


Abstract
An important goal of evolutionary biology is to understand the constraints that shape the dynamics and outcomes of evolution. Here, we address the extent to which the structure of the standard genetic code constrains evolution by analyzing adaptive mutations of the antibiotic resistance gene TEM-1 β-lactamase and the fitness distribution of codon substitutions in two influenza hemagglutinin inhibitor genes. We find that the architecture of the genetic code significantly constrains the adaptive exploration of sequence space. However, the constraints endow the code with two advantages: the ability to restrict access to amino acid mutations with a strong negative effect and, most remarkably, the ability to enrich for adaptive mutations. Our findings support the hypothesis that the standard genetic code was shaped by selective pressure to minimize the deleterious effects of mutation yet facilitate the evolution of proteins through imposing an adaptive mutation bias.
INTRODUCTION
The genetic code plays a central role in evolutionary processes defining the relationship between DNA and protein sequences. The genetic code limits the mutational exploration of sequence space (1), as single-base changes in codons can access only about six of the 19 possible amino acid substitutions and simultaneous multiple-base changes in a codon are rare (2). Furthermore, the genetic code is biased toward conservative amino acid mutations (3). As a result, most mutational trajectories have a low probability, and probable mutational trajectories tend to be conservative in nature. Thus, very similar genes may follow different evolutionary trajectories in part because the genes’ mutational neighborhoods are different (4–6) (i.e. the likely amino acid substitutions are different owing to the two genes having different but synonymous codons). What has not been experimentally addressed is the extent to which the evolution of a single gene is constrained or facilitated by the architecture of the genetic code. For a particular evolutionary outcome, how many superior fitness peaks are nearby that could have been reached if only the genetic code was arranged differently?
To the extent that the genetic code restricts a gene from evolving to higher fitness peaks, one may wonder about the possible benefits of alternative codes. However, the standard genetic code’s organization makes it apparent that the relationship between DNA triplets and amino acids was not arrived at randomly. The code’s arrangement has been proposed to result from the inherent interactions between amino acids and their cognate nucleotide triplets (7), the biochemical pathways through which amino acids are synthesized (8), selective pressure to minimize the deleterious effects of mutations and mistranslations (3,9) and the difficulty in changing the code once it is established (10). These basic theories have been further developed primarily through theoretical and simulation approaches (11). However, mutational bias, such as that arising from how the code is arranged or the nature of spontaneous mutations, may shape evolution (12,13). Thus, the architecture of the genetic code may facilitate the evolution of genes and proteins (14). For example, a code arranged to make adaptive mutations more likely would have provided an adaptive advantage over one that did not early in evolution when there may have been competing genetic codes. If so, the standard genetic code might still exhibit this property today. Does the standard genetic code enrich for adaptive mutations?
Here we experimentally address the extent to which the genetic code restricts access to beneficial alleles and whether the code’s constraints provide advantages for the evolution of proteins. We find that although the code’s architecture significantly limits evolutionary outcomes, it minimizes the deleterious cost of mutation and enriches for beneficial mutations—two properties that facilitate evolution.
MATERIALS AND METHODS
TEM-1 β-lactamase libraries and constructs
All libraries and variants of the TEM-1 gene were created using PFunkel mutagenesis as previously described (15).
Library selections
All antibiotics and chemical reagents for selections and MIC assays were obtained from Sigma-Aldrich. For the selection of alleles conferring cefotaxime resistance equivalent to that of GKTS, a previously described library (15) in which the codons for A42, E104, M182 and G238 were randomized (NNN) was plated on LB-agar plates containing 50 μg/ml spectinomycin, 50 μM IPTG and 8 μg/ml or 16 μg/ml cefotaxime. The library was plated at a cell density of 1900 or 19 000 CFU/cm2 on the 8 μg/ml cefotaxime plates and 190 000 CFU/cm2 on the 16 μg/ml cefotaxime plate. Plates were incubated at 37°C for 17 h. Forty colonies of ∼1000 that grew on the 8 μg/ml plate and the largest 10 colonies on the 16 μg/ml plate were selected for individual screening by plate MIC assay. For colonies that passed the screen, plasmid DNA was isolated from overnight cultures using the Qiagen QIAprep Spin Miniprep kit (27106) and the TEM-1 allele was sequenced. Unique plasmids were retransformed into fresh DH5α E. coli cells, and the MIC determined by both plate and liquid MIC assays.
For the selection of TEM-1 alleles with a single amino acid substitution that provides increased resistance to cefotaxime, two previously described TEM-1 comprehensive codon libraries were used: CCM1 and CCM2 (15). DH5α cells bearing CCM1 were plated at a density of 100–300 CFU/cm2 on LB-agar plates containing 50 μg/ml spectinomycin, 50 μM IPTG and 0.04 μg/ml cefotaxime. NEB 5-alpha F’Iq cells bearing CCM2 were plated as above except the cell density was 150–600 CFU/cm2, the cefotaxime was 0.02 μg/ml and the IPTG was 300 μM. The concentrations of cefotaxime used correspond to the MIC conferred by TEM-1 in DH5α and NEB 5-alpha F’Iq cells. Plates were incubated at 37°C for 17 h. The TEM-1 gene of randomly selected colonies was sequenced. Because any particular amino acid substitution is relatively rare in the libraries, we used the criteria that an amino acid substitution had to be observed twice for us to categorize it as adaptive.
