

Summary
NAD+ is an important co-factor regulating metabolic homeostasis and a rate-limiting substrate for sirtuin deacylase. We show that NAD+ levels are reduced in aged mice and C. elegans and that decreasing NAD+ levels results in a further reduction in worm lifespan. Conversely, genetic or pharmacological restoration of NAD+ prevents age-associated metabolic decline and promotes longevity in worms. These effects are dependent upon the protein deacetylase sir-2.1 and involve the induction of mitonuclear protein imbalance as well as activation of stress signaling via the mitochondrial unfolded protein response (UPRmt) and the nuclear translocation and activation of FOXO transcription factor DAF-16. Our data suggest that augmenting mitochondrial stress signaling through the modulation of NAD+ levels may be a target to improve mitochondrial function and prevent or treat age-associated decline.
Introduction
Alterations in NAD+ levels have a powerful metabolic impact, since it serves as an obligatory substrate for the deacetylase activity of the sirtuin proteins (Guarente, 2008; Haigis and Sinclair, 2010; Houtkooper et al., 2010a). The best-characterized mammalian sirtuin is SIRT1, which controls mitochondrial function through the deacetylation of targets that include PGC-1α and FOXO (Chalkiadaki and Guarente, 2012; Houtkooper et al., 2012). The administration of NAD+ precursors, such as nicotinamide mononucleotide (Yoshino et al., 2011) or nicotinamide riboside (NR) (Canto et al., 2012), has proven to be an efficient way to increase NAD+ levels and SIRT1 activity, improving metabolic homeostasis in mice. Furthermore, the NAD+-consuming poly(ADP-ribose) polymerase proteins—with PARP1 and PARP2 representing the main PARP activities in mammals—were classically described as DNA repair proteins (Gibson and Kraus, 2012; Schreiber et al., 2006), but recent studies have linked these proteins to metabolism (Asher et al., 2010; Bai et al., 2011a; Bai et al., 2011b; Erener et al., 2012). Indeed, genetic or pharmacological inactivation of PARP1 increased tissue NAD+ levels and activated mitochondrial metabolism (Bai et al., 2011b). An association between PARPs and lifespan has been postulated (Grube and Burkle, 1992; Mangerich et al., 2010), but a causal role remained unclear. A final line of evidence in support of a role for NAD+ in metabolic control came from the deletion of an alternative NAD+-consuming protein, CD38, which also led to NAD+ accumulation and subsequent SIRT1 activation in mice, and proved protective against high-fat diet-induced obesity (Barbosa et al., 2007).
Considering the intimate link between metabolism and longevity (Guarente, 2008; Houtkooper et al., 2010b), we hypothesized that increasing NAD+ levels may be sufficient to increase mitochondrial activity and extend lifespan (Houtkooper and Auwerx, 2012). Here we show how supplementation of PARP inhibitors or NAD+ precursors led to improved mitochondrial homeostasis through the activation of the worm sirtuin homolog, sir-2.1. This improvement involved the disturbed balance between OXPHOS subunits encoded by mitochondrial DNA (mtDNA) and nuclear DNA (nDNA), a state we termed mitonuclear protein imbalance. This associates with the activation of the mitochondrial unfolded protein response (UPRmt)—a mitochondrial proteostasis pathway promoting longevity (Durieux et al., 2011; Yoneda et al., 2004; Zhao et al., 2002)—and subsequent translocation and activation of the FOXO transcription factor daf-16—triggering an antioxidant protection program (Honda and Honda, 1999). Together, our results expose a novel temporal regulation network for sirtuins on eukaryotic lifespan, and pharmacological approaches to control this pathway and prevent age-related physiological decline.
Results
Disturbed NAD+ metabolism as a core biochemical phenotype of aging
To establish the role of NAD+ metabolism in aging, we compared PARP activity (global PARylation), NAD+ levels and sirtuin activation in young versus old mice (24 and 103 weeks (Houtkooper et al., 2011)). Both in liver and muscle of aged mice, PARylation was markedly increased (Figure S1A). In line with the hypothesis that PARP proteins are major NAD+ consumers, NAD+ levels were robustly decreased in older mice (Figure S1B), confirming recent data (Braidy et al., 2011; Yoshino et al., 2011). Changes in NAD+ are generally translated into altered SIRT1 activity (reviewed in (Houtkooper et al., 2012)). The lower NAD+ levels in aged mice were indeed reflected in hyperacetylation of the SIRT1 substrate PGC-1α, indicative of reduced SIRT1 activity (Figure S1C). To evaluate the possible contribution of PARP activity and NAD+ metabolism in the aging process, we turned to the worm, C. elegans, where it is easier to evaluate the impact of genetic or pharmacological manipulations on lifespan. The aging-associated changes in PARylation and NAD+ levels were evolutionarily conserved as PARylation was also markedly increased with age in nematodes (Figure 1A), and NAD+ levels were lower (Figure 1B). Changes in PARylation and NAD+ were attenuated in worms in which the PARP1 homolog—pme-1 (Gagnon et al., 2002)—was mutated (Figure 1A–B). The residual PARylation is consistent with the presence of a second PARP gene, pme-2, the worm homolog of the less active PARP2 protein (Canto and Auwerx, 2012). We further analyzed the natural aging process in worms by monitoring the accumulation of the aging-associated lipid peroxidation product lipofuscin, which was robustly reduced in pme-1 worms (Figure 1C). We then tested whether reduced NAD+ levels are causally linked to aging. First, we depleted NAD+ chemically using paraquat (Figure 1D), and this is associated with shortened lifespan (Figure 1E). One could argue, however, that the premature death could be caused by excessive DNA damage. Therefore, we also depleted NAD+ genetically. We treated rrf-3(pk1426) worms with RNAi targeting qns-1, encoding the enzyme NAD+ synthase that catalyzes the final step in NAD+ biosynthesis. Knockdown of qns-1 indeed depleted NAD+ and shortened lifespan (Figure 1F–G). Together, these data suggest that disturbance of the PARP/NAD+-signaling network in aging is evolutionarily conserved and causally involved.
Figure 1. NAD+ is causally involved in aging.

(A) Aged C. elegans displayed higher total protein PARylation levels, which were largely attenuated in pme-1 mutants. Ponceau staining is used as a loading control.
