

Abstract
Endocrine disrupting chemicals that are structurally similar to steroid or amine hormones have the potential to mimic endocrine endpoints at the receptor level. However, more recently, epigenetic-induced alteration in gene expression has emerged as an alternative way in which environmental compounds may exert endocrine effects. We review concepts related to environmental epigenetics and relevance for endocrinology through three broad examples, 1) effect of early-life nutritional exposures on future obesity and insulin resistance, 2) effect of lifetime environmental exposures such as ionizing radiation on endocrine cancer risk, and 3) potential for compounds previously classified as endocrine disrupting to additionally or alternatively exert effects through epigenetic mechanisms. The field of environmental epigenetics is still nascent, and additional studies are needed to confirm and reinforce data derived from animal models and preliminary human studies. Current evidence suggests that environmental exposures may significantly impact expression of endocrine-related genes and thereby affect clinical endocrine outcomes.
Keywords: Epigenetics, DNA methylation, histone modifications, toxicology, endocrine disrupters
Introduction
In the 1970s, diethylstilbestrol (DES), an estrogen agonist used to prevent miscarriages, was associated with vaginal clear cell adenocarcinoma in adult female offspring exposed in utero (Robboy, et al. 1977). Mothers who took DES, however, did not appear to be at increased risk for this adenocarcinoma, suggesting that in utero programming may be a mechanism for this observation. Since then, interest in studying endocrine effects of exogenous agents, and their role in epigenetic programming, has increased dramatically.
Exogenous compounds that alter “hormonal and homeostatic systems” (n.— endocrine endpoints) have been termed “endocrine disrupting chemicals” and were officially recognized in an Endocrine Society statement in 2009 (Diamanti-Kandarakis, et al. 2009). The majority of currently identified endocrine disruptors are synthetic industrial chemicals, rather than drugs. In the US, although the Toxic Substances Control Act (TSCA) of 1976 provides a legislative framework to limit the spread of toxic chemicals, companies are not required to perform monitoring for adverse health effects of product chemicals. Toxicity is typically discovered only after the product is widely used. For this reason, many chemicals with suspected or even confirmed endocrine disruption properties are in current daily use.
Many endocrine disrupting chemicals have structural similarity to steroid or amine hormones. Acting as ligands, they can either activate or antagonize the hormone’s receptor, leading to altered endocrine endpoints (Diamanti- Kandarakis et al. 2009). However, more recently, the classification of endocrine disrupting chemicals has expanded. Agents such as heavy metals that do not act directly on steroid receptors have been shown to alter hormone metabolism via epigenetic alterations. Also, compounds such as DES that are structurally similar to hormones (Fig. 1) may induce epigenetic changes through interaction with hormone receptors.
Figure 1.

Many endocrine disrupting chemicals such as DES have structural similarity to steroid hormones such as estrogen.
What is Epigenetics?
Epigenetics is defined as changes in gene expression that occur without changes in DNA sequence (Wolffe and Guschin 2000) and can be transmitted through mitosis and/or meiosis. Given that all cells in the human body contain the same DNA sequence, epigenetics can be thought of as those processes that regulate gene expression in a given cell leading to its cellular phenotype, a definition first proposed by CH Waddington (Waddington 1942). One of the more intriguing facets of epigenetics is the responsiveness of many epigenetic marks to environmental factors.
The hereditary component of complex, multifactorial processes such as obesity (Lindgren, et al. 2009), diabetes (Schafer, et al. 2011; Voight, et al. 2010), osteoporosis (Duncan, et al. 2011), and pubertal timing (He, et al. 2009; Perry, et al. 2009), cannot be fully explained by DNA sequence alone. Epigenetics offers a complementary mechanism for heredity that is independent of DNA sequence variation.
