C-3 angles are 177.0° and 176.7°, respectively.
FIGURE 3.

Crystal structure of fluoropropargyl sulfone 3 (C, black; H, grey; F, green; N, blue; O, red; S, orange; Si, yellow).
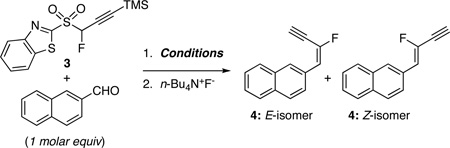
Conditions for condensation reactions with the TMS-protected 1,3-benzothiazol-2-yl fluoropropargyl sulfone 3 were screened with 2-naphthaldehyde as substrate. In KHMDS, LHMDS, and Cs2CO3-mediated reactions, the base was added to 1 molar equiv of 2-naphthaldehyde, 2.0–2.5 molar equiv of BT-sulfone 3, and additive, when used (Barbier conditions). In DBU-mediated reactions, 2.5 molar equiv of sulfone 3 was added to solution of aldehyde and base. Since partial loss of the TMS group from the fluoro enyne product was observed in some of the olefinations, in all cases TBAF was added to the reaction mixture when complete consumption of 2-naphthaldehyde had occurred. This led to the isolation of the deprotected fluoro enyne 4 in a two-step, one-pot operation. The results from these experiments are displayed in Table 1.
TABLE 1.
Screening of olefination conditions
 | |||||
|---|---|---|---|---|---|
| entry | basea | solvent | Additive | T (°C); time | (%) E/Z;b yield (%)c |
| 1d | KHMDS | THF | -- | −55; 10 min | 82/18; 86 |
| 2d | LHMDS | THF | -- | −78; 10 min | 88/12; 97 |
| 3d | LHMDS | THF | MgBr2Et2Oe | rt; 4 h | 89/11; 78 |
| 4d | LHMDS | DMF | DMPUf | −78; 10 min | 63/37; 74 |
| 5d | LHMDS | DMPU | -- | 0; 10 min | 58/42; NAg |
| 6h | DBU | CH2Cl2 | -- | rt; 10 min | 69/31; 91 |
| 7h | DBU | CH2Cl2 | -- | 0; 10 min | 70/30; 92 |
| 8h | DBU | CH2Cl2 | -- | −55; 10 min | 74/26; 95 |
| 9h | DBU | CH2Cl2 | -- | −78; 10 min | 74/26; NAg |
| 10h | DBU | CH2Cl2 | MgBr2Et2Oe | rt; 30 h | 60/40; Inci |
| 11h | DBU | THF | MgBr2Et2Oe | rt; 30 h | 90/10; Inci |
| 12h | Cs2CO3 | CH2Cl2 | -- | rt; 6 h | 74/26; NAg |
KHMDS and LHMDS: 2.4 molar equiv. DBU: 4.0 molar equiv. Cs2CO3: 3.0 molar equiv.
E/Z ratio of diastereomers in the crude reaction mixture was determined by 19F NMR, prior to isolation.
Yields are of isolated and purified products.
Sulfone 3, 2.0 molar equiv.
3.0 molar equiv.
Cosolvent, ratio of DMF/DMPU 1:1 (v/v).
Products were not isolated.
Sulfone 3, 2.5 molar equiv.
Reaction was incomplete after 30 h.
Synthesis of TMS-protected enynes, via Julia-Kocienski olefination, using TMS-protected propargyl BT-sulfone 2 has been reported.19 Yields were typically moderate, but high Z-selectivity was obtained in KHMDS-mediated reactions at −55 °C.19 In the present case, KHMDS-mediated reaction of 3 with 2-naphthaldehyde at −55 °C gave product 4 in 82/18 E/Z ratio and high 86% yield (entry 1). The use of LHMDS at −78 °C resulted in an increased E/Z ratio (88/12) and 97% yield (entry 2). Similarly, a high E/Z ratio was obtained when the reaction was performed at rt and in the presence of MgBr2·Et2O, but the yield was significantly lower (entry 3). The use of polar and/or complexing solvents substantially decreased both the selectivity and the yield (entries 4, 5). We were also curious to assess whether reactions would proceed with a weaker base. The use of DBU at room temperature resulted in a high yield of 4, but with lowered selectivity (E/Z 69/31, entry 6). Lowering the temperature to −55 °C marginally increased the E-selectivity and yield (entry 8), whereas further lowering of temperature to −78 °C did not improve the selectivity (entry 9). The effect of MgBr2·Et2O on stereoselectivity of DBU-mediated olefinations depended on the solvent used. In THF, a high 90/10 E/Z ratio was observed (entry 11), whereas a substantially lower 60/40 ratio was obtained in CH2Cl2 (entry 10). However, reactions under these conditions were very slow and were incomplete even after 30 h. Condensation could also be conducted with Cs2CO3 as base, but the reaction was much slower than with DBU.
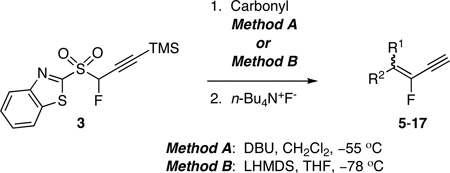
The scope of the condensation reactions of fluoropropargyl BT-sulfone 3 with carbonyl compounds was next tested with eleven aldehydes and two ketones under mild DBU-mediated conditions (Method A), and/or under LHMDS-mediated conditions (Method B), Table 2.
TABLE 2.
Condensation reactions of 3 with aldehydes and ketones
 | |||
|---|---|---|---|
| product: yield,a E/Zb | |||
| entry | carbonyl | method Ac | method Bd |
| 1 | 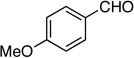 |
5: 92%, 81/19 | 5: 97%, 95/5 |
| 2 | 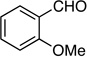 |
6: 92%, 78/22 | 6: 87%, 90/10 |
| 3 | 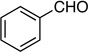 |
7: 90%, 76/24 | 7: 59%, 94/6 |
| 4 | 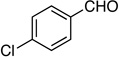 |
8: 81%, 70/30 | 8: 95%, 94/6 |
| 5 | 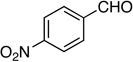 |
9: 59%, 51/49 | 9: 81%, 72/28 |
| 6 | 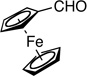 |
10: 95%, 83/17 | 10: 98%, 90/10 |
| 7 | 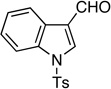 |
11: 95%, 75/25 | 11: 92%, 91/9 |
| 8 | 12: 78%, 75/25 | 12: 78%, 91/9 | |
| 9 | 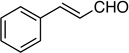 |
13: 95%, 72/28 | 13: 96%, 80/20 |
| 10 | 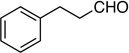 |
14: 54% (63%e), 80/20 |
14: 63%, 70/30 |
| 11 |
15: 79% (91%e), 85/15 |
-- | |
| 12 | 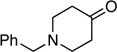 |
-- | 16: 77% |
| 13 |  |
-- | 17: 88%, E onlyf |
Yields of isolated and purified products.
E/Z ratio of diastereomers in the crude reaction mixture was determined by 19F NMR, prior to isolation. No change in ratio was observed after purification.
Method A: sulfone 3, 2.0–2.5 molar equiv; DBU, 4.0 molar equiv; TBAF, 0.20 molar equiv. For 11 and 13, 3.0 molar equiv of 3 was used. Sulfone was added to solution of aldehyde and base.
Method B for aldehydes: sulfone 3, 2.0 molar equiv; LHMDS, 2.4 molar equiv; TBAF, 2.0 molar equiv. Method B for ketones: sulfone 3, 3.0 molar equiv; LHMDS, 5.0 molar equiv; TBAF, 3.0 molar equiv. LHMDS was added to a solution of sulfone and aldehyde.
Yield was calculated using octafluoronaphthalene as an internal standard.
Determined by NOESY experiment on the diene obtained by Lindlar reduction of 17 (see Figure 5).
Yields of products obtained via two methods were generally comparable for all substrates, except for entries 3 and 5, where there is no obvious reason for the differences observed. Partial loss of the TMS group in the enyne products was observed in DBU-mediated reactions prior to addition of TBAF, but only marginal in the LHMDS-mediated ones. We did not observe any allene formation in the condensation reactions with TMS-protected 1,3-benzothiazol-2-yl fluoropropargyl sulfone 3. In our previous communication on the synthesis of fluorovinyl triazoles via a click-olefination sequence using TMS-protected 1,3-benzothiazol-2-yl fluoropropargyl sulfone, we also did not see any allene formation upon in situ TMS-deprotection, and click reaction.17g However, TMS-deprotected 1,3-benzothiazol-2-yl fluoropropargyl sulfone produced a mixture of alkyne and the corresponding rearranged allene upon chromatographic purification.17g A similar influence of a trialkylsilyl group on the reactivities of terminal alkynes has recently been reported.20a Whereas allene formation was not observed in Cu(I)-catalyzed cross-coupling of N-tosylhydrazones with terminal alkynes bearing trialkylsilyl groups,20a allene formation occurred with terminal alkynes.20b
Both methods gave alkenes with selectivity for a cis arrangement of the bulkier groups at the double bond (E-fluoro enyne). In DBU-mediated condensations with substituted benzaldehydes, this cis selectivity decreased with increased electron-withdrawing power of the substituents, ranging from E/Z 81/19 for methoxy substituent, to no selectivity in the case of nitro substituent (entries 1–5, Method A).
As proposed by Julia, anti and syn adducts, formed via addition of the sulfone carbanion to the aldehyde, undergo spirocyclization/elimination to olefins. Syn adduct undergoes faster Smiles rearrangement and gives cis olefin (Figure 4).13,15 In the present case, reversible addition of carbanion of sulfone 3 to aldehyde, with faster collapse of syn adduct, could explain the preference for cis selectivity.21 The decrease in cis selectivity, observed with increase in electron-withdrawing power of substituent in substituted benzaldehydes (Method A), is likely the result of nonstereoselective formation of anti/syn adducts, combined with a less favored/slower retroaddition of initial adducts of less stable electron-deficient aromatic aldehydes.22 Consistent with this, employing conditions that favor syn addition of the metalated carbanion of 3 to the carbonyl, via a chelated transition state (Method B), led to higher cis selectivity than when Method A was used, ranging from excellent to moderate.
FIGURE 4.

Mechanistic basis for the observed selectivity.
Ketones also reacted under LHMDS-mediated conditions (entries 12, 13), and single isomer was formed in reaction with acetophenone (17, entry 13). For determination of stereochemistry in 17, the triple bond was subjected to partial hydrogenation with Lindlar catalyst (Figure 5). The NOESY data of the resulting diene showed a correlation between the ortho protons in the phenyl ring and the internal vinylic proton, supporting the assignment of E-configuration in 17.
FIGURE 5.

Determination of stereochemistry in 17.
