

Abstract
Background and Purpose
Activation of α7 nicotinic acetylcholine receptors (nAChRs) can be neuroprotective. However, endogenous choline and ACh have not been regarded as potent neuroprotective agents because physiological levels of choline/ACh do not produce neuroprotective levels of α7 activation. This limitation may be overcome by the use of type-II positive allosteric modulators (PAMs-II) of α7 nAChRs, such as 1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methylisoxazol-3-yl)-urea (PNU-120596). This proof-of-concept study presents a novel neuroprotective paradigm that converts endogenous choline/ACh into potent neuroprotective agents in cerebral ischaemia by inhibiting α7 nAChR desensitization using PNU-120596.
Experimental Approach
An electrophysiological ex vivo cell injury assay (to quantify the susceptibility of hippocampal neurons to acute injury by complete oxygen and glucose deprivation; COGD) and an in vivo middle cerebral artery occlusion model of ischaemia were used in rats.
Key Results
Choline (20–200 μM) in the presence, but not absence of 1 μM PNU-120596 significantly delayed anoxic depolarization/injury of hippocampal CA1 pyramidal neurons, but not CA1 stratum radiatum interneurons, subjected to COGD in acute hippocampal slices and these effects were blocked by 20 nM methyllycaconitine, a selective α7 antagonist, thus, activation of α7 nAChRs was required. PNU-120596 alone was ineffective ex vivo. In in vivo experiments, both pre- and post-ischaemia treatments with PNU-120596 (30 mg·kg−1, s.c. and 1 mg·kg−1, i.v., respectively) significantly reduced the cortical/subcortical infarct volume caused by transient focal cerebral ischaemia. PNU-120596 (1 mg·kg−1, i.v., 30 min post-ischaemia) remained neuroprotective in rats subjected to a choline-deficient diet for 14 days prior to experiments.
Conclusions and Implications
PNU-120596 and possibly other PAMs-II significantly improved neuronal survival in cerebral ischaemia by augmenting neuroprotective effects of endogenous choline/ACh.
Keywords: PNU120596, PNU-120596, neuroprotection, anoxic depolarization, choline, α7 nAChR, hippocampus, cortex, ischaemia
Introduction
Stroke remains the third leading cause of death in the United States and over 85% of all strokes are ischaemic strokes (Beal, 2010; Roger et al., 2012). Over one third of stroke victims do not survive the incidence of stroke; while over 90% of stroke survivors suffer from permanent neurological deficits (Jorgensen et al., 1999; Roger et al., 2012). Clinical management of neuronal damage resulting from ischaemic stroke generally involves only palliative treatments. Currently, the only Food and Drug Administration-approved drug therapy for ischaemic stroke involves the i.v. use of tissue plasminogen activator (tPA) to dissolve clots (Furlan, 2002). This strategy appears to be effective in ischaemic stroke, but only within the first 3 h after the onset of ischaemic stroke (Brott and Bogousslavsky, 2000; Adams et al., 2007). These strict limitations reduce the percent of stroke patients eligible for tPA to as low as ∼2% (Katzan et al., 2000). Although in the last two decades, substantial efforts have been invested in developing neuroprotective medicine, these efforts have not resulted in clinically efficacious therapies for ischaemic stroke (Richard Green et al., 2003). These failures highlight the need for development of new concepts, therapeutic ideas and approaches for neuronal protection and prevention of neuronal damage secondary to ischaemia. In this search, strategies that are based on recruiting and activating endogenous pathways receive special attention as these approaches are expected to be highly efficacious and cause fewer adverse effects than approaches that utilize exogenous agents (Lo, 2008; Uteshev, 2012a). Development of these drug therapies would be expected to specifically benefit individuals from high-risk groups for ischaemic stroke (Roger et al., 2012).
There is a growing pool of supportive evidence linking activation of α7 nicotinic acetylcholine receptors (nAChRs) to enhanced neuronal survival and resistance to ischaemia and other insults (Meyer et al., 1998; Shimohama et al., 1998; Li et al., 1999; Rosa et al., 2006; Guseva et al., 2008), as well as improved cognitive performance in patients and animal models of schizophrenia (Olincy et al., 2006; Olincy and Stevens, 2007), dementia (Kem, 2000; Ren et al., 2007; Bitner et al., 2010) and traumatic brain injury (Guseva et al., 2008). This study is a proof of concept that introduces a novel approach to reduction of neuronal injury secondary to ischaemia by augmenting neuroprotective effects of endogenous choline/ACh. Choline is a full selective agonist of α7 nAChRs (Papke et al., 1996; Alkondon et al., 1997), a ubiquitous cell membrane-building material and a precursor metabolite of ACh. However, at physiological levels (i.e. <20 μM) and pathophysiological levels (i.e. >20 μM) expected in the penumbra of brain injury (Klein et al., 1998; Sarter and Parikh, 2005), choline alone is ineffective as an α7 nAChR agonist because of its low potency for α7 activation (EC50∼0.5 mM; Papke and Papke, 2002) and tendency to induce α7 desensitization (IC50∼40 μM; Uteshev et al., 2003). As a result, choline has not previously garnered much attention as a potential neuroprotective agent. These limitations may be overcome by the use of a previously reported novel class of compounds, type-II positive allosteric modulators (PAMs-II) of α7 nAChRs, such as 1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methylisoxazol-3-yl)-urea (PNU12596) (Hurst et al., 2005; Gusev and Uteshev, 2010; Kalappa et al., 2010; Uteshev, 2012a,b). PNU-120596 does not activate α7 nAChRs, but it inhibits desensitization and enhances activation of α7 nAChRs by nicotinic agonists, including choline and ACh (Hurst et al., 2005; Gusev and Uteshev, 2010; Kalappa et al., 2010). In the presence of PNU-120596, endogenous levels of choline/ACh may become neuroprotective by producing a weak persistent and tunable modality of α7 nAChR activation (Gusev and Uteshev, 2010; Kalappa et al., 2010; Sitzia et al., 2011; Uteshev, 2012b). However, excessive activation of α7 nAChRs can be neurotoxic (Del Barrio et al., 2011; Dinklo et al., 2011) because of the high permeability of α7 nAChR-mediated ion channels to Ca2+ ions (Vernino et al., 1992; Castro and Albuquerque, 1995; Fucile, 2004; Uteshev, 2010).
This study tests the hypothesis that PNU-120596 converts physiological/endogenous choline/ACh into potent neuroprotective agents in both ex vivo and in vivo experimental models of ischaemic stroke. To conduct these tests, an electrophysiological neuronal injury assay was developed and applied to hippocampal CA1 pyramidal neurons and CA1 interneurons in whole-cell current-clamp patch-clamp experiments in rat acute hippocampal slices subjected to complete oxygen and glucose deprivation (COGD). The COGD protocol was used in acute hippocampal slices (ex vivo) to model conditions that are likely present in the core (as opposed to the penumbra) of the ischaemic injury where neuronal injury and death can be delayed, but not eliminated (Lo, 2008). The effects of s.c. and i.v. administration of PNU-120596 on the ischaemic infarct volume within cortical and subcortical brain regions of the forebrain were evaluated in in vivo experiments in rats using the middle cerebral artery occlusion (MCAO) model of stroke. Some in vivo experiments were done in rats that were subjected to a choline-deficient diet for 14 days prior to experimentation. The results of this study support the hypothesis that PAMs-II can recruit and activate endogenous agonists of α7 nAChRs (i.e. choline and ACh) to significantly enhance neuronal survival in cerebral ischaemia.
Materials and methods
Animals
Young adult male Sprague-Dawley (S.-D.) rats (a total of 181 animals) were used for ex vivo (∼150 g) and in vivo (∼280 g) experiments. The animal use was in accordance with the Guide for the Care and Use of Laboratory Animals (NIH 865-23, Bethesda, MD), and all experimental protocols were approved by the Animal Care and Use Committee of Southern Illinois University School of Medicine, Springfield, IL and the Institutional Animal Care and Use Committee of University of North Texas Health Science Center at Fort Worth, TX. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Choline-deficient diet
Male S.-D. rats (∼150 g) were purchased from Charles Rivers Laboratories International, Inc. (Wilmington, MA, USA) and subjected to an ad libitum choline-deficient diet (TD.88052; Harlan Laboratories, Inc., Indianapolis, IN, USA) for 14 days upon arrival to the UNTHSC. Water was given ad libitum. Animal's weight was measured every 3 days. After 14 days, animals whose weight exceeded 260 g were selected for experimentation. Underweight animals were used for other projects. The average weight gain of choline-deficient rats (∼5.5% per day) was comparable with that reported for choline-sufficient rats of the same age (∼5.8% per day) (http://www.criver.com/SiteCollectionDocuments/rm_rm_c_sprague_dawley_rats.pdf).
Tissue preparation
Coronal whole-brain slices of 250–300 μm thickness containing hippocampal CA1 region were cut in a sucrose-rich solution at 3°C using Vibratome-1000 + slicer (Leica Microsystems, Wetzlar, Germany) upon euthanizing via rapid decapitation. The sucrose-rich solution was of the following composition (in mM): sucrose 250, KCl 3, NaH2PO4 1.23, MgCl2 5, CaCl2 0.5, NaHCO3 26, glucose 10 (pH = 7.4, when bubbled with carbogen: 95% O2 and 5% CO2). Slices were then transferred to a temporary storage chamber where they were maintained for ∼30 min at 30°C in an oxygenated artificial cerebrospinal fluid (ACSF) of the following composition (in mM): NaCl 125, KCl 3, NaH2PO4 1.23, MgCl2 1, CaCl2 2, NaHCO3 26, glucose 10 (pH = 7.4, when bubbled with carbogen). Slices were then transferred to the long-term storage chamber and maintained at room temperature for up to 6 h bubbled with carbogen.
Patch-clamp electrophysiology in acute brain slices
The recording chamber (volume 2 mL) was perfused with oxygenated ACSF at a rate of 1 mL·min−1 using a perfusion pump (Model73160-20, Cole-Parmer, Vernon Hills, IL, USA). Hippocampal CA1 pyramidal neurons and CA1 stratum radiatum interneurons in brain slices were selected on the basis of their morphology and location within the slice as reported previously (Kalappa et al., 2010). Neurons were visually selected for electrophysiological recordings using an Olympus BX-51WI microscope (Olympus Inc., Center Valley, PA, USA). Conventional whole-cell patch-clamp electrophysiological recordings were conducted using a MultiClamp-700B amplifier equipped with Digidata-1440A A/D converter (Molecular Devices, Sunnyvale, CA, USA). The membrane resting potential was continuously recorded in the current-clamp configuration. Data were filtered at 2–4 kHz, sampled at 10 kHz and stored on a personal computer. Patch pipettes were pulled using a Sutter P-97 horizontal puller (Sutter Instruments, Novato, CA, USA). The pipette resistance was 4–6 MΩ when filled with the internal solution. The access resistances (<30 MΩ) were not compensated. The extracellular recording solution was identical to ACSF used for the tissue preparation. The patch electrode solution contained (in mM): K-gluconate 140, NaCl 1, MgCl2 2, Mg-ATP 2, Na-GTP 0.3, HEPES 10, KOH 0.42 (pH 7.4). Membrane voltages were not corrected for the liquid junction potential: VLJ(K-gluconate) = 16.2 mV. All ex vivo experiments were conducted at 30–32°C.
