

Abstract
CD146, an endothelial biomarker, has been shown to be aberrantly upregulated during pathological angiogenesis and functions as a coreceptor for vascular endothelial growth factor receptor 2 (VEGFR-2) to promote disease progression. However, the regulatory mechanisms of CD146 expression during angiogenesis remain unclear. Using a microRNA screening approach, we identified a novel negative regulator of angiogenesis, microRNA 329 (miR-329), that directly targeted CD146 and inhibited CD146-mediated angiogenesis in vitro and in vivo. Endogenous miR-329 expression was downregulated by VEGF and tumor necrosis factor alpha (TNF-α), resulting in the elevation of CD146 in endothelial cells. Upregulation of CD146 facilitated an endothelial response to VEGF-induced SRC kinase family (SKF)/p38 mitogen-activated protein kinase (MAPK)/NF-κB activation and consequently promoted endothelial cell migration and tube formation. Our animal experiments showed that treatment with miR-329 repressed excessive CD146 expression on blood vessels and significantly attenuated neovascularization in a mouse model of pathological angiogenesis. Our findings provide the first evidence that CD146 expression in angiogenesis is regulated by miR-329 and suggest that miR-329 could present a potential therapeutic tool for the treatment of angiogenic diseases.
INTRODUCTION
Angiogenesis, the formation of new capillaries from preexisting ones, plays a critical role in vertebrate development as well as in disease progression (1). Physiological angiogenesis, including embryogenesis, organogenesis, and wound repair, requires a balance between pro- and antiangiogenic factors to maintain vascular homeostasis (2). During pathological angiogenesis, this balance is broken as excessive proangiogenic factors, such as vascular endothelial growth factor (VEGF) and tumor necrosis factor alpha (TNF-α), are produced, resulting in aberrant neovascularization. Uncontrolled angiogenesis has been observed for a variety of pathologies, including tumors, inflammation, and retinal neovascularization (3–5). Hence, targeting angiogenesis has long been considered a promising therapeutic avenue for angiogenic diseases (6, 7).
CD146 is an inducible adhesion molecule belonging to the immunoglobulin superfamily. Our and others' work has shown that CD146 is an endothelial biomarker and plays a fundamental role in angiogenesis (8). The importance of CD146 in physiological angiogenesis was illustrated by an earlier study of its regulation of intersegmental vessel development in zebrafish (9). During pathological angiogenesis, CD146 expression is upregulated in the endothelial cells involved in angiogenic diseases, including tumors (8), chronic renal failure (10), inflammatory bowel disease (11), and rheumatoid arthritis (12). The expression level of endothelial CD146 directly correlates with endothelial cell proliferation, migration, and tube formation (13, 14). Our previous studies showed that CD146 promotes p38/IκB kinase (IKK)/NF-κB signaling as well as angiogenic gene expression when induced by tumor cell-conditioned medium (14). Recently, we found that CD146 can function as a coreceptor of VEGF receptor 2 (VEGFR-2), with CD146 endothelial expression augmenting VEGF-induced signal transduction and angiogenesis (15). Both CD146-specific small interfering RNA (siRNA) and an anti-CD146 antibody, AA98, could block CD146-mediated angiogenesis (15). In zebrafish, morpholino knockdown of CD146 impaired tumor angiogenesis (9). In xenografted mice, the monoclonal antibody (MAb) AA98 targeting CD146 reduced the blood vessel density in tumors and inhibited tumor growth (17). Moreover, a combination of the AA98 MAb and the anti-VEGF agent bevacizumab showed an additive inhibitory effect on xenografted tumors (15). Thus, CD146 has emerged as a promising molecule for the targeting of angiogenesis-related diseases. However, the understanding of the regulatory mechanisms of CD146 expression, especially at the posttranscriptional level, remains incomplete. Here, we investigated the involvement of microRNAs (miRNAs) in the regulation of both expression and function of CD146.
MicroRNAs are small, endogenous noncoding RNAs that negatively regulate gene expression either through blocking mRNA translation or by inducing mRNA degradation (18). Importantly, evidence is accumulating that miRNAs also play a key role in angiogenesis and angiogenesis-related diseases (19, 20). All miRNAs that regulate angiogenesis have been classified as “angiomiRs” (21). Multiple angiomiRs have been identified across different species, including zebrafish, mice, and humans. For instance, miR-126, an endothelial cell-specific miRNA, was shown to govern angiogenesis and vascular integrity in mice and zebrafish (22). Another angiomiR, miR-296, promotes tumor angiogenesis by regulating the expression of VEGFR-2 in human brain microvascular endothelial cells (23). Most of the angiomiRs identified so far are positive regulators of angiogenesis.
In this study, we describe a novel member of the angiomiR family, miR-329, which regulates angiogenesis in a negative manner by directly targeting the proangiogenic CD146 gene. The expression of miR-329 is downregulated by VEGF and TNF-α, resulting in upregulation of CD146 and promotion of angiogenesis. Treatment with miR-329 significantly impaired neovascularization in a mouse model of oxygen-induced retinopathy (OIR). Our findings provide the first evidence for the regulation of CD146 by miR-329 in angiogenesis, and we propose that treatments involving miR-329 have potential therapeutic implications for the cure of vascular disorders.
MATERIALS AND METHODS
Cells and animals.
Human umbilical vein endothelial cells (HUVECs) were purchased from Cell Systems Biotechnology Vertrieb and cultured in RPMI 1640 medium (Gibco Life Technologies) supplemented with 10% fetal calf serum (FCS). Human microvascular endothelial cells (HMECs) were a gift from Yongzhang Luo. HMECs and 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco Life Technologies) with 1 g/liter glucose and 10% FCS.
All animal experiments were performed in compliance with the guidelines of the Biomedical Research Ethics Committee of the Institute of Biophysics, Chinese Academy of Sciences. SCID/Beige female mice and C57BL/6J mice 4 weeks of age were obtained from Vital River Laboratories, Beijing, China.
Bioinformatics.
miRNA target site prediction of CD146 was performed by online bioinformatics software: TargetScan, miRanda, mirBase and RNA22 (24–26). We integrated the results for all four databases and detected the common miRNAs for further biochemical validation.
The transcription start site (TSS) of miR-329 was predicted by miRStart software. An algorithmic prediction of putative NF-κB binding sites in a 3-kb sequence upstream of the miR-329 TSS was performed using online bioinformatics tools, namely, JASPAR and ECR Browser software.
Constructs.
The construction of p3×FLAG-CD146 containing the open reading frame (ORF) of CD146 has been described previously (27). The 3′ untranslated region (UTR) of CD146 was amplified and constructed downstream of the firefly luciferase gene in the modified pGL3-Control vector (Promega) (28) to generate Luc-3′UTR. The pCR3.1-CD146-3′UTR construct was generated by a PCR-based approach using p3×FLAG-CD146 and Luc-3′UTR as the templates. The mutants of the seed region of putative miR-329 binding sites in the CD146 3′ UTR (Luc-Mut-N1, Luc-Mut-N2, Luc-Mut-N3, Luc-Mut-N4, Luc-Mut-N14, Luc-Mut-P4, and pCR3.1-CD146-Mut-N14) were generated by point mutation PCR. All primers used are described in Table S1 in supplemental material.
