

Abstract
The autophagic degradation pathway is a powerful tool in the host cell arsenal against cytosolic pathogens. Contents trapped inside cytosolic vesicles, termed autophagosomes, are delivered to the lysosome for degradation. In spite of the degradative nature of the pathway, some pathogens are able to subvert autophagy for their benefit. In many cases, these pathogens have developed strategies to induce the autophagic signaling pathway while inhibiting the associated degradation activity. One surprising finding from recent literature is that some viruses do not impede degradation but instead promote the generation of degradative autolysosomes, which are the endpoint compartments of autophagy. Dengue virus, poliovirus, and hepatitis C virus, all positive-strand RNA viruses, utilize the maturation of autophagosomes into acidic and ultimately degradative compartments to promote their replication. While the benefits that each virus reaps from autophagosome maturation are unique, the parallels between the viruses indicate a complex relationship between cytosolic viruses and host cell degradation vesicles.
INTRODUCTION TO AUTOPHAGY
While many viruses avoid or suppress host immune responses, several subvert the host immune machinery to promote their own replication (1). The autophagic pathway is one well-characterized effector mechanism of the innate immune response, resulting in a highly regulated lysosomal degradation mechanism by which a cell degrades its own contents. The pathway has been shown to be essential for cellular clearance of several intracellular pathogens, including Mycobacterium tuberculosis, Toxoplasma gondii, and herpes simplex virus 1 (HSV-1) (2–4). Despite the role of autophagic signaling in innate immunity, several pathogens are capable of subverting autophagy for their own benefit (5, 6). In particular, the replication cycle of positive-strand RNA viruses, which are the causative agents of many diseases, including myocarditis, encephalitis, and hand, foot, and mouth disease (7–9), can be promoted by some aspect of the autophagic pathway. However, there is a difference between a pathogen benefitting from autophagic signaling or the machinery from the autophagic pathway and a pathogen benefitting from the endpoint activity of autophagy, which is the degradation of cytosolic contents. Both are of interest, but this review will focus exclusively on relatively new findings that several pathogens can actually benefit from the degradative activity of autophagy.
Autophagy is a constitutive degradation pathway with important roles in development, differentiation, and stress responses (10). By facilitating the removal of damaged organelles and cytoplasmic protein aggregates, autophagy has proven to be essential for maintaining cellular homeostasis (6). Several signaling pathways induce autophagic signaling, although the mechanism by which these pathways cooperate to promote vesicle formation remains unknown (11). Inhibition of the Akt/mTOR pathway has long been considered essential for induction of autophagic signaling, although recent reports have demonstrated the existence of mTOR-independent autophagy (12, 13). Induction of a ubiquitin-like conjugation system promotes lipidation of the cytosolic microtubule-associated light chain 3 (LC3) protein with phosphotidylethanolamine, generating a membrane-associated species known as LC3-II (14–16). LC3-II is associated with both the inner and the outer membrane of the growing autophagosome, and this association is essential for autophagosome formation (14, 17, 18). LC3-II remains the only protein known to stably associate with completed autophagosomes, and as such it is an invaluable marker for monitoring autophagy. The initial events in autophagosome biogenesis have been well-described elsewhere (18–20).
Autophagosomes are unique vesicles composed of two lipid bilayers which, during their formation, engulf cytosolic contents, including long-lived proteins, intracellular pathogens, and damaged organelles. This cargo is then transported to the lysosome for degradation (10). Double-membraned autophagosomes undergo a stepwise maturation process culminating in their fusion with lysosomes to form degradative autolysosomes (Fig. 1). Autophagosomes mature into amphisomes, a change primarily characterized by the acidification of the lumen of the vesicle and the acquisition of proteins associated with late endosomes and lysosomes (21). Mature amphisomes then fuse with lysosomes to form autolysosomes, in the process losing one lipid bilayer through an unknown mechanism (22, 23). The autolysosome is the compartment in which actual autophagy, the degradation of cytosolic contents, takes place.
Fig 1.
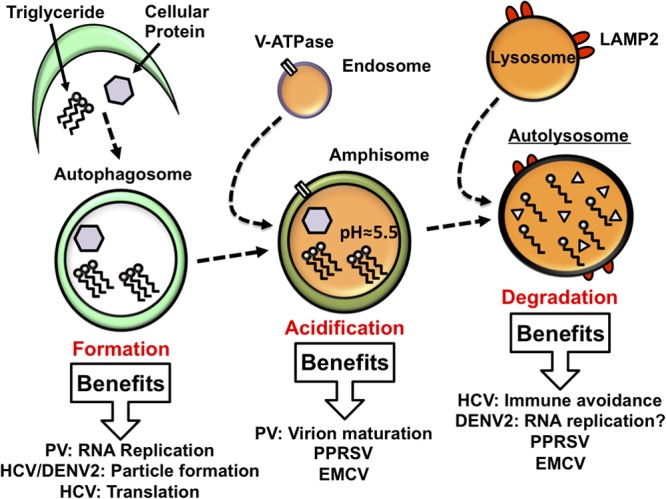
How viruses utilize autophagosome formation and maturation during infection. As the immature autophagosome forms, it captures portions of the cytoplasm. The lumen of the autophagosome acidifies, likely through fusion with late endosomes carrying vacuolar ATPases, to form the amphisome. The amphisome then fuses with the lysosome to form the autolysosome. The replication of viruses that subvert the autophagic pathway is attenuated when autophagosome formation is inhibited. Vesicle acidification is required for efficient PV virion maturation, while inhibition of degradation has no effect on the virus. Degradation of cellular triglycerides by autophagy benefits dengue virus replication. Autolysosome degradation decreases IFN activation following HCV infection. Both PPRSV and EMCV require autophagosome maturation; however, it is not clear if this is due to a requirement for vesicle acidification or autolysosome degradation. V-ATPase, vacuolar (H+)-ATPase.