Oxygen-glucose deprivation and pretreatment of acute hippocampal slices with PNU-120596 and choline
CA1 pyramidal neurons and CA1 interneurons in acute hippocampal slices subjected to COGD (i.e. no reperfusion) were used in all experiments. COGD was achieved by a continuous perfusion of slices with an oxygen-glucose deprived ACSF (i.e. COGD-ACSF) containing 10 mM sucrose instead of 10 mM glucose and bubbled with 95% N2 + 5% CO2 instead of carbogen (i.e. 95% O2 + 5% CO2). Hippocampal slices were prepared as described above and then, randomly separated into two groups, ‘control’ or ‘treatment’, and stored for ∼40 min in a chamber perfused with a standard ACSF bubbled with carbogen at room temperature. ‘Treatment’ slices were then transferred to ‘treatment’ chamber and subjected to a specific treatment for 3 h: for example, 20 μM choline +1 μM PNU-120596. ‘Control’ slices were transferred to ‘control’ chamber and incubated in standard ACSF for the same duration. After treatments, slices were transferred into the recording chamber where they remained perfused in the corresponding solutions during recordings. To initiate COGD, standard ACSF was replaced with COGD-ACSF by switching solutions that enter the recording chamber. The solution exchange delay time (i.e. time necessary to replace the system's ‘dead’ volume after a solution switch) was estimated using high concentrations of a food dye (203 ± 13 s, n = 3) and not subtracted from data.
Electrophysiological cell injury assay
Hippocampal CA1 pyramidal neurons and CA1 stratum radiatum interneurons in acute coronal slices subjected to COGD were used in all experiments. In total, 107 hippocampal CA1 pyramidal neurons and 16 stratum radiatum interneurons were used in this study. Each hippocampal slice was used only for one experiment utilizing COGD and only one neuron was tested in each slice. Each treatment with nicotinic agents had its own control data. This was achieved by using slices obtained from the same animal and preparation for both control (i.e. COGD only) and treatment (i.e. COGD + PNU + choline) sets of experiments to ensure the presence of control data sets for each animal and preparation.
An electrophysiological cell injury assay was developed and used in this study to achieve a reliable and continuous quantification of the evolution of neuronal anoxic depolarization and injury. Similar approaches have been used previously to study the effects of OGD on the mechanisms of cell death (Tanaka et al., 1997; Dijkhuizen et al., 1999; Rossi et al., 2000). The effects of choline + PNU-120596 pretreatment on the status of neuronal health or injury were determined by continuous recordings of the neuronal membrane potential before and during the insult (i.e. COGD) or treatment (i.e. choline + PNU-120596) using the conventional current-clamp whole-cell patch-clamp electrophysiology: healthy cells were defined by the normal resting membrane potential (i.e. near-60 mV), while injured cells were defined by depolarized membrane potentials (i.e. more positive than-50 mV) and dead cells were assumed to have the resting membrane potential near 0 mV. Therefore, the evolution of acute neuronal depolarization/injury by COGD was monitored in real time and injury curves were built and used for analysis. To build averaged injury curves, data points pertaining to individual injury curves obtained under identical experimental conditions (e.g. control or treatment) were averaged point-by-point across all neurons in a given group. However, the mean T50 values presented in Figures 3 were the averages of T50 obtained from the corresponding individual injury curves, not the T50 values obtained from the averaged injury curves. While the end result of OGD may be the same (i.e. neuronal depolarization/injury or death defined as a loss of the ionic gradient and the resting membrane potential), the injury curves may be different for different cell types and experimental treatments. Reperfusion of neurons defined as dead (i.e. the neuronal membrane potential was near 0 mV) with normal ACSF did not hyperpolarize the neuronal membrane potential supporting the definition. Transient or partial oxygen-glucose deprivation models where oxygen and/or glucose were briefly or only partially removed from ACSF were not investigated in this study.
Figure 3.
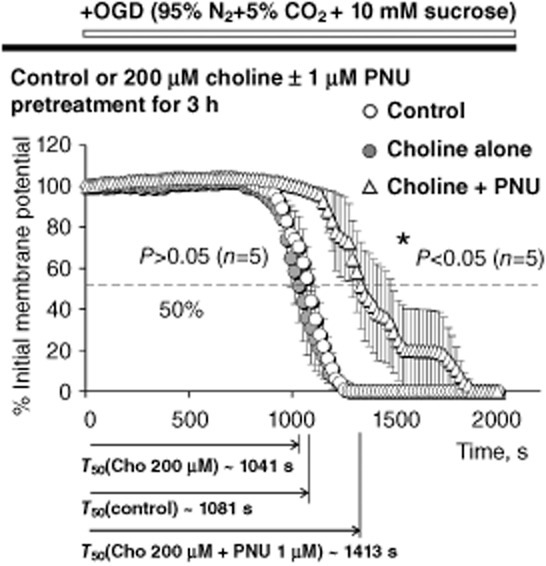
Pretreatment with 200 μM choline + 1 μM PNU-120596. The results of ex vivo experiments that employed 3 h pretreatment of acute hippocampal slices in the ACSF (open circles; control), 200 μM choline alone (closed circles) and 200 μM choline + 1 μM PNU-120596 (open triangles). The analysis and illustration were conducted and built as described for other figures. Pretreatment with 200 μM choline + 1 μM PNU-120596 (open triangles) significantly delayed COGD-induced depolarization/injury of hippocampal CA1 pyramidal neurons as compared to control (open circles; P = 0.0199, n = 5) or 200 μM choline alone (closed circles; P = 0.0118, n = 5). By contrast, pretreatment with 200 μM choline alone (closed circles) did not significantly delay injury in comparison with controls (open circles, P = 0.4766, n = 5). The unpaired two-tailed Student's t-test was used for analysis (Table 2).
Electrophysiological data analysis
The neuronal membrane potential was recorded in 10 s episodes without any delays between episodes to generate the mean membrane potential every 10 s. Thus, six means were generated every minute of recordings and the resulting injury curves were used for analysis (Figure 1). Action potentials were ignored during analysis and the membrane potential in between the neighbouring action potentials was used for analysis. For clarity, some illustrations were made using only every other means (i.e. three means per minute of recordings). The neuronal depolarization/injury was quantified by half-time to the membrane voltage 0 mV (i.e. T50). Neurons not subjected to COGD maintained their resting membrane potentials relatively constant (n = 8; Figures 1A,B and 2A). By contrast, all tested neurons subjected to COGD depolarized to 0 mV as a result and thus, were defined as dead (Figures 1C–F and 2B–F). For each neuron, T50 was estimated as duration from the moment of solution switch (i.e. t = 0) to the point where the membrane voltage drops 50% from its initial value (i.e. at t = 0). The T50 was then estimated by assuming that the membrane potential between the data points just above (T1, V1) and just below (T2, V2) the point of 50% drop (i.e. within a maximum of <10 s of recordings) was a linear function of time: T50 = T1 + (T2 − T1)*(V1 − 50)/(V1 − V2). In this analysis, V was normalized to the resting membrane potential at t = 0 to reduce interneuronal variability in the membrane resting potentials. The T50 obtained under control (i.e. COGD only) and treated (i.e. COGD + treatments) conditions and averaged over the corresponding sets of neurons were compared using the standard unpaired two-tailed Student's test (GraphPad Software, Inc., La Jolla, CA, USA). Data fitting was done using ProStat analysis packages (Pearl River, NY, USA). Statistical significance was defined by the P-value (i.e. P < 0.05). Graphs were built using GraphPad and ProStat data analysis software packages. Data were presented as means ± SEM.
Figure 1.
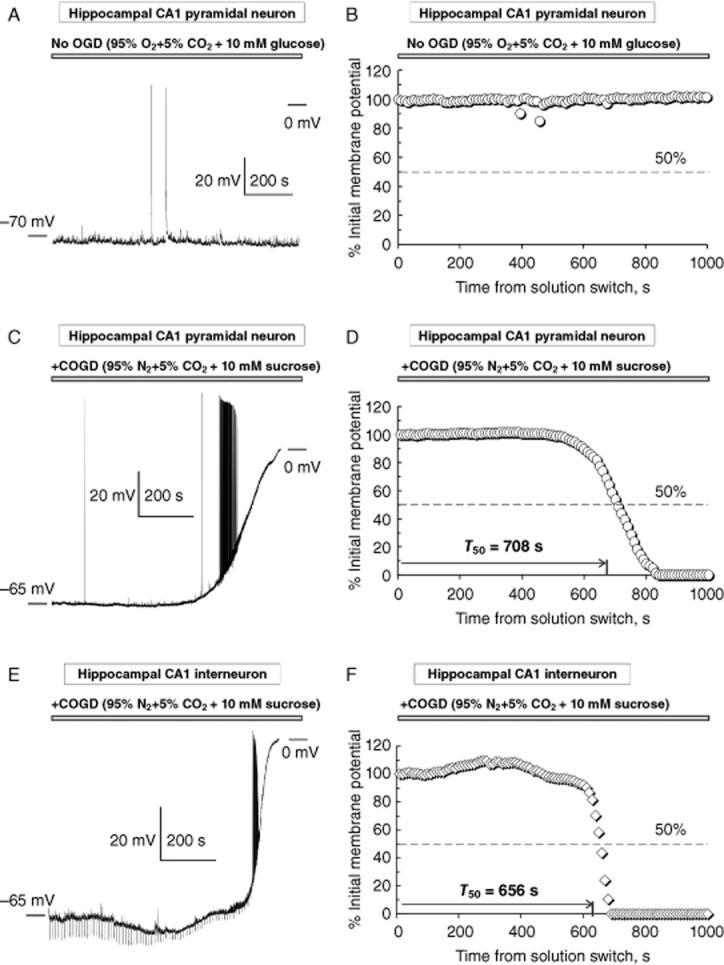
Examples of COGD-induced injury curves. Typical examples of the raw (A, C and E) and the normalized (B, D and F) injury curves recorded and built for two representative hippocampal CA1 pyramidal neurons (no OGD, A–B and COGD, C–D) and a hippocampal CA1 interneuron (E–F) in ex vivo experiments in acute hippocampal slices. As expected, no injury was detected in the absence of OGD (A–B; n = 5). Each data point in the injury curves (i.e. D and F) indicates the mean normalized membrane potential calculated over a 10 s episode as a function of time from solution switch (from ACSF to COGD-ACSF). There was no solution switch in experiments without OGD (i.e. A–B). Each injury curve (i.e. C–D and E–F) is quantified by the value of T50 indicating the half-time to 0 mV. The T50 was not defined in experiments without OGD (i.e. A–B). The dashed lines in D) and F) indicate 50% drop from the initial membrane potential. For clarity of illustrations, only every other data point is shown in D and F. All data points were used in data analysis to determine T50. Neurons used in these examples were obtained from different preparations and investigated on different experimental days. Regular downward voltage deviations in E were hyperpolarizing (−20 pA) steps used in some recordings to determine the neuronal input resistance. Depolarization-induced action potentials clearly seen in the raw recordings (C and E) were ignored during analysis and thus, are not represented in the normalized injury curves (D and F). Two strong spontaneous bursts of action potentials in A are represented as small downward deviations in B.
