

Abstract
We have designed β-strand peptides (BP) that stabilize integral membrane proteins (IMP). BPs self-assemble in solution as filaments and become restructured upon association with IMPs; the resulting IMP/BP complexes resist aggregation when diluted in detergent-free buffer and are examined as stable, single particles with low detergent background by electron microscopy. This enables clear visualization of a spectrum of flexible conformations in the highly dynamic ATP-binding cassette (ABC) transporter MsbA.
Integral membrane proteins (IMPs), a third of encoded proteins in genomes, perform essential functions as receptors, transporters and channels. The hydrophobic nature of IMPs requires their solubilization as isolated stable and functional particles, but also presents a significant challenge for many biochemical and biophysical studies. The development of novel amphiphilic reagents, including protein-based nanodiscs1, amphiphilic polymers2, peptide-based detergents3–5, and MNG detergents6, among others7, has greatly facilitated the functional and structural studies of IMPs. However, each type of amphiphile has its own limitations, and no universal reagent has been developed to facilitate general application in biophysical studies (e.g. X-ray crystallography, NMR, EM etc.).
β-sheet peptide assemblies, such as β-barrel proteins in the outer membrane of Gram-negative bacteria8, are characterized with high thermodynamic stability due to extensive hydrogen bond (H-bond) interactions between neighboring strands. This exceptional structural stability has attracted interest in de novo design of β-sheet peptides or proteins to address specific biotechnological applications. Here we demonstrate the successful design of short amphiphilic β-strand peptides (BP) that improve the stability of IMPs in solution. The designed BPs self-assemble into filamentous structures that disperse when mixed with IMPs and detergents, resulting in highly stabilized IMPs which are readily visualized as individual particles by electron microscopy (EM). This new approach allowed us to directly visualize multiple conformations of the ATP-binding cassette (ABC) exporter MsbA in a single EM preparation, thus providing new opportunities to investigate the structural dynamics of this family of proteins.
Toward the goal of stabilization and structural studies of IMPs, we designed three BPs, with the general sequence Ac-(Oct)Gly-Ser-Leu-Ser-Leu-Asp-(Oct)Gly-Asp-NH2 (Fig. 1a), to include the following notable features: (1) alternate hydrophobic and hydrophilic residues to confer facial amphiphilicity in an extended, β-strand conformation9; (2) a short, eight amino acid sequence of appropriate length to span the central nonpolar region of membrane bilayers (~3 nm)8; (3) elongated alkyl side chains at each end of the peptide to increase hydrophobicity, a modification important for IMP stabilization as implicated in lipopeptide detergent design3; and (4) incorporation of N-methyl (N-Me) amino acids that diminish H-bond donors to regulate interstrand association and avoid the formation of insoluble macroscopic fibril structures10, 11. It is notable that the same sequence lacking N-Me amino acids was barely soluble in aqueous solution and most organic solvents, making it difficult to purify as a result of its tendency to aggregate due to strong H-bond interactions, whereas the three analogous peptides (BP-1, BP-2 and BP-3, with one, two and three N-Me substituents, respectively) showed moderate water solubility (~3 mM for BP-1 and BP-2, and ~1 mM for BP-3). Overall, these BPs, distinct from a majority of amphiphiles, were designed to sequester IMP hydrophobic surfaces by forming an ordered, stabilizing β-barrel-like structure, and to decrease dynamic dissociation from IMP surfaces by introducing intermolecular H-bond interactions (Fig. 1b).
Figure 1.