Epigenetics involves several types of ‘marks’ added to either DNA or chromatin leading to transient or persistent changes in gene transcription. Each organism has a unique epigenetic signature that is partially inherited and partially created in utero and continues to be modified throughout adult life. Epigenetic marks may arise from environmental exposures that in turn interact with genotype to produce changes in gene expression, or epigenetics may explain health outcomes independent of genotype as these marks alone can either turn off or turn on gene expression, functioning as an “epimutation”. Two of the most widely studied epigenetic changes are DNA methylation and histone modifications.
DNA Methylation
DNA methylation is the addition of a methyl group to a cytosine (C)nucleotide at position 5 and typically occurs when a cytosine is positioned next to a guanine(G). Phosphates(P) link nucleosides in DNA, and, thus, this particular arrangement is termed a CpG dinucleotide. Regions of the genome dense in CpG dinucleotides are termed CpG islands. Although CpG islands have the potential for methylation and are frequently overrepresented at gene promoters, the majority of the time, they are not methylated (Jirtle and Skinner 2007). Regions with lower CpG density bordering the CpG islands are termed CpG shores and have been proposed to occur with increased frequency in regulatory sites involved in tissue differentiation (Doi, et al. 2009).
Tissue-specific methylation of CpG dinucleotides by DNA methyltransferases (DNMTs) can lead to gene silencing (Orphanides and Reinberg 2002; Wolffe and Matzke 1999). The simplest way to understand DNA methylation is to think of DNA not as strand, but as a coil wrapped tightly around a histone. Methylation leads to gene silencing because it alters DNA’s three-dimensional structure such that the coil becomes tighter at the locus corresponding to the promoter region. Within this tightened coil, transcription factors can no longer be recruited to their binding sites. Also, methyl binding proteins (MBPs) can interact with methylated CpGs and actively repress gene transcription (Bird and Wolffe 1999).
There is evidence that physiologic DNMT activity is under hormonal control. For example, DNMT1 and MBP levels vary with menstrual cycle phase and with estrogen and progesterone secretion in endometrial explant tissues (van Kaam, et al. 2011). Thus, based on their analogy to steroid hormones, some endocrine disrupting compounds have the potential to affect DNMT activity and consequently affect epigenetic marks.
Histone Modifications
Histones are globular proteins around which DNA is packaged to make chromatin. Enzyme modifications such as acetylation and methylation of lysine residues in the amino terminus lead to a histone conformational change. Acetylation leads to increased DNA accessibility, and methylation can either increase or decrease DNA accessibility depending upon the specific type of methylation and histone affected (Yan and Boyd 2006). DNA methylation and histone modification often work in tandem, as MBPs recruited by DNA methylation may exert their effects through recruitment of histone deacetylases (HDACs) resulting in chromatin condensation and transcriptional inactivation (Jones, et al. 1998; Nan, et al. 1998).
Epigenetic Reprogramming and Inheritance
Epigenetic patterns undergo erasure and reprogramming two times during the human lifecycle. One important result of epigenetic reprogramming is the correct establishment of imprinting at sites with allele-specific methylation. Independent of gender, human adult somatic cells contain one haploid set of chromosomes inherited from the mother that carry female-specific imprints, and a second haploid set of chromosomes inherited from the father that carry male-specific imprints. The first phase of erasure and reprogramming occurs during gametogenesis. In the primordial germline, DNA methylation – including methylation at imprinted loci – is erased and later reestablished. At imprinted loci, DNA methylation is reprogrammed so that it will take female-specific imprints in oocytes and male-specific imprints in spermatozoa. The second phase of epigenetic erasure and programming occurs during pre-implantation when the genome, with the possible exception of imprinted genes and some retrotransposons, becomes demethylated. After implantation, DNA methylation is restored de novo and rapidly acquires cell lineage-specific patterns to drive cell differentiation. This is the basis for the tissue-specific gene methylation pattern seen after birth and through adulthood. (Perera and Herbstman 2011; Reik, et al. 2001; Shi and Wu 2009). Folate and B12 serve as exogenous methyl donors, and their influence on DNA methylation during the in utero time period will be discussed in more detail later in this manuscript.