Figure 2.
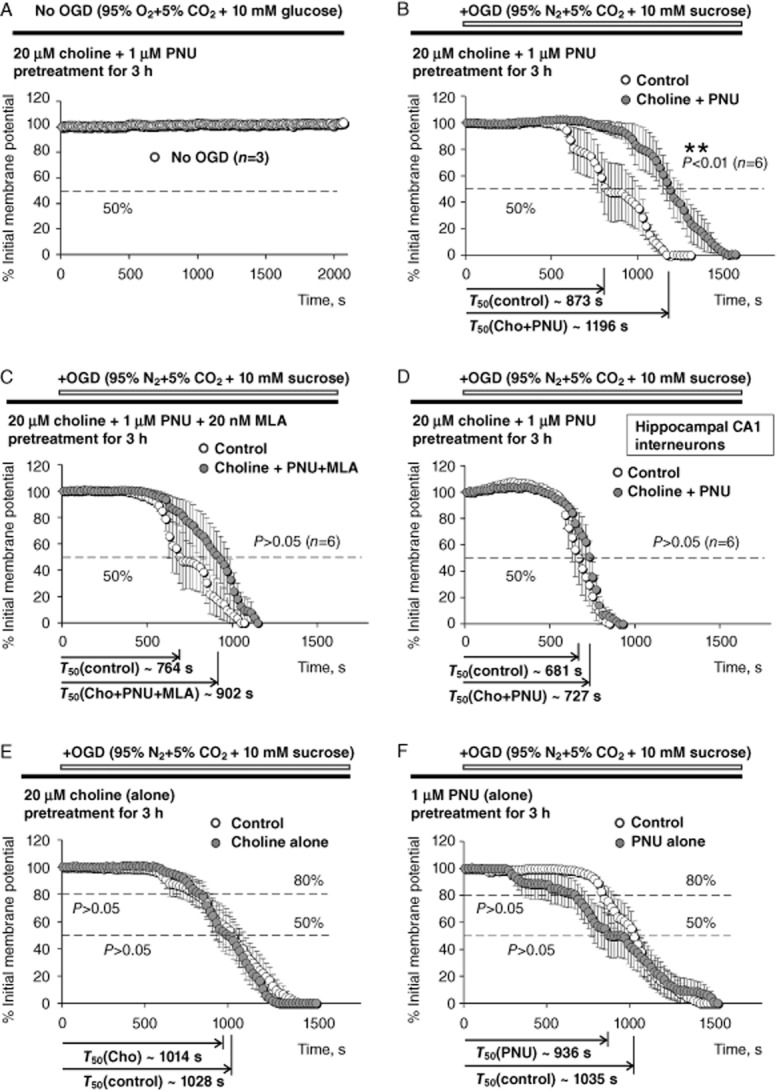
Pretreatment with 20 μM choline and/or 1 μM PNU-120596. Results of ex vivo experiments that employed pretreatment (3 h) of acute hippocampal slices with 20 μM choline and/or 1 μM PNU-120596. Averaged injury curves were built for four sets of controls and treatments: (A) no OGD, but after 3 h of pretreatment in 20 μM choline + 1 μM PNU-120596 (n = 3); (B) control (COGD, no pretreatment, open circles) versus COGD + pretreatment of CA1 pyramidal neurons with 20 μM choline + 1 μM PNU-120596 (closed circles); (C) control (COGD, no pretreatment, open circles) versus COGD + pretreatment of CA1 pyramidal neurons with 20 μM choline + 1 μM PNU-120596 + 20 nM MLA (closed circles); and (D) control (COGD, no pretreatment, open circles) versus COGD + pretreatment of CA1 interneurons with 20 μM choline + 1 μM PNU-120596 (closed circles). As expected, no injury was detected in the absence of OGD (A). Pretreatment with 20 μM choline + 1 μM PNU-120596 significantly delayed injury of hippocampal CA1 pyramidal neurons (P = 0.0097, n = 6) (B), but not CA1 interneurons (D) to COGD (P = 0.2936, n = 6). These effects were eliminated by 20 nM MLA (P = 0.1841, n = 6), a selective α7 nAChR antagonist, supporting the requirement for α7 nAChR activation (C). (E–F) The results of ex vivo experiments that employed 3 h pretreatment of acute hippocampal slices with 20 μM choline alone (E) or 1 μM PNU-120596 alone (F). The analysis and illustration were conducted and built as described for other panels, except that in addition to T50, T80 was also used to compare the early phase of injury curves under control (open circles) and pretreated (closed circles) conditions. Pretreatment with 20 μM choline alone (P = 0.8690, n = 10–11) (E) or 1 μM PNU-120596 alone (P = 0.4045, n = 9–11) (F) did not significantly delay depolarization/injury of hippocampal CA1 pyramidal neurons induced by COGD supporting the requirement for α7 nAChR activation and synergistic action of choline and PNU-120596. For illustrations and to build the averaged injury curves, data points pertaining to individual injury curves obtained under identical experimental conditions (e.g. control or treatments) were averaged point-by-point across all relevant neurons. For analysis, the T50 values obtained from the corresponding individual injury curves (e.g. Figure 1) were averaged to determine the mean T50. For clarity of illustrations, only every other data point was shown in B-D). All data points were shown in A, E–F) However, all data points were always used in data analysis to determine the mean T50. The unpaired two-tailed Student's t-test was used for analysis (Tables 2).
MCAO and infarct volume measurement
The results obtained in ex vivo experiments (Figures 4) suggested that PNU-120596 may produce significant neuroprotection in vivo. To test this hypothesis, transient focal cerebral ischaemia was induced using the suture occlusion technique as previously described (Jin et al., 2001). Animals (n = 64; Charles River) were anaesthetized with 4% isoflurane mixed with 67% N2O and 29% O2 and delivered by a mask. After a midline incision in the neck, the left external carotid artery (ECA) was carefully exposed and dissected. A 19 mm, 4-0 monofilament nylon suture was inserted from the ECA into the left internal carotid artery to occlude the origin of left middle cerebral artery. After 90 min of occlusion, the thread was removed to allow reperfusion. The ECA was ligated, and the wound was closed. Rectal temperature was maintained at ∼37°C using a heating pad.
Figure 4.
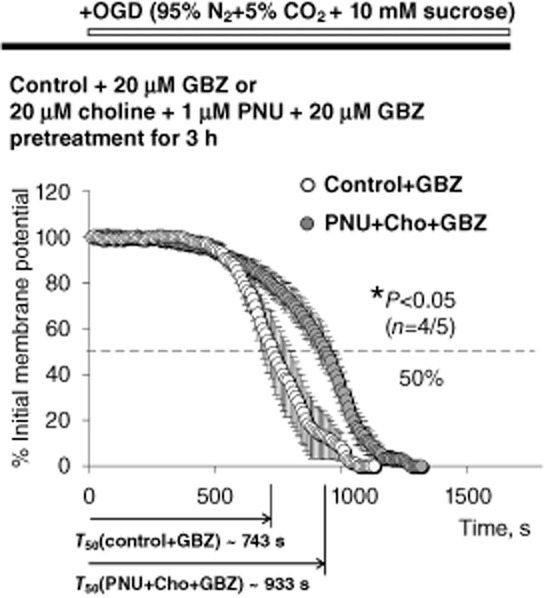
Lack of involvement of GABAergic neurotransmission. Experiments similar to those illustrated in Figures 2B and 3 were conducted using CA1 pyramidal neurons in the continuous presence of 20 μM GBZ in the ACSF during pretreatment of slices in 20 μM choline + 1 μM PNU-120596 and COGD. Inhibition of GABAergic neurotransmission did not alter the neuroprotective effects of choline + PNU-120596 because neuronal injury remained significantly delayed (P = 0.0198, n = 5) by choline + PNU-120596 in the presence of GBZ [T50 (cho + PNU + GBZ) = 933.3 ± 36.2 s, closed circles] as compared to control conditions [i.e. no pretreatment, T50 (control + GBZ) = 742.7 ± 55.0 s, n = 4, open circles]. The unpaired two-tailed Student's t-test was used for analysis.
Rats were anaesthetized and decapitated 24 h after MCAO. Brains were removed and coronal sections (2 mm thickness) immersed in 2% 2,3,5-triphenyltetrazolium chloride in saline for 20 min at 37°C, then fixed for 2 h in 4% paraformaldehyde (Bederson et al., 1986). Infarct area, left hemisphere area and total brain area were measured by a blinded observer using the Image J software, and areas were multiplied by the distance between sections to obtain the respective volumes. Infarct volume was calculated as a percentage of the volume of the contralateral hemisphere, as described previously (Swanson et al., 1990). Statistical significance was defined by the P-value (i.e. P < 0.05) using the two-tailed unpaired Student's t-test.
MS analysis of PNU-120596 amounts in brain/blood samples
Animals were anaesthetized with 4% isoflurane mixed with 67% N2O and 29% O2 (delivered by a mask) and perfused with ACSF. Samples of blood and hippocampal, frontal cortical and striatal brain tissues were collected, flash-frozen using liquid nitrogen, stored at -80°C at UNTHSC (Fort Worth, TX, USA) and shipped on dry ice overnight to Carbon Dynamics Institute, LLC (Springfield, IL, USA) for MS analyses. Subsamples were macerated with methanol for 3 min and the resultant slurry was centrifuged at 4°C for 15 min at 15 000× g. Blood samples were centrifuged at 4°C for 15 min at 19 000× g. The resultant supernatant for both preparations was further purified using a mixed-mode polymeric exchange solid phase extraction (SPE). The eluate was lyophilized and reconstituted in the initial mobile phase. PNU-120596 was analysed by a one microliter injection of each sample aliquot into a Waters 2795 Alliance equipped with a XBridge BEH130 C18 2.1 mm × 150 mm, 3.5 μm particulate column (110 Å) at a flow rate of 0.150 mL·min−1; a binary mobile phase gradient at 50:50 (Δ2.5%) A : B in 20 min (Mobil phase A = 0.01% formic acid; Mobil phase B = acetonitrile) until proper peak shape, separation and reduction of interferences were obtained. Waters Quattro Ultima triple stage quadrupole mass spectrometer equipped with an electrospray ionization source was calibrated with methionine-arginine-phenylalanine-alanine peptide for both the single and double charge state (m/z 524.2 and 262.6, respectively) to provide a 0.1 amu mass accuracy for each [M + H]+ parent ion (mp+). The mp+ ion 312 from Q1 was passed through a collision chamber (Q2), operating in radio-frequency-only mode (23 V), and focused for subsequent ionization as a consequence of the collision of the rapidly moving mp+ with an inert gas (Ar) at ∼1.8 mTorr to produce the unique product fragmentation spectrum that was subsequently scanned through a third mass filter (Q3) where selected daughter ions (md+) were collected in multiple resonance monitoring (transitions 312 > 188 173.99).
Drugs
PNU-120596 was obtained from the National Institute of Drug Addiction through the Research Resources Drug Supply Program and purchased from Tocris Bioscience (Ellisville, MO, USA) and Selleck Chemicals (Houston, TX, USA). Methyllycaconitine citrate (MLA) was purchased from Ascent Scientific Ltd. (Bristol, UK). Other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). Nomenclature of drugs and molecular targets conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2011).