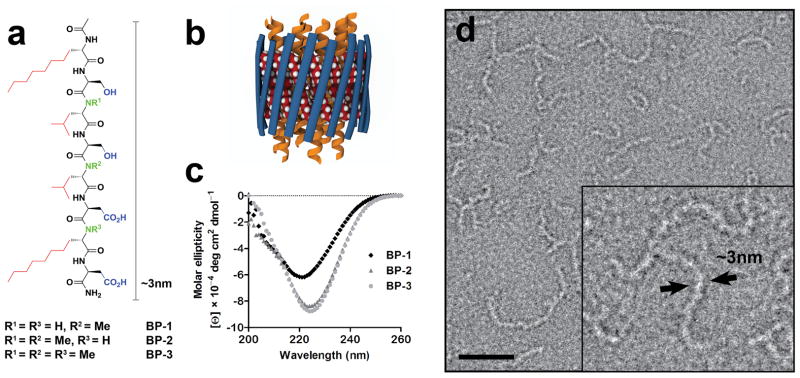
Structures of β-strand peptides designed to stabilize IMPs. (a) Designed BP sequences (BP-1, 2, and 3) feature facial amphiphilicity with alternate hydrophobic (red) and hydrophilic (blue) residues, and differ in the number of N-Me amino acids (green). (b) A cartoon representation of proposed β-barrel architecture assembled from BPs (blue strands) by interstrand H-bonding in which the hydrophobic alkyl chains (space filling spheres) associate with and sequester the IMP surfaces (yellow α-helices). (c) CD spectra of BP-1, 2 and 3 indicate secondary structures with β-sheet character. (d) Electron micrographs of negatively stained BP-1 (BP-2 and BP-3 shown in Supplementary Fig. 2) show self-assembled filamentous structures ~3 nm in diameter (inset). The scale bar represents 30 nm.
Circular dichroism (CD) spectroscopic analysis of the BPs indicated the formation of β-sheet secondary structures, with ellipticity minima at ~222 nm for BP-1, and ~225 nm for BP-2 and BP-3 (Fig. 1c). These apparent redshifts from the typical β-sheet ellipticity minimum at ~218 nm can be attributed to a conformational twist or distortion due to the N-methylated peptide backbone11, 12. Fourier transform infrared spectroscopic analysis of BP-1 also indicated mainly β-sheet secondary structures (Supplementary Fig. 1). Electron micrographs of negatively stained BPs revealed elongated, flexible filaments ~3 nm across (Fig. 1d and Supplementary Fig. 2), consistent with the dimensions of the designed peptides. EM micrographs of serial dilutions of BP-1 showed that the number of filaments correlated with the dilution, consistent with the BP-1 filaments being present in solution (Supplementary Fig. 3), and the filamentous structures also remained in supernatant after ultracentrifugation (300,000 × g for 1 h). It is also interesting to note that filament formation by BPs occurred immediately upon dissolution, even at very low concentration (<1 μM), and no evident growth or aggregation of these filaments was observed even after incubation at room temperature (RT) for more than two months. Thus, BP filaments have features distinct from protein amyloids which are typically bundles of β-sheets and subject to aggregation through a nucleation-dependent polymerization process13.
We solubilized the ABC exporter MsbA14 in BPs by a complete detergent exchange through dialysis (Online Methods). MsbA is a bacterial homolog to human multidrug resistance protein 1(MDR1/P-glycoprotein)15, 16, and functions natively to transport lipid A using energy generated by ATP hydrolysis. As in liposomes17 and stabilizing detergents14, 18, the ATPase activity of MsbA in BP-1 remained high (~4 μmol/min/mg), comparable to the measured activity of MsbA reconstituted in lipid nanodiscs (Supplementary Fig. 4). The activity of MsbA in BP-1 had little change throughout 40 days of incubation at RT, demonstrating both protein stability and retention of functionality (Fig. 2a). In contrast, measured ATPase activity of MsbA in most commercial detergents, including β-D-undecylmaltoside (UDM) commonly used for MsbA purification and crystallization14, was relatively lower (2.2 μmol/min/mg). Additionally, MsbA in UDM lost ~80% of its ATPase activity within the first day of incubation at RT, and this loss was accompanied by protein precipitation. BP-1 was also found to substantially improve the protein stability of three other IMPs, including bacteriorhodopsin (bR), the tetrameric voltage-gated potassium channel KcsA, and the full-length glucagon receptor, a class-B G-protein coupled receptor (Supplementary Fig. 5–7). The enhanced stability of IMPs in BP-1, relative to BP-2 and BP-3, as well as conventional detergents that form micelles driven mainly by hydrophobic effects, may be attributed to the less dynamic association of BP-1 to the hydrophobic surfaces of IMPs, which is likely strengthened by H-bonding among surface-bound BP-1 molecules.