(B) Aging decreased worm NAD+ levels, in both wildtype and in pme-1 mutant worms, with a higher level of NAD+ in the pme-1 mutant during aging. Two-way ANOVA indicated significant difference with age (p<0.008) and genotype (p=0.02).
(C) pme-1 mutant worms accumulated less of the aging pigment lipofuscin compared to wild type worms.
(D–E) Supplementation of N2 wild type worms with 4 mM paraquat depletes NAD+ levels (D) and shortens lifespan (E).
(F–G) RNAi against qns-1, encoding NAD+ synthase, depletes NAD+ levels (F) and shortens lifespan in worms (G)
(H) pme-1(ok988) mutant worms show a 29% mean lifespan extension (left panel) while pme-1 RNAi in the rrf-3(pk1426) strain extends lifespan by 20% (right panel).
(I–J) PARP inhibition by either AZD2281 (100 nM) or ABT-888 (100 nM) extended worm lifespan respectively by 22.9% (I) and 15% (J).
(K) The lifespan extension of AZD2281 is pme-1-dependent.
Bar graphs are expressed as mean±SEM, * p≤0.05; ** p≤0.01; *** p≤0.001. For lifespan curves, p-values are shown in the graph (n.s. = not significant).
See also Figure S1 and S2A–B. See Table S1 and S2 for additional detail on the lifespan experiments.
Increasing NAD+ levels extends lifespan through sir-2.1
We next aimed to determine whether the age-related NAD+ depletion could be reverted and thereby aging prevented. Strikingly, pme-1 deficient worms, either by mutation or RNAi, displayed respectively a 29% or 20% mean lifespan extension (Figure 1H, see Table S1 for statistics). To consolidate these results, we also examined the lifespan of worms upon inhibition of PARP activity with two distinct pan-PARP inhibitors representing different chemical scaffolds (Ferraris, 2010), i.e. AZD2281 (KU59436, olaparib) (Menear et al., 2008), or ABT-888 (veliparib) (Penning et al., 2009). Feeding of worms from eggs until death with different concentrations of PARP inhibitors resulted in a 15–23% lifespan extension (Figure 1I–J, Figure S2A–B, Table S1), with a maximum extension at 100 nM (Figure S2A; Table S2), which is why we chose this concentration for further experiments. Importantly, the lifespan of the pme-1 mutant was not further extended by AZD2281, confirming that pme-1 is the major worm PARP activity (Figure 1K).
Besides inhibiting NAD+ breakdown we also focused on supplying NAD+ precursors, notably the salvage pathway precursors nicotinamide (NAM) and NAM riboside (NR). NAM is the end-product of the sirtuin and PARP reaction, whereas NR is a recently discovered vitamin B3. Both can serve as precursors of NAD (re-)synthesis (reviewed in (Houtkooper et al., 2010a)). Similar to AZD2281 and ABT-888, lifespan extension was observed when the worms were supplemented with either NAM or NR (Figure 2A–B, Table S1). Based on the dose-dependent effects on lifespan (Figure S2C–D; Table S2), we selected 500 μM NR and 200 μM NAM as optimal concentrations for further experiments. Importantly, combined supplementation of AZD2281 and NR at their optimal concentration did not synergistically extend lifespan, compared to either compound alone (Figure 2C; Table S1). However, when both compounds were added at a suboptimal dose, each not sufficient to induce longevity (100 μM NR + 10 nM AZD2281), lifespan was synergistically extended (Figure 2D; Table S1).
Figure 2. PARP inhibitors and NAD+ precursors increase mitochondrial function.

(A–B) Supplementation of the NAD+ precursor NR (500 μM; A) or NAM (200 μM; B) in wild type N2 worms increases lifespan.
(C) Combined treatment using optimal concentrations of AZD2281 (100 nM) and NR (500 μM) extends lifespan (+16%/p=0.02), but not further than the individual compounds (+22%/p=0.0004 and +16%/p=0.01, respectively). Mean lifespans: vehicle: 16.1±0.6 days; 100 nM AZD2281: 19.7±0.8 days; 500 μM NR: 18.7±0.8 days; 100 nM AZD2281+500 μM NR: 18.6±0.7 days.
(D) A combination of sub-optimal doses of AZD2281 (10 nM) and NR (100 μM) extended lifespan (11%/p=0.01), while the individual compounds at these concentrations had no effect on lifespan (+7%/ns and +5%/ns, respectively). Mean lifespans: vehicle: 20.1±0.6 days; 10 nM AZD2281: 21.5±0.7 days; 100 μM NR: 21.1±0.7 days; 10 nM AZD2281+100 μM NR: 22.3±0.7 days.
(E) AZD2281 and NR supplementation increased NAD+ levels in C. elegans.
(F) PARP inhibition by AZD2281 (100 nM) and NR supplementation (500 μM) do not extend lifespan in the sir-2.1(ok434) mutant (n.s = not significant).
(G) Oxygen consumption was increased in day 3 and day 10 adult worms after AZD2281 (AZD; 100 nM) or NR (500 μM) treatment.
(H) AZD2281 and NR increased mitochondrial biogenesis at day 3 and day 10 of adulthood, as evidenced by the increased mtDNA/nDNA ratio.
(I) AZD2281 and NR increased worm ATP levels at day 3 of adulthood.
(J) AZD2281 and NR increased gene expression (day 3 adults) of key metabolic genes cts-1 (TCA cycle), hxk-1 (glycolysis) and pyc-1 (gluconeogenesis), but not that of cox-4.
(K) AZD2281 and NR improved worm fitness at day 3 and 10 of adulthood, as evidenced by measuring worm motility.
Bar graphs are expressed as mean±SEM, * p≤0.05; ** p≤0.01.
See also Figure S2C–D and S3. See Table S1 and S2 for additional detail on the lifespan experiments.
As observed in pme-1 worms (Figure 1B), supplementation with PARP inhibitors or NAD+ precursors significantly increased NAD+ levels (Figure 2E, S3A–B). Although the role of SIRT1 or its homologs in lifespan extension under basal, unstressed, conditions is subject of debate (Burnett et al., 2011; Rogina and Helfand, 2004; Tissenbaum and Guarente, 2001; Viswanathan and Guarente, 2011), it holds a central position in healthspan regulation in the context of disease or cellular stress (Houtkooper et al., 2012). Given the NAD+ dependence of sirtuin enzymes (Imai et al., 2000), we analyzed epistasis by treating sir-2.1(ok434) mutant worms with the PARP inhibitor AZD2281 or with NR. The effect of both these compounds on longevity was abrogated in the sir-2.1(ok434) mutant (Figure 2F), confirming sir-2.1 dependence of the lifespan extension induced by two distinct strategies to raise NAD+ levels.