PNU-120596
In ex vivo experiments, 1 μM PNU-120596 was used. This concentration lies near the EC50 for potentiating effects of PNU-120596 in heterologous systems (EC50 ∼1.5 μM; Gronlien et al., 2007; Young et al., 2008). Intravenous administration of 1 mg·kg−1 PNU-120596 has been shown to elevate the concentration of PNU-120596 in the brains of rats to similar values (∼1.5 μM; Hurst et al., 2005). In in vivo experiments, 1 mg·kg−1 (in vivo, i.v.). and 30 mg·kg−1 (in vivo, s.c.) were used. Similar doses have been used in other studies (Hurst et al., 2005; McLean et al., 2011). PNU-120596 is a highly lipophilic compound and thus, can be trapped by the tissue near the s.c. injection site. Therefore, the dose used for s.c. injections was chosen higher than that for i.v. injections to compensate for that possibility. To make a 50 mM stock solution (maximal achievable concentration is ∼200 mM), PNU-120596 was dissolved in dimethyl sulfoxide (DMSO). However, the amount of DMSO injected in each animal did not exceed 2 mL·kg−1. The appropriate amounts of 50 mM stock solution PNU + DMSO or DMSO alone (i.e. control) were injected as a bolus s.c. (30 mg·kg−1) or i.v. (1 mg·kg−1).
Results
This study used ex vivo (i.e. COGD) and in vivo (i.e. MCAO) experimental approaches to test the hypothesis that PNU-120596, a type-II PAM of α7 nAChRs, converts physiological/endogenous choline/ACh into potent neuroprotective agents via activation of α7 nAChRs. All ex vivo patch-clamp experiments were conducted at 30–32°C.
Injury curves and quantification of the evolution of COGD-induced acute neuronal injury
Subjecting acute hippocampal slices to COGD resulted in a permanent loss of the ionic gradient and depolarization of the neuronal membrane potential leading to neuronal injury and death. The stages of neuronal injury were quantified by the time-dependent decrease in the neuronal membrane potential (i.e. anoxic depolarization), a key parameter defining the status of neuronal health and the evolution of neuronal injury (i.e. injury curves). Figure 1 illustrates typical examples of the raw (recorded in current-clamp) and the normalized individual injury curves obtained from two hippocampal CA1 pyramidal neurons subjected to either control (no COGD, no pretreatment with choline + PNU-120596, Figure 1A,B) or COGD conditions (COGD, but no pretreatment with choline + PNU-120596, Figure 1C,D) and one hippocampal CA1 stratum radiatum interneuron subjected to COGD without pretreatment with choline + PNU-120596 (Figure 1E,F). As expected, neuronal depolarization/injury did not occur in the absence of COGD (Figure 1A,B) and thus, the value of half-time to 0 mV (i.e. T50, see Methods) was not defined. Similar results were obtained in five control experiments and three preparations. By contrast, neurons illustrated in Figure 1C,E were subjected to COGD and exhibited injury curves with the following T50 values: T50 (pyramidal) = 707.5 s (Figure 1D) and T50 (inter) = 655.7 s (Figure 1F). These experiments were conducted using neurons obtained from different preparations.
Pretreatment with 20 μM choline + 1 μM PNU-120596
Two groups of acute hippocampal slices obtained from the same animal and preparation were pretreated for 3 h either in standard ACSF (i.e. control group) or ACSF containing 20 μM choline + 1 μM PNU-120596 (i.e. treatment group). The T50 estimated from individual injury curves were then averaged for each neuronal sub-type and experimental condition and the mean values were compared across the corresponding groups (unpaired, two-tailed Student test). Figure 2A–D illustrates averaged injury curves built for four sets of controls and treatments: (i) No OGD after pretreatment with 20 μM choline + 1 μM PNU-120596 (n = 3; Figure 2A; the error bars are invisible); (ii) control (COGD, no pretreatment, open circles, Figure 2B) versus pretreatment of CA1 pyramidal neurons with 20 μM choline + 1 μM PNU-120596 (closed circles, Figure 2B); (iii) control (COGD, no pretreatment, open circles, Figure 2C) versus pretreatment of CA1 pyramidal neurons with 20 μM choline + 1 μM PNU-120596 + 20 nM MLA (closed circles, Figure 2C) and (iv) control (COGD, no pretreatment, open circles, Figure 2D) versus pretreatment of CA1 interneurons with 20 μM choline + 1 μM PNU-120596 (closed circles, Figure 2D).
The results of analysis of these experiments (unpaired, two-tailed Student's t-test) were summarized in Table 1 and demonstrated that pretreatment of acute hippocampal slices with 20 μM choline + 1 μM PNU-120596 significantly delayed the COGD-induced acute injury and the corresponding depolarization of hippocampal CA1 pyramidal neurons (Figure 2B), but not CA1 interneurons (Figure 2D). However, these beneficial effects of 20 μM choline + 1 μM PNU-120596 were lost when 20 nM MLA was present in the ACSF during pretreatment and recordings (Figure 2C) indicating that activation of α7 nAChRs was required for the effects of choline + PNU-120596.
Table 1.
Choline + PNU-120596 significantly delay COGD-induced depolarization/injury of hippocampal CA1 pyramidal neurons, but not CA1 stratum radiatum interneurons and activation of α7 nAChRs is required
| Hippocampal CA1 pyramidal neurons | Hippocampal CA1 interneurons | |||||
|---|---|---|---|---|---|---|
| Control | 20 μM Cho + 1 μM PNU | Control | 20 μM Cho + 1 μM PNU + 20 nM MLA | Control | 20 μM Cho + 1 μM PNU | |
| T50 (s) (#cells) | 873.4 ± 76.6 (6) | 1195.8 ± 65.9 (6) | 764.4 ± 63.8 (6) | 902.0 ± 72.3 (6) | 680.7 ± 36.0 (6) | 726.6 ± 20.4 (6) |
| P-value | 0.0097 | 0.1841 | 0.2936 | |||
Pretreatment with 20 μM choline + 1 μM PNU-120596 significantly delays COGD-induced depolarization/injury of hippocampal CA1 pyramidal neurons, but not CA1 stratum radiatum interneurons, and these beneficial effects are eliminated by 20 nM MLA, a selective α7 nAChR antagonist. Significant differences are shown in bold. Each experimental condition is presented in a separate column and has its own independent control.
Pretreatment of CA1 pyramidal neurons with 20 μM choline alone or 1 μM PNU-120596 alone
When applied alone, 20 μM choline or 1 μM PNU-120596 do not activate α7 nAChRs (Gusev and Uteshev, 2010; Kalappa et al., 2010). Therefore, to test the hypothesis that neuroprotection by choline + PNU-120596 requires α7 nAChR activation, acute hippocampal slices were pretreated for 3 h in 20 μM choline alone or 1 μM PNU-120596 alone. As expected, choline alone (closed circles; Figure 2E) and PNU-120596 alone (closed circles; Figure 2F) did not significantly delay neuronal depolarization/injury ( Table 2) when compared to control conditions (open circles; Figure 2E,F), as evidenced by insignificant differences in both T50 and T80 (Table 2). The T80 was used to test for significant differences among the earlier phases of injury curves. Therefore, activation of α7 nAChRs was absolutely required for significant neuroprotection of hippocampal CA1 pyramidal neurons subjected to COGD ex vivo.
Table 2.
Enhanced neuroprotection of hippocampal CA1 pyramidal neurons by synergistic action of choline + PNU-120596 in COGD
| 20 μM choline alone | 1 μM PNU alone | 200 μM choline ± 1 μM PNU | |||||
|---|---|---|---|---|---|---|---|
| Control | 20 μM Cho alone | Control | 1 μM PNU alone | 200 μM Cho alone | Control | 200 μM Cho + 1 μM PNU | |
| T50 (s) (# cells) | 1027.8 ± 64.9 (11) | 1013.9 ± 49.8 (10) | 1035.3 + 50.2 (11) | 935.5 + 114.2 (9) | 1041.2 ± 37.5 (5) | 1080.7 ± 37.4 (5) | 1412.6 ± 108.1 (5) |
| P-value | 0.8690 | 0.4045 | 0.4766 | 0.0199 | |||
| T80 (s) (# cells) | 933.4 ± 72.4 (11) | 903.1 ± 51.5 (10) | 960.7 + 51.4 (11) | 790.0 + 110.7 (9) | |||
| P-value | 0.7414 | 0.154 | |||||
Pretreatment with 20 μM choline alone, 200 μM choline alone or 1 μM PNU-120596 alone does not significantly delay COGD-induced depolarization/injury in hippocampal CA1 pyramidal neurons supporting the requirements for α7 activation and synergistic action of choline + PNU-120596. Significant differences are shown in bold. Each experimental condition is presented in a separate column and has its own independent control.
Pretreatment with 200 μM choline + 1 μM PNU-120596
Although physiologically relevant concentrations of choline (i.e. 20 μM) were found to delay the COGD-induced depolarization/injury of hippocampal CA1 pyramidal neurons in the presence, but not absence, of 1 μM PNU-120596 (Figure 2B,E and Table 1), activation of α7 nAChRs by higher concentrations of choline in the presence of PNU-120596 may result in neurotoxicity (Del Barrio et al., 2011; Dinklo et al., 2011) due to the high permeability of α7 ion channels to Ca2+ ions. To evaluate the resilience of the effects of choline + PNU-120596, experiments similar to those described above were conducted using hippocampal CA1 pyramidal neurons and a 10-fold higher concentration of choline (i.e. 200 μM). At 200 μM, choline may activate α7 nAChRs in the absence of PNU-120596 (Uteshev et al., 2003). Therefore, three groups of pretreatments were used ( Figure 3): (i) control (ACSF only, no choline, no PNU-120596; open circles); (ii) 200 μM choline alone (no PNU-120596; closed circles); and (iii) 200 μM choline + 1 μM PNU-120596 (open triangles). The T50 for individual injury curves were estimated and averaged to calculate the mean T50 for each group.
The results of these experiments (Table 2) demonstrated that pretreatment of acute hippocampal slices with 200 μM choline alone produced neither significant benefits, nor harm (P = 0.4766 vs. control; open and closed circles, Figure 3); while pretreatment with 200 μM choline + 1 μM PNU-120596 still significantly delayed neuronal depolarization/injury of CA1 pyramidal neurons subjected to COGD (P = 0.0199 vs. control sand P = 0.0118 vs. 200 μM choline alone; open triangles, Figure 3). Thus, in the presence of 1 μM PNU-120596, a broad range of choline concentrations (i.e. 20–200 μM) significantly delay depolarization/injury of hippocampal CA1 pyramidal neurons subjected to COGD ex vivo.
Susceptibility of CA1 pyramidal neurons and CA1 stratum radiatum interneurons to acute injury by COGD
Injury curves obtained from subjecting CA1 pyramidal neurons and interneurons to COGD ex vivo in the absence of any pretreatments were compared. CA1 interneurons were found to be significantly more susceptible to COGD than CA1 pyramidal neurons because T50 (inter) = 680.7 ± 36.0 s (n = 6) estimated for CA1 interneurons were significantly smaller (P = 0.0004; t = 3.95; unpaired, two-tailed Student's t-test) than T50 (pyramidal) = 984.7 ± 35.1 s (n = 27) estimated for CA1 pyramidal neurons.