We have shown in the past that α-fluoro-substituted Julia-Kocienski reagents are more reactive in olefination reactions compared to their protio analogs.16,17b,c To test the relative reactivities of the protio and fluoro analogs in the present case (2 and 3, respectively), a competitive experiment was performed under DBU-mediated conditions (Scheme 2). Reaction of 2.5 molar equiv of each 2 and 3 with p-methoxybenzaldehyde gave fluoro enyne (E/Z)-5 as the only product.
SCHEME 2.

Competitive reactivity of the fluoro and the protio Julia-Kocienski analogs.
To assess whether size of the silyl protecting group had any influence on stereoselectivity of the condensation reactions, triisopropylsilyl (TIPS)-protected fluoropropargyl BT-sulfone was synthesized (19, Scheme 3).
SCHEME 3.

Synthesis of TIPS-protected fluoropropargyl BT-sulfone 19.
Briefly, TIPS-protected propargyl bromide23 was reacted with the sodium salt of BT-thiol followed by oxidation to furnish TIPS-protected BT-sulfone 18 (30% yield over three steps, Scheme 3). Metalation-electrophilic fluorination of 18 under heterogeneous conditions afforded TIPS-protected fluoropropargyl BT-sulfone 19 in 69% yield. Sulfone 19 was more stable as compared to the TMS-derived reagent 3, and after workup the crude reaction mixture could be directly loaded to a silica gel column for purification. Higher stability of TIPS-protected sulfone 19 compared to 3 is also reflected in the lower molar excess of sulfone required for complete conversion of the aldehyde in the condensation reaction (1.3 versus 2.0–2.5 molar equiv). Unlike 3, reagent 19 did not decompose under reaction conditions.
Fluoropropargyl BT-sulfone 19 was reacted with 2-naphthaldehyde and 4-nitrobenzaldehyde using Method A and/or Method B (Scheme 4). Since no loss of the TIPS-protection was observed in the condensation reactions, products were isolated as the TIPS-protected enynes. In all cases tested, yields with the TIPS-protected reagent 19 and the TMS-protected 3 were comparable. Stereoselectivities were also comparable for the LHMDS-mediated reaction with 4-nitrobenzaldehyde and for the DBU-mediated reaction with 2-naphthaldehyde. However, in the DBU-mediated reaction with 4-nitrobenzaldehyde, 19 gave moderate E-selectivity, whereas no selectivity was observed with 3 (compare to Table 2, entry 5). It is plausible that upon addition of the carbanion from 19 to the aldehyde the bulk of triisopropylsilyl group decreases the rate of spirocyclization of the adducts as compared to spirocyclization of adducts resulting from 3. In the case of reversible addition, this would allow for some equilibration between syn/anti adducts of the less stable22 4-nitrobenzaldehyde. Since spirocyclization of the syn adduct occurs faster than that of anti, this would lead to higher cis selectivity.
SCHEME 4.

Condensation reactions of TIPS-protected fluoropropargyl BT-sulfone 19
The Horner-Wadsworth-Emmons analog, TIPS-protected (α-fluoropropargyl)phosphonate, has been studied in reactions with aldehydes and ketones. High yields of TIPS-protected fluoro enynes were obtained, with zero to moderate E-selectivities.10c On the other hand, reactions of carbonyl compounds and (α-fluoropropargyl)phosphonates, with phenyl- or alkyl-substituted alkynes, gave internal enynes in poor yields and typically with no selectivity.10a,b Since Sonogashira coupling offers a convenient route to internal alkynes, we wanted to explore conversions of the deprotected terminal fluoro enynes, obtained from the condensations, to internal fluoro enynes (Scheme 5). Fluoro enynes 4 and 16, when subjected to Sonogashira couplings with phenyl iodide, gave phenyl-derived fluoro enynes in good yields. Although some minor change in the E/Z ratio was observed in the reaction of E/Z-4, this may be due to the loss of the minor isomer in the workup/purification. No traces of products due to [4+2] homobenzannulation of enynes were observed in the Pd-catalyzed reactions, but a small amount (ca. 5%) of alkyne dimerization occurred.24
SCHEME 5.

Sonogashira coupling of terminal fluoro enynes
Utility of the terminal fluoro enynes for the synthesis of regiospecifically fluorinated dienes was demonstrated by further conversions of fluoro enynes 16 and 23 to stereoisomeric fluoro dienes (Scheme 6). Lindlar reduction of 16, followed by Heck reaction of the resulting terminal fluoro diene 24, furnished diene 25 with trans geometry at the newly formed double bond. Its geometric isomer 26 was synthesized via Lindlar reduction of fluoro enyne 23 (Scheme 6).
SCHEME 6.

Synthesis of isomeric dienes
CONCLUSIONS
Synthesis of conjugated 2-fluoro 1,3-enynes has been developed, using Julia-Kocienski reagents. The requisite reagents, TMS and TIPS-protected BT fluoropropargyl sulfones were synthesized from the corresponding sulfones via heterogeneous metalation-electrophilic fluorination using N-fluorobenzenesulfonimide. Both reagents reacted with aldehydes under mild, DBU-mediated conditions or with LHMDS. High yields of conjugated 2-fluoro 1,3-enynes were obtained in both cases, with moderate to excellent E-stereoselectivity. TMS-protected fluoro enynes were deprotected in situ due to lability of the TMS group. This was not the case with the TIPS-protected products, which were isolated with the silyl group present. Higher E-stereoselectivity was obtained when LHMDS was used as base. Ketones were shown to react as well.
To demonstrate the synthetic utility of terminal fluoro enynes, two were converted to internal fluoro enynes via Sonogashira reaction. Further, the use of fluoro enynes for the preparation of stereoisomeric fluoro dienes was shown. For this, 1-benzyl-4-(1-fluoroprop-2- ynylidene)piperidine was subjected to Sonogashira coupling and partial reduction of the alkyne to yield the (Z)-1-benzyl-4-(1-fluoro-3-phenylallylidene)piperidine. The E-isomer was synthesized by conversion of 1-benzyl-4-(1-fluoroprop-2-ynylidene)piperidine to diene via partial reduction of the triple bond followed by Heck reaction at the terminal alkene.
EXPERIMENTAL SECTION
General Experimental Considerations
THF was distilled over LiAlH4 and then over sodium, toluene was distilled over sodium, and CH2Cl2 was distilled over CaCl2. For reactions performed under a nitrogen atmosphere, glassware was dried with heat gun under vacuum. LDA (2.0 M solution in heptane/THF/EtPh) and LHMDS (1.0 M in THF) were obtained from commercial sources. All other reagents were obtained from commercial sources and used without further purification. N-fluorobenzenesulfonimide (NFSI) is commercially available. Thin layer chromatography was performed either on aluminum foil-backed silica gel plates (200 µm), or glass-backed silica gel plates (250 µm). Column chromatographic purifications were performed on 200–300 mesh silica gel. 1H NMR spectra were recorded at 500 MHz and were referenced to residual solvent. 13C NMR spectra were recorded at 125 MHz and are referenced to the carbon resonance of the deuterated solvent. 19F NMR spectra were recorded at 282 MHz with CFCl3 as internal standard. Proton assignments have been made only for the acetylenic and some of the vinylic protons in the E/Z mixtures of enynes and/or dienes. Because the compounds are closely related, assignments of these protons are supported by the data obtained from NOESY, HMQC, and HMBC of one representative enyne (compound E-9). On the basis of these 2D data, we have also made carbon assignments to E-9, but not to other analogs. Assignment of stereochemistry in 17 is based upon the 2D data of enyne 17 and its reduction product (E)-(3-fluoropenta-2,4-dien-2-yl)benzene. Chemical shifts (δ) are reported in parts per million and coupling constants (J) are in hertz (Hz). HRMS data were gathered using a TOF analyzer, the ionization modes are specified under each compound heading.
Improved Procedure for Synthesis of 2-[1-Fluoro-3-(trimethylsilyl)prop-2-ynylsulfonyl]benzo[d]thiazole (3).17g
A stirring solution of sulfone 217g (2.00 g, 6.46 mmol, 1 molar equiv) in dry toluene (45.0 mL) was cooled to −78 °C (dry ice/isoPrOH) under nitrogen. LDA (3.23 mL, 6.46 mmol, 1.0 molar equiv) was added and after 12 min, solid NFSI (2.24 g, 7.11 mmol, 1.1 molar equiv) was added. The reaction mixture was allowed to stir at −78 °C for 50 min, then warmed to room temperature and the stirring was continued for an additional 50 min. Saturated aq NH4Cl (20 mL) was added to the reaction mixture and the layers were separated. The aqueous layer was extracted with EtOAc (25–30 mL, 3 x) and the combined organic layer was washed with saturated aq NaHCO3, water, and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. The crude reaction mixture (brown oil) was suspended in toluene (7 mL) and passed through a short silica column (100–200 mesh, 6 cm × 3 cm) using toluene (100 mL) as eluent. The solvent was removed under reduced pressure and the crude product (colorless oil) was purified by column chromatography (19 cm × 2.8 cm, 10% EtOAc in hexanes) to yield 1.32 g (62%) of 317g as an off-white solid. Crystallization from hexanes gave white crystals, m.p. 87–89 °C.
Condensations of Sulfone 3 with Aldehydes, General Procedures
Method A
A solution of aldehyde (0.500 mmol, 1.0 molar equiv) and DBU (2.00 mmol, 4.0 molar equiv) in CH2Cl2 (6.8 mL per mmol of aldehyde) was cooled under nitrogen to −55 °C (dry ice/isoPrOH). A solution of fluoropropargyl sulfone 3 (1.00 mmol, 2.0 molar equiv) in CH2Cl2 (2.0 mL per mmol of sulfone) was added via a syringe and the reaction mixture turned dark. The reaction mixture was stirred for 5 min and checked by TLC (10% EtOAc in hexanes). If TLC showed unreacted aldehyde, fluoropropargyl sulfone 3 (0.25 or 0.50 mmol, 0.5 or 1.0 molar equiv) in CH2Cl2 (2.0 mL per mmol of sulfone) was added via a syringe and the reaction mixture was stirred for another 5 min. TBAF (1.0 M solution in THF, 0.100 mL, 0.20 molar equiv; for E/Z-5, 0.090 mL, 0.18 molar equiv was used) was added and the reaction mixture was concentrated under reduced pressure. The crude reaction mixture was analyzed by 19F NMR, and the combined E/Z product mixture was isolated by column chromatography (10% EtOAc in hexanes, unless stated otherwise).
Method B
A solution of aldehyde (0.500 mmol, 1.0 molar equiv) and sulfone 3 (1.00 mmol, 2.0 molar equiv) in dry THF (26.0 ]mL per mmol of aldehyde) was cooled under nitrogen to −78 °C (dry ice/isoPrOH). LHMDS (1.0 M solution in THF, 1.2 mL, 2.4 molar equiv) was added and upon addition, the reaction mixture turned intense orange. The reaction mixture was stirred at −78 °C for 5 min and checked by TLC (10% EtOAc in hexanes), which showed disappearance of the aldehyde. TBAF (1.0 M solution in THF, 1.00 mL, 2.0 molar equiv) was added and the reaction mixture was allowed to stir for another 5 min. Saturated aq NH4Cl was added and the mixture was poured into EtOAc. The organic layer was separated and the aqueous layer was extracted with EtOAc (3x). The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. The crude reaction mixture was analyzed by 19F NMR, and the combined E/Z product mixture was isolated by column chromatography (10% EtOAc in hexanes, unless stated otherwise).