MIC assays
For MIC assay on agar plates, cultures of variants were prepared in LB broth at 37°C with shaking at 250 rpm until all cultures reached saturation, ∼24 h. Cultures were diluted 100-fold in LB broth, and incubated for ∼2.5 h until the OD was about 0.3. The cultures were diluted to 104 CFU/μl, and 1 μl was spotted on Mueller–Hinton agar plates containing 50 μg/ml spectinomycin, 50 µM IPTG and √2-fold increasing concentrations of cefotaxime. The plates were incubated at 35°C for 20 h. The MIC was determined as the minimal concentration at which no growth was observed.
For liquid MIC assays, the initial cultures were prepared as above and then diluted to a concentration of 1 × 106 CFU/μl in Mueller–Hinton broth. A total of 150 μl of this diluted culture was added to wells of a 96-well assay plate along with 150 μl of Mueller–Hinton broth containing 100 μg/ml spectinomycin, 100 µM IPTG and 2-fold increasing concentrations of cefotaxime. The plate was covered and sealed in a plastic bag and incubated at 35°C for 20 h. The MIC was determined as the minimal concentration at which no visible growth was observed.
The above two MIC tests used different temperatures and media than the selections. The MIC assay conditions used are standardized conditions for quantifying beta-lactam resistance that allow comparisons with other studies (16).
Enrichment values of experimentally observed codon substitutions
Enrichment values for each codon substitution introduced in HB36.4 and HB80.3 were determined by using custom Matlab scripts to analyze the Illumina deep sequencing data on the libraries before and after selection for hemagluttinin binding. The data were filtered as in the study by Whitehead et al. (17) to include sequencing reads with only one codon substitution, and the final list to include only codon substitutions with at least 100 sequencing counts in the reference library. The enrichment value E was calculated as
 |
(1) |
Thus, E quantifies the relative prevalence of an allele in the selected library compared with the reference library (the naïve library). The enrichment value of the wild-type sequence was determined by averaging the enrichment values of all codons synonymous with the wild type. Codon substitution counts, enrichment values and the wild-type enrichment values were consistent with values for amino acid substitutions presented by Whitehead et al. (17).
The genetic code’s enrichment of adaptive mutations and meta-analysis
The percent enrichment and P-values were determined as described in Supplementary Table S6. Meta-analysis on the P-values was performed using the Stouffer’s Z-trend method (weighted Z score) using the program MetaP (http://people.genome.duke.edu/∼dg48/metap.php).
RESULTS
The natural and in vitro evolution of TEM-1 β-lactamase for conferring cefotaxime resistance converges on the same set of mutations
We chose to examine the genetic code’s constraints on evolution with the antibiotic resistance TEM-1 gene encoding TEM-1 β-lactamase—a gene that has provided many insights into how epistasis constrains evolution (16,18–20). TEM-1 hydrolytically inactivates β-lactam drugs such as penicillin, but has very low activity on the third-generation cephalosporin β-lactam cefotaxime. Clinically isolated alleles of TEM-1 conferring elevated antibiotic resistance arise through accumulation of point mutations (i.e. 1-bp substitutions). For example, TEM-52 differs from TEM-1 by three point mutations resulting in the E104K/M182T/G238S mutations (21) that increase cefotaxime resistance ∼4000-fold (16). The in vitro evolution of TEM-1 mimics its natural evolution (22). Six independent in vitro evolution studies that applied selective pressure for increased cefotaxime resistance found the E104K/M182T/G238S combination of mutations in the best alleles (20,22–26). A fourth mutation (A42G) (25) that also arises from a point mutation increases cefotaxime resistance to about 33 000-fold over TEM-1 (16). The high fitness of the gene bearing the A42G/E104K/M182T/G238S mutations (referred to here as GKTS) has not been surpassed by increasing the mutation rate (20,26), using mutator strains of bacteria (24), forcing explorations of alternative trajectories through use of bottlenecks (20), or using computational protein optimization strategies (which are not constrained by the genetic code) (27). This suggests that the evolutionary outcome of GKTS is largely reproducible and inevitable, given a strong selective pressure for cefotaxime resistance (16). Among the accessible local optima for cefotaxime resistance on the β-lactamase fitness landscape, GKTS may be the global optimum.
To what extent did the architecture of the genetic code direct this outcome? There are 204 − 1 = 159 999 possible amino acid combinations at these four positions in TEM-1 (including combinations with up to three wild-type amino acids). However, only 2743 (i.e. 7 × 8 × 7 × 7 − 1) or 1.7% of these are readily accessible combinations, as they do not require simultaneous multiple mutations in any one codon, which is a rare occurrence. Synonymous mutations followed by a second point mutation can expand the accessible amino acid combinations, but only to some extent. Also, subsequent second mutations in codons with previously accumulated beneficial mutations can occur, but this requires that both mutations be beneficial and that the second mutation increases the fitness of the gene. This significant constraint will keep the readily accessible combination of mutations at these four codons low. More to the point, such double mutations are not present in the best cefotaxime resistance alleles arrived at by natural or in vitro evolution of TEM-1. The rarity of natural adaptive mutations with multiple-base substitutions in a single codon is exemplified by a recent study that examined 516 spontaneous ceftazidime-resistant isolates of Burkholderia thailandensis and found 29 different codon substitutions in the penA β-lactamase gene that provided this resistance—all of which were point mutations (28). In addition, the occurrence of reciprocal sign epistasis will further constrain which combination of amino acids are accessible by evolution (29).