The prenatal erasure and reprogramming of DNA methylation patterns makes the in utero time period a window of potential vulnerability for epigenetic dysregulation from environmental exposures. This is particularly relevant in endocrinology where there is burgeoning evidence that the fetal environment may program adult outcomes such as obesity and type 2 diabetes mellitus (Law, et al. 1992; Stocker, et al. 2005).
Other potential vulnerable windows for epigenetic dysregulation that might affect endocrine systems include puberty, during which time there is an overall rapid increase in DNA turnover and cell growth, as well as old age, which has been associated with progressive age-related changes in DNA methylation (Bjornsson, et al. 2008) (Fig. 2).
Figure 2.
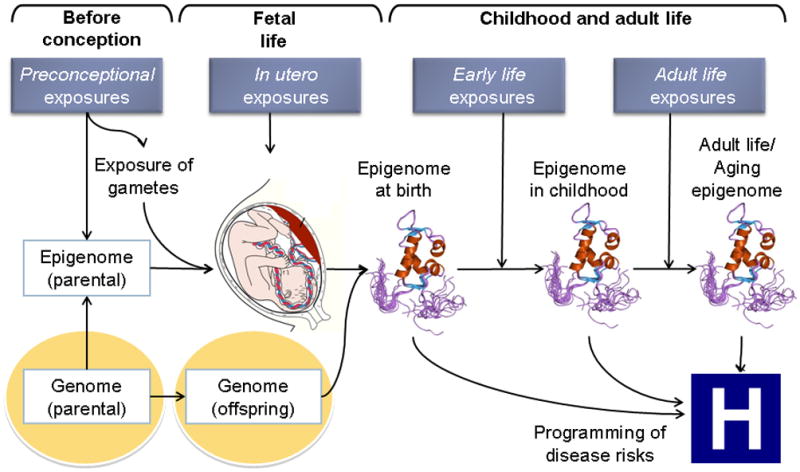
Exposures that occur preconception, in utero, in early life, and in adult life may result in epigenetic dysregulation.
Epigene-Environment Interaction
Epigenetic dysregulation can result from environmental exposures including dietary factors, physical activity, social stressors, and environmental toxicants (Alegria-Torres, et al. 2011; Mathers, et al. 2010). However, there is a paucity of human studies that document the causal pathway from environmental exposure to epigenetic modification to clinical outcome (Fig. 3).
Figure 3.

There are multiple potential causal pathways through which an environmental exposure may lead to epigenetic modifications which may, in turn, impact clinical outcomes. Additional human studies that extend over the entire causal pathway are needed to establish cause-and-effect relationships between exposures and outcomes.
We have chosen to demonstrate these themes by reviewing three broad examples where environmental epigenetics has impacted endocrinology. These include 1) effect of early-life nutritional exposures on future obesity and insulin resistance, 2) effect of lifetime environmental exposures such as ionizing radiation on endocrine cancer risk, and 3) potential for endocrine disrupting compounds to affect endocrine endpoints through epigenetic modifications. We review available data and suggest avenues for future research.
Nutritional status and epigenetic changes
Poor nutrition during pregnancy has been associated with DNA hypomethylation in offspring, which may result from a decrease in dietary sources of methyl group donors such as folate, methionine, and choline in conjunction with decreased availability of B vitamins (B2, B6, and B12)(Mathers et al. 2010). Plasma homocysteine level, an inverse marker of folate supplementation, has been associated with LINE-1 hypomethylation, gene-specific CpG island methylation patterns, and lower birthweight percentile (Fryer, et al. 2011; Fryer, et al. 2009). Also, offspring of humans exposed to famine in early gestation had hypomethylation of the imprinted IGF2 gene as compared to unexposed siblings (Heijmans, et al. 2008). Parental overfeeding has also been associated with epigenetic modifications in offspring. In humans, paternal insulin resistance, presumably a result of overeating and obesity, was associated with low infant birth weight and increased risk of diabetes in offspring (Hypponen, et al. 2003; Lindsay, et al. 2000). Similarly, in rodents, female offspring of high fat diet (HFD) fathers had lower birth weight and developed glucose intolerance, impaired insulin secretion, and decreased pancreatic islet and β–cell mass as compared to control offspring. Il13ra2, part of the Jak-Stat signaling pathway, was hypomethylated and upregulated in HFD offspring, suggesting that a HFD may have affected the epigenetic profile of the paternal germ cells (Ng, et al. 2010).