Oligonucleotides and transfection.
siRNA duplexes targeting coding sequence (CDS) positions 410 to 428 for CD146 or CDS positions 1562 to 1580 for green fluorescent protein (GFP) were synthesized by Invitrogen using previously reported sequences (14).
The miR-329 mimics, 2′-O-methyl antisense oligonucleotide targeted toward miR-329 (anti-miR-329) and negative control (miR ctrl and anti-miR ctrl), were purchased from GenePharma. 2′-O-Methylation, 5′-cholesterol, thiosulfate phosphorylation-modified agomir mmu-miR-329, and control miRNA synthesized by Ribobio Co. were used for the treatment of OIR in a murine model.
HUVECs were transfected with 50 nM miRNA, anti-miRNA, or siRNA using the Fugene HD method and then incubated for 36 h before analysis.
Activation of angiogenic signals in HUVECs.
Serum-starved HUVECs (12 h) were incubated with NF-κB inhibitor BAY11-7082 (200 ng/ml) or transfected with different small RNAs or vectors before induction with VEGF (Upstate Biotechnology) or TNF-α (Peprotech) (50 ng/ml each). The HUVECs then were stimulated for 20 min, 12 h, and 24 h for analysis of the activation of SKF/p38, NF-κB, or downstream gene expression, respectively.
Dual luciferase reporter assays.
Firefly luciferase reporter vectors containing wild-type (wt) or mutant CD146 3′ UTR were cotransfected with 50 nM miR-329 or anti-miR-329 into HUVECs. Thirty-six hours posttransfection, firefly luciferase activities were measured with the Dual-Luc reporter assay system (Promega) and normalized to the Renilla luciferase levels of pRLTK (Promega).
ChIP assays.
For the chromatin immunoprecipitation (ChIP) assays, HUVECs were treated with 50 ng/ml VEGF and TNF-α for 12 h, cross-linked with 1% formaldehyde, and lysed. The chromatin was sheared to a 300- to 1,500-bp length by sonication. Each immunoprecipitation was performed using 2 μg of rabbit anti-p65 antibody (ab7970; Abcam) or control rabbit IgG (sc-2027; Santa Cruz). Immunocomplexes were captured using protein A-agarose beads blocked by salmon sperm DNA (16-157; Millipore) and washed extensively. Cross-links were reversed overnight, followed by treatment with RNase A at 37°C for 1 h and proteinase K at 65°C for 1 h. The purified DNA was analyzed by real-time PCR using a series of PCR primers (see Table S1 in the supplemental material) covering the genomic region of interest and then normalized to input DNA.
RNA isolation and real-time qPCR.
Total RNA from cells or retinas of mice (two retinas per sample) was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. Two micrograms of RNA was reverse transcribed into cDNA and subjected to SYBR green real-time quantitative PCR (qPCR) to determine the mRNA expression levels. All measurements were normalized to the expression of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA employing the 2−ΔCT (threshold cycle) method. The real-time PCR primers are shown in Table S1 in the supplemental material. We used the TaqMan reverse transcription kit and TaqMan microRNA assay kit (Ambion) to assess expression of mature miR-329. The ABI 7300 system was used for quantification. miR-329 expression was normalized to U6 using the 2−ΔCT threshold cycle method. Relative levels of expression of mature miR-329 and mRNA were determined by the comparative 2−ΔΔCT method.
Western blotting.
Western blotting was performed as described previously (27). Antibodies against CD146 (AA1) (29), NF-κB p65, IκBα (Santa Cruz Biotechnology), phospho-NF-κB p65, phospho-p38 mitogen-activated protein kinase (MAPK), p38, phospho-SKF, SRC (Cell Signaling Technology), or GAPDH (Abcam) were detected with horseradish peroxidase (HRP)-conjugated secondary antibodies (GE Healthcare). Protein bands were quantified by Quantity One software.
Cell migration assay.
Cell migration was assayed using a modified Boyden chamber assay (8-μm-pore size; Corning Costar) as described previously (30). After the appropriate treatments, cells were trypsinized, washed, and resuspended with serum-free medium. A total of 8,000 cells in 100 μl were added to the upper chamber in the presence or absence of VEGF and TNF-α (50 ng/ml each). The lower chambers contained complete medium. After incubation at 37°C for 12 h, cells that migrated to the lower membrane surface were fixed with 4% paraformaldehyde and stained with crystal violet. Pictures were taken with an UPlanFL N digital camera mounted onto an Olympus BX51 microscope and acquired by Olympus DP controller software. Migrated cells were counted by Image J software.
Tube formation.
The tube formation assay was carried out as described by Colorado et al. (31). In brief, 96-well plates were coated with 70 μl Matrigel (BD Biosciences). After the appropriate treatments, cells resuspended in complete medium (10,000 cells) were added to each well and induced with or without VEGF and TNF-α (50 ng/ml each) overnight. Images were taken with a charge-coupled device (CCD) color camera (model KP-D20AU; Hitachi) mounted onto an inverted microscope (Eclipse model TS100; Nikon) and acquired using Image Pro Plus software. Tube lengths were measured using Image Pro Plus software.
Immunohistochemistry.
For 3,3′-diaminobenzidine (DAB) staining, paraffin-embedded tissue sections (5 μm in thickness) were deparaffinized and stained first with an antibody specific for endothelial marker CD31 (6C5; Abcam) and then with biotin-conjugated secondary antibodies (1:1,000) (Roche, CA), followed by HRP-conjugated streptavidin (Roche, CA). The sections were finally counterstained with hematoxylin. Images were taken with an UPlanFL N digital camera mounted onto an inverted microscope (Olympus BX51) and acquired by Olympus DP controller software.
For immunohistofluorescence, retinal sections were deparaffinized and stained with antibodies specific for CD31 or CD146 (AA4) (29), followed by goat anti-rabbit Alexa Fluor 488- or goat anti-mouse Alexa Fluor 555-labeled secondary antibodies (Invitrogen). Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) (Roche, CA). Coverslips were subsequently examined with an Olympus IX81 digital camera mounted onto a confocal laser scanning microscope (Olympus FV 1000), and images were acquired using Olympus Fluoview Viewer 1.6 software. Vessels in the retinal layers were enumerated.
In vivo Matrigel plug assay.
The Matrigel plug assay was carried out as described previously (32). Twenty hours after transfection, 5 × 106 HUVECs resuspended in 60 μl phosphate-buffered saline (PBS) were mixed with 500 μl Matrigel containing 15 U of heparin (Sigma-Aldrich), with or without VEGF and TNF-α (50 ng/ml each). The cell-Matrigel mixture was injected subcutaneously into 4-week-old SCID/Beige female mice along the abdominal midline. After 6 days, the animals were killed and the plugs were removed for immunohistochemical analysis. The blood vessels were counted.
Oxygen-induced retinopathy, intravitreal injections, and quantitative assessment of retinal neovascularization.
Retinal neovascularization was induced in mouse pups using a C57BL/6J oxygen-induced retinopathy (OIR) model (33). In brief, mouse pups at postnatal day 7 (P7) and their mothers were exposed to a 75% ± 2% oxygen atmosphere for 5 days and then removed to normal air until P17. With the exception of the control solution (0.9% NaCl)-injected group, each pup received an intravitreal injection of 1 μl control microRNA in the left eye and modified mouse miR-329 in their right eye (5 μg/μl each) at P12.