Lack of involvement of GABAergic neurotransmission
One possible mechanism that could explain the observed delay in injury of CA1 pyramidal neurons treated with choline + PNU-120596 is enhanced GABAergic inhibition of these neurons in the presence of choline + PNU-120596. CA1 pyramidal neurons are the prime target of GABAergic CA1 interneurons that express high densities of α7 nAChRs sensitive to PNU-120596 (Kalappa et al., 2010). To test this hypothesis, experiments similar to those illustrated in Figure 2B were conducted in the continuous presence of 20 μM gabazine (GBZ), a selective GABAAR antagonist. In these experiments, GBZ was present in the ACSF during both pretreatment and COGD. Inhibition of GABAARs did not alter the neuroprotective effects of choline + PNU-120596 on CA1 pyramidal neurons: T50 (control + GBZ) = 742.7 ± 55.0 s (n = 4) versus T50 (choline + PNU + GBZ) = 933.3 ± 36.2 s (n = 5) (P = 0.0198, Figure 4) suggesting that GABAergic inhibition is not a key mechanism of neuroprotection by choline + PNU-120596 in COGD ex vivo.
PNU-120596-mediated protection in an in vivo ischaemia model
To test the hypothesis that PNU-120596 produces significant neuroprotection in vivo, the MCAO model of stroke was employed in young adult S.-D. rats (∼280 g). The effects of s.c. administration of PNU-120596 (30 mg·kg−1) 3 h and 24 h prior to MCAO (i.e. will be referred to as ‘3 h pre-MCAO’ and ‘24 h pre-MCAO’, respectively) as well as i.v. administration of PNU-120596 (1 mg·kg−1) 30 min after the start of MCAO (i.e. will be referred to as ‘post-MCAO’) on the infarct volume measured 24 h after MCAO were evaluated. The expectation was that PNU-120596 will enhance activation of α7 nAChRs by endogenous nicotinic agonists to significantly enhance neuronal resistance to MCAO and thus, significantly reduce the infarct volume caused by MCAO in both pre- and post-treatment paradigms. In each of these experiments, only the left MCA was occluded. In matching control groups of animals, only vehicle (i.e. DMSO) was injected s.c. or i.v. respectively. The results of these experiments demonstrated significant reduction in the infarct volume in 3 h pre-MCAO as well as post-MCAO treatment groups (unpaired, two-tailed, Student's t-test): P = 0.0051 (n = 4, 3 h pre-MCAO; Figure 5A–C) and P = 0.0413 (n = 5, post-MCAO; Figure 5D–F). The infarct volume was not significantly affected by PNU-120596 in the 24 h pre-MCAO treatment group (unpaired, two-tailed, Student's t-test): P = 0.5199 (n = 5, 24 h pre-MCAO; Figure 6).
Figure 5.
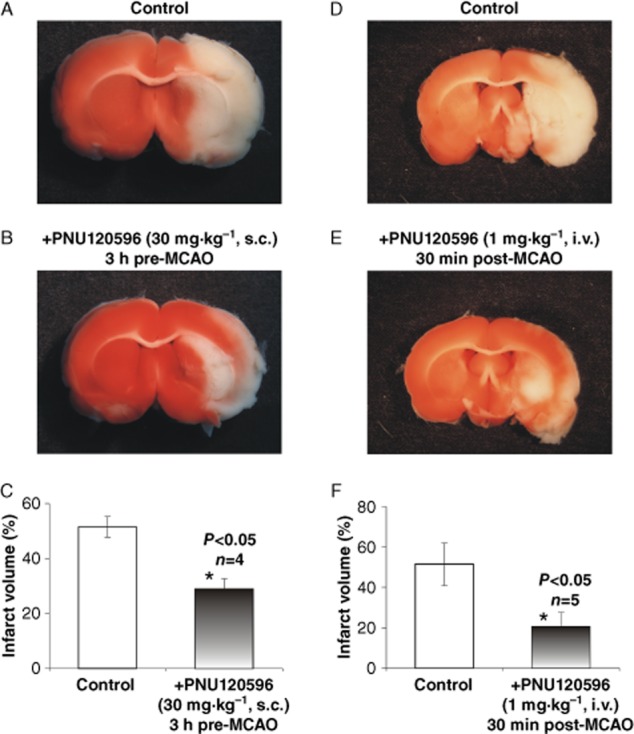
PNU-120596 enhances resistance of cortical/subcortical brain regions to injury induced by focal cerebral ischaemia. Focal cerebral ischaemia was induced by a transient (90 min) MCAO. Typical examples of injured whole-brain coronal sections (2 mm thickness) treated with vehicle (i.e. DMSO) only (A and D), PNU-120596 (30 mg·kg−1, s.c., 3 h pre-MCAO, n = 4) (B) or PNU-120596 (1 mg·kg−1, i.v., 30 min post-MCAO, n = 5) (E) respectively. Twenty-four hours after MCAO, brain sections were prepared for analysis of infarct volume within cortical and subcortical brain regions from control and treated groups. The results indicate that both 3 h pre-MCAO treatment with PNU-120596 (30 mg·kg−1, s.c.) and 30 min post-MCAO treatment with PNU-120596 (1 mg·kg−1, i.v.) significantly (unpaired, two-tailed, Student's t-test) reduced infarct volume (P = 0.0051, n = 4; C and P = 0.0413, n = 5; F respectively).
Figure 6.
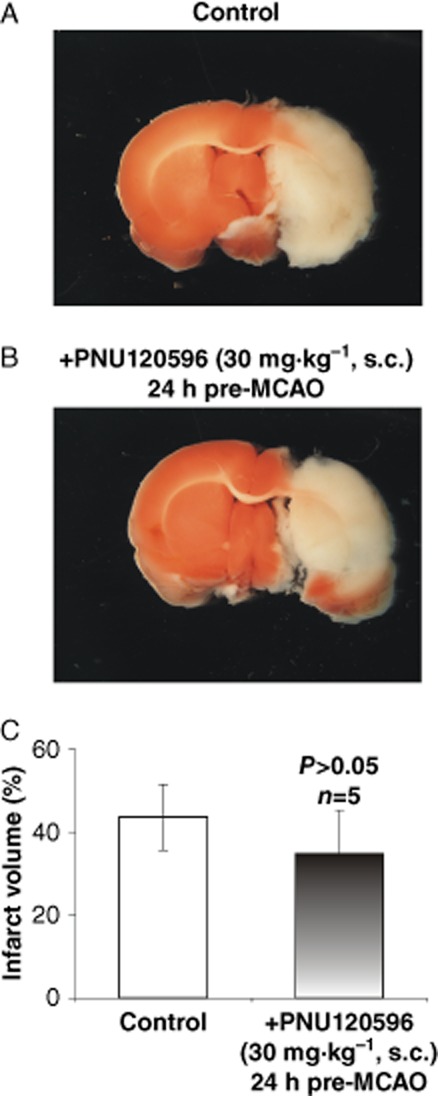
PNU-120596 administered s.c. 24 h prior to focal cerebral ischaemia failed to enhance resistance of cortical/subcortical brain regions to ischaemic injury. To test the hypothesis that PNU-120596 preconditions cortical and subcortical brain regions by enhancing neuronal resistance to ischaemia, PNU-120596 was injected s.c. 24 h prior to MCAO. The half-life time of PNU-120596 is ∼8 h (McLean et al., 2011). It was expected that PNU-120596 preconditions neurons and thus, its presence is not required at the time of ischaemic injury (i.e. 24 h after PNU-120596 injection). However, the effects of 24 h pre-MCAO s.c. treatment with PNU-120596 were found to be statistically insignificant (unpaired, two-tailed, Student's t-test) suggesting that the presence of PNU-120596 and thus, the enhanced activation of α7 nAChRs around the time of ischaemic injury are required for enhanced resistance to ischaemic injury.
PNU-120596-mediated protection in animals subjected to a choline-deficient diet
Although choline-deficient diet has been shown to significantly reduce only the plasma and not the cerebrospinal levels of choline (Klein et al., 1998), the effectiveness of recruiting and activation of endogenous choline/ACh by PNU-120596 in MCAO experiments may be reduced once the animals are challenged with choline deficiency. To determine whether choline deficiency can reduce neuroprotection by PNU-120596 in MCAO experiments, S.-D. rats were subjected to a choline-deficient diet for 14 days prior to experimentation (see Methods). At the time of MCAO, the animals (>260 g) were divided into two groups: control (DMSO only) and treated (PNU-120596, 1 mg·kg−1, i.v., 30 min post-MCAO). Other experimental details and data analysis were identical to those used in choline-sufficient animals (see above and Methods). In choline-deficient animals, PNU-120596 remained effective and produced significant (P = 0.0343; unpaired, two-tailed, Student's t-test) reduction in the MCAO-induced infarct volume in treated (n = 4) vs. control (n = 4) animals (Figure 7).
Figure 7.
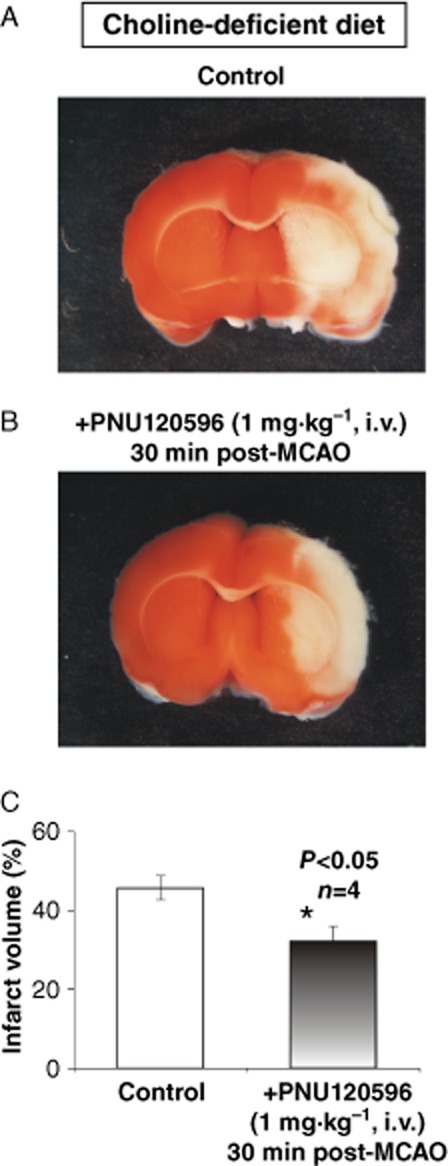
Neuroprotective effects of PNU-120596 remain in choline-deficient rats. Experiments similar to those illustrated in Figure 5D–E were conducted in rats subjected to a choline-deficient diet to determine whether choline deficiency can reduce the neuroprotective effects of PNU-120596 in MCAO experiments. (A–B) Typical examples of injured whole-brain coronal sections (2 mm thickness) treated with vehicle (i.e. DMSO) only (A) or PNU-120596 (1 mg·kg−1, i.v., 30 min post-MCAO, n = 4) (B). The results indicate that PNU-120596 remained neuroprotective and significantly reduced MCAO-induced infarct volume (P = 0.0343, n = 4; C) in rats subjected to a choline-deficient diet (unpaired, two-tailed, Student's t-test).