More recently, in a related study in humans, hypermethylation of the RXRA gene in umbilical cord tissue was associated with lower maternal carbohydrate intake during early pregnancy and later childhood adiposity. RXRA may be involved in insulin sensitivity, adipogenesis, and fat metabolism based on its interaction with PPAR when serving as a transcription factor (Godfrey, et al. 2011). These rodent and human studies suggest that parental nutritional status affects the DNA methylation profile and subsequent obesity and insulin resistance in offspring.
Epigenetics and Endocrine Cancers
Abundant literature exists linking environmentally-induced epigenetic modifications to tumor formation and progression in non-endocrine cancers through altered expression of tumor-suppressor genes and proto-oncogenes. For example, benzene exposure has been associated with increased risk of acute myelogenous leukemia (AML)(2005). Individuals with higher than average benzene exposure had a significant reduction in methylation of selected genomic repetitive elements, in conjunction with hypermethylation of tumor-suppressor gene p15 and hypomethylation of the MAGE-1 gene, suggesting epigenetic modification as a possible pathogenic mechanism (Bollati, et al. 2007).
With regard to endocrine-related cancers, epigenetic modifications have also been implicated in thyroid carcinoma via hypermethylation and inactivation of tumor suppressor genes including cyclin-dependent kinase inhibitor p16INK4A, microtubule stabilizer RASSF1A, GTPase-activating protein Rap1GAP, and PI3K/akt pathway modulator PTEN, (Russo, et al. 2011). The PTEN promoter is hypermethylated in 50% of papillary carcinomas and almost 100% of follicular carcinomas, which is notable because a deleterious mutation in this gene causes PTEN hamartoma tumor syndrome leading to increased risk of papillary and thyroid carcinomas (Hobert and Eng 2009). As far as a potential environmental trigger for this epigenetic profile, ionizing radiation exposure has traditionally been thought to increase risk of thyroid cancer as a result of DNA mutagenesis, for example, through RET/PTC rearrangements (Caudill, et al. 2005; Christodouleas, et al. 2011). However, ionizing radiation has also been associated with global DNA hypomethylation that may be genotype-specific (Giotopoulos, et al. 2006) and that even occurs in cells not directly irradiated (Tamminga, et al. 2008). Prior ionizing radiation has also been associated with gene-specific hypermethylation in cancer patients (Bennett, et al. 2010; Figueroa, et al. 2009; Mathers et al. 2010). Additional research is needed to establish whether ionizing radiation leads to specific epigenetic changes seen in thyroid carcinoma and subsequent thyroid carcinoma development.
Pituitary adenomas have also been associated with hypermethylation-related silencing of tumor-suppressor genes such as RB1 and cyclin-dependent kinase inhibitors p15 and p16 (reviewed in Vandeva, et al. 2010). In some studies, growth hormone- secreting adenomas have resulted in tissue-specific loss of imprinting at the paternal stimulatory G-protein allele, which leads to increased expression of GNAS and constitutive activation of adenylyl cyclase (Hayward, et al. 2001; reviewed in Mantovani, et al. 2010). As imprinting is an early embryonic process, in utero exposure may be responsible for dysregulated imprinting and subsequent development of growth hormone- secreting adenomas; however, this has not yet been evaluated.