Figure 2.

MsbA stability and negative staining EM images. (a) MsbA solubilized in BPs retained high ATPase activity (measured in triplicate at 37 °C) throughout prolonged incubation at RT, demonstrating the protein’s stability. Data represent means ± standard error. (b–f) EM images of MsbA particles. (b) MsbA was mixed with BP-1 with concurrent removal of UDM by dialysis, then diluted 100-fold in detergent-free buffer. Individual particles were readily discernible against a clean background and structural details of the particles were evident. (c) Twofold magnification of (b), with representative 2D class averages (bottom). (d) 3D reconstructions of averages in (c). Dashed lines represent approximate position of the lipid membrane. See Online Methods and Supplementary Fig. 10–11 for more details on the 2D and 3D analyses. (e) MsbA preparation in UDM, diluted 100-fold in the same detergent-containing buffer. While particles were well separated on a relatively clean background, no protein structural details were discernible. (f) Preparation of MsbA in UDM (as in (e)), diluted 20-fold with detergent-free buffer, showing highly aggregated protein. Scale bar represents 35 nm in all images except in (d) where the scale bar represents 5 nm.
An intriguing question is how IMPs are stabilized by BPs pre-assembled into filamentous structures. Negative stain EM imaging confirmed the disappearance of BP-1 filaments upon addition of UDM-purified MsbA (Supplementary Fig. 8). Subsequent removal of UDM by dialysis lowered detergent background and decreased the particle clustering observed in the EM images, allowing us to visualize single MsbA particles (Supplementary Fig. 8). CD spectroscopic analysis of the MsbA/BP-1 sample indicated that the β-sheet secondary structures of BP-1 were preserved (Supplementary Fig. 9). We conclude that the BP-1 filaments undergo a restructuring process directed by the IMP template; a recent study showed a cylindrical β-barrel crystal structure for an 11-residue amyloid-forming protein fragment19, supporting the feasibility of transformation between the two types of structures. However, a detailed understanding of the process and binding modes involved, whether by β-barrel formation or through other types of structures, will require higher resolution structural studies.
BP-1 proves to be an excellent tool for EM-based single particle analysis for MsbA. When MsbA/BP-1 samples were diluted as much as 100-fold using detergent-free buffer, EM analysis showed apparently stable and well-preserved particles (Fig. 2b). It is significant that we were able to clearly identify multiple conformations of MsbA in BP-1 even in individual particles in the unprocessed EM micrographs. Two dimensional (2D) class-average images of thousands of particles from a single MsbA preparation clearly showed the two nucleotide binding domains (NBD) separated at varied distances in the absence of nucleotides (Fig. 2c and Supplementary Fig. 10). Random conical tilt (RCT) analysis provided unbiased 3D reconstructions (~26 Å resolution for three selected conformations), further confirming MsbA’s conformational heterogeneity (Fig. 2d and Supplementary Fig. 10–11). In contrast, it is difficult to discern MsbA structures in UDM due to the pronounced particle clustering and high detergent background (Supplementary Fig. 8 and Fig. 2e), and dilution of MsbA/UDM complexes with detergent-free buffer resulted in immediate protein aggregation (Fig. 2f). An additional advantage of BP-1 for EM analysis, particularly for small IMPs (<150 kDa), is that it contributes very little background clutter as compared, for example, to detergent micelles (Fig. 2e, and Supplementary Fig. 12) or to the relatively large nanodiscs (10–15 nm) commonly used in IMP preparations20. Thus, single particle analysis enabled by the new BP reagents provides a new approach to obtaining dynamic structural information of MsbA, and is potentially applicable to other ABC transporters and IMPs.