Condensations of Sulfone 3 with Ketones: General Procedure
A stirred solution of ketone (0.300 mmol, 1.0 molar equiv) and sulfone 3 (294 mg, 0.900 mmol, 3.0 molar equiv) in dry THF (2.5 mL) was cooled under nitrogen to −78 °C (dry ice/isoPrOH). LHMDS (1.0 M solution in THF, 1.5 mL, 5.0 molar equiv) was added, the reaction mixture was stirred at −78 °C for 2 h and checked by TLC (20% EtOAc in hexanes for 16 and 10% EtOAc in hexanes for 17), which showed disappearance of the ketone. TBAF (1.0 M solution in THF, 0.900 mL, 3.0 molar equiv) was added, and the reaction mixture was allowed to stir for another 5 min. Saturated aq NH4Cl was added and the mixture was poured into EtOAc. The organic layer was separated and the aqueous layer was extracted with EtOAc (3x). The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The crude reaction mixture was analyzed by 19F NMR, and the product was isolated by column chromatography (10% EtOAc in hexanes).
Synthesis of (E/Z)-2-(2-Fluorobut-1-en-3-ynyl)naphthalene (E /Z -4)
Method B. 2-Naphthaldehyde: 78.0 mg (0.500 mmol); sulfone 3: 327 mg (1.00 mmol, 2.0 molar equiv); LHMDS: 1.2 mL (1.0 M, 1.2 mmol, 2.4 molar equiv); THF: 13.0 mL; TBAF: 1.00 mL (1.0 M, 1.0 mmol, 2.0 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of E/Z-4: 95.2 mg (97%), as a white solid, E/Z ratio: 88/12. Rf (10% EtOAc in hexanes) = 0.41. 1H NMR (500 MHz, CDCl3):δ 8.01 (s, 1H, Ar-H, E-isomer), 7.95 (s, 1H, Ar-H, Z-isomer), 7.85-7.80 (m, 3H, Ar-H, both E and Z-isomers and 1H, Ar-H, E-isomer), 7.66 (d, 1H, Ar-H, J = 8.5 Hz, Z-isomer), 7.49-7.48 (m, 2H, Ar-H, both E and Z-isomer), 6.84 (d, 1H, =CH, 3JFH = 17.4 Hz, E-isomer), 6.27 (d, 1H, =CH, 3JFH = 35.1 Hz, Z-isomer), 3.67 (d, 1H, ≡CH, 4JFH = 4.3 Hz, E-isomer), 3.34 (d, 1H, ≡CH, 4JFH = 3.7 Hz, Z-isomer). 19F NMR (282 MHz, CDCl3): δ −104.69 (d, 3JFH = 15.3 Hz, E-isomer), −107.17 (d, 3JFH = 36.6 Hz, Z-isomer). HRMS (APPI) calcd for C14H9F [M]+ 196.0683, found 196.0682.
Synthesis of 1-(2-Fluorobut-1-en-3-ynyl)-4-methoxybenzene (E /Z-5)
Method A. 4-Methoxybenzaldehyde: 68.0 mg (0.500 mmol); sulfone 3: 409 mg (1.25 mmol, 2.5 molar equiv); DBU: 305 mg (2.0 mmol, 4.0 molar equiv); CH2Cl2: 5.9 mL; TBAF: 0.090 mL (1.0 M, 0.090 mmol, 0.18 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of E/Z-5: 80.5 mg (92%), as a light-yellow liquid, E/Z ratio: 81/19. Rf (10% EtOAc in hexanes) = 0.47. 1H NMR (500 MHz, CDCl3): δ 7.58 (d, 2H, Ar-H, J = 8.5 Hz, E-isomer), 7.46 (d, 2H, Ar-H, J = 8.0 Hz, Z-isomer), 6.89-6.88 (m, 2H, Ar-H, both E and Z-isomers), 6.63 (d, 1H, =CH, 3JFH = 17.5 Hz, E-isomer), 6.06 (d, 1H, ArCH, 3JFH = 35.5 Hz, Z-isomer), 3.82 (s, 3H, OCH3, both E and Z-isomers), 3.63 (d, 1H, ≡CH, 4JFH = 3.7 Hz, E-isomer), 3.29 (d, 1H, ≡CH, 4JFH = 2.8 Hz, Z-isomer). 19F NMR (282 MHz, CDCl3): δ −108.90 (d, 3JFH = 18.3 Hz, E-isomer), −110.93 (d, 3JFH = 33.6 Hz, Z-isomer). HRMS (APPI) calcd for C11H9FO [M]+ 176.0632, found 176.0629.
Synthesis of 1-(2-Fluorobut-1-en-3-ynyl)-2-methoxybenzene (E/Z-6)
Method B. 2-Methoxybenzaldehyde: 68.0 mg (0.500 mmol); sulfone 3: 327 mg (1.00 mmol, 2.0 molar equiv); LHMDS: 1.2 mL (1.0 M, 1.2 mmol, 2.4 molar equiv); THF: 13.0 mL; TBAF: 1.00 mL (1.0 M, 1.0 mmol, 2.0 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of E/Z-6: 76.7 mg (87%), as a colorless liquid, E/Z ratio: 90/10. Rf (10% EtOAc in hexanes) = 0.47. 1H NMR (500 MHz, CDCl3): δ 7.94 (d, 1H, Ar-H, J = 7.4 Hz, E-isomer), 7.79 (d, 1H, Ar-H, J = 7.4 Hz, Z-isomer), 7.29-7.28 (m, 1H, Ar-H, both E and Z-isomers), 7.05 (d, 1H, =CH, 3JFH = 18.0 Hz, E-isomer), 6.98-6.94 (m, 1H, Ar-H, both E and Z-isomers), 6.89-6.88 (m, 1H, Ar-H, both E and Z-isomers), 6.62 (d, 1H, =CH, 3JFH = 36.8 Hz, Z-isomer), 3.85 (s, 3H, OCH3, both E and Z-isomers), 3.52 (d, 1H, ≡CH, 4JFH = 4.1 Hz), 3.28 (d, 1H, ≡CH, 4JFH = 3.7 Hz). 19F NMR (282 MHz, CDCl3): δ −105.33 (d, 3JFH = 18.3 Hz, E-isomer), −109.16 (d, 3JFH = 36.6 Hz, Z-isomer). HRMS (APPI) calcd for C11H9FO [M]+ 176.0632, found 176.0635.
Synthesis of (2-Fluorobut-1-en-3-ynyl)benzene (E/Z-7)
Method B. Benzaldehyde: 53.0 mg (0.500 mmol); sulfone 3: 327 mg (1.00 mmol, 2 molar equiv); LHMDS: 1.2 mL (1.0 M, 1.2 mmol, 2.4 molar equiv); THF: 13.0 mL; TBAF: 1.00 mL (1.0 M, 1.0 mmol, 2.0 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of E/Z-7: 43.0 mg (59%), as a colorless liquid, E/Z ratio: 94/6. Rf (10% EtOAc in hexanes) = 0.54. 1H NMR (500 MHz, CDCl3): δ 7.63 (d, 2H, Ar-H, J = 7.4 Hz, E-isomer), 7.51 (d, 2H, Ar-H, J = 7.4 Hz, Z-isomer), 7.37-7.34 (m, 2H, Ar-H, both E and Z-isomers), 7.32-7.28 (m, 1H, Ar-H, both E and Z-isomers), 6.68 (d, 1H, =CH, 3JFH = 17.0 Hz, E-isomer), 6.11 (d, 1H, =CH, 3JFH = 35.0 Hz, Z-isomer), 3.61 (d, 1H, ≡CH, 4JFH = 4.6 Hz, E-isomer), 3.29 (d, 1H, ≡CH, 4JFH = 3.7 Hz, Z-isomer). 19F NMR (282 MHz, CDCl3): δ −105.32 (d, 3JFH = 15.3 Hz, E-isomer), −107.42 (d, 3JFH = 33.6 Hz, Z-isomer). HRMS (APPI) calcd for C10H7F [M]+ 146.0526, found 146.0525.
Synthesis of 1-Chloro-4-(2-fluorobut-1-en-3-ynyl)benzene (E/Z-8)
Method A. 4-Chlorobenzaldehyde: 70.3 mg (0.500 mmol); sulfone 3: 327 mg (1.00 mmol, 2.0 molar equiv); DBU: 305 mg (2.00 mmol, 4.0 molar equiv); CH2Cl2: 5.4 mL; TBAF: 0.100 mL (1.0 M, 0.10 mmol, 0.20 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of E/Z-8: 73.4 mg (81%), as an orange liquid, E/Z ratio: 70/30. Rf (10% EtOAc in hexanes) = 0.47. 1H NMR (500 MHz, CDCl3): δ 7.56 (d, 2H, Ar-H, J = 8.8 Hz, E-isomer), 7.44 (d, 2H, Ar-H, J = 8.3 Hz, Z-isomer), 7.33-7.31 (m, 2H, Ar-H, both E and Z-isomers), 6.62 (d, 1H, =CH, 3JFH = 17.1 Hz, E-isomer), 6.06 (d, 1H, =CH, 3JFH = 34.2 Hz, Z-isomer), 3.63 (d, 1H, ≡CH, 4JFH = 3.9 Hz, E-isomer), 3.31 (d, 1H, ≡CH, 4JFH = 3.9 Hz, Z-isomer). 19F NMR (282 MHz, CDCl3): δ −104.25 (d, 3JFH = 15.3 Hz, E-isomer), −106.45 (d, 3JFH = 33.6 Hz, Z-isomer). HRMS (APPI) calcd for C10H6ClF [M]+ 180.0137, found 180.0139.
Synthesis of 1-(2-Fluorobut-1-en-3-ynyl)-4-nitrobenzene (E/Z-9)
Method A. 4-Nitrobenzaldehyde: 75.6 mg (0.500 mmol); sulfone 3: 409 mg (1.25 mmol, 2.5 molar equiv); DBU: 305 mg (2.0 mmol, 4.0 molar equiv); CH2Cl2: 5.9 mL; TBAF: 0.100 mL (1.0 M, 0.10 mmol, 0.20 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of E/Z-9: 56.8 mg (59%), as a yellow solid, E/Z ratio: 51/49. Rf (20% EtOAc in hexanes) = 0.53. 1H NMR (500 MHz, CDCl3): δ 8.21 (d, 2H, Ar-H, J = 8.6 Hz, both E and Z-isomers), 7.79 (d, 2H, Ar-H, J = 8.9 Hz, E-isomer), 7.65 (d, 2H, Ar-H, J = 8.8 Hz, Z-isomer), 6.72 (d, 1H, =CH, 3JFH = 15.9 Hz, E-isomer), 6.18 (d, 1H, =CH, 3JFH = 33.6 Hz, Z-isomer), 3.70 (d, 1H, ≡CH, 4JFH = 4.3 Hz, E-isomer), 3.38 (d, 1H, ≡CH, 4JFH = 4.0 Hz, Z-isomer). 19F NMR (282 MHz, CDCl3): δ −97.63 (d, 3JFH = 15.3 Hz, E-isomer), −100.80 (d, 3JFH = 33.6 Hz, Z-isomer). HRMS (ESI) calcd for C10H7FNO2 [M+H]+ 192.0455, found 192.0444.