PARP inhibitors and NAD+ precursors boost mitochondrial function
As NAD+ and SIRT1/sir-2.1 are thought to influence oxidative metabolism (Guarente, 2008; Houtkooper et al., 2012), we functionally characterized mitochondrial activity in AZD2281- and NR-treated worms by measuring oxygen consumption rates. While respiration strongly decreased with age in control worms, worms treated with AZD2281, NR and NAM maintained or even increased respiration at young adult level (Figure 2G, S3C). This was corroborated by increased mitochondrial abundance evaluated by the mtDNA/nDNA ratio (Figure 2H), increased ATP levels (Figure 2I), and increased gene expression of enzymes controlling key metabolic pathways, e.g. the TCA cycle gene citrate synthase (cts-1), the glycolysis gene hexokinase (hxk-1) and the gluconeogenesis gene pyruvate carboxylase (pyc-1) (Figure 2J). Functionally, the improved metabolic state was accompanied by better worm motility at later age (Figure 2K), a sign of improved fitness with age.
Mitochondrial theories of aging
Two pathways have been described to link mitochondrial activity and aging, UPRmt and the reactive oxygen species (ROS) defense pathway. UPRmt is activated when protein balance in mitochondria is disturbed. This occurs for instance upon accumulation of misfolded or unfolded proteins (Haynes and Ron, 2010). Alternatively, disturbance of the delicate balance between mitochondrial protein production from nDNA and mtDNA—a state we termed mitonuclear protein imbalance—is associated with UPRmt activation (Houtkooper et al., 2013). This triggers a mitochondrial-to-nuclear stress-signaling pathway, which induces the transcription of nDNA-encoded mitochondrial molecular chaperones, such as mtHSP70, HSP60 and HSP10 and the protease CLPP (Durieux et al., 2011; Yoneda et al., 2004; Zhao et al., 2002). On the other hand, the defense against the accumulation of ROS is activated when altered mitochondrial activity causes formation of excessive oxygen radicals, with the ultimate goal of preventing damage of the (sub)cellular microenvironment, e.g. lipids, DNA and proteins. This pathway involves the nuclear translocation of the FOXO transcription factor daf-16 (Berdichevsky et al., 2006), which, amongst other targets, induces the transcription of the mitochondrial antioxidant sod-3 (Honda and Honda, 1999). The mammalian daf-16 homolog FOXO3A has been described as a deacetylation target for SIRT1 in mammals (Brunet et al., 2004; Motta et al., 2004; van der Horst et al., 2004), and daf-16 was shown to interact with sir-2.1 (Berdichevsky et al., 2006; Hashimoto et al., 2010), providing a possible link between NAD+ and this pathway.
It is unclear whether, and if so how, these two “mitochondrial aging” pathways are activated by NAD+ and how they are intertwined, although it was already shown that ROS do not cause UPRmt (Durieux et al., 2011). We hence set out to perform a detailed temporal analysis of the activation of both stress pathways, focusing on day 1 and day 3 of adulthood. These days were selected based on the premise that they are in different phases of the mitohormesis response (Mouchiroud et al., 2011; Schulz et al., 2007; Zarse et al., 2012). Mitohormesis refers to the concept that an adaptive and protective antioxidant response is activated in mitochondria by an initial accumulation of moderate levels of potentially toxic ROS. While day 1 is the first day of adulthood and coincides with a burst in ROS production, day 3 is the late reproductive phase (but still pre-aging) and is characterized by activated ROS defense (Schulz et al., 2007).
Early phase activation of UPRmt by NAD+ boosters
By using confocal microscopy in the pmyo-3::mito::GFP reporter, which expresses mitochondria-targeted GFP in the muscle, we determined the morphology and intensity of the mitochondrial network. At day 1 of adulthood, AZD2281 or NR treatment led to a dense mitochondrial network that appears to be more fragmented compared to vehicle control worms (Figure 3A). This reticular network was likely caused by reduced expression of the mitochondrial fusion genes fzo-1 and opa-1, without changes in the fission regulator drp-1 (Figure 3B). We then measured the two prime mitochondrial pathways involved in aging, ROS and UPRmt. ROS was measured using the mitoSOX probe, which indicates specific mitochondrial superoxide production. Both AZD2281 and NR induced a strong superoxide burst at day 1 of adulthood, although at this stage no protective activation of sod-3 expression was observed (Figure 3C). When we measured at the same age UPRmt activation using hsp-6::GFP reporter worms (Yoneda et al., 2004), both compounds robustly induced UPRmt (Figure 3D), without activating the ER-specific unfolded protein response (hsp-4::GFP; Figure 3E). Importantly, the induction of UPRmt, but not sod-3, was confirmed in worms treated with pme-1 RNAi, confirming that the effect is not due to off-target effects of the drugs (Figure S4A). The precocious activation of UPRmt after AZD2281 or NR treatment was associated with mitonuclear protein imbalance, as evidenced by the decreased ratio between the nDNA-encoded ATP5A and the mtDNA-encoded MTCO1 (Figure 3F). We next tested whether UPRmt was essential for longevity induced by NAD+ boosters or PARP inhibitors. We used worms treated with RNAi directed against the UPRmt gene, ubl-5, an essential component of the mitochondria-to-nucleus feedback (Durieux et al., 2011). Indeed, ubl-5 RNAi abrogated the lifespan extension conferred by both AZD2281 and NR treatment (Figure 3G–H). This coincided with the attenuation of UPRmt activation in ubl-5 RNAi worms (Figure 3I). These data suggest that UPRmt is an early-phase response induced by increased NAD+ levels.
Figure 3. Early phase response of NAD+ boosters on mitochondrial aging pathways.

(A) The effects of AZD2281 (100 nM) and NR (500 μM) on mitochondrial content and morphology in body wall muscle. At day 1 of adulthood, mitochondria of AZD2281- or NR-treated worms appear more fragmented. Stars represent nuclei, insets show higher magnification of a small section of the image, marked by dashed rectangle.