The acidification of the amphisome is believed to be the result of fusion with late endosomes bearing vacuolar ATPases (21, 24). Treatment of cells with inhibitors of vesicular acidification, including bafilomycin A1, chloroquine, and ammonium chloride, prevents autolysosome formation (24–26). This indicates that acidification of either the autophagosome, the lysosome, or both is a prerequisite for fusion of the autophagosome with the lysosome. Two proteins, lysosomal-associated membrane protein 2 (LAMP-2) and Rab7, have been reported to be required for autophagosome maturation. Rab7 is a small GTP-binding protein that has functions in late endosomal transport (27, 28). Depletion of endogenous Rab7 or expression of a dominant negative form results in decreased autophagosome maturation (29, 30). LAMP-2, a lysosomal transmembrane protein, is one of the most abundant lysosomal components (31). Depletion of LAMP-2 prevents autophagosome fusion with lysosomes (32–34). These data indicate that multiple steps of the autophagosome formation and maturation pathway are regulated by the cell and may be subverted by pathogens.
VIRAL SUBVERSION OF THE AUTOPHAGIC PATHWAY
As an obligate intracellular parasite, a virus depends on its ability to evade the host cell's antiviral defenses, as well as its ability to regulate cellular processes that facilitate its own replication, for its success. Subversion of the autophagic pathway, which aids both of these goals, has been the most extensively studied in positive-strand RNA viruses (35, 36). Optimal production of positive-strand RNA viruses depends on the initiation of the autophagic pathway during infection. This is counterintuitive, because the autophagic pathway promotes degradation of cytosolic contents and positive-strand RNA viruses replicate in the cytosol.
The physical hallmark of the autophagic pathway is the formation of cytosolic double-membraned vesicles (19, 37). Positive-strand RNA viruses replicate their genomes in association with cytosolic membranes (38, 39). Therefore, by inducing autophagy, these viruses may be facilitating the creation of scaffolds for their own replication. However, these vesicles are part of a degradative pathway, and if this pathway is unaltered, the vesicles will fuse with lysosomes and their contents will be degraded. Coxsackievirus B3 (CVB3), an enterovirus in the Picornaviridae family, appears to have developed a strategy to prevent this. CVB3 relies on autophagosome formation for optimal virus replication (40, 41). However, there is evidence that during both in vitro and in vivo infections amphisome maturation and autophagic protein degradation are inhibited (40, 41). The mechanism by which CVB3 upregulates autophagosome formation while restricting autophagic degradation is unknown. Treatment of CVB3-infected cells with inhibitors of autophagosome maturation results in increased virus production, indicating that at least a portion of the virus remains sensitive to autophagic degradation (41). Recently it was also shown that rotavirus induces autophagic signals to promote virus replication but that the virus blocks protein degradation (42). As with CVB3, the mechanism by which rotaviruses specifically inhibit autophagosome degradation is not yet known. Further work to identify the specific virus or host cell proteins used by these viruses to prevent degradation will help our understanding of autophagic regulation in general.
A similar antidegradative phenomenon has been observed in bacterial infection models, with the best-characterized being Legionella pneumophila infection, which induces replication vesicles that resemble autophagosomes (43). The vacuoles become acidic; however, the bacteria secrete a factor that delays their fusion with lysosomes (44). A recent report indicates that Legionella can interfere with the formation of LC3-II, although the significance of this to autophagosome maturation is unclear (45). Inhibition of autophagosome maturation has been observed for several other nonviral pathogens (46).
In the following sections, we discuss recent advances in understanding the interaction of three positive-strand RNA viruses with the late stages of the autophagic pathway. Replication of all three viruses is reduced when autophagy is inhibited. Conversely, stimulation of autophagy increases infectious virus production (47–52). To date, this subset of viruses are the only pathogens shown to induce autophagosome formation to promote their own replication while allowing maturation of the vesicles, fusion of amphisomes with lysosomes, and subsequent cargo degradation. For reference, a brief description of assays used to monitor autophagosome maturation and autophagic degradation is provided in Table 1, and more detail is available in reference 56.
Table 1.