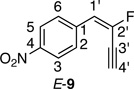
For the purpose of proton and carbon assignments, a small amount of E/Z-9 was separated by TLC (500 µm glass-backed silica gel plate, mobile phase 5% EtOAc in hexanes) and 1H, 13C and 2D spectra (NOESY, HMQC, HMBC) of E-9 were recorded. 1H NMR (500 MHz, CDCl3) of E-9: δ 8.21 (d, 2H, Ar-H, J = 8.8 Hz, H-3, H-5), 7.79 (d, 2H, Ar-H, J = 8.8 Hz, H-2, H-6), 6.72 (d, 1H, 3JFH = 16.1 Hz, H-1’), 3.70 (d, 1H, 4JFH = 3.9 Hz, H-4’). 13C NMR (125 MHz, CDCl3) of E-9: δ 147.5 (s, 1C, C-4), 142.6 (d, 1C, 1JCF = 240.3 Hz, C-2’), 138.4 (d, 1C, 3JCF = 10.1 Hz, C-1), 128.9 (d, 2C, 4JCF = 3.2 Hz, C-2, C-6), 124.1 (s, 2C, C-3, C-5), 117.1 (d, 1C, 2JCF = 33.4 Hz, C-1’), 87.5 (d, 1C, 3JCF = 6.0 Hz, C-4’), 75.1 (d, 1C, 2JCF = 41.6 Hz, C-3’).
Synthesis of (2-Fluorobut-1-en-3-ynyl)ferrocene (E /Z-10)
Method A. Ferrocenecarboxaldehyde: 107 mg (0.500 mmol); sulfone 3: 409 mg (1.25 mmol, 2.5 molar equiv); DBU: 305 mg (2.0 mmol, 4.0 molar equiv); CH2Cl2: 5.9 mL; TBAF: 0.100 mL (1.0 M, 0.10 mmol, 0.20 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of E/Z-10: 121 mg (95%), as an orange solid, E/Z ratio: 83/17. Rf (10% EtOAc in hexanes) = 0.53. 1H NMR (500 MHz, CDCl3): δ 6.41 (d, 1H, =CH, 3JFH = 15.6 Hz, E-isomer), 5.94 (d, 1H, =CH, 3JFH = 35.1 Hz, Z-isomer), 4.62 (s, 2H, Ar-H, E-isomer), 4.49 (d, 2H, Ar-H, J = 1.5 Hz, Z-isomer), 4.30-4.29 (m, 2H, Ar-H, both E and Z-isomers), 4.17 (s, 5H, Ar-H, E-isomer), 4.16 (s, 5H, Ar-H, Z-isomer), 3.66 (d, 1H, ≡CH, 4JFH = 4.0 Hz, E-isomer), 3.34 (d, 1H, ≡CH, 4JFH = 3.4 Hz, Z-isomer). 19F NMR (282 MHz, CDCl3): δ −111.06 (d, 3JFH = 15.3 Hz, E-isomer), −111.97 (d, 3JFH = 33.6 Hz, Z-isomer). HRMS (ESI) calcd for C14H11FFe [M]+ 254.0189, found 254.0191.
Synthesis of 3-(2-Fluorobut-1-en-3-ynyl)-1-Tosyl-1H-Indole (E/Z-11)
Method A. 1-Tosyl-1H-indole-3-carboxaldehyde:25 150 mg (0.500 mmol); sulfone 3: 490 mg (1.50 mmol, 3.0 molar equiv); DBU: 305 mg (2.0 mmol, 4.0 molar equiv); CH2Cl2: 6.4 mL; TBAF: 0.100 mL (1.0 M, 0.10 mmol, 0.20 molar equiv). Eluting solvent for column chromatography: CH2Cl2. Yield of E/Z-11: 162 mg (95%), as an off-white solid, E/Z ratio: 75/25. Rf (CH2Cl2) = 0.74. 1H NMR (500 MHz, CDCl3): δ 8.12 (s, 1H, Ar-H, E-isomer), 8.00-7.97 (m, 1H, Ar-H, both E and Z-isomers), 7.94 (s, 1H, Ar-H, Z-isomer), 7.80-7.77 (m, 2H, Ar-H, both E and Z-isomers), 7.55 (d, 1H, Ar-H, J = 8.0 Hz, Z-isomer), 7.51 (d, 1H, Ar-H, J = 7.5 Hz, E-isomer), 7.36-7.33 (m, 1H, Ar-H, both E and Z-isomers), 7.29-7.23 (m, 3H, Ar-H, both E and Z-isomers), 6.75 (d, 1H, =CH, 3JFH = 14.3 Hz, E-isomer), 6.31 (d, 1H, =CH, 3JFH = 35.0 Hz, Z-isomer), 3.83 (d, 1H,≡CH, 4JFH = 3.7 Hz, E-isomer), 3.39 (d, 1H, ≡CH, 4JFH = 3.7 Hz, Z-isomer), 2.35 (s, 3H, CH3, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ −106.89 (d, 3JFH = 15.3 Hz, E-isomer), −100.96 (d, 3JFH = 33.6 Hz, Z-isomer). HRMS (ESI) calcd for C19H15FNO2S [M+H]+ 340.0802, found 340.0806.
Synthesis of (E/Z)-2-(2-Fluorobut-1-en-3-ynyl)thiophene (E/Z-12)
Method A. 2-Thiophenecarboxaldehyde: 56.1 mg (0.500 mmol); sulfone 3: 409 mg (1.25 mmol, 2.5 molar equiv); DBU: 305 mg (2.00 mmol, 4.0 molar equiv); CH2Cl2: 5.9 mL; TBAF: 0.100 mL (1.0 M, 0.10 mmol, 0.20 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of E/Z-12: 59.0 mg (78%), as a light-brown liquid, E/Z ratio: 75/25. Rf (10% EtOAc in hexanes) = 0.42. 1H NMR (500 MHz, CDCl3): δ 7.39 (d, 1H, Ar-H, J = 4.6 Hz, Z-isomer), 7.31(d, 1H, Ar-H, J = 5.1 Hz, E-isomer), 7.24 (d, 1H, Ar-H, J = 2.8 Hz, E-isomer), 7.15 (d, 1H, Ar-H, J = 3.2 Hz, Z-isomer), 7.04-7.02 (m, 1H, Ar-H, both E and Z-isomers), 6.89 (d, 1H, =CH, 3JFH = 14.3 Hz, E-isomer), 6.42 (d, 1H, =CH, 3JFH = 33.6 Hz, Z-isomer), 3.85 (d, 1H, ≡CH, 4JFH = 4.1 Hz, E-isomer), 3.38 (d, 1H, ≡CH, 4JFH = 3.7 Hz, Z-isomer). 19F NMR (282 MHz, CDCl3): δ −111.82 (d, 3JFH = 15.3 Hz, E-isomer), −106.40 (d, 3JFH = 33.6 Hz, Z-isomer). HRMS (APPI) calcd for C8H5FS [M]+ 152.0091, found 152.0089.
Synthesis of (4-Fluorohexa-1,3-dien-5-ynyl)benzene (E/Z-13)
Method A. Cinnamaldehyde: 66.1 mg (0.500 mmol); sulfone 3: 490 mg (1.50 mmol, 3.0 molar equiv); DBU: 305 mg (2.0 mmol, 4.0 molar equiv); CH2Cl2: 6.4 mL; TBAF: 0.100 mL (1.0 M, 0.10 mmol, 0.20 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of E/Z-13: 82.1 mg (95%), as a deep-orange liquid, E/Z ratio: 72/28. Rf (30% EtOAc in hexanes) = 0.60. 1H NMR (500 MHz, CDCl3): δ 7.45-7.43 (m, 2H, Ar-H, both E and Z-isomers), 7.35-7.32 (m, 2H, Ar-H, both E and Z-isomers), 7.28-7.26 (m, 1H, Ar-H, both E and Z-isomers), 7.07 (dd, 1H, =CH, J = 15.7; 11.1 Hz, Z-isomer), 6.88 (dd, 1H, =CH, J = 15.7; 11.5 Hz, E-isomer), 6.68 (d, 1H, ArCH, J = 15.7 Hz, E-isomer), 6.63 (d, 1H, ArCH, J = 15.7 Hz, Z-isomer), 6.47 (t, 1H, =CH, 3JFH, 3JHH = 12.0 Hz, E-isomer), 6.04 (dd, 1H, =CH, 3JFH = 30.9 Hz; 3JHH = 11.1 Hz, Z-isomer), 3.68 (d, 1H, ≡CH, 4JFH = 3.2 Hz, E-isomer), 3.35 (d, 1H, ≡CH, 4JFH = 3.2 Hz, Z-isomer). 19F NMR (282 MHz, CDCl3): δ −110.55 (d, 3JFH = 12.2 Hz, E-isomer), −111.43 (d, 3JFH = 30.5 Hz, Z-isomer). HRMS (APPI) calcd for C12H9 F [M]+ 172.0683, found 172.0683.
Synthesis of (4-Fluorohex-3-en-5-ynyl)benzene (E/Z-14)
Method A. 3-Phenylpropanal: 67.1 mg (0.500 mmol); sulfone 3: 327 mg (1.0 mmol, 2.0 molar equiv); DBU: 305 mg (2.0 mmol, 4.0 molar equiv); CH2Cl2: 5.4 mL; TBAF: 0.100 mL (1.0 M, 0.10 mmol, 0.20 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of E/Z-14: 46.6 mg (54%), as a light-greenish liquid, E/Z ratio: 80/20. Rf (10% EtOAc in hexanes) = 0.59. 1H NMR (500 MHz, CDCl3): δ 7.31-7.18 (m, 5H, Ar-H, both E and Z-isomers), 5.69 (dt, 1H, =CH, 3JFH = 14.3 Hz, 3JHH = 8.3 Hz, E-isomer), 5.31 (dt, 1H, =CH, 3JFH = 33.6 Hz, 3JHH = 7.8 Hz, Z-isomer), 3.34 (d, 1H, ≡CH, 4JFH = 3.2 Hz, E-isomer), 3.08 (d, 1H, ≡CH, 4JFH = 3.7 Hz, Z-isomer), 2.74-2.69 (m, 2H, CH2, both E and Z-isomers), 2.53-2.45 (m, 2H, CH2, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ −110.43 (d, 3JFH = 12.2 Hz, E-isomer), −112.51 (d, 3JFH = 33.6 Hz, Z-isomer). HRMS (CI) calcd for C12H11 F [M]+ 174.0839, found 174.0860.