(B) At day 1 of adulthood, AZD2281 and NR reduced the expression of mitochondrial fusion genes fzo-1 and opa-1, without affecting the fission gene drp-1.
(C) At day 1, AZD2281 and NR cause a burst of ROS, as measured using the MitoSOX probe. This was not accompanied by an induction of the antioxidant gene sod-3 (measured using a GFP-coupled sod-3 reporter).
(D–E) At day 1, AZD2281 and NR induced the mitochondrial unfolded protein response (UPRmt) (hsp-6 reporter; D), without activating the ER unfolded protein response (hsp-4 reporter; E). In panel D, representative images are shown on the left, while quantification is shown in the bar graph on the right.
(F) AZD2281 and NR induced mitonuclear protein imbalance, as evidenced by the decreased ratio between nDNA-encoded ATP5A and mtDNA-encoded MTCO1. Representative Western blot shown on the left, quantification of the ratio in three independent experiments is shown on the right.
(G–H) RNAi of the UPRmt regulator ubl-5 abrogated the lifespan extension induced by AZD2281 (G; at 100 nM) and NR (H; at 500 μM).
(I) The UPRmt induction by AZD2281 and NR at day 1 is also ubl-5 dependent.
Bar graphs are expressed as mean±SEM, * p≤0.05; ** p≤0.01.
See also Figure S4. See Table S1 for additional detail on the lifespan experiments.
Late phase activation of ROS defense by NAD+ boosters
To test for potential adaptive responses with respect to ROS and UPRmt, we measured similar parameters at day 3 of adulthood. While fragmented at day 1, mitochondria appear hyperfused at day 3 (Figure 4A), which is now at this time associated with increased gene expression of fzo-1 and opa-1 (Figure 4B). When we analyzed the activation of UPRmt and ROS defense at day 3 of adulthood, we not only observed a continued activation of UPRmt, but now, unlike the situation at day 1, also a strong activation of the sod-3 promoter in worms treated with AZD2281, NR (Figure 4C), or pme-1 RNAi (Figure S4B). We then characterized the ROS defense mechanisms in more detail. At day 3, the higher sod-3::GFP levels after AZD2281 or NR treatment were not due to increased daf-16 gene expression, a key transcriptional regulator of antioxidant defense, was not affected (Figure 4D). The absence of an induction of the daf-16 gene suggests that daf-16 activity might be regulated by AZD2281 and NR via changes in subcellular distribution rather than transcription (Alam et al., 2010). Strikingly, the stress responses were specific for sod-3 and UPRmt, as expression of cytosolic or ER UPR markers, as well as other antioxidants did not change, or even decreased (Figure 4E).
Figure 4. Late phase effects of NAD+ boosters on mitochondrial aging pathways.

(A) The effects of AZD2281 (100 nM) and NR (500 μM) on mitochondrial content and morphology in body wall muscle. At day 3 of adulthood, mitochondria of AZD2281- or NR-treated worms appear more fused. Stars represent nuclei, insets show higher magnification of a small section of the image, marked by dashed rectangle.
(B) At day 3 of adulthood, AZD2281 and NR increased the expression of mitochondrial fusion genes fzo-1 and opa-1, without affecting the fission gene drp-1.
(C) At day 3 of AZD2281 (100 nM) or NR (500 μM) treatment, UPRmt is still activated (left panel), but now accompanied by an induction of the sod-3::GFP reporter (right panel).
(D–E) AZD2281- and NR-treated worms showed no change in daf-16 expression (D) or activation of expression of other stress genes (E).
(F) Supplementation of PARP inhibitor AZD2281 (100 nM) or NAD+ precursor NR (500 μM) increases mean lifespan of wild type N2 worms treated with 4 mM paraquat. p-values are shown in the graph.
(G) Representative images of daf-16::GFP reporter worms treated with either vehicle or AZD2281 (top)/NR (bottom), showing nuclear accumulation of daf-16 following treatment, indicated by arrowheads.
(H) Quantification of daf-16 nuclear translocation following treatment with AZD2281 (top), NR (bottom). Localization is shown as percentage of worms that shows nuclear (yellow or light blue) or cytosolic (red or dark blue) localization
(I) Lifespan extension following AZD2281 or NR treatment is dependent on daf-16.
(J) The induction of sod-3::GFP reporter following AZD2281 or NR treatment is dependent on the UPRmt regulator ubl-5.
Bar graphs are expressed as mean±SEM, * p≤0.05; ** p≤0.01; *** p≤0.001.
See also Figure S4–5. See Table S1 for additional detail on the lifespan experiments.
Considering the strong induction of sod-3 expression, we next tested whether AZD2281 and NR could protect against the premature aging caused by the ROS-inducer paraquat. While vehicle-treated worms die young (median = 6 days), AZD2281, NR or NAM treated worms survive significantly longer (median = 7 days) (Figure 4F, S5A). As mentioned above, DAF-16 activity is primarily regulated by its subcellular localization (Alam et al., 2010). We hence used the daf-16::GFP reporter strain to establish DAF-16 localization at day 3 following AZD2281, NR or NAM exposure. In line with the increased sod-3 transcriptional activation (Figure 4C), daf-16 appeared in a more punctate pattern consistent with its nuclear localization that is required for it to activate transcription of its antioxidant target genes (Figure 4G, S5B). Quantitative analysis of this daf-16 translocation after these various stimuli shows that about 2–3 times more daf-16 is localized in the nucleus (Figure 4H, S5C). Confirming the crucial role of daf-16, we observed that the lifespan extension induced by compounds that enhance NAD+ levels was strictly dependent on the expression of daf-16 (Figure 4I), suggesting that the induction of antioxidant defense is key for AZD2281 and NR to grant longevity. Finally, the induction of sod-3 expression was dependent on the presence of the UPRmt regulator ubl-5 (Figure 4J), suggesting that this UPRmt is involved in the transcriptional activation of antioxidants as well.