The retinal neovascularization was examined by immunohistofluorescence, retinal fluorescein angiography, and real-time PCR at P17. For retinal fluorescein angiography, mouse pups were anesthetized and perfused with 600 μl 50-mg/ml fluorescein isothiocyanate-labeled dextran (Sigma-Aldrich). The eyes were fixed in 4% paraformaldehyde for 1 h, and the retina was dissected from the eyecup. Retinal flat mount images were acquired with a DS-U2 digital camera mounted onto a stereo microscope (Nikon SMZ 1500) and acquired using Nis-Elements F3.0 software. Areas of retinal vaso-obliteration and neovascularization from OIR mice were processed and quantified using Adobe PhotoShop 7.0 according to Conner et al. (34).
Statistical analysis.
All experiments were performed in triplicate. Results were presented as means ± standard deviations (SD). The statistical differences between the miR-329 and control miRNA treatment groups in the OIR mouse model were determined by paired Student's t test. Statistical significance of other experiments was determined by unpaired Student's t test. P values of <0.05 were considered statistically significant.
RESULTS
miR-329 targets CD146 in endothelial cells.
To determine whether there are potential miRNAs targeting CD146 in the endothelium, we combined two different strategies (see Fig. S1 in the supplemental material). First, we performed an in silico search to screen for microRNAs binding to the 3′ untranslated region (3′ UTR) of CD146. Ten miRNAs were predicted by three out of four bioinformatic algorithms. Among all the predicted miRNAs, only one, namely, miR-329, was represented in all four algorithms (Fig. 1A). Second, we analyzed miRNAs that were reported earlier as being associated with angiogenesis using the RNA22 algorithm and obtained 25 miRNAs that have putative binding sites in the CD146 3′ UTR. Altogether, 35 miRNAs were selected for further investigation (see Table S2 in the supplemental material).
Fig 1.

miR-329 downregulates CD146 in endothelial cells. (A) Venn diagram displaying the prediction of microRNAs targeting CD146. (B and C) miR-329 at different concentrations was transfected into HUVECs, and the expression levels of CD146 were determined by Western blotting (B) or quantitative real-time PCR (C). GAPDH served as an internal control. CD146 siRNA was used as a positive control. For mRNA expression, CD146 was normalized to GAPDH first, and expression levels were compared to that of the control transfection (miR ctrl, set as 1). (D and E) miR-329 antisense inhibitor (anti-miR-329) or control anti-miRNA (anti-miR ctrl) (50 nM) was transfected into HUVECs. The protein expression and mRNA expression of CD146 were analyzed by Western blotting (D) or quantitative real-time PCR (E), respectively. GAPDH was used as an internal control. Normalization for mRNA expression was performed as in panel C. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data are presented as means ± SD from three independent experiments.
We then performed an initial screening of the effects of 35 predicted miRNAs on CD146 expression using two biochemical methods (see Fig. S1 in the supplemental material). First, to test whether these miRNAs downregulated endothelial CD146, we transfected each of the 35 miRNAs into human umbilical vein endothelial cells (HUVECs) and analyzed endogenous CD146 expression using Western blotting. Second, we used a luciferase assay to test whether these miRNAs downregulate the expression of the 3′ UTR of CD146. A luciferase reporter construct containing the CD146 3′ UTR (Luc-3′UTR) was cotransfected with each of the 35 miRNAs into 293T cells, and the luciferase activity was measured. Among all 35 miRNAs, miR-329 induced the most pronounced downregulation of both endothelial CD146 and CD146 3′-UTR expression (see Table S2 in the supplemental material).
To further investigate the effect of miR-329 on CD146 expression, we overexpressed miR-329 in HUVECs (Fig. 1B and C) and human microvascular endothelial cells (HMECs) (see Fig. S2A and B in the supplemental material). The results showed that miR-329 downregulated CD146 expression at both the protein and mRNA levels in a dose-dependent manner. To study whether endogenous miR-329 regulates CD146 expression, an antisense oligonucleotide targeted toward miR-329 was introduced into endothelial cells. Inhibition of miR-329 in HUVECs or HMECs resulted in increased expression of CD146 at both the protein and mRNA levels (Fig. 1D and E; see Fig. S2C and D), suggesting that endogenous miR-329 indeed participates in the regulation of endothelial CD146.
In order to determine whether miR-329 takes effect through direct binding to the CD146 3′ UTR, we first evaluated the alignment predicted by the TargetScan algorithm and found four potential miR-329 binding sites within the 3′ UTR of CD146 (Fig. 2A). We utilized a construct carrying a luciferase gene upstream of the wild-type CD146 3′ UTR (Luc-3′UTR wt) or the mutant 3′ UTR containing mismatches in the seed region of each binding site (Luc-Mut-N1, -N2, -N3, and -N4) (see Table S1 in the supplemental material). As shown in Fig. 2B, the reporter activity of Luc-3′UTR was significantly inhibited by miR-329. However, in the presence of miR-329, the activities of the Luc-Mut-N1 and Luc-Mut-N4 mutants were not reduced to the same extent as that observed for the Luc-3′UTR wt construct. Moreover, mutations in both binding sites 1 and 4 (Luc-Mut-N14) caused a complete loss of luciferase sensitivity to miR-329 inhibition (Fig. 2B). The luciferase results were confirmed by using the CD146 cDNA sequence combined with the 3′ UTR. At the protein level, miR-329 could inhibit the expression of CD146 with its wild-type 3′ UTR but not the mutant 3′ UTR at binding sites 1 and 4 (Fig. 2C). Together, these data suggest that miR-329 targets CD146 mRNA directly, namely through binding to the 3′-UTR sites 1 and 4.
Fig 2.

miR-329 targets CD146 by directly binding to its 3′ UTR. (A) Sequence alignment of miR-329 with the CD146 3′ UTR from different species: Homo sapiens, Pan troglodytes, Macaca mulatta, and Mus musculus. nt, nucleotides. (B) The luciferase assay identified binding sites of miR-329 within the 3′ UTR of CD146 mRNA. The luciferase activity of indicated reporter vectors was analyzed after cotransfection with miR-329 (50 nM) in 293T cells. Luc-Mut-P4 was used as a positive control. The luciferase activity in each experiment that used transfection with control miRNA (miR ctrl) was set as 1. Luc-empty, reporter gene without a 3′ UTR; Luc-3′UTR, reporter gene with the wild-type CD146 3′ UTR; Luc-Mut-N1, Luc-Mut-N2, Luc-Mut-N3, and Luc-Mut-N4, CD146 3′ UTRs with mismatches in the seed region of the respective miR-329-binding site; Luc-Mut-N14, CD146 3′ UTR with mutations in both binding sites 1 and 4; Luc-Mut-P4, CD146 3′ UTR with the fourth binding site completely complemented to miR-329. (C) A construct containing the CD146 3′ UTR or a mutant 3′ UTR (Mut-N14) downstream of the CD146 gene was transfected into 293T cells in the presence of miR-329. GADPH served as an internal control. (D) HUVECs were cotransfected with anti-miR-329 (50 nM) and Luc-3′UTR or Luc-Mut-N14. The luciferase activity of the reporter genes was measured and normalized to that of Luc-empty cotransfected with control anti-miRNA (anti-miR ctrl). NS., not significant; * or #, P < 0.05; ##, P < 0.01; ***, P < 0.001. Error bars represent SD. Data are representative of three independent experiments.