The genetic code constrains the evolution of TEM-1
To test the extent to which the genetic code constrains the evolution of TEM-1, we asked whether there exist other amino acid combinations at these four positions that provide fitness equal to or better than GKTS. We used a library in which these four codons were randomized at all three base positions (15). We placed the mutated gene downstream from the IPTG-inducible tac promoter on a plasmid with the p15A origin (copy number ∼10), as in previous in vitro evolution experiments with TEM-1 (20,22). Our library consisted of 5.8 million transformants (short of the theoretical 412 = 16.8 million DNA variants, but in excess of the possible 204 = 160 000 protein variants), and the majority of library members contained mutations at all four positions (15). Although we subsequently determined in separate experiments that the degenerate oligos used to make the library were enriched for G’s by about 2.2-fold (15), a large fraction of the protein variants are likely to be present in the library.
We subjected this library to selections for cefotaxime resistance equivalent or superior to that achieved by GKTS. We performed a secondary screen on 50 of the resulting ∼1000 colonies for those with a MIC at or above that conferred by GKTS. Clones passing this screen were sequenced. The plasmid DNA from unique clones was retransformed into fresh DH5α E. coli and the cefotaxime MIC determined by solid and liquid media growth assays.
The sequences and corresponding MICs revealed that there are many alleles with equivalent or superior combinations of amino acids at these four positions (Table 1). Of the 17 identified alleles (11 unique amino acid combinations), only four were identified more than once, indicating that there are additional high fitness alleles yet to be found. Although we observe small differences (up to two-fold) between synonymous alleles in the plate MIC assay (Table 1), this assay showed variability in replicate experiments of up to two-fold in some instances (Supplementary Table S1). Six unique amino acid sequences differed from GKTS at two of the four positions. Most strikingly, 55% (6 of 11) of the amino acid combinations identified require more than one point mutation in at least one codon (i.e. Hamming distance >4). We find the existence of numerous equivalent or superior alleles nearby what appeared to be a dominant resistance allele quite striking. This experiment shows that there are many alleles equivalent or superior to GKTS nearby in sequence space that are not readily accessed by natural or in vitro evolution
Table 1.
Cefotaxime resistance of selected TEM-1 β-lactamase alleles
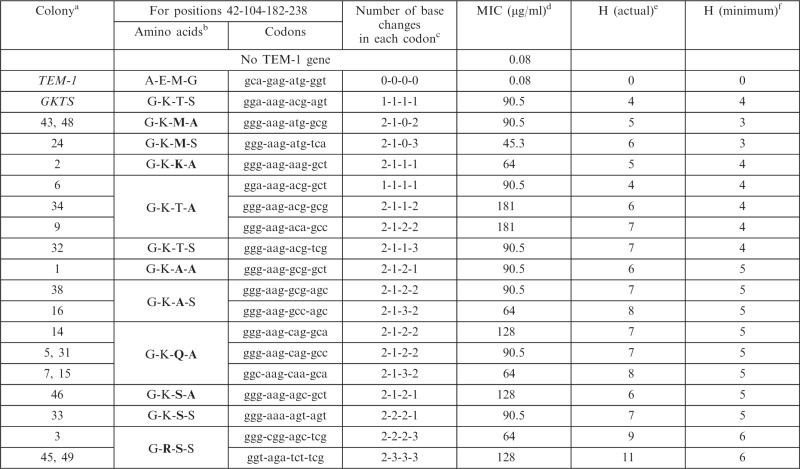 |
aTwo numbers indicate that the allele was found twice.
bBold indicates amino acids differing from those in GKTS.
cRelative to TEM-1.
dMedian value of three replicates. Assays performed in √2 increments of cefotaxime (Mueller–Hinton-agar, 104 CFU/spot, 35°C for 20 h). Data for all replicates are in Supplementary Table S1. MICs determined by Mueller–Hinton broth liquid growth assay at 35°C can be found in Supplementary Table S2.
eHamming distance between the allele and TEM-1.
fMinimum Hamming distance to achieve same amino acid sequence.
To address whether there is a mutational pathway to any of the alleles with a Hamming distance >4, we chose GKQA (codons: ggg-aag-cag-gca) as a representative allele and constructed the 14 combinations of these four codon substitutions. We considered each codon substitution (whether a point mutation or a multi-bp substitution) as a single mutational step in order to ask whether GKQA could be reached if the genetic code were arranged differently such that each of the required amino acid substitutions were possible with a point mutation. We tested the cefotaxime resistance of these variants and assessed the feasibility of the 24 possible trajectories from TEM-1 to GKQA. We assumed that the evolution of TEM-1 fits the strong selection/weak mutation model of evolution by which the time to fixation or loss of a mutation is much shorter than the time between mutations. Thus, we required that mutations accumulate one at a time with increasing fitness at each step for a trajectory to be deemed feasible, as in a previous study (16). Nine of the 24 possible trajectories were feasible (Figure 1). Four trajectories ended at an intermediate (GKMA) with equivalent resistance to GKQA. Like the feasible trajectories for evolving GKTS (16), the first mutation necessarily occurs at positions 104 or 238, and the fittest double mutant has mutations at both positions (the difference is that 238 is mutated to A instead of S for GKQA). A lack of a mutational trajectory cannot explain why GKQA has not been found in the natural or in vitro evolution of TEM-1. Instead, we posit that the requirement for multiple mutations in a single codon is one reason that makes this allele’s occurrence unlikely. Thus, the architecture of the genetic code constrains evolution by making some viable mutational trajectories improbable.