The design of nanostructured BPs, based on a bottom-up design approach, represents a new strategy for stabilizing IMPs. The short BP monomers (~8 residues) are engineered to span the thickness of the membrane lengthwise while also forming an ordered secondary structure. In addition, controlled H-bonding between BP strands decreases dynamic dissociation from IMP surfaces as compared to more loosely bound and less ordered detergent molecules. Finally, EM imaging of the ABC transporter MsbA illustrates a clear advantage of using BP-1 as the stabilization reagent in enabling the visualization of dynamic conformations. These unique features of BPs confer enhanced IMP stability without constraining protein functionality, demonstrating their strong potential as broadly useful reagents for IMP research. We have noted that MsbA and bR samples in BP-1 can be concentrated to >10 mg/mL, and the enhanced IMP stability and potential to form well-ordered IMP/BP complexes substantiate their utility for high-resolution structural studies such as by X-ray crystallography. We anticipate that BPs will be further engineered to introduce additional novel features, and to enable challenging IMP studies thus far limited by the currently available reagents.
Online Methods
Peptide synthesis
The peptides were synthesized by Fmoc strategy using Rink Amide AM resin (EMD). HCTU was used as the amino acid coupling reagent, except that coupling of N-Me amino acids used HATU. Fmoc-L-octylglycine ((Oct)Gly) was custom-synthesized or purchased directly from Acros. Peptides were acetylated at the N-terminus, cleaved from resin in the presence of 95% trifluoroacetic anhydride (TFA) and 2.5% triisopropylsilane, then purified by reverse-phase HPLC (CH3CN-H2O, 0.1% TFA). Molecular weight of each peptide was verified by electrospray mass spectrometry.
Circular dichroism spectroscopy
Far-UV CD spectra were recorded on an AVIV 202-SF CD spectrometer (AVIV Instruments) at 25 °C using a quartz sample cell with 1 mm path length (1 nm step size, 3 sec integration per step, average of three scans). BP samples were prepared at concentrations of ~0.1 – 1 mM in buffer containing 100 mM Tris (pH 7.35) and 40 mM NaCl. Molar ellipticity [θ] was calculated from the equation [θ] = (100*θ)/(c*l), where θ = observed ellipticity (deg), c = concentration (mol/L), l = cell path length (cm).
Fourier transfer infrared spectroscopy
FTIR spectra were collected on a Bruker Equinox 55 FTIR spectrometer equipped with liquid N2 cooled HgCdTe infrared detector, which was continuously purged with dry N2 gas. BP-1 (2.5 mM) was dissolved in D2O solution containing 100 mM Tris (pH7.5) and 40 mM NaCl. Approximately 8 μL of sample volume was loaded into a liquid cell with CaF2 windows and a 75 μm Teflon spacer. 4,000 scans with 2 cm−1 resolution were averaged and a Blackman-Harris 3-term apodization function was used in the Fourier transform. Absorbance of BP-1 was corrected by subtracting absorbance from buffer. Absorbance in the Amide I band (1700–1600 cm−1) was used for the assignment of peptide secondary structures21.
Preparation of MsbA, bR and KcsA
E. coli MsbA14 and Halobacterium bR22 were prepared as described and purified in UDM and OG (Affymetrix), respectively. Full length KcsA was prepared as described23, except that the step for cleavage of C-terminal fragment was eliminated and DM was used throughout membrane solubilization and protein purification. BPs were mixed with the purified protein typically in a 50:1–100:1 molar ratio. However, we have found that a ~25-fold molar excess of BP-1 sufficiently stabilized MsbA. The primary detergents were removed by dialysis against detergent-free buffer (6 kDa cutoff membrane, 4 °C, 1–2 days), monitored by thin layer chromatography. Complete detergent exchange of UDM to BP-1 in MsbA samples was achieved in ~ 8 hours, and E. coli lipids in the protein sample were stained by iodine vapor and molybdenum blue, which gave the lipid:MsbA ratio ~ 0.03:1 wt/wt. IMPs solubilized in BPs remained in supernatant after centrifugation at 300,000 × g for 2 h at 4°C, and were analyzed by SDS-PAGE. Extraction and purification of MsbA in lauryl maltose neopentyl glycol (LMNG, Affymetrix) was conducted similarly as in UDM. MsbA was also solubilized in Amphipol A8–35 (Affymetrix) at a weight ratio of 5:1 (A8–35: MsbA) according to a literature procedure24. Reconstitution of MsbA and KcsA into nanodiscs comprised of MSP1D1 and dimyristoylphosphatidylcholine (DMPC) was carried out following published procedures1.