To further investigate whether endogenous miR-329 directly binds to the CD146 3′ UTR at sites 1 and 4, we transfected Luc-3′UTR or Luc-Mut-N14 into HUVECs. As shown in Fig. 2D, the luciferase activity of Luc-3′UTR was inhibited in comparison with that of the luciferase reporter gene construct without a 3′ UTR, Luc-empty, presumably due to the binding of CD146-associated miRNAs to the transfected CD146 3′ UTR. In contrast, the luciferase activity of Luc-Mut-N14 was significantly increased compared to the activity associated with the Luc-3′UTR construct. In addition, inhibition of endogenous miR-329 with anti-miR-329 partially rescued luciferase activity of Luc-3′UTR but not that of the mutant 3′ UTR (Fig. 2D), implying that endogenous miR-329 could not bind to Luc-Mut-N14. These experiments further support the direct negative effect of endogenous miR-329 on the CD146 3′ UTR via binding sites 1 and 4.
miR-329 is downregulated by angiogenic factors resulting in increased CD146 expression.
To determine whether the function of miR-329 in the endothelium is regulated by angiogenic factors, we stimulated HUVECs with two proangiogenic factors, VEGF and TNF-α, before analyzing the expression of miR-329 and CD146. Our results showed that exposure to either VEGF or TNF-α resulted in downregulation of miR-329 expression and simultaneous upregulation of CD146 expression in HUVECs (Fig. 3A and B). When the two proangiogenic factors were combined, the inverse correlation between CD146 and miR-329 expression levels was even more marked (Fig. 3A and B). However, overexpression of miR-329 attenuated CD146 expression induced by the proangiogenic factors, suggesting that miR-329 contributed to the VEGF- and TNF-α-mediated CD146 regulation (Fig. 3C).
Fig 3.

miR-329 is downregulated by angiogenic factors resulting in increased CD146 expression. (A and B) HUVECs were cultured in the presence of VEGF (50 ng/ml) or TNF-α (50 ng/ml) or coincubated with NF-κB pathway inhibitor BAY11-7082 (200 ng/ml) as indicated. Endogenous miR-329 expression (A) and the mRNA level of CD146 (B) were determined by real-time PCR. U6 or GAPDH served as an internal control, respectively. Normalized expression in HUVECs without stimulation was set as 1. (C) Western blot analysis of CD146 expression after HUVECs were transfected with miR-329 (50 nM) in the presence of VEGF and TNF-α (50 ng/ml each). CD146 siRNA was used as a positive control. GAPDH served as an internal control. (D) ChIP was performed on HUVECs treated or not treated with VEGF and TNF-α (50 ng/ml each). Immunoprecipitation was performed using an NF-κB p65 antibody, followed by real-time PCR amplification of the genomic DNA fragments covering each of the predicted binding sites. The site position from the miR-329 transcription start site is shown. Enrichment of NF-κB was normalized to a total input control. (E) HUVECs were transfected with anti-miR-329 (50 nM) and incubated with VEGF and TNF-α (50 ng/ml each) or BAY11-7082 (200 ng/ml). CD146 expression was analyzed by Western blotting. GAPDH was used as an internal control. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Bar graphs (means ± SD) present normalized values from at least three independent experiments.
Since both VEGF and TNF-α promote the activation of NF-κB signaling (16, 35), we next explored whether miR-329 is regulated by the NF-κB pathway. We used the inhibitor BAY11-7082 to prevent NF-κB activation and found that the expression of miR-329 was rescued by BAY11-7082 following VEGF or TNF-α treatment (Fig. 3A). These results suggested that miR-329 expression was downregulated by NF-κB. To determine whether NF-κB directly binds to the promoter region of miR-329, we analyzed the miR-329 promoter region using the JASPAR and ECR browser software. Interestingly, nine putative NF-κB binding sites were identified within 3 kb upstream of the miR-329 transcription start site (TSS) (see Table S3 in the supplemental material). Using this information, we designed nine primer pairs across the different binding sites and performed chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR) experiments using an anti-NF-κB p65 antibody. We found significant enrichment for NF-κB p65 binding at binding site 6 (bp −2005 to −1996 upstream of miR-329 TSS) in VEGF- and TNF-α-treated HUVECs compared with nontreated cells (Fig. 3D). These results suggest that NF-κB-mediated downregulation of miR-329 may occur through direct binding to the miR-329 promoter.
To investigate whether VEGF and TNF-α regulated CD146 expression by way of NF-κB-mediated downregulation of miR-329, we abolished NF-κB activation using the inhibitor BAY11-7082 and observed an increase in miR-329 expression, concomitant with a decrease in CD146 expression. Furthermore, we inhibited endogenous miR-329 expression and found that anti-miR-329 partly abrogated the inhibition of CD146 expression by BAY11-7082 (Fig. 3E). These data suggest that miR-329 is downregulated by VEGF- and TNF-α-mediated NF-κB activation, resulting in the upregulation of CD146.
miR-329 inhibits angiogenic signaling by targeting CD146.
Having established that NF-κB activation results in an increase of CD146 levels via repression of miR-329, we in turn investigated whether an increase in miR-329 expression can reduce CD146-dependent angiogenic signaling. As we reported recently, CD146 can function as a coreceptor for the proangiogenic VEGFR-2, an interaction that enhances VEGF-induced signaling (15). This raised the question as to whether miR-329 is a crucial component of a positive, VEGF/CD146-dependent signaling feedback loop. Here, as shown in Fig. 4, VEGF induced SRC kinase family (SKF) phosphorylation and activated the p38 MAPK/NF-κB signaling cascade. However, the VEGF-induced downstream signals were impaired by miR-329 and reactivated by restoring CD146 expression with an miRNA-resistant CD146 lacking its 3′ UTR (Fig. 4A to C). In contrast, neither miR-329 nor CD146 restoration had any effect on TNF-α-induced NF-κB activation (see Fig. S3 in the supplemental material), suggesting that miR-329 may mainly inhibit VEGF-mediated signaling. This could be explained by the role of CD146 as a coreceptor for VEGFR-2.
Fig 4.

miR-329 inhibits angiogenic signaling by targeting CD146. (A to C) Activation of SRC kinase family (SKF) (A), p38 (B), and NF-κB (NF-κB p65 phosphorylation and IκBα degradation) (C) induced by VEGF was determined by Western blotting after HUVECs were transfected with miR-329 or cotransfected with the CD146 ORF (open reading frame without 3′ UTR). Total SKF, p38, GAPDH, or p65 was used as an internal control. (D) NF-κB activation induced by VEGF and TNF-α was determined in HUVECs transfected with miR-329 or cotransfected with the CD146 ORF. Normalization was performed as in panel C. (E) HUVECs under indicated treatments were subjected to real-time PCR for the analysis of mRNA levels of proangiogenic genes. GAPDH was used as an internal control. The mRNA level in each experiment without stimulation was set as 1. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Error bars represent SD. Data are representative of three experiments.