Synthesis of 3-Fluoroundec-3-en-1-yne (E/Z-15)
Method A. Octanal: 64.1 mg (0.500 mmol); sulfone 3: 327 mg (1.00 mmol, 2.0 molar equiv); DBU: 305 mg (2.0 mmol, 4.0 molar equiv); CH2Cl2: 5.4 mL; TBAF: 0.100 mL (1.0 M, 0.10 mmol, 0.20 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of E/Z-15: 66.6 mg (79%), as a colorless liquid, E/Z ratio: 85/15. Rf (10% EtOAc in hexanes) = 0.73. 1H NMR (500 MHz, CDCl3): δ 5.67 (dt, 1H, =CH, 3JFH = 15.7 Hz, 3JHH = 8.3 Hz, E-isomer), 5.29 (dt, 1H, =CH, 3JFH = 33.2 Hz, 3JHH = 7.8 Hz, Z-isomer), 3.34 (d, 1H, ≡CH, 4JFH = 3.2 Hz, E-isomer), 3.09 (d, 1H, ≡CH, 4JFH = 3.7 Hz, Z-isomer), 2.19-2.11 (m, 2H, CH2, both E and Z-isomers), 1.42-1.29 (m, 10H, 5CH2, both E and Z-isomers), 0.86 (m, 3H, CH3, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ −111.45 (d, 3JFH = 15.3 Hz, E-isomer), −114.05 (d, 3JFH = 33.6 Hz, Z-isomer). HRMS (CI) calcd for C11H17 F [M]+ 168.1309, found 168.1302.
Synthesis of 1-Benzyl-4-(1-fluoroprop-2-ynylidene)piperidine (16)
N-benzylpiperidin-4-one: 57.0 mg (0.300 mmol); sulfone 3: 294 mg (0.900 mmol, 3.0 molar equiv); LHMDS: 1.5 mL (1.0 M, 1.5 mmol, 5.0 molar equiv); THF: 2.5 mL; TBAF: 0.900 mL (1.0 M, 0.90 mmol, 3.0 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of 16: 52.7 mg (77%), as a colorless liquid. Rf (20% EtOAc in hexanes) = 0.52. 1H NMR (500 MHz, CDCl3): δ 7.34-7.25 (m, 5H, Ar-H), 3.53 (s, 2H, CH2), 3.35 (d, 1H, ≡CH, 4JFH = 3.7 Hz), 2.50 (t, 2H, CH2, 4JFH = 5.5 Hz), 2.45 (s, 4H, 2CH2), 2.40 (t, 2H, CH2, 4JFH = 5.1 Hz). 13C NMR (125 MHz, CDCl3): δ 138.5, 134.9 (d, 1JCF = 227.0 Hz), 129.2 (2C), 128.4 (2C), 127.3, 127.0 (d, 2JCF = 18.3 Hz), 83.4 (d, 3JCF = 7.8 Hz), 74.7 (d, 2JCF = 44.4 Hz), 62.9, 53.8 (d, 4JCF = 1.8 Hz), 53.3 (d, 4JCF = 1.4 Hz), 29.0 (d, 3JCF = 1.8 Hz), 26.0 (d, 3JCF = 3.2 Hz). 19F NMR (282 MHz, CDCl3): δ −121.4 (s). HRMS (EI) calcd for C15H16FN [M]+ 229.1261, found 229.1250.
(3E)-(3-Fluoropent-2-en-4-yn-2-yl)benzene (17)
Acetophenone: 36.0 mg (0.300 mmol); sulfone 3: 294 mg (0.900 mmol, 3.0 molar equiv); LHMDS: 1.5 mL (1.0 M, 1.5 mmol, 5.0 molar equiv); THF: 2.5 mL; TBAF: 0.900 mL (1.0 M, 0.90 mmol, 3.0 molar equiv). Eluting solvent for column chromatography: 10% EtOAc in hexanes. Yield of 17: 42.3 mg (88%), as a colorless liquid. Rf (10% EtOAc in hexanes) = 0.76. 1H NMR (500 MHz, CDCl3): δ 7.47 (d, 2H, Ar-H, J = 7.4 Hz), 7.37 (t, 2H, Ar-H, J = 7.4 Hz), 7.32 (t, 1H, Ar-H, J = 7.4 Hz), 3.18 (d, 1H, ≡CH, 4JFH = 3.7 Hz), 2.14 (d, 3H, CH3, 4JFH = 4.1 Hz). 13C NMR (125 MHz, CDCl3): δ 138.4 (d, 1JCF = 232.1 Hz), 137.6 (d, 3JCF = 4.7 Hz), 128.4 (2C), three resonances: 128.27, 128.25, 128.23 (2C, one d and one s), 127.4 (d, 2JCF = 22.4 Hz), 82.5 (d, 3JCF = 7.3 Hz), 75.9 (d, 2JCF = 42.1 Hz), 16.3 (d, 3JCF = 4.1 Hz). 19F NMR (282 MHz, CDCl3): δ −110.0 (s). HRMS (EI) calcd for C11H9F [M]+ 160.0683, found 160.0667. Stereochemical Assignment. Fluoro enyne 17 (7.0 mg, 0.044 mmol) and Lindlar catalyst (5% Pd on CaCO3 poisoned with lead, 2.5 mg) were suspended in anhydrous hexanes (0.600 mL) in a dry vial and one small drop of quinoline was added. The vial was evacuated, filled with hydrogen gas via a hydrogen balloon, and the reaction was conducted at atmospheric pressure for 2 h. The mixture was filtered through Celite and the residue was washed with EtOAc (10.0 mL). The filtrate was evaporated under reduced pressure and the crude mixture was purified by column chromatography (10% EtOAc in hexanes). Yield of (E)-(3-fluoropenta-2,4-dien-2-yl)benzene: 3.0 mg (42%), as a colorless liquid. Rf (10% EtOAc in hexanes) = 0.78. 1H NMR (500 MHz, CDCl3): δ 7.35 (t, 2H, Ar-H, J = 7.3 Hz), 7.28 (t, 1H, Ar-H, J = 7.3 Hz), 7.23 (d, 2H, Ar-H, J = 7.6 Hz), 6.25 (ddd, 1H, =CH, J = 28.3, 17.1, 11.2 Hz), 5.51 (d, 1H, =CH2, J = 17.1 Hz), 5.07 (d, 1H, =CH2, J = 10.7 Hz), 2.10 (d, 3H, J = 3.4 Hz). 19F NMR (282 MHz, CDCl3): δ −124.4 (d, 3JHF = 27.5 Hz). HRMS (ESI) calcd for C11H12F [M + H]+ 163.0918, found 163.0947. NOESY correlation between the vinylic proton at the C-2 and the ortho protons in the phenyl ring confirmed the E stereochemistry of the double bond in fluoroenyne 17.
Synthesis of 2-[3-(Triisopropylsilyl)prop-2-ynylsulfonyl]benzo[d]thiazole (18)
Step 1:23
A solution of 1.00 g of propargyl bromide (80 wt% in toluene: 6.72 mmol) in THF (20.0 ml) was cooled to −78 °C (dry ice/isoPrOH) and 4.20 mL (6.72 mmol, 1.0 molar equiv) of n-BuLi (1.6 M in hexane) was added dropwise. After stirring the mixture at −78 °C for 10 min, 1.42 mL (6.72 mmol, 1.0 molar equiv) of TIPS-Cl was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for another 2 h. The reaction was quenched by addition of saturated aq NH4Cl and the mixture was poured into EtOAc. The organic layer was separated and the aqueous layer was extracted with EtOAc (3x). The combined organic layer was washed with water and brine, and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was used in the next step without purification.
Step 2
The yellow oil obtained in step 1 was diluted with DMF (10.0 mL) and the sodium salt of 2-mercapto-1,3-benzothiazole (1.65 g, 8.74 mmol) was added. After stirring for 2 h at room temperature, 10.0 mL of water was added and the mixture was poured into EtOAc. The organic layer was separated and the aqueous layer was extracted with EtOAc (3x). The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure.
Step 3
The crude product obtained in step 2 (1.23 g) was dissolved in CHCl3 (15.0 mL) and cooled to −10 °C (ice/salt). A solution of m-CPBA (1.85 g, 10.7 mmol) in CHCl3 (28 mL) was slowly added via an addition funnel. After the addition was complete, the mixture was allowed to warm to room temperature and the stirring was continued for 12 h. Saturated aq NaHCO3 (10.0 mL) was added, the organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2x). The combined organic layer was washed with water and brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure. The crude product was purified by column chromatography (20% EtOAc in hexanes). Yield of 18: 800 mg (30% over three steps), as a white solid. Rf (10% EtOAc in hexanes) = 0.23. 1H NMR (500 MHz, CDCl3): δ 8.22 (d, 1H, Ar-H, J = 8.3 Hz), 8.00 (d, 1H, Ar-H, J = 7.8 Hz), 7.65-7.58 (m, 2H, Ar-H), 4.48 (s, 2H, CH2), 0.878-0.873 (m, 21H, Si(iso-Pr)3). 13C NMR (125 MHz, CDCl3): δ 164.3, 152.8, 137.3, 128.3, 127.8, 125.8, 122.3, 92.4, 92.3, 48.5, 18.5, 11.1. HRMS (ESI) calcd for C19H27NO2S2Si [M + H]+ 394.1325, found 394.1330.
Synthesis of 2-[1-Fluoro-3-(triisopropylsilyl)prop-2-ynylsulfonyl]benzo[d]thiazole (19)
A stirring solution of sulfone 18 (500 mg, 1.27 mmol, 1.0 molar equiv) in dry toluene (10.0 mL) was cooled under nitrogen to −78 °C (dry ice/isoPrOH). LDA (0.700 mL, 1.1 molar equiv of a 2.0 M solution in heptane/THF/EtPh) was added and after 15 min, solid NFSI (479 mg, 1.52 mmol, 1.2 molar equiv) was added. The reaction mixture was allowed to stir at −78 °C for 50 min then warmed to room temperature, and stirring was continued for an additional 50 min. Saturated aq NH4Cl was added to the reaction mixture and the layers were separated. The aqueous layer was extracted with EtOAc (3 x), and the combined organic layer was washed with water, saturated aq NaHCO3 and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography (10% EtOAc in hexanes). Yield of 19: 359 mg (69%), as a white solid. Rf (20% EtOAc in hexanes) = 0.50. 1H NMR (500 MHz, CDCl3): δ 8.25 (d, 1H, Ar-H, J = 8.2 Hz), 8.02 (d, 1H, Ar-H, J = 7.6 Hz), 7.66-7.60 (m, 2H, Ar-H), 6.19 (d, 1H, CHF, 2JFH = 48.5 Hz), 1.04-1.01 (m, 21H, Si(iso-Pr)3). 13C NMR (125 MHz, CDCl3): δ 161.4, 152.9, 137.7, 128.7, 128.0, 126.1, 122.4, 101.9 (d, 3JCF = 8.2 Hz), 91.9 (d, 1JCF = 222.5 Hz), 91.6 (d, 2JCF = 23.3 Hz), 18.48, 18.47, 11.0. 19F NMR (282 MHz, CDCl3): δ −164.46 (d, 2JFH = 48.8 Hz). HRMS (ESI) calcd for C19H27FNO2S2Si [M + H]+ 412.1231, found 412.1204.