Endocrine Disruptors and Epigenetics
A growing number of animal models have linked prenatal endocrine disruptor exposure to offspring epigenetic modifications. Bisphenol A (BPA), a synthetic chemical and weak estrogen agonist found in food and beverage containers (Le, et al. 2008), baby bottles (Nam, et al. 2010), and dental materials (Fleisch, et al. 2010), induced hypomethylation and increased expression of the Agouti gene in prenatally-exposed mice leading to yellow rather than brown fur, as well as obesity, diabetes, and tumorigenesis (Dolinoy 2008). Furthermore, rodent mothers with the agouti phenotype were more likely to have offspring with that phenotype in the second generation. This important study serves as proof of principle that prenatal exposure to synthetic estrogen agonists such as BPA can affect the epigenome and thereby lead to endocrinologic sequelae.
Diethylstilbestrol (DES), an estrogen agonist found to cause vaginal clear-cell adenocarcinoma in the female offspring of users, has been associated with epigenetic modifications in offspring uterine tissues. Specifically, in mouse models of prenatal DES exposure and resultant genital tract neoplasia, hypomethylation occurred along with increased uterine expression of estrogen-sensitive LTF and proto-oncogenes including EGF and SRF (Falck and Forsberg 1996; Li, et al. 1997; Nelson, et al. 1994).
Exposure to vinclozolin, a fungicide and an antiandrogen, during embryogenesis decreased adult sperm motility and concentration and increased the rate of kidney and prostate disease, immune system abnormalities, hypercholesterolemia, and tumorigenesis in rat first-generation male offspring. This phenotype was transferred through four generations (F1–F4) of male offspring and was associated with alterations in the sperm methylation profile of the F1–F3 generations (Anway, et al. 2005). This is one of the first studies to suggest the potential for transgenerational inheritance of epigenetic marks presumably resulting, in this case, from incomplete erasure of epigenetic marks during gametogenesis. However, similar studies attempting to replicate the above findings in vinclozoilin-exposed mice demonstrated changes in sperm methylation profile in the F1 and F2, but not the F3 generation (Inawaka, et al. 2009; Schneider, et al. 2008; Stouder and Paoloni-Giacobino 2010) suggesting that the phenotype of the F2 generation may be due to exposure of the F1 germline to the maternal environment rather than true transgenerational inheritance.
Future Directions
Environmental epigenetics offers several clear research opportunities, particularly as it relates to endocrinology, including the following:
Human studies of fetal epigenetic vulnerability to environmental exposures at physiologically relevant doses will confirm existing animal models.
Human observational studies able to show the relationship from exposure to epigenetic modification to outcome over a lifespan will be required to differentiate causality from mere association.
Additional endocrine tissue- and gene-specific studies of epigenetic modifications will further delineate causality.
Finally, integrated teamwork including clinical endocrinologists, environmental toxicologists and epidemiologists will strengthen study design and thereby lead to heightened understanding of the impact of environmental epigenetics on endocrinology.
Conclusions
Although the field of environmental epigenetics is just moving out of its infancy, it has already begun to demonstrate the breadth of potential impact of environmental exposures on the expression of endocrine-related genes. These results not only suggest the need for increased biomonitoring of synthetic compounds, but also point out specific windows of human susceptibility as well as potential mechanisms that could represent the substrate for future preventive interventions.
Acknowledgments
Funding
The authors have received support from the following grants from the US National Institutes of Health: P30ES000002, R21ES019773, R21ES020010, R01ES020268, R01ES013744, and P42ES016454; and have received support from the following grant from AHRQ: T32 HS00063.