Excessive amounts of VEGF and TNF-α are coexpressed during pathological angiogenesis. Under conditions more similar to pathological angiogenesis (i.e., when used in combination), VEGF- and TNF-α-induced NF-κB activation was still dampened by miR-329 and reactivated by restoring CD146 expression (Fig. 4D). Notably, miR-329 could reduce the expression of a panel of downstream effector genes, including those coding for VEGF, intercellular adhesion molecule 1 (ICAM-1), interleukin-8 (IL-8), and matrix metalloproteinase 9 (MMP-9), except for the gene coding for MMP-2 (Fig. 4E). This observed reduction was abrogated after restoring CD146 expression to baseline levels. In addition, miR-329 impaired VEGF- and TNF-α-induced reorganization of the cytoskeleton (data not shown). Taking these results together, we could show that when overexpressed above physiological levels, miR-329 can suppress the expression of effector genes that promote angiogenesis, predominantly through targeting of CD146.
miR-329 suppresses angiogenesis.
To address the function of miR-329 in angiogenesis, we performed endothelial cell migration and tube formation assays. As shown in Fig. 5A and B, we observed that inhibition of endogenous miR-329 in HUVECs significantly upregulated CD146 expression (see Fig. S4A in the supplemental material) and promoted endothelial cell migration and tube formation, while CD146-specific siRNA abolished anti-miR-329-induced angiogenesis. Next, we examined the function of miR-329 in proangiogenic factor-induced angiogenesis. The results showed that ectopic expression of miR-329 reduced CD146 expression (see Fig. S4B) and impaired HUVEC migration and tube formation induced by either VEGF or TNF-α (see Fig. S5A to D in the supplemental material), or by the two in combination (Fig. 5C and D). This effect, however, was rescued by expression of an miR-329-resistent CD146 ORF construct (open reading frame without 3′ UTR). These findings indicated that miR-329 inhibited endothelial cell function mainly through targeting of CD146.
Fig 5.

miR-329 suppresses angiogenesis. (A) The migration capability of HUVECs was measured using a Transwell system after HUVECs were transfected with anti-miR-329 or cotransfected with CD146 siRNA. The migrated cells were counted by Image J software and are represented in the graph below. (B) Tube formation was analyzed after HUVECs were transfected with anti-miR-329 or cotransfected with CD146 siRNA. The quantification of the tube length was done by Image Pro Plus software and is presented in the histogram below. (C and D) VEGF- and TNF-α-induced cell migration (C) or tube formation (D) was measured after HUVECs were transfected with miR-329 or cotransfected with the CD146 ORF (open reading frame without 3′ UTR). The number of migrated cells or the length of the vessels was measured from 3 independent assays. (E and F) Matrigel plugs containing HUVECs transfected with anti-miR-329 (E) or miR-329 (F) were implanted in SCID mice. The microvessel formation in the explants was analyzed after immunostaining with anti-CD31 antibody. The average vessel number in each section of Matrigel plugs was quantified by Image J software (n = 5). The bar represents 50 μm. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Error bars represent SD. Data are representative of at least three experiments.
To explore the potential role of miR-329 in angiogenesis in vivo, we performed a Matrigel plug assay in SCID mice, using a HUVEC line that we transfected with either miR-329, anti-miR-329, or control miRNA. Six days postimplantation, endothelial cells formed capillary networks in the explants, and blood vessels were counted after immunohistochemistry using the endothelial marker CD31. Consistent with our in vitro experiments, we found that inhibition of miR-329 by anti-miR-329 resulted in more vessel lumen formation in the Matrigel plugs (Fig. 5E), while ectopic expression of miR-329 significantly impaired the blood vessel formation in response to proangiogenic factors (Fig. 5F). Taken together, these data indicate that miR-329 functions as an angiogenesis suppressor both in vitro and in vivo.
miR-329 inhibits retinal neovascularization in an OIR mouse model.
To further address the role of miR-329 during pathological angiogenesis in vivo, we employed a murine model of oxygen-induced retinopathy (OIR), a widely used pathological neovascularization model (36). In this model, mouse pups at postnatal stage day 7 (P7) were exposed to 75% oxygen incubation and were then transferred to room air at P12 to induce retinal ischemia (Fig. 6A). From P12 to P17, proangiogenic factors, including VEGF and TNF-α, were produced in greatly increased amounts, causing severe pathological retinal vascular growth at P17 (4, 37). To investigate the correlation of CD146 and miR-329 expression under pathological conditions in vivo, we analyzed their expression in normal and OIR-affected retinas, using quantitative real-time PCR. Importantly, we observed significantly elevated levels of CD146 mRNA, correlating well with the observed decrease of miR-329, when comparing the OIR retinas at P17 with both OIR retinas before ischemia induction at P12, as well as with normal retinas at P17 (Fig. 6B). These inversely correlated levels of miR-329 and CD146 suggest a regulatory role of miR-329 for CD146 expression in vivo.
Fig 6.

miR-329 inhibits retinal neovascularization in an OIR mouse model. (A) Schematic representation for OIR induction and miR-329 injection. Modified miR-329, control miRNA (miR ctrl) (5 μg, respectively), or control solution (0.9% NaCl) was injected intravitreously in the OIR mice at postnatal day 12 (P12). Retinal samples were collected at P17 for RNA isolation, flat mount staining, and immunohistochemical analysis. (B) Real-time PCR analysis of the endogenous CD146 and miR-329 expression in the retinas of normal or OIR mice at P12 and P17. GAPDH or U6 was used as an internal control. CD146/GAPDH or miR-329/U6 was normalized to that of normal retinas at P12 (n = 8 to 14). (C) The expression of endothelial CD146 in the normal retinas and in OIR retinas treated with control solution, control miRNA, or miR-329 (5 μg) was measured at P17. The relative fluorescence density of CD146/CD31 was analyzed in the confocal images of panel E using Image Pro Plus software (n = 5). (D) Flat-mounted retinas under indicated treatments were perfused with FITC-dextran and observed by fluorescence microscopy to assess retinal vasculature (green) at P17. Images were processed, and the areas of vaso-obliteration (VO) (yellow) and neovascularization (NV) (red) were added by using Adobe PhotoShop 7.0 software. The ratios of VO or NV to total retinal area were quantified and are presented in the graphs below (n = 8). The bar represents 500 μm. (E and F) The vascularization of the retinas in normal or OIR mice treated as indicated was analyzed by immunofluorescence. Staining of blood vessels was performed with anti-CD146 antibody (red) and anti-CD31 antibody (green). Nuclei were stained with DAPI (blue). The numbers of vessels in the inner retinas (ganglion cell layer [GCL] and inner nuclear layer [INL]) (E), and the migrated vessels to the vitreous cavity (F) were quantified (CD31 positive) and are presented in the histograms (9 to 14 mice per group). Arrows show longitudinal and transverse aberrant microvessels. VC, vitreous cavity. The bar represents 50 μm. NS., not significant, *, P < 0.05; **, P < 0.01; ***, P < 0.001. Error bars represent SD. Data are representative of at least three experiments.