Condensations of Sulfone 19 with Aldehydes
Synthesis of (3-Fluoro-4-(naphthalen-2-yl)but-3-en-1-ynyl)triisopropylsilane (E/Z-20)
Method A. A solution of 2-naphthaldehyde (15.0 mg, 0.096 mmol) and DBU (58.5 mg, 0.385 mmol, 4.0 molar equiv) in CH2Cl2 (0.600 mL) was cooled under nitrogen to −55 °C (dry ice/isoPrOH). A solution of fluoropropargyl sulfone 19 (51.5 mg, 0.125 mmol, 1.3 molar equiv) in CH2Cl2 (0.300 mL) was added via a syringe. The dark reaction mixture was stirred for 15 min and checked by TLC (10% EtOAc in hexanes), which showed disappearance of the aldehyde. Reaction mixture was analyzed by 19F NMR, and the combined E/Z product mixture was isolated by column chromatography (10% EtOAc in hexanes). Yield of E/Z-20: 29.4 mg (87%), as a white solid, E/Z ratio: 71/29. Rf (10% EtOAc in hexanes) = 0.81. 1H NMR (500 MHz, CDCl3): δ 8.08 (s, 1H, Ar-H, E-isomer), 7.94 (s, 1H, Ar-H, Z-isomer), 7.91 (d, 1H, Ar-H, J = 8.5 Hz, E-isomer), 7.82-7.76 (m, 3H, Ar-H, both E and Z-isomers), 7.66 (d, 1H, Ar-H, J = 8.5 Hz, Z-isomer), 7.48-7.46 (m, 2H, Ar-H, both E and Z-isomers), 6.74 (d, 1H, =CH, 3JFH = 17.4 Hz, E-isomer), 6.23 (d, 1H, =CH, 3JFH = 35.1 Hz, Z-isomer), 1.17-1.14 (m, 21H, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ −101.66 (d, 3JFH = 18.3 Hz, E-isomer), −104.65 (d, 3JFH = 33.6 Hz, Z-isomer). HRMS (ESI) calcd for C23H30FSi [M+H]+ 353.2095, found 353.2070.
Synthesis of (E/Z)-(3-Fluoro-4-(4-nitrophenyl)but-3-en-1-ynyl)triisopropylsilane (E/Z-21)
Method A. A solution of 4-nitrobenzaldehyde (20.0 mg, 0.132 mmol) and DBU (80.4 mg, 0.528 mmol, 4.0 molar equiv) in CH2Cl2 (0.900 mL) was cooled under nitrogen to −55 °C (dry ice/isoPrOH). A solution of fluoropropargyl sulfone 19 (70.8 mg, 0.172 mmol, 1.3 molar equiv) in CH2Cl2 (0.400 mL) was added via a syringe. The dark reaction mixture was stirred for 5 min and checked by TLC (10% EtOAc in hexanes), which showed disappearance of the aldehyde. The reaction mixture was analyzed by 19F NMR, then loaded onto a silica gel column and the combined E/Z olefin mixture was eluted with 10% EtOAc in hexanes. Yield of E/Z-21: 29.2 mg (64%), as a light-yellow solid, E/Z ratio: 67/33. Rf (10% EtOAc in hexanes) = 0.58. 1H NMR (500 MHz, CDCl3): δ 8.20 (d, 2H, Ar-H, J = 8.8 Hz, Z-isomer), 8.17 (d, 2H, Ar-H, J = 8.8 Hz, E-isomer), 7.86 (d, 2H, Ar-H, J = 8.8 Hz, E-isomer), 7.64 (d, 2H, Ar-H, J = 8.8 Hz, Z-isomer), 6.63 (d, 1H, =CH, 3JFH = 15.6 Hz, E-isomer), 6.13 (d, 1H, =CH, 3JFH = 33.2 Hz, Z-isomer), 1.15-1.12 (m, 21H, both E and Z-isomers). 19F NMR (282 MHz, CDCl3): δ −94.43 (d, 3JFH = 15.3 Hz, E-isomer), −98.03 (d, 3JFH = 33.6 Hz, Z-isomer). HRMS (ESI) calcd for C19H27FNO2Si [M+H]+ 348.1790, found 348.1774. Method B. A solution of 4-nitrobenzaldehyde (20.0 mg, 0.132 mmol, 1.0 molar equiv) and sulfone 19 (70.8 mg, 0.172 mmol, 1.3 molar equiv) in dry THF (3.4 mL) was cooled under nitrogen to −78 °C (dry ice/isoPrOH). LHMDS (1.0 M solution in THF, 0.317 mL, 2.4 molar equiv) was added, the reaction mixture was stirred at −78 °C for 10 min and checked by TLC (10% EtOAc in hexanes), which showed disappearance of the aldehyde. Saturated aq NH4Cl was added and the mixture was poured into EtOAc. The organic layer was separated and the aqueous layer was extracted with EtOAc (3x). The combined organic layer was washed with water and brine, dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The crude reaction mixture was analyzed by 19F NMR, and the combined E/Z product mixture was then isolated by column chromatography (10% EtOAc in hexanes). Yield of E/Z-21: 39.7 mg (87%), as a light-yellow solid, E/Z ratio: 76/24.
Sonogashira Cross-Coupling of Fluoro Enynes
Synthesis of 2-(2-Fluoro-4-phenylbut-1-en-3-yn-1-yl)naphthalene (E/Z-22)
A solution of fluoro enyne E/Z-4 (E/Z 88/12, 17.0 mg, 0.09 mmol, 1.00 molar equiv), iodobenzene (35.5 mg, 0.18 mmol, 2.0 molar equiv), [PdCl2(PPh3)2] (3.2 mg, 5 mol%) and CuI (1.7 mg, 10 mol%) in Et3N (0.30 mL) was stirred at room temperature. After 2 h, TLC (hexanes) showed complete disappearance of fluoro enyne 4. The reaction mixture was diluted with EtOAc and washed with water and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. The crude reaction mixture was analyzed by 19F NMR, and then purified by column chromatography (hexanes). Yield of E/Z-22: 16.0 mg (68%), as a white solid, E/Z ratio: 93/7. Rf (hexanes) = 0.28. 1H NMR (500 MHz, CDCl3): δ 8.12 (s, 1H, Ar-H, E-isomer), 7.91 (s, 1H, Ar-H, Z-isomer), 7.90 (dd, 1H, Ar-H, J = 8.5, 1.5 Hz, E-isomer), 7.85-7.82 (m, 3H, Ar-H, both E and Z-isomers), 7.72 (dd, 1H, Ar-H, J = 8.5, 1.5 Hz, Z-isomer), 7.61-7.55 (m, 2H, Ar-H, both E and Z-isomers), 7.51-7.39 (m, 5H, Ar-H, both E and Z-isomers), 6.82 (d, 1H, =CH, 3JFH = 16.5 Hz, E-isomer), 6.27 (d, 1H, =CH, 3JFH = 35.1 Hz, Z-isomer). 19F NMR (282 MHz, CDCl3): δ −102.43 (d, 3JFH = 15.3 Hz, E-isomer), −104.41 (d, 3JFH = 36.6 Hz, Z-isomer). HRMS (ESI) calcd for C20H14F [M+H]+ 273.1074, found 273.1057.
Synthesis of 1-Benzyl-4-(1-fluoro-3-phenylprop-2-ynylidene)piperidine (23)
A solution of fluoro enyne 16 (50.0 mg, 0.220 mmol, 1.00 molar equiv), iodobenzene (90.0 mg, 0.440 mmol, 2.0 molar equiv), [PdCl2(PPh3)2] (7.7 mg, 5 mol%) and CuI (4.0 mg, 10 mol%) in Et3N (0.750 mL) was stirred at room temperature. After 2 h, TLC (20% EtOAc in hexanes) showed complete disappearance of fluoro enyne 16. The reaction mixture was diluted with EtOAc and washed with water and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. The crude reaction mixture was analyzed by 19F NMR, and then purified by column chromatography (20% EtOAc in hexanes). Yield of 23: 53.0 mg (79%), as a pale-yellow oil. Rf (SiO2, 20% EtOAc in hexanes) = 0.34. 1H NMR (500 MHz, CDCl3): δ 7.48-7.46 (m, 2H, Ar-H), 7.34-7.27 (m, 8H, Ar-H), 3.54 (s, 2H), 2.55-2.47 (m, 8H). 13C NMR (125 MHz, CDCl3): δ 138.5, 135.6 (d, 1JCF = 227.0 Hz), 131.7 (2C, d, 5JCF = 1.8 Hz), 129.3 (2C), 129.1, 128.6 (2C), 128.4 (2C), 127.3, 125.8 (d, 2JCF = 19.7 Hz), 122.1 (d, 4JCF = 2.3 Hz), 94.9 (d, 3JCF = 7.8 Hz), 80.2 (d, 2JCF = 43.5 Hz), 63.0, 54.0 (d, 4JCF = 1.8 Hz), 53.4 (d, 4JCF = 1.4 Hz), 29.3 (d, 3JCF = 1.3 Hz), 26.2 (d, 3JCF = 3.2 Hz). 19F NMR (282 MHz, CDCl3): δ −118.85 (s). HRMS (ESI) calcd for C21H21FN [M + H]+ 306.1653, found 306.1626.