Figure 1.
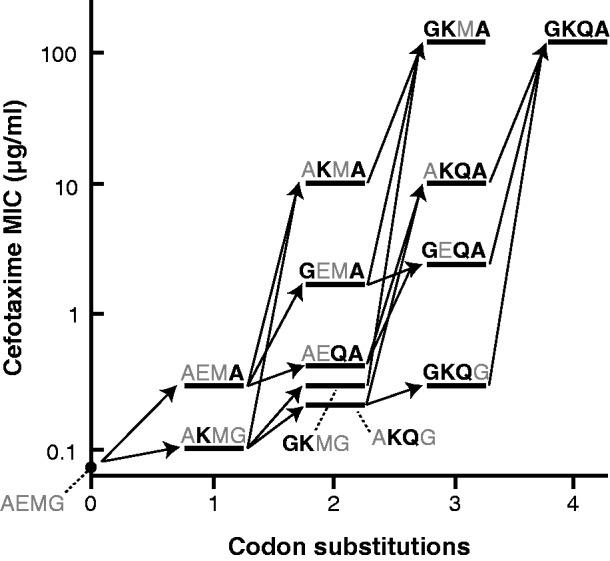
Feasible trajectories for evolving GKQA (colony 14) from TEM-1 (i.e. AEMG) by accumulation of codon substitutions one at a time. Mutations are shown in black, bold letters. Of the 24 possible trajectories, five end with GKQA and four end with GKMA, an allele with equivalent fitness to GKQA. Cefotaxime resistance was measured by plate assay as in Table 1, and the value reported represents the median of three replicates. Data for all replicates are provided in Supplementary Table S3.
Epistasis and mutational bias also constrain the exploration of sequence space
Alleles such as GKMA and GKTA provide resistance equivalent to GKTS (Table 1) and have amino acid combinations that can be reached by three or four point mutations, respectively, yet these combinations have not been previously identified in laboratory evolution experiments. We speculate that there are two reasons why such these alleles have not been previously identified. The first reason is epistasis. For example, G238A was more common in the identified alleles than G238S and is present in both GKMA and GKTA. However, when occurring as the only mutation in TEM-1, G238S provides a 4-fold higher kcat/Km and a 4-fold higher MIC than G238A (30). Because G238S is the amino acid substitution that, by itself, increases cefotaxime resistance the most, it is most likely to be fixed first. The apparent equivalence of G238A and G238S among our selected alleles is illustrative of the epistatic nature of mutations. The second reason is mutational bias. For example, the G:C → C:G mutation necessary for G238A is considerably less common than the G:C → A:T mutation for G238S in error-prone PCR reactions (31) and spontaneously in E. coli (32).
The genetic code minimizes the fitness cost of mutations
There is no arrangement of a 20 amino acid / 64 codon genetic code that would not significantly limit the types of amino acid substitutions that are readily accessible, as a single codon can be mutated only to nine other codons with a point mutation. Thus, different genetic codes will constrain evolutionary paths in different ways. The adaptive theory of the origin of the genetic code postulates that the code’s conservative architecture is a result of selective pressure to minimize the deleterious effects of point mutations and mistranslation errors (3,9). The adaptive theory predicts that for non-synonymous mutations, the average fitness cost of point mutations should be less than that of 2-bp and 3-bp substitutions. However, such a test of the adaptive theory (and of the conservative nature of the code) has never been systematically applied to any gene. How does the mutational fitness distribution partition between 1-, 2- and 3-bp codon substitutions of a gene?
To address this question, we examined the distribution of fitness effects of 1896 unique single amino acid substitutions in two genes that were previously modified through a combination of computational design and directed evolution to inhibit H1N1 influenza hemagglutinin (17,33). Inhibitor HB36.4 derives from Apc36109 from Bacillus stearothermophilus and HB80.3 from the Myb domain of the Rad transcription factor from Antirrhinum majus (33). Because the natural proteins are not inhibitors of hemagglutinin, we suggest that HB36.4 and HB80.3 should be viewed as genes that are not evolutionarily mature for hemagglutinin inhibition. Whitehead et al. (17) created NNK degenerate codon libraries consisting of all possible single amino acid substitutions in all 51 positions in HB80.3 and 53 of 93 positions of HB36.4. In NNK libraries, the first two nucleotides in a codon can be any base, but the third nucleotide is limited to G or T to reduce the frequency of nonsense codons while still allowing all possible amino acids. The libraries were subjected to deep sequencing before and after selection for hemagglutinin binding in a yeast display format. In their study, the base 2 logarithm of the ratio of the frequencies of each amino acid substitution in the selected versus unselected libraries—referred to as the enrichment value—served as a proxy for the change in free energy of binding. Several lines of evidence support the suitability of this proxy (17).