In order to evaluate the antiangiogenic properties of miR-329 in vivo, miR-329 or control miRNA was injected intravitreously into the eyes of the OIR mice at P12, and the retinas were isolated at P17 for analysis (Fig. 6A). The downregulation of mouse CD146 by miR-329 was observed in the blood vessels of injected OIR retinas (Fig. 6C), once more confirming the negative effect of miR-329 on CD146 in vivo. Next, to assess the retinal vasculature treated with miR-329, flat-mounted retinas were perfused with fluorescein isothiocyanate (FITC)-dextran and observed by fluorescence microscopy. Interestingly, miR-329 treatment significantly reduced the area of neovascularization tufts (>40%) and also suppressed the formation of tortuous and dilated vessels, thus preserving a better structure of the retinal vasculature, although there was no significant difference in the area of vaso-obliteration (Fig. 6D). Histological analysis also showed that miR-329 significantly reduced the number of vessels in the retinas and the migrated vessels to the vitreous cavity. In contrast, eyes injected with control solution or control miRNA exhibited abundant longitudinal migrating vessels (Fig. 6E and F). Overall, these results indicate the potential therapeutic efficacy of miR-329 in pathological retinal angiogenesis.
DISCUSSION
Although CD146 has been known to play a critical role in angiogenesis for some time (38), the exact mechanism of how CD146 expression is regulated during angiogenesis is still poorly understood. In the present study, we identified a novel angiomiR, miR-329, as a negative regulator of endothelial CD146 and suppressor of CD146-mediated angiogenesis. This conclusion about the role of miR-329 is supported by several lines of evidence. First, we provide in vitro and in vivo experimental results indicating that miR-329 directly downregulates CD146 levels. This regulation of CD146 was shown to be through direct binding of miR-329 to two specific binding sites within the 3′ UTR of the CD146 mRNA. Overexpression of miR-329 inhibited CD146 mRNA and protein levels, while inhibition of endogenous miR-329 by an anti-miR-329 resulted in increased levels of CD146. Our in vivo experiments also suggest that miR-329 can downregulate CD146 expression in retinal blood vessels of mice. Second, we have demonstrated that miR-329 expression can be regulated by proangiogenic factors. The reciprocal expression of miR-329 and CD146 was observed not only in activated endothelial cells but also in neovascularized retinas. We found that in response to VEGF and TNF-α, miR-329 was downregulated by NF-κB signaling, a process that resulted in the elevation of CD146. Third, when targeting CD146, miR-329 diminished VEGF-induced SKF/p38 MAPK/NF-κB activation as well as the expression of multiple angiogenic genes, including the VEGF, ICAM-1, IL-8, and MMP-9 genes. Importantly, miR-329 inhibited endothelial cell migration and tube formation in vitro and repressed the blood formation in Matrigel plugs in vivo. Conversely, inhibition of miR-329 resulted in the promotion of angiogenesis. Finally, miR-329 significantly protected against pathological angiogenesis in a mouse model of oxygen-induced retinopathy. Based on all these findings, we propose a role for miR-329 as an angiogenesis suppressor by way of direct targeting of CD146 (Fig. 7).
Fig 7.
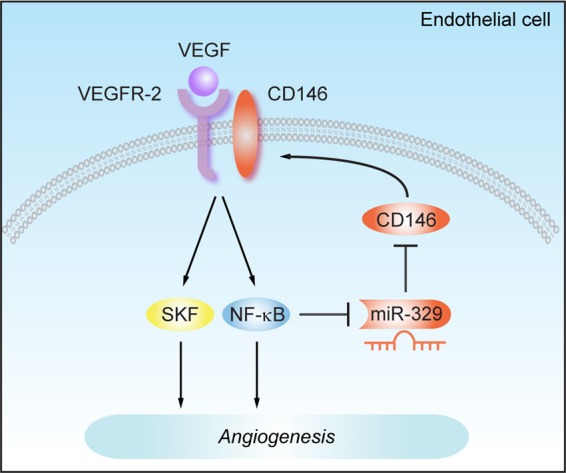
A schematic model for the function of miR-329 in angiogenesis. Binding of VEGF to VEGFR-2 activates NF-κB signaling, resulting in the downregulation of miR-329 in endothelial cells. The decreased miR-329 expression causes an increase in levels of CD146, a proangiogenic effector and a coreceptor for VEGFR-2. Thus, miR-329 functions as an angiogenesis suppressor by targeting CD146.
CD146 is known to be aberrantly upregulated in vascular disorders. Previously, it has been shown that CD146 overexpression is regulated at the level of transcription in certain tumor cells. For instance, dysregulated CD146 gene transcription appears to occur via epigenetic modifications and binding of transcription factors to its promoter sequence (39, 40). Here, we reveal for the first time, the importance of sequences other than the promoter, namely, the 3′ UTR of the CD146 mRNA for its regulation in the endothelial system. We propose that this regulatory point could provide a novel potential therapeutic route for targeting pathogenic angiogenesis. In particular, our results indicate that miR-329 regulates CD146 expression through direct binding to its 3′ UTR. Moreover, the levels of CD146 appear to be under the combinatorial control of the VEGF and TNF-α signaling pathways, which through activation of NF-κB signaling suppress miR-329. This, in turn, results in the accumulation of CD146, creating a positive-feedback loop crucial for promoting a proangiogenic microenvironment. This may help to explain the observed correlation between an increase in CD146 expression and pathological angiogenesis. Our findings indicated that miR-329 plays an important role in the VEGF- and TNF-α-induced CD146 expression. However, alternative pathways may also be involved in the regulation of CD146. In support of this assumption, our screening data (see Table S2 in the supplemental material) suggest that additional miRNA candidates, such as miR-221, miR-632, and miR-143, may play a part in inhibiting endothelial CD146 expression. Understanding the precise function of these miRNAs warrants future investigation.
Although increasing evidence indicates the important role miRNAs play in angiogenesis, the exact targets and function of only a few miRNAs have been identified in the endothelium so far (21). In addition, most of these angiomiRs are reported as positive effectors for angiogenesis, such as miR-27a/b, miR-296, and miR-130a (21, 23, 41, 42). In this study, we identified miR-329 as a new member of the angiomiR family that negatively regulates angiogenesis. Gain- and loss-of-function experiments showed that miR-329 regulated many aspects of endothelial cell activities, including capillary network formation, cell migration, and organization of the cytoskeleton. In concordance with these results, overexpression of CD146 enhanced angiogenesis, rescuing the miR-329 gain-of-function phenotype, whereas knockdown of CD146 impaired angiogenesis and reversed the miR-329 loss-of-function phenotype in endothelial cells. These results indicated that CD146 plays a major role for the antiangiogenic function of miR-329.
We hypothesize that miR-329 inhibits angiogenesis by suppression of multiple functions of CD146. CD146 is a coreceptor for VEGFR-2 (15); thus, miR-329-mediated knockdown of CD146 could impair VEGF-induced signaling and angiogenesis. In particular, we found that miR-329 and CD146 are involved in the regulation of VEGF-mediated SKF activation, a point not addressed previously. In addition, although miR-329 had no effect on TNF-α-activated NF-κB signaling, miR-329 could significantly suppress TNF-α-induced angiogenesis, which was rescued by CD146 restoration. One of the possible explanations for this phenomenon is that CD146 is an endothelial adhesion molecule, which is located in the endothelial cell junctions, where it mediates endothelial cell adhesion (13, 43). Thus, inhibition of angiogenesis by miR-329 may result from the direct inhibition of the proangiogenic function of CD146. On the other hand, while our results indicate that the antiangiogenic function of miR-329 was mainly through targeting of CD146, it is likely that the actions of miR-329 reflect the combined regulation of multiple targets that modulate angiogenesis. In our study, we noted that miR-329 indirectly impaired the expression of multiple angiogenic genes, suggesting a global effect of miR-329 on growth factor signaling. Further systematic investigation should shed light on the exact nature of additional targets regulated by miR-329.