Synthesis of Stereoisomeric Fluoro Dienes
Synthesis of Fluoro Diene 25: Reduction of 16 to Diene 24 and Heck Coupling of 24. Synthesis of 1-Benzyl-4-(1-fluoroallylidene)piperidine (24)
Fluoro enyne 16 (25.0 mg, 0.110 mmol) and Lindlar catalyst (5% Pd on CaCO3 poisoned with lead, 5.0 mg) were suspended in anhydrous hexanes (1.0 mL) in a dry vial and one drop of quinoline was added. The vial was evacuated and filled with hydrogen gas via a hydrogen balloon, and the reaction was conducted at atmospheric pressure for 2 h. The mixture was filtered through Celite and the residue was washed with EtOAc (10.0 mL). The filtrate was evaporated under reduced pressure and the crude mixture was purified by column chromatography (20% EtOAc in hexanes). Yield of 24: 22.0 mg (87%), as a pale-yellow oil. Rf (20% EtOAc in hexanes) = 0.40. 1H NMR (500 MHz, CDCl3): δ 7.33-7.24 (m, 5H, Ar-H), 6.44 (ddd, 1H, =CH, J = 28.1, 17.0, 11.1 Hz), 5.47 (d, 1H, =CH2, J = 17.0 Hz), 5.13 (d, 1H, =CH2, J = 11.1 Hz), 3.52 (s, 2H), 2.46 (br s, 6H), 2.31 (t, 2H, J = 5.5 Hz). 13C NMR (125 MHz, CDCl3): δ 149.9 (d, 1JCF = 238.9 Hz), 138.6, 129.3 (2C), 128.4 (2C), 127.2, 124.9 (d, 2JCF = 27.5 Hz), 117.8 (d, 2JCF = 17.9 Hz), 113.5 (d, 3JCF = 6.0 Hz), 63.1, 54.1 (d, 4JCF = 1.8 Hz), 53.8 (d, 4JCF = 1.4 Hz), 27.5 (d, 3JCF = 5.0 Hz), 26.0 (d, 3JCF = 7.8 Hz). 19F NMR (282 MHz, CDCl3): δ −130.47 (d, 3JHF = 27.5 Hz). HRMS (ESI) calcd for C15H19FN [M + H]+ 232.1496, found 232.1519.
(E)-1-Benzyl-4-(1-fluoro-3-phenylallylidene)piperidine (25) via Heck Coupling of 24
Et3N (0.045 mL, 5 molar equiv) was added to a solution of fluoro diene 24 (15 mg, 0.065 mmol), iodobenzene (26.5 mg, 0.130 mmol, 2 molar equiv), Pd(OAc)2 (1.5 mg, 10 mol%), and P(o-tol)3 (4.0 mg, 20 mol%) in anhydrous DMF (0.650 mL), in a dry vial.26 The vial was flushed with nitrogen gas and sealed with a Teflon-lined cap. The reaction mixture was stirred at 100 °C for 15 h, then diluted with EtOAc (10 mL), and washed with water and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure. The crude reaction mixture was analyzed by 19F NMR and then purified by column chromatography (10% EtOAc in hexanes). Yield of 25: 14.0 mg (70%), as a brown oil. Rf (20% EtOAc in hexanes) = 0.33. 1H NMR (500 MHz, CDCl3): δ 7.42 (d, 2H, J = 7.8 Hz, Ar-H), 7.35-7.23 (m, 8H, Ar-H), 6.84-6.75 (m, 2H), 3.54 (s, 2H), 2.52-2.40 (m, 6H), 2.42 (t, 2H, J = 5.4 Hz). 13C NMR (125 MHz, CDCl3): δ 150.2 (d, 1JCF = 238.0 Hz), 138.5, 137.0, 129.3 (2C), 128.9 (2C), 128.4 (2C), 128.1 (d, 3JCF = 5.5 Hz), 128.0, 127.3, 126.8 (2C), 118.4 (d, 2JCF = 18.8 Hz), 116.5 (d, 2JCF = 25.2 Hz), 63.1, 54.2 (d, 4JCF = 1.8 Hz), 53.9 (d, 4JCF = 1.5 Hz), 27.8 (d, 3JCF = 5.0 Hz), 26.3 (d, 3JCF = 8.2 Hz). 19F NMR (282 MHz, CDCl3): δ −128.10 to −128.20 (m). HRMS (ESI) calcd for C21H23FN [M + H]+ 308.1809, found 308.1794.
Synthesis of Fluoro Diene 26 via Reduction of Sonogashira Product 23. Synthesis of (Z)-1-Benzyl-4-(1-fluoro-3-phenylallylidene)piperidine (26)
Fluoro enyne 23 (10 mg, 0.033 mmol) and Lindlar catalyst (5% Pd on CaCO3 poisoned with lead, 2.5 mg) were suspended in CH3OH (0.80 mL) in a dry vial and one drop of quinoline was added. The vial was evacuated, filled with hydrogen gas via a hydrogen balloon, and the reaction was conducted at atmospheric pressure for 10 h. The mixture was filtered through Celite and the residue was washed with EtOAc (10.0 mL). The filtrate was evaporated under reduced pressure and the crude mixture was purified by column chromatography (20% EtOAc in hexanes). Yield of 26: 6.8 mg (68%), as a pale-yellow oil. Rf (20% EtOAc in hexanes) = 0.32. 1H NMR (500 MHz, CDCl3): δ 7.34-7.21 (m, 10H, Ar-H), 6.49 (d, 1H, =CH, J = 12.2 Hz), 6.12 (dd, 1H, =CH, J = 23.7, 12.5 Hz), 3.50 (s, 2H) 2.45-2.43 (m, 4H), 2.37 (t, 2H, J = 5.1 Hz), 2.21 (t, 2H, J = 5.6 Hz). 13C NMR (125 MHz, CDCl3): δ 149.2 (d, 1JCF = 240.8 Hz), 138.6, 137.3, 131.4, 129.3 (2C), 129.2 (d, 3JCF = 6.0 Hz), 128.4 (2C), 128.0 (2C), 127.6 (2C), 127.2, 118.9 (d, 2JCF = 18.0 Hz), 117.8 (d, 2JCF = 26.1 Hz), 63.2, 53.9 (d, 4JCF = 1.7 Hz), 53.8 (d, 4JCF = 1.9 Hz), 28.1 (d, 3JCF = 4.6 Hz), 25.9 (d, 3JCF = 7.8 Hz). 19F NMR (282 MHz, CDCl3): δ −115.90 (d, 3JHF = 24.4 Hz). HRMS (ESI) calcd for C21H23FN [M + H]+ 308.1809, found 308.1812.
Single-Crystal X-ray Diffraction for 3
The intensity data for 3 were measured on a KappaCCD diffractometer (graphite-monochromated Mo Ka radiation, λ = 0.71073 Å, f-w scans) at 100 (1) K. The data were corrected for absorption. Details of the solution and refinements for C13H14FNO2S2Si (3) are as follows. The crystals of 3, with approximate dimensions 0.060 × 0.24 × 0.40 mm, were triclinic with space group P-1. The final unit-cell constants of 3 were a = 6.9930(14) b = 8.7110(17), c = 13.512(3) Å, α = 74.66(3), β= 75.88(3), y = 85.75(3)°, V = 769.8(3) Å3, Z = 2, p = 1.413 g cm−1, μ= 0.433 mm−1, formula weight = 327.46. The structure of 3 was solved with SHELXS-97 and refined by full-matrix least squares on F2 with SHELXL-97. The hydrogen atoms were calculated with the riding model in the structure-factor calculations, but their parameters were not refined. The final discrepancy indices, 3.00 < q < 27.62°, were R = 0.0487 (calculated on F for 2681 reflections) and Rw = 0.1215 (calculated on F2 for all 3520 reflections) with 184 parameters varied. The major peaks of the final difference map are −0.46 and + 0.64 e Å3.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NSF Grant CHE-1058618 and by PSC CUNY awards. Infrastructural support was provided by NIH Grants 2G12RR03060-26A1 from the National Center for Research Resources and by 8G12MD007603-27 from the National Institute on Minority Health and Health Disparities. We thank Dr. Andrew Poss (Honeywell) for a sample of NFSI, Dr. Louis J. Todaro (The CUNY X-Ray Facility, Hunter College) for X-ray analysis, Ms. Nonka Sevova and Dr. Bill Boggess (University of Notre Dame, Mass Spectrometry and Proteomics Facility) for HRMS analyses supported by NSF Grant CHE-0741793.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Copies of 1H and 13C NMR spectra of 1–26, NOESY of the reduced derivative of 17 [(E)-(3-fluoropenta-2,4-dien-2-yl)benzene] and the cif file for 3. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- 1.For examples in natural product synthesis, see the following reviews: Thirsk C, Whiting A. J. Chem. Soc., Perkin Trans 1. 2002:999–1023. Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. Int. Ed. 2005;44:4442–4489. doi: 10.1002/anie.200500368. Shi Shun ALK, Tykwinski RR. Angew. Chem. Int. Ed. 2006;45:1034–1057. doi: 10.1002/anie.200502071.
- 2.(a) Meinwald J, Meinwald YC, Chalmers AM, Eisner T. Science. 1968;160:890–892. doi: 10.1126/science.160.3830.890. [DOI] [PubMed] [Google Scholar]; (b) Eisner T, Hill D, Goetz M, Jain S, Alsop D, Camazine S, Meinwald J. J. Chem. Ecol. 1981;7:1149–1158. doi: 10.1007/BF00987634. [DOI] [PubMed] [Google Scholar]
- 3.(a) Guerrero A, Camps F, Coll J, Riba M, Einhorn J, Descoins Ch, Lallemand JY. Tetrahedron Lett. 1981;22:2013–2016. [Google Scholar]; (b) Quero C, Malo EA, Fabriàs G, Camps F, Lucas P, Renou M, Guerrero A. J. Chem. Ecol. 1997;23:713–726. [Google Scholar]
- 4.Kinnel RB, Dieter RK, Meinwald J, Van Engen D, Clardy J, Eisner T, Stallard MO, Fenical W. Proc. Natl. Acad. Sci. USA. 1979;76:3576–3579. doi: 10.1073/pnas.76.8.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Estevez-Braun A, Estevez-Reyes R, Moujir LM, Ravelo AG, Gonzalez AG. J. Nat. Prod. 1994;57:1178–1182. doi: 10.1021/np50110a009. [DOI] [PubMed] [Google Scholar]; (b) Ashour ML, Wink M. J. Pharm. Pharmacol. 2011;63:305–321. doi: 10.1111/j.2042-7158.2010.01170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Gupta AK, Shear NH. J. Am. Acad. Dermatol. 1997;37:979–988. doi: 10.1016/s0190-9622(97)70076-8. [DOI] [PubMed] [Google Scholar]; (b) Abdel-Rahman SM, Nahata MC. Ann. Pharmacother. 1997;31:445–456. doi: 10.1177/106002809703100412. [DOI] [PubMed] [Google Scholar]; (C) Iverson SL, Uetrecht JP. Chem. Res. Toxicol. 2001;14:175–181. doi: 10.1021/tx0002029. [DOI] [PubMed] [Google Scholar]
- 7.(a) Negishi E-i, Anastasia L. Chem Rev. 2003;103:1979–2017. doi: 10.1021/cr020377i. [DOI] [PubMed] [Google Scholar]; (b) Doucet H, Hierso J-C. Angew. Chem. Int. Ed. 2007;46:834–871. doi: 10.1002/anie.200602761. [DOI] [PubMed] [Google Scholar]; (c) Chinchilla R, Nájera C. Chem Rev. 2007;107:874–922. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]; (d) Weibel J-M, Blanc A, Pale P. Chem Rev. 2008;108:3149–3173. doi: 10.1021/cr078365q. [DOI] [PubMed] [Google Scholar]; (e) Yamamoto Y. Chem Rev. 2008;108:3199–3222. doi: 10.1021/cr078359u. [DOI] [PubMed] [Google Scholar]; (f) Satoh T, Tsurugi H, Miura M. The Chemical Record. 2008;8:326–336. doi: 10.1002/tcr.20158. [DOI] [PubMed] [Google Scholar]
- 8.See for example the following for some applications of fluoroorganics; In agrochemistry: Jeschke P. ChemBioChem. 2004;5:570–589. doi: 10.1002/cbic.200300833. In bioorganic, medicinal and pharmaceutical chemistry. Berkowitz DB, Karukurichi KR, de la Salud-Bea R, Nelson DL, McCune CD. J. Fluorine Chem. 2008;129:731–742. doi: 10.1016/j.jfluchem.2008.05.016. Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008;37:320–330. doi: 10.1039/b610213c. Welch JT, editor. Selective Fluorination in Organic and Bioorganic Chemistry. Washington, DC: American Chemical Society; 1991. Bégué J-P, Bonnet-Delpon D. Bioorganic and Medicinal Chemistry of Fluorine. Hoboken, NJ: John Wiley & Sons, Inc; 2008. In materials chemistry. Berger R, Resnati G, Metrangolo P, Weber E, Hulliger J. Chem. Soc. Rev. 2011;40:3496–3508. doi: 10.1039/c0cs00221f.