Because differences in enrichment values for synonymous mutations were considerably smaller than differences between non-synonymous mutations (17), we assigned the enrichment values of each amino acid substitution to its respective codon substitutions and used this value as a proxy for the change in gene fitness caused by the 5857 codon substitutions (i.e. the number of unique codon substitutions that can code for the 1896 unique amino acid substitutions observed). The distribution of these fitness effects (Figure 2A–D) indicates that the standard genetic code’s architecture asymmetrically partitions fitness effects of amino acid substitutions between point mutations and multi-bp codon substitutions and minimizes the fitness cost of point mutations. The average cost of mutation for point mutations is substantially less than for 2-bp substitutions, and 3-bp substitutions have the highest average fitness cost (Figure 2E). Mutations causing a smaller decrease in fitness are enriched in point mutations, and mutations with the largest negative effect are almost exclusively 2- and 3-bp changes. The same distribution trends are observed when we determined the enrichment values for the 2813 experimentally observed codon substitutions (Supplementary Figure S1). Among beneficial mutations, the median effect of mutations for multi-bp mutations was marginally higher than that of point mutations (Supplementary Table S4).
Figure 2.

Distribution of fitness effects of non-synonymous codon substitutions in (A and B) HB36.4 and (C and D) HB80.3. The distribution is partitioned into codon changes with 1-, 2- and 3-base changes. The red dashed vertical line indicates the enrichment value of the parental genes, and the blue dashed horizontal bar indicates the fraction of all possible mutations of the gene that are point mutations. Enrichment values for parental genes are slightly greater than zero because most mutations have a negative effect on fitness. (E) Median enrichment values for types of codon substitutions. Distributions based on codon enrichment values instead of amino acid enrichment values are provided in Supplementary Figure S1.
We interpret these results (Figure 2E) as evidence that the code’s arrangement minimizes the fitness cost of amino acid substitutions. An alternative explanation is that the genes are products of evolution under the standard genetic code, and thus their make-up is such that point mutations will cause minimal deleterious effects. However, this viewpoint is mitigated somewhat by the fact that HB36.4 and HB80.3 were not evolved by nature for hemagglutinin binding, but rather are a product of codon optimization for yeast expression, computational design and limited laboratory evolution. The genes may better represent ones in the process of evolving rather than ‘evolutionarily mature’ genes.
The genetic code is biased towards adaptive mutations
Figure 2 shows that readily accessible amino acid substitutions (i.e. those from point mutations) have smaller deleterious effects on fitness. How does this affect the evolution of proteins? For a gene with L codons, there are 19L possible amino acid substitutions. The standard genetic code imposes a restriction on which of these 19L are likely (i.e. on average, only about 6L occur with a point mutation; the exact number for a particular gene will depend on the gene’s DNA sequence). In other words, the genetic code provides a set of codon-based rules governing which amino acid substitutions readily occur. Are these rules biased toward adaptive mutations? If so, then the code’s arrangement would provide an advantage because accessible amino acid substitutions would be more likely to be adaptive than randomly chosen amino acid substitutions (Figure 3).
Figure 3.
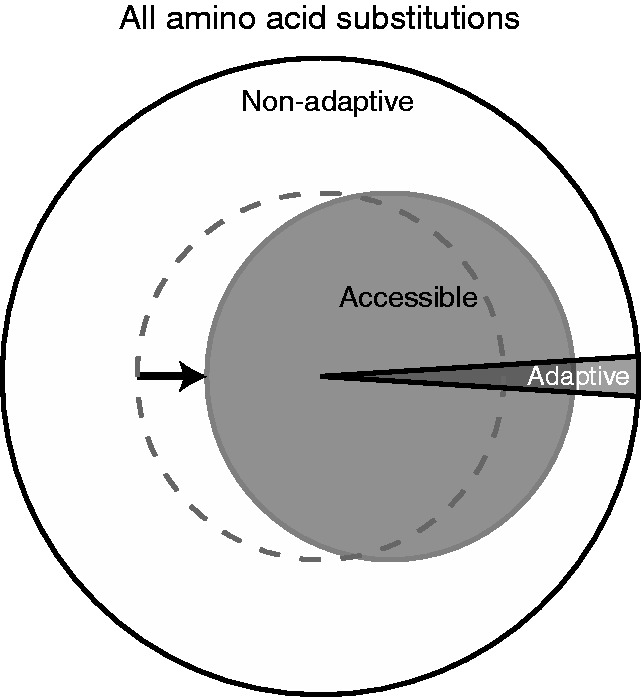
Enrichment of adaptive amino acid substitutions of genes by the standard genetic code. The filled gray circle depicts a code in which point mutations preferentially access adaptive amino acid substitutions while the dotted circle depicts a non-enriching code that randomly samples amino acid substitutions.