UPRmt controls lifespan extension by sir-2.1 overexpression
The role of sir-2.1 in lifespan regulation is debated. While original reports claimed that overexpression of sir-2.1 led to strong lifespan extension (Tissenbaum and Guarente, 2001), this was later shown to be partly due to an unlinked mutation affecting sensory perception (Burnett et al., 2011; Viswanathan and Guarente, 2011). Interestingly, when the unintended mutation was lost by genetic outcrossing, the resulting MV389 sir-2.1 transgenic worms strains maintained ~10% lifespan extension (Viswanathan and Guarente, 2011). We used this sir-2.1 transgenic worm strain to test whether increased lifespan due to enhanced sirtuin expression involves UPRmt. The MV389 worms lived 25% longer compared to isogenic controls (Figure 5A) and displayed a robust sir-2.1 overexpression that could be abrogated using RNAi (Figure 5B). The fact that the MV389 worms lived slightly longer in our hands compared to previous data (Viswanathan and Guarente, 2011) is due to small differences in our technical approach, as we now censored worms with a protruding vulva phenotype. Similar to compounds that boost NAD+ levels, sir-2.1 overexpression did not affect the daf-16 gene expression, but increased expression of mitochondrial regulators cts-1 and fzo-1, the daf-16 target sod-3, and the expression of the UPRmt gene hsp-6, without changes in other stress markers, such as hsp-4, hsf-1, sod-1, sod-2, gst-4, and prdx-2 (Figure 5C). MV389 worms also displayed mitonuclear protein imbalance with a relative decrease in the ratio between nDNA-encoded ATP5A and mtDNA-encoded MTCO1 (Figure 5D). The longevity of MV389 worms was fully dependent on sir-2.1 and daf-16, as RNAi against these genes completely abrogated the lifespan extension (Figure 5E–F). Importantly, sir-2.1 knockdown in MV389 was also sufficient to block the induction of the UPRmt gene hsp-6 (Figure 5G). In line with this observation, crossing the sir-2.1 transgenic worms with the hsp-6::GFP UPRmt reporter strain confirmed the robust induction of UPRmt (Figure 5H–I), which was attenuated by RNAi targeting sir-2.1 or the UPRmt regulator ubl-5 (Figure 5I), indicating an epistatic link between sir-2.1 expression and the UPRmt pathway. Consistent with this link, ubl-5 RNAi almost completely restored the lifespan of the MV389 strain to wild type levels, showing that the effects of sir-2.1 overexpression were also fully dependent on UPRmt (Figure 5J).
Figure 5. sir-2.1 overexpression extends lifespan through UPRmt.

(A) Outcrossed sir-2.1 transgenic worms live significantly longer compared to control worms. MV389 represents the outcrossed sir-2.1 overexpressing strain. p-value is shown in the graph.
(B) sir-2.1 is robustly overexpressed in sir-2.1 transgenic worms. This induction is almost completely blocked by sir-2.1 RNAi demonstrating specificity.
(C) sir-2.1 overexpression induced the gene expression of cts-1 (TCA cycle), fzo-1 (mitochondrial fusion), hsp-6 (UPRmt) and sod-3 (ROS defense), but not of other stress genes or of the ROS defense regulator daf-16.
(D) sir-2.1 overexpression in MV389 induced mitonuclear protein imbalance, as evidenced by the reduction in the ratio between nDNA-encoded ATP5A and mtDNA-encoded MTCO1. The Western blot depicts three independent samples for each strain, while the right panel shows a quantification of the mitonuclear protein imbalance.
(E–F) (E) sir-2.1, and (F) daf-16 RNAi abrogated the lifespan extension of MV389
(G) sir-2.1 overexpression in the MV389 strain induces the UPRmt gene hsp-6, an effect that is attenuated upon sir-2.1 RNAi.
(H–I) In MV389 worms that were crossed with the UPRmt (hsp-6::GFP) reporter worms, UPRmt was markedly increased compared to hsp-6::GFP control worms. Panel H shows GFP expression in sir-2.1 wild type (top) and sir-2.1 overexpressing (bottom) worms. Panel I shows the quantification of this GFP signal and that this induction was attenuated upon ubl-5 or sir-2.1 RNAi.
(J) ubl-5 RNAi abrogated the lifespan extension of MV389.
Bar graphs are expressed as mean±SEM, * p≤0.05; ** p≤0.01; *** p≤0.001.
See Table S1 for additional detail on the lifespan experiments.
Increased NAD+/SIRT1 signaling induces UPRmt in mammalian cells
We next aimed to confirm the physiological relevance of the NAD+/SIRT1/UPRmt/SOD signaling axis in mammalian cells. In line with our previous results in mice (Bai et al., 2011b; Canto et al., 2012), both PARP inhibition and supplying NAD+ precursors for 48h in the hepatocyte cell line AML12 increased the mtDNA/nDNA ratio (Figure 6A–B, top panel), demonstrating mitochondrial biogenesis. Similar to worms at day 3, biogenesis was accompanied by induction of the mitochondrial fusion gene Mfn2 (Figure 6A–B, bottom panel). Furthermore, AZD2281 and NR dose-dependently induced gene expression of the mammalian UPRmt homolog Hsp60, as evidenced using a luciferase reporter controlled by the human Hsp60 promoter (Figure 6C–D). At the protein level, AZD2281 and NR induced the UPRmt protease CLPP (Figure 6E–H). A detailed time course analyses showed that this UPRmt response was induced after 12h (Figure 6E–H), and coincides with the induction of mitonuclear protein imbalance, reflected by the ratio between nDNA-encoded SDHA versus the mtDNA-encoded MTCO1 (Figure 6E–H). Interestingly, the expression of SOD2—the mammalian ortholog of worm sod-3—also increased, but while SOD2 protein expression trended up 6h after starting the treatment (Figure 6E–H), it was only after 24h that both protein level and SOD activity were significantly increased (Figure 6I–J), possibly because activity of SOD2 is also regulated by post-translational modifications such as acetylation (Houtkooper et al., 2012). The late response of SOD2 is reminiscent of the induction of sod-3 in worms, which is also trailing the primary induction of UPRmt (Figure 3C–D, 4C).
Figure 6. NAD+ boosters activate UPRmt in mammalian cells.

(A–B) Treatment with NR (A; 1 mM) or AZD2281 (B; 1 μM) increases the ratio between mitochondrial DNA (mtDNA) and nuclear DNA (nDNA), a common marker for mitochondrial abundance in the AML12 hepatocyte cell line (upper panels). This increase reflects induced mitochondrial biogenesis. At the same time, NR and AZD2281 induce the expression of the mitochondrial fusion gene Mfn2 (lower panels).