Evaluation of blood/brain levels of PNU-120596
To confirm the presence of PNU-120596 in the brain tissue and blood plasma, samples of blood plasma and hippocampal, frontal cortical and striatal tissues of five animals (S.-D. rats, ∼280 g) subjected to a protocol utilizing 3 h pre-MCAO were evaluated using MS (see Methods). The results of MS analyses demonstrated that the amount of PNU-120596 in the hippocampus, frontal cortex and striatum collected 3 h after s.c. injections of 30 mg·kg−1 PNU-120596 in S.-D. rats were comparable (in ng per g of tissue): 189.5 ± 67.8 (n = 5), 157.5 ± 49.0 (n = 5) and 149.0 ± 42.7 (n = 5), respectively, and somewhat higher than the amounts of PNU-120596 remaining in the blood plasma of injected animals (in nM): 63.4 ± 19.3 (n = 5). These values are comparable to those reported previously (McLean et al., 2011) as well as the concentrations (i.e. 1 μM) used in our ex vivo experiments (Figures 4). These results suggest that sub-μM to low-μM concentrations of PNU-120596 and the corresponding doses of systemically administrated PNU-120596 significantly enhance neuronal survival in ischaemic injury.
Discussion
A deficient blood supply to the whole brain, as in global ischaemia, and specific cortical and subcortical brain regions, as in focal ischaemia, may irreversibly damage brain tissues and cause a partial or complete loss of cognitive, autonomic and motor functions. Although in the last two decades, substantial efforts have been invested in developing neuroprotective medicine, these efforts have not resulted in clinically efficacious therapies for ischaemic stroke (Roger et al., 2012). These failures highlight the need for development of new therapeutic concepts, approaches and drug treatments for neuronal protection and prevention of neuronal damage secondary to ischaemic stroke. The results of this proof-of-concept study support the hypothesis that PAMs-II, such as PNU-120596, can convert endogenous agonists of α7 nAChRs (i.e. choline and ACh) into potent neuroprotective agents in post-ischaemic neuronal injury in cortical and subcortical brain regions. The proposed strategy incorporates the neuroprotective capacity of α7 nAChR activation by endogenous choline/ACh enhanced by PNU-120596 (Hurst et al., 2005; Gusev and Uteshev, 2010).
α7 nAChRs are commonly expressed in the brain including the hippocampus, cortex and striatum (Breese et al., 1997; Whiteaker et al., 1999; Quik et al., 2009; Woodruff et al., 2011) – brain regions that are selectively vulnerable to ischaemia; and activation of these receptors can enhance neuronal resistance to ischaemic and other types of insults (Meyer et al., 1998; Shimohama et al., 1998; Li et al., 1999; Rosa et al., 2006; Guseva et al., 2008; Quik et al., 2009) as well as cognitive performance in patients and animal models of schizophrenia (Olincy et al., 2006; Olincy and Stevens, 2007), dementia (Kem, 2000; Ren et al., 2007; Bitner et al., 2010) and traumatic brain injury (Guseva et al., 2008). Although choline is a full selective endogenous α7 nAChR agonist, at physiological levels (i.e. <20 μM) and pathophysiological levels expected in the penumbra of brain injury sites (i.e. >20 μM), choline alone is ineffective as an α7 nAChR agonist (Alkondon and Albuquerque, 2006; Djuricic et al., 1991; Klein et al., 1993, 1998; Sarter and Parikh, 2005; Scremin and Jenden, 1989; Uteshev et al., 2003) because of its low potency for α7 activation (EC50∼0.5 mM; Papke and Papke, 2002) and tendency to induce α7 desensitization (IC50∼40 μM) (Uteshev et al., 2003). As a result, choline has not previously garnered much attention as a potential neuroprotective agent. However, these limitations may be overcome by the use of PAMs-II, such as previously reported PNU-120596 (Hurst et al., 2005). PNU-120596 inhibits α7 desensitization and thus, can produce a persistent and tunable activation modality of α7 nAChRs (Gusev and Uteshev, 2010; Kalappa et al., 2010), which may be fine-tuned and optimized to achieve neuroprotection (Li et al., 1999; Papke et al., 2000; Uteshev et al., 2003; Uteshev, 2012a,b). Activation of α7 nAChRs can also be achieved by exogenous α7 agonists (e.g. DMXBA; Kem, 2000) or inhibitors of ACh hydrolysis (e.g. donepezil; Kawakami et al., 1996). However, neuroprotection produced by endogenous nicotinic agonists recruited by a PAM-II may be more efficacious and with fewer adverse effects (Lo, 2008; Uteshev, 2012a) because choline is well tolerated and ubiquitously present in the brain, while PNU-120596 (the first PAM-II available on the market) is highly selective for α7 nAChRs and to date, non-α7-mediated effects of PNU-120596 have not been reported. Therefore, this study tested the hypothesis that PNU-120596 enhances survival of neurons subjected to complete oxygen-glucose deprivation (ex vivo) and focal cerebral ischaemia (in vivo). The rationale for this study was that because activation and desensitization of α7 nAChRs are delicately balanced, the proposed study provided an important proof-of-concept for the development and future clinical trials of PAMs-II, such as PNU-120596 or other functionally related compounds (Faghih et al., 2009; Dinklo et al., 2011; McLean et al., 2011; Freitas et al., 2012; Parada et al., 2013). These factors highlight the main advantage of the PAM-II-based neuroprotective paradigm – the ability of PAMs-II to recruit endogenous choline as a well-tolerated neuroprotective agent ubiquitously present in the extracellular fluid. Although endogenous ambient ACh, along with choline, may also contribute to activation of α7 nAChRs and neuroprotection in the presence of PNU-120596, the extracellular levels of ACh in vivo are extremely low (<10 nM) due to ACh hydrolysis (Hartmann et al., 2008) and thus, potential neuroprotective effects of ACh in vivo would be expected to arise primarily from synaptic release of ACh. However, PNU-120596 alone did not significantly delay COGD-induced neuronal injury ex vivo (Figure 2F) suggesting that at least in acute hippocampal slices, the potential contribution of synaptic ACh to neuroprotection is limited, if any.
The results of ex vivo experiments support the need for activation of α7 ion channels as a requirement for α7-mediated enhanced neuroprotection in COGD. This is evidenced by the inhibitory effects of 20 nM MLA on the effects of choline + PNU-120596 (Figure 2C) and the lack of effects by 20 μM choline alone (Figure 2E), 200 μM choline alone (Figure 3) and 1 μM PNU-120596 alone (Figure 2F). By inhibiting α7 nAChR desensitization and enhancing the potency of choline/ACh for α7 activation, PNU-120596 and other PAMs-II allow endogenous choline/ACh to produce a weak persistent level of α7 activation (Gusev and Uteshev, 2010; Kalappa et al., 2010; Uteshev, 2012b) – an activation modality of α7 nAChRs that can be neuroprotective as discussed in this and other studies (Meyer et al., 1998; Shimohama et al., 1998; Li et al., 1999; Rosa et al., 2006). However, the involvement of α7 nAChR-mediated ion channels in the effects of choline + PNU-120596 introduces concerns for neurotoxicity because α7 channels are highly permeable to Ca2+ ions (Vernino et al., 1992; Castro and Albuquerque, 1995; Fucile, 2004; Uteshev, 2010) and thus, excessive activation of α7 nAChRs, especially in the presence of PNU-120596, can become toxic for α7-expressing cells, as previously discussed (Li et al., 1999; Del Barrio et al., 2011; Dinklo et al., 2011; Uteshev, 2012a). Despite these valid concerns, our ex vivo data demonstrate that neurotoxicity does not result from synergistic action of physiologically relevant 20 μM choline in the presence of 1 μM PNU-120596 (Figure 2A). On the contrary, activation of α7 nAChRs by 20 μM choline + 1 μM PNU-120596 significantly delayed COGD-induced depolarization/injury of CA1 pyramidal neurons ex vivo (Figure 2B). These results are further supported by data obtained in vivo (Figures 7). Moreover, the neuroprotective effects of choline + PNU-120596 are found to be remarkably resilient and somewhat insensitive to the extracellular levels of choline because a 10-fold increase in the choline concentration (i.e. from 20 μM to 200 μM) did not significantly alter the effects of choline + PNU-120596 on injury curves (Figure 3). In addition, the neuroprotective effects of PNU-120596 remained significant in choline-deficient rats (Figure 7). These findings may translate into an enhanced safety and a high therapeutic index of potential PAM-II-based therapies. These properties may allow PAM-II-based therapies to remain effective even under conditions of a highly variable dietary choline intake. Although the molecular mechanisms of neuroprotection by choline + PNU-120596 are currently not known, the Ca2+-dependent and/or -independent JAK2/PI3K/AKT-dependent pathways are likely candidates (Thomsen et al., 2010; Del Barrio et al., 2011) and these and other possible mechanisms will be investigated in our future studies.
The MS analysis of the amounts of PNU-120596 in blood/brain samples demonstrated that PNU-120596 concentrations (sub- to low-μM) achieved and used in our in vivo and ex vivo studies were comparable and similar to those reported in similar previous studies (McLean et al., 2011). Therefore, it is conceivable that significant neuroprotection after ischaemic insults and the corresponding mechanisms engaged by PNU-120596 in our ex vivo and in vivo experiments were driven by similar cellular and molecular mechanisms. However, although α7-expressing neurons are the main focus of this study, α7 nAChRs are expressed in a broad variety of non-neuronal tissues including glia (Sharma and Vijayaraghavan, 2001; Parada et al., 2013; Shytle et al., 2004) and immune cells (Wang et al., 2003; De Rosa et al., 2009). Thus, the significant reduction of the infarct volume observed in our in vivo experiments may have resulted from a combined net effect of α7 nAChR activation in multiple neuronal and non-neuronal tissues. The impact and contributions of these individual sources of brain protection remain unknown and present a great interest.
The results of our in vivo experiments suggest that pretreatment with PNU-120596 does not produce a true long-term preconditioning: that is, neuroprotection does not remain after PNU-120596 is cleared from the system; because neuroprotection by PNU-120596 was significant only in the 3 h pre-MCAO protocol, but not the 24 h pre-MCAO protocol with PNU-120596 (30 mg·kg−1; s.c.) treatments. This is consistent with ∼8 h half-life time of PNU-120596 reported previously (McLean et al., 2011). Therefore, the brain does not appear to possess a long-term memory for neuroprotective effects of PNU-120596 and thus, a true preconditioning is ineffective and the presence of PNU-120596 appears to be required for significant neuroprotection.