Our study also revealed the potential role of miR-329 in pathological angiogenesis. Oxygen-induced retinopathy has long served as a useful model for studying pathological angiogenesis (36). Although recent studies reported the expression profiles of many miRNAs in the normal retinas of mice, including miR-329, its targets and function were unknown (44, 45). Moreover, only a few miRNAs have been identified to regulate OIR (46, 47). Here, we provide the first evidence that endogenous miR-329 is decreased in OIR retinas compared with those of healthy eyes. Intravitreal injection of miR-329 resulted in downregulation of CD146 expression and significantly dampened aberrant neovascularization, thus alleviating the severity of OIR disease. These findings suggest that treatment with miR-329 could provide a potential therapeutic approach to curing this disease.
In addition, OIR replicates several key features not only of tumor angiogenesis but also of inflammation and diabetic retinopathy. Because angiogenic signaling is a common feature in neovascularization, we propose that miR-329 could play a common regulatory role in other pathological processes. We are currently investigating the role of miR-329 in tumor-related angiogenesis, and initial data seem to suggest that miR-329 can also regulate CD146 expression in endothelial cells cultured with tumor cell-conditioned medium. Thus, our findings will have implications that extend beyond our OIR model and should be of great relevance for other angiogenesis disorders as well.
In comparison to siRNA therapy, endogenous microRNA therapeutics provide a natural means of manipulating disease genes, potentially avoiding issues related to immune responses or drug resistance (48, 49). The ability of miRNAs to regulate a broad range of genes may make them a suitable pleiotropic tool for disease treatment. It has been shown that miR-23 and miR-27 enhance angiogenesis by targeting antiangiogenic genes, such as those coding for SPROUTY2 and SEMA6A, while inhibition of these miRNAs exhibits a promising therapeutic effect for choroidal neovascularization (50). It is therefore of particular interest to determine if miR-329 can regulate additional gene targets in angiogenesis.
Taken together, our findings provide novel insights into the regulatory mechanisms of CD146 at the posttranscriptional level in angiogenesis. Importantly, treatment with miR-329 may provide a potential therapeutic avenue for the treatment of vascular system-related diseases.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (no. 91029732, 81272409, and 31270908), the National Basic Research Program of China (973 Program; 2009CB521704), the Strategic Priority Research Program of the Chinese Academy of Sciences, Stem Cell and Regenerative Medicine Research (XDA01040409), and the National Important Science and Technology Specific Projects (2012ZX10002009-016).
We thank Yongzhang Luo for providing the HMECs, Wenzhen Yu for instruction on the intravitreal injection of mice, and Zhenwei Yang for analyzing the data from real-time PCR. We thank Irene Gramaglia, Pengcheng Bu, and Tao Tu for critical comments on the manuscript.
Footnotes
Published ahead of print 22 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00343-13.
REFERENCES
- 1.Carmeliet P, Jain RK. 2011. Molecular mechanisms and clinical applications of angiogenesis. Nature 473:298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chung AS, Ferrara N. 2011. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 27:563–584 [DOI] [PubMed] [Google Scholar]
- 3.Potente M, Gerhardt H, Carmeliet P. 2011. Basic and therapeutic aspects of angiogenesis. Cell 146:873–887 [DOI] [PubMed] [Google Scholar]
- 4.Sapieha P, Joyal JS, Rivera JC, Kermorvant-Duchemin E, Sennlaub F, Hardy P, Lachapelle P, Chemtob S. 2010. Retinopathy of prematurity: understanding ischemic retinal vasculopathies at an extreme of life. J. Clin. Invest. 120:3022–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griffioen AW, Molema G. 2000. Angiogenesis: potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol. Rev. 52:237–268 [PubMed] [Google Scholar]
- 6.Grothey A, Galanis E. 2009. Targeting angiogenesis: progress with anti-VEGF treatment with large molecules. Nat. Rev. Clin. Oncol. 6:507–518 [DOI] [PubMed] [Google Scholar]
- 7.Folkman J. 1971. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285:1182–1186 [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Yan X. 2013. CD146, a multi-functional molecule beyond adhesion. Cancer Lett. 330:150–162 [DOI] [PubMed] [Google Scholar]
- 9.So JH, Hong SK, Kim HT, Jung SH, Lee MS, Choi JH, Bae YK, Kudoh T, Kim JH, Kim CH. 2010. Gicerin/Cd146 is involved in zebrafish cardiovascular development and tumor angiogenesis. Genes Cells 15:1099–1110 [DOI] [PubMed] [Google Scholar]
- 10.Daniel L, Bardin N, Moal V, Dignat-George F, Berland Y, Figarella-Branger D. 2005. Tubular CD146 expression in nephropathies is related to chronic renal failure. Nephron Exp. Nephrol. 99:e105–e111 [DOI] [PubMed] [Google Scholar]
- 11.Tsiolakidou G, Koutroubakis IE, Tzardi M, Kouroumalis EA. 2008. Increased expression of VEGF and CD146 in patients with inflammatory bowel disease. Dig. Liver Dis. 40:673–679 [DOI] [PubMed] [Google Scholar]
- 12.Neidhart M, Wehrli R, Bruhlmann P, Michel BA, Gay RE, Gay S. 1999. Synovial fluid CD146 (MUC18), a marker for synovial membrane angiogenesis in rheumatoid arthritis. Arthritis Rheum. 42:622–630 [DOI] [PubMed] [Google Scholar]
- 13.Kang Y, Wang F, Feng J, Yang D, Yang X, Yan X. 2006. Knockdown of CD146 reduces the migration and proliferation of human endothelial cells. Cell Res. 16:313–318 [DOI] [PubMed] [Google Scholar]
- 14.Zheng C, Qiu Y, Zeng Q, Zhang Y, Lu D, Yang D, Feng J, Yan X. 2009. Endothelial CD146 is required for in vitro tumor-induced angiogenesis: the role of a disulfide bond in signaling and dimerization. Int. J. Biochem. Cell Biol. 41:2163–2172 [DOI] [PubMed] [Google Scholar]
- 15.Jiang T, Zhuang J, Duan H, Luo Y, Zeng Q, Fan K, Yan H, Lu D, Ye Z, Hao J, Feng J, Yang D, Yan X. 2012. CD146 is a coreceptor for VEGFR-2 in tumor angiogenesis. Blood 120:2330–2339 [DOI] [PubMed] [Google Scholar]
- 16.Zhuang J, Jiang T, Lu D, Luo Y, Zheng C, Feng J, Yang D, Chen C, Yan X. 2010. NADPH oxidase 4 mediates reactive oxygen species induction of CD146 dimerization in VEGF signal transduction. Free Radic. Biol. Med. 49:227–236 [DOI] [PubMed] [Google Scholar]
- 17.Yan X, Lin Y, Yang D, Shen Y, Yuan M, Zhang Z, Li P, Xia H, Li L, Luo D, Liu Q, Mann K, Bader BL. 2003. A novel anti-CD146 monoclonal antibody, AA98, inhibits angiogenesis and tumor growth. Blood 102:184–191 [DOI] [PubMed] [Google Scholar]