We examined this question in HB36.4, HB80.3 and TEM-1 by first identifying an extensive set of adaptive amino acid substitutions in these genes. For our analysis of HB36.4 and HB80.3, we used the set of amino acid substitutions enriched over wild-type as determined by Whitehead et al. (17). For TEM-1, we previously constructed comprehensive codon mutagenesis libraries that consisted of ≥97% of all 18 081 possible single codon substitutions in TEM-1 (i.e. 63 possible codons × 287 positions in TEM-1) (15). From these libraries, we previously identified 38 tazobactam resistance alleles (19 unique amino acid substitutions)—tazobactam being an inhibitor of TEM-1 (15). Here, in an analogous manner, we identified 77 cefotaxime resistance alleles (30 unique amino acid substitutions) by sequencing the TEM-1 gene of 500 colonies that formed when the library was challenged to grow on plates with elevated levels of cefotaxime (Supplementary Table S5). Whereas the adaptive amino acid substitutions identified in HB36.4 and HB80.3 span the range from smallest to largest beneficial effect, those identified in TEM-1 are the amino acid substitutions with the largest adaptive effect (see Supplementary Text).
We then calculated the fraction of adaptive amino acid mutations that could be reached with a point mutation and compared that with the fraction of all amino acid substitutions that are possible with a point mutation. For these genes, the standard genetic code enriched for adaptive amino acid substitutions up to 40% (Table 2). We assessed the significance of this result by comparing the experimentally determined number of adaptive mutations accessible by a point mutation with the distribution of that value expected if adaptive mutations were chosen at random from all possible mutations (a hypergeometric distribution). The P-value obtained reflects the probability of arriving at that enrichment value or higher by chance under the null hypothesis that adaptive amino acid substitutions are no more likely to be accessible by a point mutation than are all possible amino acid substitutions. The relatively small number of adaptive amino acid substitutions identified for TEM-1 makes obtaining low P-values unlikely unless the enrichment is very large. If the null hypothesis was true, we would expect to find that enrichment would be found as often and to the same extent as depletion; however, depletion was not observed in any of the four sets of adaptive mutations examine. Considering the P-values of all four experiments collectively by meta-analysis (34), we find that the enrichment observed in our experiments is significant (P = 0.0027). This result supports our hypothesis that the standard genetic code, by its limitations on which amino acid substitutions are accessible by a point mutation, facilitates the evolution of proteins by enriching for adaptive mutations.
Table 2.
Enrichment for adaptive mutations provided by the standard genetic code
| Gene | Adaptive advantage | % Enrichment of adaptive amino acidsa |
|---|---|---|
| TEM-1 | Cefotaxime resistance | 39.6 (P = 0.106) |
| TEM-1 | Tazobactam resistance | 35.6 (P = 0.210) |
| HB36.4 | Hemagglutinin binding | 30.6 (P = 0.0066) |
| HB80.3 | Hemagglutinin binding | 0.51 (not significant) |
aDetails on this calculation provided in Supplementary Table S6. The P-values provide the probabilities that the observed enrichment was arrived at by chance under the null hypothesis that adaptive mutations are as likely to be accessible by point mutations as non-adaptive mutations.
DISCUSSION
To the extent our results with these three genes can be generalized, our results indicate that the standard genetic code possesses a remarkable feature—it provides the advantage of reducing the negative effects of mutations while selectively enriching for adaptive mutations. Thus, although the code’s structure limits the exploration of sequence space, it does so in a manner that benefits the evolution of proteins and is a molecular-level example of how constraints can facilitate evolution (35). We speculate that the architecture of the code results in part from selective pressure for a code that facilitates the evolution of proteins. This evolvability theory on the origin of the genetic code is not mutually exclusive with existing theories and offers additional insight into the origin of the standard genetic code.
Our use of the term ‘evolvability’ and our proposal that the code’s enrichment for adaptive mutations provided an adaptive advantage requires further clarification. When compared with other genetic codes that do not enrich for adaptive mutations, the standard genetic code imposes a bias toward adaptive mutations in the standing genetic variation. Of course this bias would not be true for all genes or in all possible environments. Rather, we contend it would be true on average. Thus, provided that adaptation is limited by the supply of adaptive mutations, the standard genetic code would confer a higher degree of evolvability than a code that does not enrich. This advantage the code possesses invokes clade selection, as it provides an advantageous backdrop for adaptive evolution. As evolution proceeds, surviving lineages would become increasingly biased toward those with this code, which experienced more beneficial mutations sooner than their competitors.
Whether this evolvability provided an adaptive advantage (as we propose) or is a byproduct of evolution is a difficult question to answer (36,37). However, our proposal does not suffer from many criticisms of evolvability involving clade selection (37,38). First, we do not need to invoke increased mutation rates or capacity to produce new variation as the source of evolvability. Rather, we contend that the standard genetic code provides a better standing genetic variation for evolution than would codes that do not enrich for adaptive mutations. Thus, early in evolutionary history, this could have provided an adaptive advantage contributing to the standard genetic code winning out over alternative codes that may have been present at the time. Second, the genetic code is not a simple ‘variability allele’ that is prone to being lost by recombination because it is subject to indirect selection. The genetic code is a manifestation of a large set of genes and is central to life. It cannot be lost, and it is difficult to think of ways in which the code would be vulnerable to ‘selfish’ alternatives. Thus, although our results indicate that the standard genetic code possesses the ability to enrich for adaptive mutations today and in the future, we are not invoking a teleological view of evolution. Rather, the adaptive advantage existed earlier in evolution in the context of other competing codes while genomes were smaller and the genetic code exhibited plasticity. We believe that the code’s retention of this feature is a testament to how difficult it is to substantially change the genetic code after its fixation in the last universal common ancestor (10).