The chance of neuronal survival after a toxic insult is determined by the neuronal susceptibility to injuries and neuronal sensitivity to therapeutic treatments. In this study, we used an electrophysiological cell injury assay and the COGD protocol to model conditions that are likely present in the core of the ischaemic injury where neuronal depolarization/injury and death can be delayed, but not eliminated (Lo, 2008). The effects of choline + PNU-120596 in hippocampal CA1 pyramidal neurons and CA1 stratum radiatum interneurons then have been compared. While highly effective in CA1 pyramidal neurons, 20 μM choline + 1 μM PNU-120596 failed to delay COGD-induced depolarization/injury of CA1 interneurons (Figure 2D). At least two factors may have contributed to these differences. CA1 interneurons express ∼20-fold greater densities of α7 nAChRs than CA1 pyramidal neurons (Kalappa et al., 2010). Thus, concentrations of choline + PNU-120596 that produce only a weak α7 activity in CA1 pyramidal neurons would be expected to produce a ∼20-fold greater level of activity in CA1 interneurons. These levels of activation may be excessive and thus, suboptimal and ineffective. However, a 10-fold increase in choline concentration from 20 μM to 200 μM did not produce any harm (Figure 3) suggesting that the beneficial effects of choline + PNU-120596 span over a wide range of choline concentrations and the level of α7 activation may not be the prime reason for the observed differences in the effects of choline + PNU-120596 in CA1 pyramidal and interneurons. Moreover, COGD-induced depolarization/injury of CA1 pyramidal neurons remained significantly delayed by choline + PNU-120596 in the presence of 20 μM GBZ (Figure 4). Therefore, GABAergic neurotransmission in the CA1 pyramidal region does not significantly contribute to neuroprotection by choline + PNU-120596 in COGD ex vivo. Another possibility is that CA1 interneurons may be intrinsically more susceptible to acute injury resulted from COGD making choline + PNU-120596 ineffective. This possibility is supported by our data. Indeed, CA1 interneurons are significantly more susceptible to acute injury by COGD than CA1 pyramidal neurons in the presence (Figure 2C) and absence of PNU-120596 (see Results). These results may seem somewhat contradictory to the results reported in some previous studies where hippocampal CA1 interneurons were found less vulnerable to ischaemic insults than hippocampal CA1 pyramidal neurons (Ferrer et al., 1995; Freund and Buzsaki, 1996; Frahm et al., 2004; Avignone et al., 2005; Zhan et al., 2006; Zhan et al., 2007). However, these conclusions were made in relation to delayed (i.e. hours) neuronal injury/death after transient ischaemia, not acute injury/death by COGD investigated in ex vivo experiments of this study. Nevertheless, the available data suggest that PAM-II-based neuroprotective protocols may need to be optimized and individualized for specific ischaemic insults, neuronal sub-types and brain sub-regions reflecting differences in the levels of α7 nAChR expression and the intrinsic neuronal susceptibility to specific ischaemic insults and injuries.
The potential clinical utility of PAMs-II may extend to certain genetic, age- and trauma-related neurodegenerative, sensorimotor and psychiatric disorders characterized by cognitive decline and attention deficits (e.g. schizophrenia, dementia and traumatic brain injury) since these pathologies are also associated with decreased cholinergic tone and a decrease, but not total disappearance, of functional α7 nAChRs (Guan et al., 2000; Olincy et al., 2006; Kelso and Pauly, 2011). Thus, in addition to providing neuroprotective effects, PAMs-II are expected to be able to improve cognitive function and attention impairments in patients and animal models by increasing and partially restoring cholinergic tone (Kem, 2000; Olincy et al., 2006; Ren et al., 2007; Guseva et al., 2008; Bitner et al., 2010; Brown et al., 2010). In this regard, treatments with PNU-120596 or functionally similar compounds may benefit individuals with ischaemic stroke and certain age- and trauma-related cognitive deficits via multiple mechanisms and routes of action.
Acknowledgments
This study was supported by the National Institutes of Health Grant DK-082625 and the start-up funds from UNTHSC to V. V. Uteshev. We thank the National Institute on Drug Abuse Research Resources Drug Supply Program for PNU-120596. We thank Dr. James Simpkins for constructive comments on the manuscript.
Glossary
- COGD
complete oxygen and glucose deprivation
- GBZ
gabazine
- MCAO
middle cerebral artery occlusion
- MLA
methyllycaconitine
- PAM-II
positive allosteric modulator
- PNU-120596
1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methylisoxazol-3-yl)-urea
Conflict of interest
None.
References
- Adams HP, Jr, del Zoppo G, Alberts MJ, Bhatt DL, Brass L, Furlan A, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;38:1655–1711. doi: 10.1161/STROKEAHA.107.181486. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. Subtype-specific inhibition of nicotinic acetylcholine receptors by choline: a regulatory pathway. J Pharmacol Exp Ther. 2006;318:268–275. doi: 10.1124/jpet.106.103135. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX. Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci. 1997;9:2734–2742. doi: 10.1111/j.1460-9568.1997.tb01702.x. [DOI] [PubMed] [Google Scholar]
- Avignone E, Frenguelli BG, Irving AJ. Differential responses to NMDA receptor activation in rat hippocampal interneurons and pyramidal cells may underlie enhanced pyramidal cell vulnerability. Eur J Neurosci. 2005;22:3077–3090. doi: 10.1111/j.1460-9568.2005.04497.x. [DOI] [PubMed] [Google Scholar]
- Beal CC. Gender and stroke symptoms: a review of the current literature. J Neurosci Nurs. 2010;42:80–87. [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Germano SM, Nishimura MC, Davis RL, Bartkowski HM. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke. 1986;17:1304–1308. doi: 10.1161/01.str.17.6.1304. [DOI] [PubMed] [Google Scholar]
- Bitner RS, Bunnelle WH, Decker MW, Drescher KU, Kohlhaas KL, Markosyan S, et al. In vivo pharmacological characterization of a novel selective alpha7 neuronal nicotinic acetylcholine receptor agonist ABT-107: preclinical considerations in Alzheimer's disease. J Pharmacol Exp Ther. 2010;334:875–886. doi: 10.1124/jpet.110.167213. [DOI] [PubMed] [Google Scholar]
- Breese CR, Adams C, Logel J, Drebing C, Rollins Y, Barnhart M, et al. Comparison of the regional expression of nicotinic acetylcholine receptor alpha7 mRNA and [125I]-alpha-bungarotoxin binding in human postmortem brain. J Comp Neurol. 1997;387:385–398. doi: 10.1002/(sici)1096-9861(19971027)387:3<385::aid-cne5>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Brott T, Bogousslavsky J. Treatment of acute ischemic stroke. N Engl J Med. 2000;343:710–722. doi: 10.1056/NEJM200009073431007. [DOI] [PubMed] [Google Scholar]
- Brown KL, Comalli DM, Biasi MD, Woodruff-Pak DS. Trace eyeblink conditioning is impaired in alpha7 but not in beta2 nicotinic acetylcholine receptor knockout mice. Front Behav Neurosci. 2010;4:166. doi: 10.3389/fnbeh.2010.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro NG, Albuquerque EX. alpha-Bungarotoxin-sensitive hippocampal nicotinic receptor channel has a high calcium permeability. Biophys J. 1995;68:516–524. doi: 10.1016/S0006-3495(95)80213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rosa MJ, Dionisio L, Agriello E, Bouzat C, Esandi Mdel C. Alpha 7 nicotinic acetylcholine receptor modulates lymphocyte activation. Life Sci. 2009;85:444–449. doi: 10.1016/j.lfs.2009.07.010. [DOI] [PubMed] [Google Scholar]
- Del Barrio L, Martin-de-Saavedra MD, Romero A, Parada E, Egea J, Avila J, et al. Neurotoxicity induced by okadaic acid in the human neuroblastoma SH-SY5Y line can be differentially prevented by alpha7 and beta2* nicotinic stimulation. Toxicol Sci. 2011;123:193–205. doi: 10.1093/toxsci/kfr163. [DOI] [PubMed] [Google Scholar]
- Dijkhuizen RM, Beekwilder JP, van der Worp HB, Berkelbach van der Sprenkel JW, Tulleken KA, Nicolay K. Correlation between tissue depolarizations and damage in focal ischemic rat brain. Brain Res. 1999;840:194–205. doi: 10.1016/s0006-8993(99)01769-2. [DOI] [PubMed] [Google Scholar]
- Dinklo T, Shaban H, Thuring JW, Lavreysen H, Stevens KE, Zheng L, et al. Characterization of 2-[[4-fluoro-3-(trifluoromethyl)phenyl]amino]-4-(4-pyridinyl)-5-thiazoleme thanol (JNJ-1930942), a novel positive allosteric modulator of the {alpha}7 nicotinic acetylcholine receptor. J Pharmacol Exp Ther. 2011;336:560–574. doi: 10.1124/jpet.110.173245. [DOI] [PubMed] [Google Scholar]
- Djuricic B, Olson SR, Assaf HM, Whittingham TS, Lust WD, Drewes LR. Formation of free choline in brain tissue during in vitro energy deprivation. J Cereb Blood Flow Metab. 1991;11:308–313. doi: 10.1038/jcbfm.1991.63. [DOI] [PubMed] [Google Scholar]
- Faghih R, Gopalakrishnan SM, Gronlien JH, Malysz J, Briggs CA, Wetterstrand C, et al. Discovery of 4-(5-(4-chlorophenyl)-2-methyl-3-propionyl-1H-pyrrol-1-yl)benzenesulfonami de (A-867744) as a novel positive allosteric modulator of the alpha7 nicotinic acetylcholine receptor. J Med Chem. 2009;52:3377–3384. doi: 10.1021/jm9003818. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Soriano MA, Vidal A, Planas AM. Survival of parvalbumin-immunoreactive neurons in the gerbil hippocampus following transient forebrain ischemia does not depend on HSP-70 protein induction. Brain Res. 1995;692:41–46. doi: 10.1016/0006-8993(95)00527-w. [DOI] [PubMed] [Google Scholar]
- Frahm C, Haupt C, Witte OW. GABA neurons survive focal ischemic injury. Neuroscience. 2004;127:341–346. doi: 10.1016/j.neuroscience.2004.05.027. [DOI] [PubMed] [Google Scholar]
- Freitas K, Carroll FI, Damaj MI. The antinociceptive effects of nicotinic receptors alpha7-positive allosteric modulators in murine acute and tonic pain models. J Pharmacol Exp Ther. 2012;344:264–275. doi: 10.1124/jpet.112.197871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fucile S. Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium. 2004;35:1–8. doi: 10.1016/j.ceca.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Furlan AJ. Acute stroke therapy: beyond i.v. tPA. Cleve Clin J Med. 2002;69:730–734. doi: 10.3949/ccjm.69.9.730. [DOI] [PubMed] [Google Scholar]
- Gronlien JH, Hakerud M, Ween H, Thorin-Hagene K, Briggs CA, Gopalakrishnan M, et al. Distinct profiles of alpha7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol. 2007;72:715–724. doi: 10.1124/mol.107.035410. [DOI] [PubMed] [Google Scholar]
- Guan ZZ, Zhang X, Ravid R, Nordberg A. Decreased protein levels of nicotinic receptor subunits in the hippocampus and temporal cortex of patients with Alzheimer's disease. J Neurochem. 2000;74:237–243. doi: 10.1046/j.1471-4159.2000.0740237.x. [DOI] [PubMed] [Google Scholar]