- 18.Carthew RW, Sontheimer EJ. 2009. Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fish JE, Srivastava D. 2009. MicroRNAs: opening a new vein in angiogenesis research. Sci. Signal. 2:pe1. 10.1126/scisignal.252pe1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urbich C, Kuehbacher A, Dimmeler S. 2008. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 79:581–588 [DOI] [PubMed] [Google Scholar]
- 21.Wang S, Olson EN. 2009. AngiomiRs—key regulators of angiogenesis. Curr. Opin. Genet. Dev. 19:205–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. 2008. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 15:261–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wurdinger T, Tannous BA, Saydam O, Skog J, Grau S, Soutschek J, Weissleder R, Breakefield XO, Krichevsky AM. 2008. miR-296 regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell 14:382–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. 2005. Combinatorial microRNA target predictions. Nat. Genet. 37:495–500 [DOI] [PubMed] [Google Scholar]
- 25.Sethupathy P, Megraw M, Hatzigeorgiou AG. 2006. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat. Methods 3:881–886 [DOI] [PubMed] [Google Scholar]
- 26.Rajewsky N. 2006. microRNA target predictions in animals. Nat. Genet. 38(Suppl):S8–S13 [DOI] [PubMed] [Google Scholar]
- 27.Luo Y, Zheng C, Zhang J, Lu D, Zhuang J, Xing S, Feng J, Yang D, Yan X. 2012. Recognition of CD146 as an ERM-binding protein offers novel mechanisms for melanoma cell migration. Oncogene 31:306–321 [DOI] [PubMed] [Google Scholar]
- 28.Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. 2006. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 38:228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Zheng C, Zhang J, Yang D, Feng J, Lu D, Yan X. 2008. Generation and characterization of a panel of monoclonal antibodies against distinct epitopes of human CD146. Hybridoma (Larchmt.) 27:345–352 [DOI] [PubMed] [Google Scholar]
- 30.Redmond EM, Cullen JP, Cahill PA, Sitzmann JV, Stefansson S, Lawrence DA, Okada SS. 2001. Endothelial cells inhibit flow-induced smooth muscle cell migration: role of plasminogen activator inhibitor-1. Circulation 103:597–603 [DOI] [PubMed] [Google Scholar]
- 31.Colorado PC, Torre A, Kamphaus G, Maeshima Y, Hopfer H, Takahashi K, Volk R, Zamborsky ED, Herman S, Sarkar PK, Ericksen MB, Dhanabal M, Simons M, Post M, Kufe DW, Weichselbaum RR, Sukhatme VP, Kalluri R. 2000. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 60:2520–2526 [PubMed] [Google Scholar]
- 32.Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, Urbich C, Zeiher AM, Dimmeler S. 2009. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 324:1710–1713 [DOI] [PubMed] [Google Scholar]
- 33.Smith LE, Wesolowski E, McLellan A, Kostyk SK, D'Amato R, Sullivan R, D'Amore PA. 1994. Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 35:101–111 [PubMed] [Google Scholar]
- 34.Connor KM, Krah NM, Dennison RJ, Aderman CM, Chen J, Guerin KI, Sapieha P, Stahl A, Willett KL, Smith LE. 2009. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat. Protoc. 4:1565–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bowie A, Moynagh PN, O'Neill LA. 1996. Mechanism of NF kappa B activation by interleukin-1 and tumour necrosis factor in endothelial cells. Biochem. Soc. Trans. 24:2S http://www.biochemsoctrans.org [DOI] [PubMed] [Google Scholar]
- 36.Stahl A, Connor KM, Sapieha P, Chen J, Dennison RJ, Krah NM, Seaward MR, Willett KL, Aderman CM, Guerin KI, Hua J, Lofqvist C, Hellstrom A, Smith LEH. 2010. The mouse retina as an angiogenesis model. Invest. Ophthalmol. Vis. Sci. 51:2813–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gardiner TA, Gibson DS, de Gooyer TE, de la Cruz VF, McDonald DM, Stitt AW. 2005. Inhibition of tumor necrosis factor-alpha improves physiological angiogenesis and reduces pathological neovascularization in ischemic retinopathy. Am. J. Pathol. 166:637–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ouhtit A, Gaur RL, Abd Elmageed ZY, Fernando A, Thouta R, Trappey AK, Abdraboh ME, El-Sayyad HI, Rao P, Raj MG. 2009. Towards understanding the mode of action of the multifaceted cell adhesion receptor CD146. Biochim. Biophys. Acta 1795:130–136 [DOI] [PubMed] [Google Scholar]
- 39.Liu JW, Nagpal JK, Jeronimo C, Lee JE, Henrique R, Kim MS, Ostrow KL, Yamashita K, van Criekinge V, Wu G, Moon CS, Trink B, Sidransky D. 2008. Hypermethylation of MCAM gene is associated with advanced tumor stage in prostate cancer. Prostate 68:418–426 [DOI] [PubMed] [Google Scholar]
- 40.Jean D, Bar-Eli M. 2000. Regulation of tumor growth and metastasis of human melanoma by the CREB transcription factor family. Mol. Cell. Biochem. 212:19–28 [PubMed] [Google Scholar]
- 41.Chen Y, Gorski DH. 2008. Regulation of angiogenesis through a microRNA (miR-130a) that down-regulates antiangiogenic homeobox genes GAX and HOXA5. Blood 111:1217–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Urbich C, Kaluza D, Fromel T, Knau A, Bennewitz K, Boon RA, Bonauer A, Doebele C, Boeckel JN, Hergenreider E, Zeiher AM, Kroll J, Fleming I, Dimmeler S. 2012. MicroRNA-27a/b controls endothelial cell repulsion and angiogenesis by targeting semaphorin 6A. Blood 119:1607–1616 [DOI] [PubMed] [Google Scholar]
- 43.Solovey AN, Gui L, Chang L, Enenstein J, Browne PV, Hebbel RP. 2001. Identification and functional assessment of endothelial P1H12. J. Lab. Clin. Med. 138:322–331 [DOI] [PubMed] [Google Scholar]
- 44.Karali M, Peluso I, Gennarino VA, Bilio M, Verde R, Lago G, Dolle P, Banfi S. 2010. miRNeye: a microRNA expression atlas of the mouse eye. BMC Genomics 11:715. 10.1186/1471-2164-11-715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hackler L, Jr, Wan J, Swaroop A, Qian J, Zack DJ. 2010. MicroRNA profile of the developing mouse retina. Invest. Ophthalmol. Vis. Sci. 51:1823–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bai Y, Bai X, Wang Z, Zhang X, Ruan C, Miao J. 2011. MicroRNA-126 inhibits ischemia-induced retinal neovascularization via regulating angiogenic growth factors. Exp. Mol. Pathol. 91:471–477 [DOI] [PubMed] [Google Scholar]
- 47.Shen J, Yang X, Xie B, Chen Y, Swaim M, Hackett SF, Campochiaro PA. 2008. MicroRNAs regulate ocular neovascularization. Mol. Ther. 16:1208–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castanotto D, Rossi JJ. 2009. The promises and pitfalls of RNA-interference-based therapeutics. Nature 457:426–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. 2003. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 21:635–637 [DOI] [PubMed] [Google Scholar]
- 50.Zhou Q, Gallagher R, Ufret-Vincenty R, Li X, Olson EN, Wang S. 2011. Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23∼27∼24 clusters. Proc. Natl. Acad. Sci. U. S. A. 108:8287–8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.