(C–D) AZD2281 and NR dose-dependently activate the transcription of a human Hsp60 promoter luciferase reporter, transfected into AML12 cells
(E–H) The NAD+ precursor NR (E, G; 1 mM) and the PARP inhibitor AZD2281 (F, H; 1 μM) induce UPRmt, as evidenced by CLPP expression, in a time-dependent fashion in AML12 cells. The mitochondrial antioxidant SOD2 also increased over time. HSP90 is a cytosolic stress marker that is not affected by treatment with AZD2281 or NR. NR and AZD2281 also induced marked mitonuclear protein imbalance, as evidenced by the ratio SDHA/MTCO1 (nDNA- and mtDNA-encoded, respectively). Actin served as a loading control. Panels G and H represent quantifications of the Western blots in E and F.
(I–J) At the later stages following NR (I) or AZD2281 (J) treatment, SOD activity was increased. The fact that activity trails behind protein expression may be due to post-translational modifications regulating SOD2 activity.
Bar graphs are expressed as mean±SEM, * p≤0.05; ** p≤0.01; *** p≤0.001.
Finally, we assessed mitonuclear protein imbalance and UPRmt activation in primary mouse hepatocytes in which SIRT1 expression was either enhanced or abrogated. Similar to sir-2.1 transgenic worms, primary hepatocytes in which Sirt1 was overexpressed by an adenoviral vector (ad-Sirt1) showed mitonuclear protein imbalance, i.e. decreased ratio between ATP5A (encoded by nDNA) and MTCO1 (by mtDNA), associated with UPRmt induction—as evidenced by the robust increase in CLPP and HSP60 protein expression—and SOD2 expression, relative to control (Ad-GFP transduced) hepatocytes (Figure 7A). We then aimed to analyze the effect of treating wild type hepatocytes with AZD2281 or NR, either in the presence or absence of Sirt1. To this end, we isolated hepatocytes from floxed Sirt1 mice (Sirt1L2/L2). These were subsequently infected with either ad-GFP (for wild type) or adenoviral-controlled Cre recombinase (ad-Cre; for Sirt1 knockout). Similar to ad-Sirt1 treated hepatocytes (Figure 7A), wild type primary hepatocytes (ad-GFP) that were treated with AZD2281 or NR displayed mitonuclear protein imbalance—i.e. decreased ratio between nDNA- and mtDNA-encoded OXPHOS proteins—increased UPRmt (CLPP and HSP60) and increased SOD2 expression, while these effects were completely blocked in the ad-Cre infected Sirt1 floxed hepatocytes (Figure 7B). Altogether, these data validate the conservation of the NAD+/SIRT1/UPRmt/SOD signaling pathway in mammals, as well as the role of mitonuclear protein imbalance therein.
Figure 7. Activation of UPRmt is Sirt1-dependent in mammals.
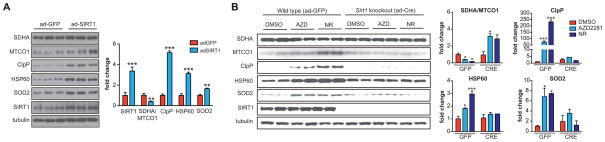
(A) Primary hepatocytes infected with Ad-mSirt1, to overexpress Sirt1, display marked mitonuclear protein imbalance (ratio MTCO1/SDHA), UPRmt activation (CLPP and HSP60) and antioxidant induction (SOD2), compared to hepatocytes from the same isolation infected with Ad-GFP, which served as controls. Sirt1 overexpression was confirmed; tubulin is used as a loading control.
(B) Primary hepatocytes of Sirt1 floxed mice were infected with adenoviral-assisted GFP (“wild type” negative control) or Cre recombinase (Sirt1 knockout). Treatment of GFP controls with NR or AZD2281 induced mitonuclear protein imbalance, and UPRmt, while these effects were attenuated when Sirt1 was knocked out. SIRT1 blots show knockout efficiency; tubulin was probed as a loading control.
Bar graphs are expressed as mean±SEM, * p≤0.05; ** p≤0.01; *** p≤0.001.
See also Figure S6.
Discussion
The progressive functional decline that happens during the normal aging process is associated with the accumulation of mutations in DNA (Hoeijmakers, 2009) and changes in metabolic rate and mitochondrial function (Houtkooper et al., 2011). How these DNA damage and mitochondrial metabolism pathways are intertwined is starting to be elucidated, but the mechanistic basis remains poorly understood. Key players in both these pathways include the sirtuin and PARP proteins, both NAD+-dependent enzymes and considerable prior knowledge to support a role of these pathways with respect to age-related metabolic diseases has accumulated. For instance, the increased NAD+ levels in Parp1−/− or Cd38−/− mice and in mice treated with the NAD+ precursors NR or NMN, led to SIRT1-dependent improved mitochondrial function, protecting them against the metabolic damage induced by high-fat diets (Bai et al., 2011b; Barbosa et al., 2007; Canto et al., 2012; Yoshino et al., 2011). Conversely, reduced levels of NAD+ metabolism were observed in aged rats and mice (Braidy et al., 2011; Yoshino et al., 2011), although no mechanistic explanation of this observation was presented.
We hence set out to investigate the causal link between NAD+ and aging, whether increasing NAD+ levels and sirtuin activity are sufficient to prevent aging and if so, which downstream aging pathways are involved. We primarily used the nematode C. elegans as it allows detailed genetic interventions coupled to full lifespan analysis. Additionally, we set up a range of mammalian, cell-based studies to complement our C. elegans results. Our data indicate that PARP activity, by modulating NAD+ availability, regulates mitochondrial biogenesis and as such plays an important role to preserve mitochondrial and organismal fitness. Not only is PARylation increased and are NAD+ levels reduced in aged worms and mice, but we also show that depleting NAD+ levels, either chemically using paraquat or genetically by inhibiting the NAD+ synthase qns-1, is sufficient to shorten worm lifespan. Conversely, interventions aimed to safeguard NAD+ levels curb the aging process and extend lifespan in C. elegans. Two independent strategies were used to boost NAD+ levels, i.e. mutation/RNAi (pme-1) or inhibition (AZD2281 or ABT-888) of the NAD+-consuming PARP enzymes and supplementation of NAD+ precursors (NR or NAM). The fact that both these approaches to raise NAD+ levels promote a similar phenotype strengthens the hypothesis that NAD+ is a critical metabolite influencing mitochondrial fitness and lifespan in a sir2.1-dependent way. Also, since NAD+ precursors and sir-2.1 overexpression are unlikely to modify the PARylation status of proteins or DNA directly, this excludes PARylation as a causal contributor of aging. Although the role of sir-2.1 in longevity is subject of debate (reviewed in (Houtkooper et al., 2012)) our data provide new and unequivocal evidence in support of its importance in lifespan regulation.