- Gusev AG, Uteshev VV. Physiological concentrations of choline activate native alpha7-containing nicotinic acetylcholine receptors in the presence of PNU-120596 [1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methylisoxazol-3-yl)-urea] J Pharmacol Exp Ther. 2010;332:588–598. doi: 10.1124/jpet.109.162099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guseva MV, Hopkins DM, Scheff SW, Pauly JR. Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. J Neurochem. 2008;25:975–983. doi: 10.1089/neu.2008.0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Kiewert C, Duysen EG, Lockridge O, Klein J. Choline availability and acetylcholine synthesis in the hippocampus of acetylcholinesterase-deficient mice. Neurochem Int. 2008;52:972–978. doi: 10.1016/j.neuint.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Hurst RS, Hajos M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, et al. A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J Neurosci. 2005;25:4396–4405. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Minami M, Lan JQ, Mao XO, Batteur S, Simon RP, et al. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci U S A. 2001;98:4710–4715. doi: 10.1073/pnas.081011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen HS, Nakayama H, Raaschou HO, Olsen TS. Stroke. Neurologic and functional recovery the Copenhagen Stroke Study. Phys Med Rehabil Clin N Am. 1999;10:887–906. [PubMed] [Google Scholar]
- Kalappa BI, Gusev AG, Uteshev VV. Activation of functional alpha7-containing nAChRs in hippocampal CA1 pyramidal neurons by physiological levels of choline in the presence of PNU-120596. Plos ONE. 2010;5:e13964. doi: 10.1371/journal.pone.0013964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzan IL, Furlan AJ, Lloyd LE, Frank JI, Harper DL, Hinchey JA, et al. Use of tissue-type plasminogen activator for acute ischemic stroke: the Cleveland area experience. JAMA. 2000;283:1151–1158. doi: 10.1001/jama.283.9.1151. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Inoue A, Kawai T, Wakita M, Sugimoto H, Hopfinger AJ. The rationale for E2020 as a potent acetylcholinesterase inhibitor. Bioorg Med Chem. 1996;4:1429–1446. doi: 10.1016/0968-0896(96)00137-x. [DOI] [PubMed] [Google Scholar]
- Kelso ML, Pauly JR. Therapeutic targets for neuroprotection and/or enhancement of functional recovery following traumatic brain injury. Prog Mol Biol Transl Sci. 2011;98:85–131. doi: 10.1016/B978-0-12-385506-0.00003-X. [DOI] [PubMed] [Google Scholar]
- Kem WR. The brain alpha7 nicotinic receptor may be an important therapeutic target for the treatment of Alzheimer's disease: studies with DMXBA (GTS-21) Behav Brain Res. 2000;113:169–181. doi: 10.1016/s0166-4328(00)00211-4. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J, Holler T, Cappel E, Koppen A, Loffelholz K. Release of choline from rat brain under hypoxia: contribution from phospholipase A2 but not from phospholipase D. Brain Res. 1993;630:337–340. doi: 10.1016/0006-8993(93)90674-c. [DOI] [PubMed] [Google Scholar]
- Klein J, Koppen A, Loffelholz K. Regulation of free choline in rat brain: dietary and pharmacological manipulations. Neurochem Int. 1998;32:479–485. doi: 10.1016/s0197-0186(97)00127-7. [DOI] [PubMed] [Google Scholar]
- Li Y, Papke RL, He YJ, Millard WJ, Meyer EM. Characterization of the neuroprotective and toxic effects of alpha7 nicotinic receptor activation in PC12 cells. Brain Res. 1999;830:218–225. doi: 10.1016/s0006-8993(99)01372-4. [DOI] [PubMed] [Google Scholar]
- Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean SL, Idris NF, Grayson B, Gendle DF, Mackie C, Lesage AS, et al. PNU-120596, a positive allosteric modulator of alpha7 nicotinic acetylcholine receptors, reverses a sub-chronic phencyclidine-induced cognitive deficit in the attentional set-shifting task in female rats. J Psychopharmacol. 2011;26:1265–1270. doi: 10.1177/0269881111431747. [DOI] [PubMed] [Google Scholar]
- Meyer EM, Tay ET, Zoltewicz JA, Papke RL, Meyers C, King M, et al. Neuroprotective and memory-related actions of novel alpha-7 nicotinic agents with different mixed agonist/antagonist properties. J Pharmacol Exp Ther. 1998;284:1026–1032. [PubMed] [Google Scholar]
- Olincy A, Stevens KE. Treating schizophrenia symptoms with an alpha7 nicotinic agonist, from mice to men. Biochem Pharmacol. 2007;74:1192–1201. doi: 10.1016/j.bcp.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, et al. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry. 2006;63:630–638. doi: 10.1001/archpsyc.63.6.630. [DOI] [PubMed] [Google Scholar]
- Papke RL, Papke JKP. Comparative pharmacology of rat and human alpha7 nAChR conducted with net charge analysis. Br J Pharmacol. 2002;137:49–61. doi: 10.1038/sj.bjp.0704833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Bencherif M, Lippiello P. An evaluation of neuronal nicotinic acetylcholine receptor activation by quaternary nitrogen compounds indicates that choline is selective for the alpha 7 subtype. Neurosci Lett. 1996;213:201–204. doi: 10.1016/0304-3940(96)12889-5. [DOI] [PubMed] [Google Scholar]
- Papke RL, Meyer E, Nutter T, Uteshev VV. alpha 7 receptor-selective agonists and modes of alpha 7 receptor activation. Eur J Pharmacol. 2000;393:179–195. doi: 10.1016/s0014-2999(00)00009-1. [DOI] [PubMed] [Google Scholar]
- Parada E, Egea J, Buendia I, Negredo P, Cunha AC, Cardoso S, et al. The microglial alpha7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2012.4671. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Huang LZ, Parameswaran N, Bordia T, Campos C, Perez XA. Multiple roles for nicotine in Parkinson's disease. Biochem Pharmacol. 2009;78:677–685. doi: 10.1016/j.bcp.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Thinschmidt J, Liu J, Ai L, Papke RL, King MA, et al. alpha7 Nicotinic receptor gene delivery into mouse hippocampal neurons leads to functional receptor expression, improved spatial memory-related performance, and tau hyperphosphorylation. Neuroscience. 2007;145:314–322. doi: 10.1016/j.neuroscience.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Richard Green A, Odergren T, Ashwood T. Animal models of stroke: do they have value for discovering neuroprotective agents? Trends Pharmacol Sci. 2003;24:402–408. doi: 10.1016/S0165-6147(03)00192-5. [DOI] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics – 2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa AO, Egea J, Gandia L, Lopez MG, Garcia AG. Neuroprotection by nicotine in hippocampal slices subjected to oxygen-glucose deprivation: involvement of the alpha7 nAChR subtype. J Mol Neurosci. 2006;30:61–62. doi: 10.1385/JMN:30:1:61. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- Sarter M, Parikh V. Choline transporters, cholinergic transmission and cognition. Nat Rev Neurosci. 2005;6:48–56. doi: 10.1038/nrn1588. [DOI] [PubMed] [Google Scholar]
- Scremin OU, Jenden DJ. Focal ischemia enhances choline output and decreases acetylcholine output from rat cerebral cortex. Stroke. 1989;20:92–95. doi: 10.1161/01.str.20.1.92. [DOI] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc Natl Acad Sci U S A. 2001;98:4148–4153. doi: 10.1073/pnas.071540198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimohama S, Greenwald DL, Shafron DH, Akaike A, Maeda T, Kaneko S, et al. Nicotinic alpha 7 receptors protect against glutamate neurotoxicity and neuronal ischemic damage. Brain Res. 1998;779:359–363. doi: 10.1016/s0006-8993(97)00194-7. [DOI] [PubMed] [Google Scholar]
- Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, et al. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem. 2004;89:337–343. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- Sitzia F, Brown JT, Randall AD, Dunlop J. Voltage- and temperature-dependent allosteric modulation of alpha7 nicotinic receptors by PNU-120596. Front Pharmacol. 2011;2:81. doi: 10.3389/fphar.2011.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Tanaka E, Yamamoto S, Kudo Y, Mihara S, Higashi H. Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J Neurophysiol. 1997;78:891–902. doi: 10.1152/jn.1997.78.2.891. [DOI] [PubMed] [Google Scholar]
- Thomsen MS, Hansen HH, Timmerman DB, Mikkelsen JD. Cognitive improvement by activation of alpha7 nicotinic acetylcholine receptors: from animal models to human pathophysiology. Curr Pharm Des. 2010;16:323–343. doi: 10.2174/138161210790170094. [DOI] [PubMed] [Google Scholar]
- Uteshev VV. Evaluation of Ca2+ permeability of nicotinic acetylcholine receptors in hypothalamic histaminergic neurons. Acta Biochim Biophys Sin (Shanghai) 2010;42:8–20. doi: 10.1093/abbs/gmp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uteshev VV. alpha7 nicotinic ACh receptors as a ligand-gated source of Ca(2+) ions: the search for a Ca (2+) optimum. Adv Exp Med Biol. 2012a;740:603–638. doi: 10.1007/978-94-007-2888-2_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uteshev VV. Somatic integration of single ion channel responses of alpha7 nicotinic acetylcholine receptors enhanced by PNU-120596. Plos ONE. 2012b;7:e32951. doi: 10.1371/journal.pone.0032951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uteshev VV, Meyer EM, Papke RL. Regulation of neuronal function by choline and 4OH-GTS-21 through alpha 7 nicotinic receptors. J Neurophysiol. 2003;89:1797–1806. doi: 10.1152/jn.00943.2002. [DOI] [PubMed] [Google Scholar]
- Vernino S, Amador M, Luetje CW, Patrick J, Dani JA. Calcium modulation and high calcium permeability of neuronal nicotinic acetylcholine receptors. Neuron. 1992;8:127–134. doi: 10.1016/0896-6273(92)90114-s. [DOI] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Davies AR, Marks MJ, Blagbrough IS, Potter BV, Wolstenholme AJ, et al. An autoradiographic study of the distribution of binding sites for the novel alpha7-selective nicotinic radioligand [3H]-methyllycaconitine in the mouse brain. Eur J Neurosci. 1999;11:2689–2696. doi: 10.1046/j.1460-9568.1999.00685.x. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Thundyil J, Tang SC, Sobey CG, Taylor SM, Arumugam TV. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener. 2011;6:11. doi: 10.1186/1750-1326-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young GT, Zwart R, Walker AS, Sher E, Millar NS. Potentiation of alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc Natl Acad Sci U S A. 2008;105:14686–14691. doi: 10.1073/pnas.0804372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan RZ, Nadler JV, Schwartz-Bloom RD. Depressed responses to applied and synaptically-released GABA in CA1 pyramidal cells, but not in CA1 interneurons, after transient forebrain ischemia. J Cereb Blood Flow Metab. 2006;26:112–124. doi: 10.1038/sj.jcbfm.9600171. [DOI] [PubMed] [Google Scholar]
- Zhan RZ, Nadler JV, Schwartz-Bloom RD. Impaired firing and sodium channel function in CA1 hippocampal interneurons after transient cerebral ischemia. J Cereb Blood Flow Metab. 2007;27:1444–1452. doi: 10.1038/sj.jcbfm.9600448. [DOI] [PubMed] [Google Scholar]