- 9.(a) Tellier F, Sauvêtre R, Normant J-F. Tetrahedron Lett. 1986;27:3147–3148. [Google Scholar]; (b) Eddarir S, Francesch C, Mestdagh H, Rolando C. Bull. Soc. Chim. Fr. 1997;134:741–755. [Google Scholar]; (c) Yoshida M, Yoshikawa S, Fukuhara T, Yoneda N, Hara S. Tetrahedron. 2001;57:7143–7148. [Google Scholar]; (d) Eddarir S, Francesch C, Mestdagh H, Rolando C. Tetrahedron Lett. 1990;31:4449–4452. [Google Scholar]; (e) Percy JM, Wilkes RD. Tetrahedron. 1997;53:14749–14762. [Google Scholar]; (f) Zhang X, Burton DJ. J. Fluorine Chem. 2001;112:317–324. [Google Scholar]; (g) Xu J, Wang Y, Burton DJ. Org. Lett. 2006;8:2555–2558. doi: 10.1021/ol060785z. [DOI] [PubMed] [Google Scholar]; (h) Wang Y, Xu J, Burton DJ. J. Org. Chem. 2006;71:7780–7784. doi: 10.1021/jo061305k. [DOI] [PubMed] [Google Scholar]; (i) Yang Z-Y, Burton DJ. Tetrahedron Lett. 1990;31:1369–1372. [Google Scholar]; (j) Yang Z-Y, Burton DJ. J. Fluorine Chem. 1991;53:307–326. [Google Scholar]; (k) Ichikawa J, Ikeura C, Minami T. J. Fluorine Chem. 1993;63:281–285. [Google Scholar]
- 10.(a) Sanders TC, Golen JA, Williard PG, Hammond GB. J. Fluorine Chem. 1997;85:173–175. [Google Scholar]; (b) Benayoud F, Chen L, Moniz GA, Zapata AJ, Hammond GB. Tetrahedron. 1998;54:15541–15554. [Google Scholar]; (c) Zapata AJ, Gu Y, Hammond GB. J. Org. Chem. 2000;65:227–234. doi: 10.1021/jo991620p. [DOI] [PubMed] [Google Scholar]
- 11.(a) Camps F, Coll J, Fabriàs G, Guerrero A, Riba M. Experientia. 1984;40:933–934. [Google Scholar]; (b) Camps F, Fabriàs G, Guerrero A. Tetrahedron. 1986;42:3623–3629. [Google Scholar]; (c) Camps F, Fabriàs G, Gasol V, Guerrero A, Hernández R, Montoya R. J. Chem. Ecol. 1988;14:1331–1346. doi: 10.1007/BF01020138. [DOI] [PubMed] [Google Scholar]
- 12.(a) Smart BE. J. Fluorine Chem. 2001;109:3–11. [Google Scholar]; (b) Lemal DM. J. Org. Chem. 2004;69:1–11. doi: 10.1021/jo0302556. [DOI] [PubMed] [Google Scholar]; (c) O’Hagan D. Chem. Soc. Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- 13.(a) Baudin JB, Hareau G, Julia SA, Ruel O. Tetrahedron Lett. 1991;32:1175–1178. [Google Scholar]; (b) Baudin JB, Hareau G, Julia SA, Ruel O. Bull. Soc. Chim. Fr. 1993;130:336–357. [Google Scholar]; (c) Baudin JB, Hareau G, Julia SA, Lorne R, Ruel O. Bull. Soc. Chim. Fr. 1993;130:856–878. [Google Scholar]
- 14.(a) Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998:26–28. [Google Scholar]; (b) Kocienski PJ, Bell A, Blakemore PR. Synlett. 2000:365–366. [Google Scholar]
- 15.For reviews on Julia-Kocienski olefination see: Blakemore PR. J. Chem. Soc., Perkin Trans. I. 2002:2563–2585. Plesniak K, Zarecki A, Wicha J. Top. Curr. Chem. 2007;275:163–250. doi: 10.1007/128_049. Aïssa C. Eur. J. Org. Chem. 2009:1831–1844.
- 16.For a recent review on fluoro Julia-Kocienski olefination, see Zajc B, Kumar R. Synthesis. 2010:1822–1836. doi: 10.1055/s-0029-1218789.
- 17.(a) Ghosh AK, Zajc B. Org. Lett. 2006;8:1553–1556. doi: 10.1021/ol060002+. [DOI] [PubMed] [Google Scholar]; (b) Zajc B, Kake S. Org. Lett. 2006;8:4457–4460. doi: 10.1021/ol0616236. [DOI] [PubMed] [Google Scholar]; (c) He M, Ghosh AK, Zajc B. Synlett. 2008:999–1004. doi: 10.1055/s-2008-1072513. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) del Solar M, Ghosh AK, Zajc B. J. Org. Chem. 2008;73:8206–8211. doi: 10.1021/jo801235x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ghosh AK, Banerjee S, Sinha S, Kang SB, Zajc B. J. Org. Chem. 2009;74:3689–3697. doi: 10.1021/jo802784w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ghosh AK, Zajc B. J. Org. Chem. 2009;74:8531–8540. doi: 10.1021/jo901313k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kumar R, Pradhan P, Zajc B. Chem. Commun. 2011;47:3891–3893. doi: 10.1039/c0cc05083k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Mandal SK, Ghosh AK, Kumar R, Zajc B. Org. Biomol. Chem. 2012;10:3164–3167. doi: 10.1039/c2ob07031f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Chevrie D, Lequeux T, Demoute JP, Pazenok S. Tetrahedron Lett. 2003;44:8127–8130. [Google Scholar]; (b) Pfund E, Lebargy C, Rouden J, Lequeux T. J. Org. Chem. 2007;72:7871–7877. doi: 10.1021/jo070994c. [DOI] [PubMed] [Google Scholar]; (c) Alonso DA, Fuensanta M, Gómez-Bengoa E, Nájera C. Adv. Synth. Catal. 2008;350:1823–1829. [Google Scholar]; (d) Calata C, Catel J-M, Pfund E, Lequeux T. Tetrahedron. 2009;65:3967–3973. [Google Scholar]; (e) Calata C, Pfund E, Lequeux T. J. Org. Chem. 2009;74:9399–9405. doi: 10.1021/jo901540c. [DOI] [PubMed] [Google Scholar]; (f) Zhao Y, Huang W, Zhu L, Hu J. Org. Lett. 2010;12:1444–1447. doi: 10.1021/ol100090r. [DOI] [PubMed] [Google Scholar]; (g) Prakash GKS, Shakhmin A, Zibinsky M, Ledneczki I, Chacko S, Olah GA. J. Fluorine Chem. 2010;131:1192–1197. [Google Scholar]; (h) Allendörfer N, Es-Sayed M, Nieger M, Bräse S. Synthesis. 2010:3439–3448. [Google Scholar]; (i) Zhu L, Ni C, Zhao Y, Hu J. Tetrahedron. 2010;66:5089–5100. [Google Scholar]; (j) Calata C, Pfund E, Lequeux T. Tetrahedron. 2011;67:1398–1405. [Google Scholar]; (k) Jacobsen CB, Nielsen M, Worgull D, Zweifel T, Fisker E, Jørgensen KA. J. Am. Chem. Soc. 2011;133:7398–7404. doi: 10.1021/ja110624k. [DOI] [PubMed] [Google Scholar]
- 19.Bonini C, Chiummiento L, Videtta V. Synlett. 2006:2079–2082. [Google Scholar]
- 20.(a) Ye F, Ma X, Li H, Zhang Y, Wang J. J. Am. Chem. Soc. 2012;134:5742–5745. doi: 10.1021/ja3004792. [DOI] [PubMed] [Google Scholar]; (b) Xiao Q, Xia Y, Li H, Zhang Y, Wang J. Angew. Chem. Int. Ed. 2011;50:1114–1117. doi: 10.1002/anie.201005741. [DOI] [PubMed] [Google Scholar]
- 21.We have previously proposed that an alternate zwitterionic mechanistic pathway, favoring trans selectivity with aromatic aldehydes, is disfavored in the case of α-fluoro substituted sulfones, due to destabilized β-fluoro carbocation (see ref 16 and 17b). Absence of a zwitterionic pathway has also been suggested for unfluorinated propargyl sulfones, which give cis selectivity (Z-isomer) (see ref 13 and 15)
- 22.Neuvonen H, Neuvonen K, Koch A, Kleinpeter E, Pasanen P. J. Org. Chem. 2002;67:6995–7003. doi: 10.1021/jo020121c. [DOI] [PubMed] [Google Scholar]
- 23.Hoogboom J, Swager TM. J. Am. Chem. Soc. 2006;128:15058–15059. doi: 10.1021/ja065662o. [DOI] [PubMed] [Google Scholar]
- 24.(a) Gevorgyan V, Yamamoto Y. J. Organomet. Chem. 1999;576:232–247. [Google Scholar]; (b) Saito S, Yamamoto Y. Chem. Rev. 2000;100:2901–2915. doi: 10.1021/cr990281x. [DOI] [PubMed] [Google Scholar]
- 25.Wu T-S, Liou M-J, Lee C-J, Jong T-T, McPhail AT, McPhail DR, Lee K-H. Tetrahedron Lett. 1989;30:6649–6652. [Google Scholar]
- 26.(a) Ziegler CB, Jr., Heck RF. J. Org. Chem. 1978;43:2941–2946. [Google Scholar]; (b) Cabri W, Candiani I. Acc. Chem. Res. 1995;28:2–7. [Google Scholar]; (c) Beletskaya IP, Cheprakov AV. Chem. Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]; (d) Lagisetty P, Zhang L, Lakshman MK. Adv. Synth. Catal. 2008;350:602–608. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.