Two aging pathways are known to play a role in the mitochondrial theory of aging, UPRmt and ROS (Durieux et al., 2011; Sena and Chandel, 2012). In worms that accumulated NAD+ or in which sir-2.1 is overexpressed, we observed a marked induction of UPRmt, as well as improved ROS defense. Based on our time course experiments in worms, and its ubl-5 dependence, we concluded that the initial phase is characterized by a burst in ROS and induction of UPRmt, which is associated with a state we termed mitonuclear protein imbalance, referring to the altered balance between nDNA- and mtDNA-encoded OXPHOS subunits. This suggests that, when nDNA-encoded protein synthesis is not matched by mtDNA-encoded protein synthesis, UPRmt is activated to provide protective capacity. We demonstrated that the UPRmt regulator ubl-5 is essential for the lifespan extension inferred by either increasing NAD+ levels or by sir-2.1 overexpression.
Besides UPRmt, the NAD+ boosting strategies, using either the PARP inhibitor AZD2281 or the NAD+ precursors NR and NAM, also induced an antioxidant response. Following an early-phase burst in ROS, DAF-16 translocated to the nucleus, and activated expression of the key mitochondrial antioxidant sod-3, and likely involves the sir-2.1/daf-16 signaling pathway (Berdichevsky et al., 2006; Hashimoto et al., 2010). Interestingly, although it was previously shown that ROS are not critically involved in the induction of UPRmt (Durieux et al., 2011), our data demonstrate that the converse may apply, i.e. blocking UPRmt through ubl-5 RNAi is sufficient to block a further induction the antioxidant response. Although further work is needed to establish how UBL-5 controls sod-3 expression, the combined activation of UPRmt and ROS defense results in protection against age-related decline and lifespan extension, at least in worms (Figure S6). We confirmed this link also in mammals. PARP inhibitors (Bai et al., 2011b) and NAD+ precursors (Canto et al., 2012; Yoshino et al., 2011) have been reported to increase NAD+ levels, activate sirtuins and induce mitochondrial biogenesis through PGC-1α in mammals (Canto et al., 2009; Rodgers et al., 2005). Here, we showed that AZD2281 and NR, but also Sirt1 overexpression, likewise induced a robust UPRmt and ROS defense in mammals, underscoring the essential and conserved nature of this dual stress response.
Our results expose a hitherto unrecognized evolutionarily conserved pathway by which NAD+/sirtuin activity leads to UPRmt, a new SIRT1 signaling pathway, that in parallel to the established daf-16/sod-3 antioxidant defense (Berdichevsky et al., 2006), improves metabolic health and extends lifespan. We expect that other sirtuin activating compounds may also induce UPRmt. Indeed, data from our lab indicates that resveratrol requires UPRmt activation to extend lifespan (Houtkooper et al., 2013). Altogether, the data in the worm assembled in this study validate a link between NAD+, sirtuins, UPRmt, antioxidants, and lifespan control. Furthermore, our results in mammalian cell-based systems suggest that the mechanisms underlying NAD+/SIRT1/UPRmt/SOD signaling pathway are conserved and may open up the opportunity for preventive and/or therapeutic use of this pathway in the context of aging and aging-related disorders.
Experimental procedures
C. elegans experiments
C. elegans strains were cultured at 20°C on nematode growth media agar plates seeded with E. coli strain OP50 unless stated otherwise. Strains were provided by the Caenorhabditis Genetics Center (University of Minnesota) and detailed in the supplemental extended experimental procedures. Worm lifespan tests were performed as described (Mouchiroud et al., 2011). Briefly, 60–100 animals were used per conditions and scored every other day.
Treatments with PARP inhibitors—AZD2281 and ABT-888—or NAD+ precursors—nicotinamide riboside (NR) and nicotinamide (NAM)—were added at the indicated concentration just before pouring the plates. Animals were exposed to compounds during the full life from eggs until death.
Mammalian experiments
Primary hepatocytes were isolated from Sirt1L2/L2 (see more detail in supplemental information) and transduced either with an Ad-GFP or an Ad-Cre virus at a MOI=5 to generate matched Sirt1+/+ (wild type) and Sirt1−/− (knock-out) hepatocytes, respectively. After overnight infection, culture medium was removed and Sirt1+/+ and Sirt1−/− hepatocytes cultures were treated or not with NR or AZD2281. To obtain gain-of-Sirt1-function hepatocytes, Ad-mSirt1 (adgene 8438) or Ad-GFP virus was used at the same MOI to infect primary hepatocytes obtained from a male C57BL/6J mouse to generate hepatocytes overexpressing Sirt1 and GFP (control hepatocytes), respectively.
Worm and cell-based assays
Worm oxygen consumption was measured using the Seahorse XF96 equipment (Seahorse Bioscience). Worms were transferred in 96-well standard Seahorse plates (10 worms/well) and oxygen consumption was measured 6 times. Respiration rates were normalized to the number of worms in each individual well. Gene expression (RT-qPCR), protein expression (Western blot), mtDNA copy number, and ROS assays were performed following routine procedures, as outlined in the supplementary extended experimental procedures.
Statistics
Survival analyses were performed using the Kaplan Meier method and the significance of differences between survival curves calculated using the log rank test. Differences between two groups were assessed using two-tailed t-tests. To compare the interaction between age and genotype, two-way ANOVA tests were performed. Analysis of variance, assessed by Bonferroni’s multiple comparison test, was used when comparing more than two groups. We used R (R software version 2.9.0) for the calculation of mean lifespan and SEM, and GraphPad Prism 5 (GraphPad Software) for all other statistical analyses. All p-values <0.05 were considered significant.
Supplementary Material
Highlights.