Preparation of glucagon receptor
Human wild-type glucagon receptor (GLR), a class-B G-protein coupled receptor (GPCR), containing a PreScission protease site, 10× His, and Flag tag at the C-terminus was expressed in Spodoptera frugiperda (Sf9) insect cells. Sf9 membranes were prepared with 1 wash cycle of hypotonic buffer (25 mM HEPES (pH 7.5), 10 mM MgCl, 20 mM KCl) in the presence of EDTA-free protease inhibitor cocktail tablets (Roche) and 4 wash cycles of high-salt buffer (25 mM HEPES (pH 7.5), 1 M NaCl, 10 mM MgCl, 20 mM KCl). Two grams of washed membranes containing the human wild-type GLR was incubated with 270 μM of antagonist NNC359 (generously provided by Novo Nordisk, Denmark) and 2 mg/ml of iodoacetamide (Sigma) for 30 min at room temperature. The receptor was solubilized with 1.00/0.20% (w/v) of n-dodecyl-β-D-maltopyranoside/cholesteryl hemisuccinate (Sigma) (DDM/CHS) or UDM/CHS for 2 h at 4 °C.
The NaCl and DDM/CHS concentrations of the solubilized protein were adjusted to 800 mM and 0.50/0.10%, respectively. The solubilized protein was bound to talon resin (Clontech) overnight in the presence of 15 mM imidazole (pH 7.5) and 100 μM of NNC359. The talon resin was washed with 10× bed volume of wash buffer (25 mM HEPES (pH 7.0), 800 mM NaCl, 10% glycerol, 0.05/0.01% DDM/CHS or 0.10/0.02% UDM/CHS, 50 μM NNC359, 40 mM imidazole (pH 7.5)). Wild-type GLR was eluted with elution buffer (25 mM HEPES (pH 7.0), 150 mM NaCl, 10% glycerol, 0.05/0.01% DDM/CHS or 0.10/0.02% UDM/CHS, 50 μM NNC359, 300 mM imidazole (pH 7.5)). The eluted GLR was concentrated using a Vivaspin centrifuge concentrator (GE Healthcare) with a 100,000 kDa molecular weight cut-off, and protein concentration was determined by analytical size exclusion chromatography. The protein was then diluted to 1.5 mg/ml with elution buffer containing no imidazole or ligand.
For stability test, GLR/BP-1 samples were prepared in two different approaches, either by 20x dilution of purified GLR (DDM/CHS preparation) into the same buffer containing BP-1 (0.50 mM) or by dialysis as described above for other IMPs. Each set of samples contains the same concentration of NNC359. To facilitate detergent exchange, GLR purified in UDM/CHS was used in the dialysis experiment in which no GLR ligand was present in the dialysis buffer, and NNC359 was added back to keep the ligand concentration as about the same as in the control sample before dialysis.
Stability assays
Stability of MsbA, bR and KcsA in BPs and control detergents was assayed at sample concentrations ranging from 0.2–0.5 mg/mL according to literature procedures. bR samples in buffer (100 mM Tris, 40 mM NaCl, pH 7.4) were incubated at 37 °C for 2 months, and bR stability was monitored by recording its characteristic UV-vis absorption spectrum5. MsbA samples prepared in 20 mM Tris (pH 7.5), 20 mM NaCl buffer were incubated at RT for 40 days, and stability was monitored by measuring MsbA ATPase activity using a standard linked enzyme ATPase assay25. KcsA samples (20 mM Tris, 20 mM NaCl, pH 7.5) were incubated at 37°C for 5 days, and then analyzed using SDS-PAGE26.