Our results support the idea that both robustness to error and improved access to adaptive mutations were selected for in the genetic code’s evolution. We speculate that there are two possible ways in which our evolvability theory can be reconciled with the adaptive theory vis-à-vis error minimization. (i) First, perhaps a code’s error minimization must be balanced by its propensity to promote the evolution of proteins. A code maximized for robustness to error would allow only the most conservative of mutations, which may not be optimal from the perspective of protein evolution. We postulate that a code that allows for the right balance between error minimization and effective exploration of sequence space would be evolutionarily advantageous. In this view, the evolvability theory provides a possible explanation for the extent to which, if any, the code is not optimized for error minimization (11). (ii) Second, it may be that the error minimization and adaptive mutation enrichment provided by the genetic code are two sides of the same coin. Potentially, a conservative genetic code increases the probability of achieving an adaptive mutation by reducing the effect of the mutations (39), consistent with Fisher’s Geometric Theorem (40). If error minimization and enrichment for adaptive mutations do come together as a package, an interesting but difficult question to address experimentally is the extent to which each contributed to the origin of the genetic code.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–6, Supplementary Figure 1, Supplementary Text and Supplementary References [15,41,42].
FUNDING
National Science Foundation (NSF) [DEB-0950939 to M.O.]. Funding for open access charge: NSF [DEB-0950939 to M.O.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Timothy A. Whitehead, Aaron Chevalier and David Baker for providing their deep sequencing data of the selected and unselected HB36.4 and HB80.3 libraries (17). We thank Jeffrey J. Gray and Stephen J. Freeland for constructive comments on the manuscript.
REFERENCES
- 1.Smith JM. Natural selection and the concept of a protein space. Nature. 1970;225:563–564. doi: 10.1038/225563a0. [DOI] [PubMed] [Google Scholar]
- 2.Terekhanova NV, Bazykin GA, Neverov A, Kondrashov AS, Seplyarskiy VB. Prevalence of multinucleotide replacements in evolution of primates and Drosophila. Mol. Biol. Evol. 2013;30:1315–1325. doi: 10.1093/molbev/mst036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woese CR. On the evolution of the genetic code. Proc. Natl Acad. Sci. USA. 1965;54:1546–1552. doi: 10.1073/pnas.54.6.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burch CL, Chao L. Evolvability of an RNA virus is determined by its mutational neighbourhood. Nature. 2000;406:625–628. doi: 10.1038/35020564. [DOI] [PubMed] [Google Scholar]
- 5.Cambray G, Mazel D. Synonymous genes explore different evolutionary landscapes. PLoS Genet. 2008;4:e1000256. doi: 10.1371/journal.pgen.1000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall AR, Griffiths VF, MacLean RC, Colegrave N. Mutational neighbourhood and mutation supply rate constrain adaptation in Pseudomonas aeruginosa. Proc. Biol. Sci. 2010;277:643–650. doi: 10.1098/rspb.2009.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gamow G. Possible relation between deoxyribonucleic acid and protein structure. Nature. 1954;173:318. [Google Scholar]
- 8.Wong JT. A co-evolution theory of the genetic code. Proc. Natl Acad. Sci. USA. 1975;72:1909–1912. doi: 10.1073/pnas.72.5.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonneborn TM. Degeneracy of the genetic code: extent, nature, and genetic implications. In: Bryson V, Voge HJ, editors. Evolving Genes and Proteins. New York: Academic Press; 1965. pp. 377–397. [Google Scholar]
- 10.Crick FH. The origin of the genetic code. J. Mol. Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 11.Koonin EV, Novozhilov AS. Origin and evolution of the genetic code: the universal enigma. IUBMB Life. 2009;61:99–111. doi: 10.1002/iub.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitch WM. An improved method of testing for evolutionary homology. J. Mol. Biol. 1966;16:9–16. doi: 10.1016/s0022-2836(66)80258-9. [DOI] [PubMed] [Google Scholar]
- 13.Stoltzfus A, Yampolsky LY. Climbing mount probable: mutation as a cause of nonrandomness in evolution. J. Hered. 2009;100:637–647. doi: 10.1093/jhered/esp048. [DOI] [PubMed] [Google Scholar]
- 14.Zhu W, Freeland S. The standard genetic code enhances adaptive evolution of proteins. J. Theor. Biol. 2006;239:63–70. doi: 10.1016/j.jtbi.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 15.Firnberg E, Ostermeier M. PFunkel: efficient, expansive, user-defined mutagenesis. PLoS One. 2012;7:e52031. doi: 10.1371/journal.pone.0052031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weinreich DM, Delaney NF, Depristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312:111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 17.Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, Myers CA, Kamisetty H, Blair P, Wilson IA, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 2012;30:543–548. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS. Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature. 2006;444:929–932. doi: 10.1038/nature05385. [DOI] [PubMed] [Google Scholar]