Abbreviations
- DES
Diethylstilbestrol
- TSCA
Toxic Substances Control Act
- DNMT
DNA methyltransferase
- MBPs
methyl binding proteins
- CpG
cytosine-phosphate-guanine
- HDACs
histone deacetylases
- HFD
high fat diet
- BPA
bisphenol A
Footnotes
Declaration of Interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Chem Biol Interact; Proceedings of the International Symposium on Recent Advances in Benzene Toxicity; Munich, Germany. 9–12 October 2004; 2005. pp. 1–270. [PubMed] [Google Scholar]
- Alegria-Torres JA, Baccarelli A, Bollati V. Epigenetics and lifestyle. Epigenomics. 2011;3:267–277. doi: 10.2217/epi.11.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett KL, Lee W, Lamarre E, Zhang X, Seth R, Scharpf J, Hunt J, Eng C. HPV status-independent association of alcohol and tobacco exposure or prior radiation therapy with promoter methylation of FUSSEL18, EBF3, IRX1, and SEPT9, but not SLC5A8, in head and neck squamous cell carcinomas. Genes Chromosomes Cancer. 2010;49:319–326. doi: 10.1002/gcc.20742. [DOI] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP. Methylation-induced repression--belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekstrom TJ, Harris TB, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67:876–880. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- Caudill CM, Zhu Z, Ciampi R, Stringer JR, Nikiforov YE. Dose-dependent generation of RET/PTC in human thyroid cells after in vitro exposure to gamma-radiation: a model of carcinogenic chromosomal rearrangement induced by ionizing radiation. J Clin Endocrinol Metab. 2005;90:2364–2369. doi: 10.1210/jc.2004-1811. [DOI] [PubMed] [Google Scholar]
- Christodouleas JP, Forrest RD, Ainsley CG, Tochner Z, Hahn SM, Glatstein E. Short-term and long-term health risks of nuclear-power-plant accidents. N Engl J Med. 2011;364:2334–2341. doi: 10.1056/NEJMra1103676. [DOI] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinoy DC. The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr Rev. 2008;66(Suppl 1):S7–11. doi: 10.1111/j.1753-4887.2008.00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan EL, Danoy P, Kemp JP, Leo PJ, McCloskey E, Nicholson GC, Eastell R, Prince RL, Eisman JA, Jones G, et al. Genome-wide association study using extreme truncate selection identifies novel genes affecting bone mineral density and fracture risk. PLoS Genet. 2011;7:e1001372. doi: 10.1371/journal.pgen.1001372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck L, Forsberg JG. Immunohistochemical studies on the expression and estrogen dependency of EGF and its receptor and C-fos proto-oncogene in the uterus and vagina of normal and neonatally estrogen-treated mice. Anat Rec. 1996;245:459–471. doi: 10.1002/(SICI)1097-0185(199607)245:3<459::AID-AR2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, Paietta E, Fernandez H, Tallman MS, Greally JM, Carraway H, et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood. 2009;114:3448–3458. doi: 10.1182/blood-2009-01-200519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisch AF, Sheffield PE, Chinn C, Edelstein BL, Landrigan PJ. Bisphenol A and related compounds in dental materials. Pediatrics. 2010;126:760–768. doi: 10.1542/peds.2009-2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer AA, Emes RD, Ismail KM, Haworth KE, Mein C, Carroll WD, Farrell WE. Quantitative, high-resolution epigenetic profiling of CpG loci identifies associations with cord blood plasma homocysteine and birth weight in humans. Epigenetics. 2011;6:86–94. doi: 10.4161/epi.6.1.13392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer AA, Nafee TM, Ismail KM, Carroll WD, Emes RD, Farrell WE. LINE-1 DNA methylation is inversely correlated with cord plasma homocysteine in man: a preliminary study. Epigenetics. 2009;4:394–398. doi: 10.4161/epi.4.6.9766. [DOI] [PubMed] [Google Scholar]