The stability of GLR was monitored using a fluorescence thermal denaturation assay27, which has been employed for several GPCRs and other classes of IMPs. Briefly, 4 μg of GLR was incubated with 0.4 μg of thiol-reactive fluorophore, N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM), for 20 minutes at 4 °C in a total volume of 60 μl. Fluorescence emission was then measured on a Cary Eclipse Fluorescence Spectrophotometer (λex = 387 nm; λem = 463 nm) from 20 – 90 °C with 1 °C intervals and a ramp rate of 2 °C/min, and the background fluorescence of buffer in the absence of protein was subtracted. Midpoints of the thermal transitions (Tm) were obtained using a least squares non-linear regression analysis (GraphPad Prism) of fluorescence signal versus T plots according to equation described previously28.
Electron microscopy
All samples were applied onto freshly glow discharged carbon-coated copper grids. The sample was diluted to distribute particles homogenously over the grid surface; in most cases a 1:20 dilution with detergent-free buffer was sufficient. Sample (3 μl) was applied to the grid and allowed to adhere for 15 s before being blotted from the side. Immediately after blotting, 3 μl of 2% uranyl formate solution was applied onto the grid and blotted off directly from the same side. This was repeated three times. 10 mL of 2% uranyl formate was freshly prepared, aliquotted into 40 μl portions, flash frozen in liquid nitrogen and stored at −80°C. For every grid a fresh aliquot was used. Data was acquired using a Tecnai F20 Twin transmission electron microscope operating at 200 kV, using a dose of ~45 e−/Å2 and nominal underfocus ranging from 1.5 to 2.5 μm. Images were collected automatically at a nominal magnification of 80,000x and pixel size 0.136 nm onto a Tietz F415 4k × 4k pixel CCD camera (15 μm pixel) using the Leginon data collection software29. Experimental data were processed by the Appion software package30 interfaced with the Leginon database infrastructure. Particles were automatically selected using a difference of Gaussian (DoG) algorithm31 and extracted with a box size of 128 pixels. XMIPP reference-free maximum likelihood alignment32 was applied to identify the best particles from the selection using 20 averages. All resulting class averages of the maximum likelihood alignment were used as reference for Spider 2D alignment33 with consecutive Coran Classification. RCT reconstructions were automatically carried out on selected 2D class averages31. Each volume contained between 100 and 200 single particles.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institute of General Medical Sciences (P50 GM073197 and 9 P41 GM103310-11). We thank J. Kelly for use of his CD instrument, F. Romesberg for use of his FTIR instrument, and G. Chang (University of California, San Diego), S. Choe (Salt Institute for Biological Studies), S. Sligar (University of Illinois, Urbana) for providing recombinant DNA of MsbA, KcsA, and MSP1D1, respectively. We thank J. Kelly, G. Chang, A. Ward, V. Cherezov, P. Dawson, and W. Chen for discussions and comments. We thank E. Hildebrandt for editorial assistance.
Footnotes
Note: Supplementary information is available in the online version of the paper.