Enhancing NAD+ availability extends lifespan in C. elegans through sir-2.1
The NAD+/sir-2.1-dependent activation of mitochondrial UPR is crucial for longevity
Higher NAD+ bioavailability protects against ROS by enhancing daf-16 activity
UPRmt is a conserved target of the NAD+/SIRT1 axis in mammalian cells
Acknowledgments
We thank Pierre Gönczy for sharing reagents and equipment, the Caenorhabditis Genetics Center for providing worm strains and the Auwerx team for discussions. LM is supported by the “Fondation pour la Recherche Médicale” and RHH by a VENI grant from ZonMw (number 91613050) and an AMC Postdoc fellowship. JA is the Nestlé Chair in Energy Metabolism. This work is supported by grants of the Ecole Polytechnique Fédérale de Lausanne, the EU Ideas program (ERC-2008-AdG-23138), the NIH (R01HL106511-01A and R01AG043930), the Velux Stiftung, the Swiss National Science Foundation (31003A-124713 and 31003A-125487 and CSRII3-136201).
Footnotes
Supplemental information includes extended experimental procedures, six figures and three tables, including detailed lifespan statistics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alam H, Williams TW, Dumas KJ, Guo C, Yoshina S, Mitani S, Hu PJ. EAK-7 controls development and life span by regulating nuclear DAF-16/FoxO activity. Cell metabolism. 2010;12:30–41. doi: 10.1016/j.cmet.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Reinke H, Altmeyer M, Gutierrez-Arcelus M, Hottiger MO, Schibler U. Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell. 2010;142:943–953. doi: 10.1016/j.cell.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Bai P, Canto C, Brunyanszki A, Huber A, Szanto M, Cen Y, Yamamoto H, Houten SM, Kiss B, Oudart H, et al. PARP-2 Regulates SIRT1 Expression and Whole-Body Energy Expenditure. Cell Metab. 2011a;13:450–460. doi: 10.1016/j.cmet.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, et al. PARP-1 Inhibition Increases Mitochondrial Metabolism through SIRT1 Activation. Cell Metab. 2011b;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, Chini EN. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. Faseb J. 2007;21:3629–3639. doi: 10.1096/fj.07-8290com. [DOI] [PubMed] [Google Scholar]
- Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell. 2006;125:1165–1177. doi: 10.1016/j.cell.2006.04.036. [DOI] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and sirt1 activity in wistar rats. PLoS ONE. 2011;6:e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Burnett C, Valentini S, Cabreiro F, Goss M, Somogyvari M, Piper MD, Hoddinott M, Sutphin GL, Leko V, McElwee JJ, et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011;477:482–485. doi: 10.1038/nature10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol Rev. 2012;64:166–187. doi: 10.1124/pr.110.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. The NAD(+) Precursor Nicotinamide Riboside Enhances Oxidative Metabolism and Protects against High-Fat Diet-Induced Obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol. 2012;8:287–296. doi: 10.1038/nrendo.2011.225. [DOI] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erener S, Mirsaidi A, Hesse M, Tiaden AN, Ellingsgaard H, Kostadinova R, Donath MY, Richards PJ, Hottiger MO. ARTD1 deletion causes increased hepatic lipid accumulation in mice fed a high-fat diet and impairs adipocyte function and differentiation. FASEB J. 2012;26:2631–2638. doi: 10.1096/fj.11-200212. [DOI] [PubMed] [Google Scholar]
- Ferraris DV. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J Med Chem. 2010;53:4561–4584. doi: 10.1021/jm100012m. [DOI] [PubMed] [Google Scholar]
- Gagnon SN, Hengartner MO, Desnoyers S. The genes pme-1 and pme-2 encode two poly(ADP-ribose) polymerases in Caenorhabditis elegans. Biochem J. 2002;368:263–271. doi: 10.1042/BJ20020669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- Grube K, Burkle A. Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc Natl Acad Sci U S A. 1992;89:11759–11763. doi: 10.1073/pnas.89.24.11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Mitochondria--a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Horikawa M, Nomura T, Sakamoto K. Nicotinamide adenine dinucleotide extends the lifespan of Caenorhabditis elegans mediated by sir-2.1 and daf-16. Biogerontology. 2010;11:31–43. doi: 10.1007/s10522-009-9225-3. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci. 2010;123:3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999;13:1385–1393. [PubMed] [Google Scholar]
- Houtkooper RH, Argmann C, Houten SM, Canto C, Jeninga EH, Andreux PA, Thomas C, Doenlen R, Schoonjans K, Auwerx J. The metabolic footprint of aging in mice. Sci Rep. 2011;1:134. doi: 10.1038/srep00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Auwerx J. Exploring the therapeutic space around NAD+ J Cell Biol. 2012;199:205–209. doi: 10.1083/jcb.201207019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010a;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Williams RW, Auwerx J. Metabolic networks of longevity. Cell. 2010b;142:9–14. doi: 10.1016/j.cell.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Mangerich A, Herbach N, Hanf B, Fischbach A, Popp O, Moreno-Villanueva M, Bruns OT, Burkle A. Inflammatory and age-related pathologies in mice with ectopic expression of human PARP-1. Mech Ageing Dev. 2010;131:389–404. doi: 10.1016/j.mad.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S, et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phth alazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem. 2008;51:6581–6591. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- Mouchiroud L, Molin L, Kasturi P, Triba MN, Dumas ME, Wilson MC, Halestrap AP, Roussel D, Masse I, Dalliere N, et al. Pyruvate imbalance mediates metabolic reprogramming and mimics lifespan extension by dietary restriction in Caenorhabditis elegans. Aging Cell. 2011;10:39–54. doi: 10.1111/j.1474-9726.2010.00640.x. [DOI] [PubMed] [Google Scholar]
- Penning TD, Zhu GD, Gandhi VB, Gong J, Liu X, Shi Y, Klinghofer V, Johnson EF, Donawho CK, Frost DJ, et al. Discovery of the Poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer. J Med Chem. 2009;52:514–523. doi: 10.1021/jm801171j. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J Biol Chem. 2004;279:28873–28879. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- Viswanathan M, Guarente L. Regulation of Caenorhabditis elegans lifespan by sir-2.1 transgenes. Nature. 2011;477:E1–2. doi: 10.1038/nature10440. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117:4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide Mononucleotide, a Key NAD(+) Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR, Ristow M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15:451–465. doi: 10.1016/j.cmet.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.