- Giotopoulos G, McCormick C, Cole C, Zanker A, Jawad M, Brown R, Plumb M. DNA methylation during mouse hemopoietic differentiation and radiation-induced leukemia. Exp Hematol. 2006;34:1462–1470. doi: 10.1016/j.exphem.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, Rodford J, Slater-Jefferies JL, Garratt E, Crozier SR, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011;60:1528–1534. doi: 10.2337/db10-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward BE, Barlier A, Korbonits M, Grossman AB, Jacquet P, Enjalbert A, Bonthron DT. Imprinting of the G(s)alpha gene GNAS1 in the pathogenesis of acromegaly. J Clin Invest. 2001;107:R31–36. doi: 10.1172/JCI11887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Kraft P, Chen C, Buring JE, Pare G, Hankinson SE, Chanock SJ, Ridker PM, Hunter DJ, Chasman DI. Genome-wide association studies identify loci associated with age at menarche and age at natural menopause. Nat Genet. 2009;41:724–728. doi: 10.1038/ng.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med. 2009;11:687–694. doi: 10.1097/GIM.0b013e3181ac9aea. [DOI] [PubMed] [Google Scholar]
- Hypponen E, Smith GD, Power C. Parental diabetes and birth weight of offspring: intergenerational cohort study. BMJ. 2003;326:19–20. doi: 10.1136/bmj.326.7379.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inawaka K, Kawabe M, Takahashi S, Doi Y, Tomigahara Y, Tarui H, Abe J, Kawamura S, Shirai T. Maternal exposure to anti-androgenic compounds, vinclozolin, flutamide and procymidone, has no effects on spermatogenesis and DNA methylation in male rats of subsequent generations. Toxicol Appl Pharmacol. 2009;237:178–187. doi: 10.1016/j.taap.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Law CM, Barker DJ, Osmond C, Fall CH, Simmonds SJ. Early growth and abdominal fatness in adult life. J Epidemiol Community Health. 1992;46:184–186. doi: 10.1136/jech.46.3.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le HH, Carlson EM, Chua JP, Belcher SM. Bisphenol A is released from polycarbonate drinking bottles and mimics the neurotoxic actions of estrogen in developing cerebellar neurons. Toxicol Lett. 2008;176:149–156. doi: 10.1016/j.toxlet.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Washburn KA, Moore R, Uno T, Teng C, Newbold RR, McLachlan JA, Negishi M. Developmental exposure to diethylstilbestrol elicits demethylation of estrogen-responsive lactoferrin gene in mouse uterus. Cancer Res. 1997;57:4356–4359. [PubMed] [Google Scholar]
- Lindgren CM, Heid IM, Randall JC, Lamina C, Steinthorsdottir V, Qi L, Speliotes EK, Thorleifsson G, Willer CJ, Herrera BM, et al. Genome-wide association scan meta-analysis identifies three Loci influencing adiposity and fat distribution. PLoS Genet. 2009;5:e1000508. doi: 10.1371/journal.pgen.1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay RS, Dabelea D, Roumain J, Hanson RL, Bennett PH, Knowler WC. Type 2 diabetes and low birth weight: the role of paternal inheritance in the association of low birth weight and diabetes. Diabetes. 2000;49:445–449. doi: 10.2337/diabetes.49.3.445. [DOI] [PubMed] [Google Scholar]
- Mantovani G, Lania AG, Spada A. GNAS imprinting and pituitary tumors. Mol Cell Endocrinol. 2010;326:15–18. doi: 10.1016/j.mce.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Mathers JC, Strathdee G, Relton CL. Induction of epigenetic alterations by dietary and other environmental factors. Adv Genet. 2010;71:3–39. doi: 10.1016/B978-0-12-380864-6.00001-8. [DOI] [PubMed] [Google Scholar]
- Nam SH, Seo YM, Kim MG. Bisphenol A migration from polycarbonate baby bottle with repeated use. Chemosphere. 2010;79:949–952. doi: 10.1016/j.chemosphere.2010.02.049. [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Nelson KG, Sakai Y, Eitzman B, Steed T, McLachlan J. Exposure to diethylstilbestrol during a critical developmental period of the mouse reproductive tract leads to persistent induction of two estrogen-regulated genes. Cell Growth Differ. 1994;5:595–606. [PubMed] [Google Scholar]
- Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature. 2010;467:963–966. doi: 10.1038/nature09491. [DOI] [PubMed] [Google Scholar]
- Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002;108:439–451. doi: 10.1016/s0092-8674(02)00655-4. [DOI] [PubMed] [Google Scholar]
- Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol. 2011;31:363–373. doi: 10.1016/j.reprotox.2010.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JR, Stolk L, Franceschini N, Lunetta KL, Zhai G, McArdle PF, Smith AV, Aspelund T, Bandinelli S, Boerwinkle E, et al. Meta-analysis of genome-wide association data identifies two loci influencing age at menarche. Nat Genet. 2009;41:648–650. doi: 10.1038/ng.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Robboy SJ, Scully RE, Welch WR, Herbst AL. Intrauterine diethylstilbestrol exposure and its consequences: pathologic characteristics of vaginal adenosis, clear cell adenocarcinoma, and related lesions. Arch Pathol Lab Med. 1977;101:1–5. [PubMed] [Google Scholar]
- Russo D, Damante G, Puxeddu E, Durante C, Filetti S. Epigenetics of thyroid cancer and novel therapeutic targets. J Mol Endocrinol. 2011;46:R73–81. doi: 10.1530/JME-10-0150. [DOI] [PubMed] [Google Scholar]
- Schafer SA, Machicao F, Fritsche A, Haring HU, Kantartzis K. New type 2 diabetes risk genes provide new insights in insulin secretion mechanisms. Diabetes Res Clin Pract. 2011;93(Suppl 1):S9–24. doi: 10.1016/S0168-8227(11)70008-0. [DOI] [PubMed] [Google Scholar]
- Schneider S, Kaufmann W, Buesen R, van Ravenzwaay B. Vinclozolin--the lack of a transgenerational effect after oral maternal exposure during organogenesis. Reprod Toxicol. 2008;25:352–360. doi: 10.1016/j.reprotox.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Shi L, Wu J. Epigenetic regulation in mammalian preimplantation embryo development. Reprod Biol Endocrinol. 2009;7:59. doi: 10.1186/1477-7827-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker CJ, Arch JR, Cawthorne MA. Fetal origins of insulin resistance and obesity. Proc Nutr Soc. 2005;64:143–151. doi: 10.1079/pns2005417. [DOI] [PubMed] [Google Scholar]
- Stouder C, Paoloni-Giacobino A. Transgenerational effects of the endocrine disruptor vinclozolin on the methylation pattern of imprinted genes in the mouse sperm. Reproduction. 2010;139:373–379. doi: 10.1530/REP-09-0340. [DOI] [PubMed] [Google Scholar]
- Tamminga J, Koturbash I, Baker M, Kutanzi K, Kathiria P, Pogribny IP, Sutherland RJ, Kovalchuk O. Paternal cranial irradiation induces distant bystander DNA damage in the germline and leads to epigenetic alterations in the offspring. Cell Cycle. 2008;7:1238–1245. doi: 10.4161/cc.7.9.5806. [DOI] [PubMed] [Google Scholar]
- van Kaam KJ, Delvoux B, Romano A, D’Hooghe T, Dunselman GA, Groothuis PG. Deoxyribonucleic acid methyltransferases and methyl-CpG-binding domain proteins in human endometrium and endometriosis. Fertil Steril. 2011;95:1421–1427. doi: 10.1016/j.fertnstert.2011.01.031. [DOI] [PubMed] [Google Scholar]
- Vandeva S, Jaffrain-Rea ML, Daly AF, Tichomirowa M, Zacharieva S, Beckers A. The genetics of pituitary adenomas. Best Pract Res Clin Endocrinol Metab. 2010;24:461–476. doi: 10.1016/j.beem.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington C. The Epigenome. Endeavour. 1942;1:18–20. [Google Scholar]
- Wolffe AP, Guschin D. Review: chromatin structural features and targets that regulate transcription. J Struct Biol. 2000;129:102–122. doi: 10.1006/jsbi.2000.4217. [DOI] [PubMed] [Google Scholar]
- Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286:481–486. doi: 10.1126/science.286.5439.481. [DOI] [PubMed] [Google Scholar]
- Yan C, Boyd DD. Histone H3 acetylation and H3 K4 methylation define distinct chromatin regions permissive for transgene expression. Mol Cell Biol. 2006;26:6357–6371. doi: 10.1128/MCB.00311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]