- 19.DePristo MA, Hartl DL, Weinreich DM. Mutational reversions during adaptive protein evolution. Mol. Biol. Evol. 2007;24:1608–1610. doi: 10.1093/molbev/msm118. [DOI] [PubMed] [Google Scholar]
- 20.Salverda ML, Dellus E, Gorter FA, Debets AJ, van der Oost J, Hoekstra RF, Tawfik DS, de Visser JA. Initial mutations direct alternative pathways of protein evolution. PLoS Genet. 2011;7:e1001321. doi: 10.1371/journal.pgen.1001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poyart C, Mugnier P, Quesne G, Berche P, Trieu-Cuot P. A novel extended-spectrum TEM-type beta-lactamase (TEM-52) associated with decreased susceptibility to moxalactam in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 1998;42:108–113. doi: 10.1128/aac.42.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barlow M, Hall BG. Predicting evolutionary potential: in vitro evolution accurately reproduces natural evolution of the tem beta-lactamase. Genetics. 2002;160:823–832. doi: 10.1093/genetics/160.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kopsidas G, Carman RK, Stutt EL, Raicevic A, Roberts AS, Siomos MA, Dobric N, Pontes-Braz L, Coia G. RNA mutagenesis yields highly diverse mRNA libraries for in vitro protein evolution. BMC Biotechnol. 2007;7:18. doi: 10.1186/1472-6750-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orencia MC, Yoon JS, Ness JE, Stemmer WP, Stevens RC. Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nat. Struct. Biol. 2001;8:238–242. doi: 10.1038/84981. [DOI] [PubMed] [Google Scholar]
- 25.Stemmer WPC. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 26.Zaccolo M, Gherardi E. The effect of high-frequency random mutagenesis on in vitro protein evolution: a study on TEM-1 beta-lactamase. J. Mol. Biol. 1999;285:775–783. doi: 10.1006/jmbi.1998.2262. [DOI] [PubMed] [Google Scholar]
- 27.Hayes RJ, Bentzien J, Ary ML, Hwang MY, Jacinto JM, Vielmetter J, Kundu A, Dahiyat BI. Combining computational and experimental screening for rapid optimization of protein properties. Proc. Natl Acad. Sci. USA. 2002;99: 15926–15931. doi: 10.1073/pnas.212627499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yi H, Cho KH, Cho YS, Kim K, Nierman WC, Kim HS. Twelve positions in a beta-lactamase that can expand its substrate spectrum with a single amino acid substitution. PLoS One. 2012;7:e37585. doi: 10.1371/journal.pone.0037585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poelwijk FJ, Kiviet DJ, Weinreich DM, Tans SJ. Empirical fitness landscapes reveal accessible evolutionary paths. Nature. 2007;445:383–386. doi: 10.1038/nature05451. [DOI] [PubMed] [Google Scholar]
- 30.Cantu C, 3rd, Palzkill T. The role of residue 238 of TEM-1 beta-lactamase in the hydrolysis of extended-spectrum antibiotics. J. Biol. Chem. 1998;273:26603–26609. doi: 10.1074/jbc.273.41.26603. [DOI] [PubMed] [Google Scholar]
- 31.Shafikhani S, Siegel RA, Ferrari E, Schellenberger V. Generation of large libraries of random mutants in Bacillus subtilis by PCR-based plasmid multimerization. Biotechniques. 1997;23:304–310. doi: 10.2144/97232rr01. [DOI] [PubMed] [Google Scholar]
- 32.Lee H, Popodi E, Tang H, Foster PL. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc. Natl Acad. Sci. USA. 2012;109:E2774–E2783. doi: 10.1073/pnas.1210309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher's approach. J. Evol. Biol. 2005;18:1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- 35.Gould SJ. The evolutionary biology of constraint. Daedalus. 1980;109:39–52. [Google Scholar]
- 36.Colegrave N, Collins S. Experimental evolution: experimental evolution and evolvability. Heredity. 2008;100:464–470. doi: 10.1038/sj.hdy.6801095. [DOI] [PubMed] [Google Scholar]
- 37.Sniegowski PD, Murphy HA. Evolvability. Curr. Biol. 2006;16:R831–R834. doi: 10.1016/j.cub.2006.08.080. [DOI] [PubMed] [Google Scholar]
- 38.Lynch M. The frailty of adaptive hypotheses for the origins of organismal complexity. Proc. Natl Acad. Sci. USA. 2007;104(Suppl. 1):8597–8604. doi: 10.1073/pnas.0702207104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freeland SJ. The Darwinian genetic code: an adaptation for adapting? Genet. Program. Evol. Mach. 2002;3:113–127. [Google Scholar]
- 40.Fisher RA. The Genetical Theory of Natural Selection. Oxford: The Clarendon Press; 1930. [Google Scholar]
- 41.Schenk MF, Szendro IG, Krug J, de Visser JA. Quantifying the adaptive potential of an antibiotic resistance enzyme. PLoS Genet. 2012;8:e1002783. doi: 10.1371/journal.pgen.1002783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salverda ML, De Visser JA, Barlow M. Natural evolution of TEM-1 beta-lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 2010;34:1015–1036. doi: 10.1111/j.1574-6976.2010.00222.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.