AUTHOR CONTRIBUTIONS
Peptides were designed by Q.Z., H.T. and synthesized by H.T. and R.S.R. Peptide assembly was characterized by H.T., S.C.L., A.M., and J.Z. Protein stabilizations were performed by H.T. and S.C.L.; F.Y.S. prepared GLR and assisted with the assay with oversight by R.C.S. MsbA samples for EM were prepared by S.C.L., and EM study was performed by A.M. with oversight by B.C. and C.P. The data were interpreted by all authors. The manuscript was written by H.T., S.C.L, A.M., B.C. and Q.Z. The study was conceived and overseen by Q.Z.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Bayburt TH, Grinkova YV, Sligar SG. Nano Lett. 2002;2:853–856. [Google Scholar]
- 2.Tribet C, Audebert R, Popot JL. Proc Natl Acad Sci USA. 1996;93:15047–15050. doi: 10.1073/pnas.93.26.15047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGregor CL, et al. Nat Biotechnol. 2003;21:171–176. doi: 10.1038/nbt776. [DOI] [PubMed] [Google Scholar]
- 4.Zhao X, et al. Proc Natl Acad Sci USA. 2006;103:17707–17712. doi: 10.1073/pnas.0607167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schafmeister CE, Miercke LJ, Stroud RM. Science. 1993;262:734–738. doi: 10.1126/science.8235592. [DOI] [PubMed] [Google Scholar]
- 6.Chae PS, et al. Nat Methods. 2010;7:1003–1008. doi: 10.1038/nmeth.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Q, Tao H, Hong WX. Methods. 2011;55:318–323. doi: 10.1016/j.ymeth.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulz GEb. Curr Opin Struct Biol. 2000;10:443–447. doi: 10.1016/s0959-440x(00)00120-2. [DOI] [PubMed] [Google Scholar]
- 9.Xiong H, Buckwalter BL, Shieh HM, Hecht MH. Proc Natl Acad Sci USA. 1995;92:6349–6353. doi: 10.1073/pnas.92.14.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doig AJ. Chem Commun. 1997:2153–2154. [Google Scholar]
- 11.Gordon DJ, Sciarretta KL, Meredith SC. Biochemistry. 2001;40:8237–8245. doi: 10.1021/bi002416v. [DOI] [PubMed] [Google Scholar]
- 12.Zhang S, Prabpai S, Kongsaeree P, Arvidsson PI. Chem Commun. 2006:497–499. doi: 10.1039/b513277k. [DOI] [PubMed] [Google Scholar]
- 13.Harper JD, Lansbury PT., Jr Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 14.Ward A, Reyes CL, Yu J, Roth CB, Chang G. Proc Natl Acad Sci USA. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aller SG, et al. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin MS, Oldham ML, Zhang Q, Chen J. Nature. 2012;490:566–569. doi: 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou P, McHaourab HS. J Mol Biol. 2009;393:574–585. doi: 10.1016/j.jmb.2009.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee SC, et al. Proc Natl Acad Sci USA. 2013;110:E1203–1211. doi: 10.1073/pnas.1221442110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laganowsky A, et al. Science. 2012;335:1228–1231. doi: 10.1126/science.1213151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi L, et al. Science. 2012;335:1355–1359. doi: 10.1126/science.1214984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adochitei A, Drochioiu G. Rev Roum Chim. 2011;56:783–791. [Google Scholar]
- 22.Landau EM, Rosenbusch JP. Proc Natl Acad Sci USA. 1996;93:14532–14535. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doyle DA, et al. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 24.Gohon Y, et al. Biophys J. 2008;94:3523–3537. doi: 10.1529/biophysj.107.121848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Q, et al. Angew Chem Int Ed Engl. 2007;46:7023–7025. doi: 10.1002/anie.200701556. [DOI] [PubMed] [Google Scholar]
- 26.Cortes DM, Perozo E. Biochemistry. 1997;36:10343–10352. doi: 10.1021/bi971018y. [DOI] [PubMed] [Google Scholar]
- 27.Alexandrov AI, Mileni M, Chien EY, Hanson MA, Stevens RC. Structure. 2008;16:351–359. doi: 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 28.Thompson AA, et al. Methods. 2011;55:310–317. doi: 10.1016/j.ymeth.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suloway C, et al. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 30.Lander GC, et al. J Struct Biol. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. J Struct Biol. 2009;166:205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheres SH, et al. J Mol Biol. 2005;348:139–149. doi: 10.1016/j.jmb.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 33.Frank J, et al. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.