Assays for autophagosome maturation and autophagic degradationa
| Assay | Description | Read out |
|---|---|---|
| Protease sensitivity of LC3-II | LC3-II is degraded by lysosomal proteases following fusion of the autophagosome with the lysosome (53). Lysosomal protease inhibitors, which inhibit LC3-II degradation by the autolysosome, increase the steady-state level of LC3 II (53, 56). | Protein degradation by the autophagic pathway |
| p62 Degradation | The p62/SQSTM1 protein directly binds LC3-II on the autophagosome membrane (54). p62 is degraded within the autolysosomes (55). A decrease in the steady state level of p62 following induction of autophagy indicates successful protein degradation through the pathway (55, 56). | Autolysosome formation |
| LC3-II-lysosome colocalization | The cellular locale of autophagosome-associated GFP-tagged LC3-II can be monitored by fluorescence microscopy (67). During the initial stages of the autophagic pathway, colocalization of LC3-II with lysosomal markers is low. As autophagosomes mature and fuse with lysosomes, colocalization with lysosomal markers increases. Cellular lysosomes can be visualized by staining for protein markers such as LAMP-2, LAMP-1, and cathepsin D (31). | Autolysosome formation |
| Tandem-tagged GFP-RFP-LC3 | Tandem-tagged RFP-GFP-LC3 localizes to the autophagosome membrane following induction of autophagy (58). Only the signal generated by the GFP protein is sensitive to the acidic and/or proteolytic conditions in the lumen of mature autophagosome and lysosomes. Colocalization of GFP and RFP signals is observed on early or immature autophagosomes. As autophagosomes mature, the GFP signal is lost, leading to only RFP fluorescence. | Autophagosome maturation |
| Transmission electron microscopy | Autophagosomes can by identified by TEM as membrane-bound structures containing cytoplasmic material. Immature autophagosomes (AVi) show a double membrane visible as two membrane bilayers separated by an electron-lucent cleft. These vacuoles contain cytosol and/or organelles that appear morphologically intact (56). Mature or degradative vesicles (AVd) typically show partial degradation of the enclosed cytoplasmic material, as well as increased electron density in the lumen of the vesicle. | Autophagosome maturation |
An extensive discussion of known assays for analysis of autophagic signaling, autophagosome formation, and all other aspects of the pathway can be found in reference 53. TEM, transmission electron microscopy.
DENGUE VIRUS
Dengue virus, a member of the Flaviviridae, is the causative agent of dengue fever, which in a small subset of the population progresses to dengue hemorrhagic fever/dengue shock syndrome (57, 58). Dengue virus (DENV) is comprised of four antigenically related but distinct viruses (DENV-1 to DENV-4) with each virus comprising many distinct genotypes (59). Thus far, only DENV-2 and DENV-3 have been shown to require autophagosome formation for maximum virus replication (52, 57, 60). During infection with either virus, viral proteins involved in translation and replication locate to autophagosomes (52, 60).
While DENV-2 and DENV-3 both subvert autophagosome formation, there are some major differences in the way these viruses interact with the late stages of the autophagic pathway. DENV-3 nonstructural proteins primarily colocalize with autolysosomes during infection, whereas DENV-2 proteins are primarily located on immature autophagosomes (52). During DENV-2 virus infection, autophagy increases the cell's degradative capacity, specifically in regard to cellular lipid droplets (51). The degradation of lipid droplets by autophagy is referred to as lipophagy (61). Increased lipophagy during DENV-2 infection results in both a decrease in triglycerides and an increase in β-oxidation. Inhibition of autophagosome formation reduces infectious virus production; however, when cells are supplied with the products of lipophagy, virus production returns to levels observed in cells capable of autophagy. Heaton and Randall speculate that the release of free fatty acids during lipophagy increases ATP generation, which is critical for viral replication (51). A role for lipophagy during DENV-3 infection has not yet been reported.
HEPATITIS C VIRUS
Hepatitis C virus (HCV), another flavivirus, is a major cause of chronic liver disease (62). The HCV RNA-dependent RNA polymerase interacts with the cellular autophagy protein Atg5, and the two proteins colocalize during early time points of infection (63). Additionally, HCV RNA and proteins cofractionate with LC3-II on a discontinuous sucrose gradient (64). Expression of HCV proteins NS5A and NS4B in isolation is sufficient to induce autophagic signaling (65, 66). While there is agreement that HCV induces autophagic signaling, the specific role of autophagy during HCV infection remains controversial. Autophagy was shown to be essential for translation of the viral genome but dispensable once the infection had begun (49). These data contrast with a report that knockdown of autophagy genes had no effect on virus translation and RNA replication but instead was essential for HCV particle formation (47).
HCV-infected cells expressing tandem-tagged green fluorescent protein-red fluorescent protein-LC3 (GFP-RFP-LC3) (Table 1) show predominantly red fluorescence, indicating maturation of autophagosomes into acidic amphisomes (67, 68). The RFP-positive puncta colocalize with LAMP-1, demonstrating fusion of the autophagosome with either late endosomes or lysosomes (68). However, there is at least one report of an incomplete autophagic response to HCV infection. No change in either p 62 levels or long-lived protein degradation was observed following transfection with the HCV replicon (69). This discrepancy may result from a difference between transfection of the viral genome and infection with live virus. However, elevated autolysosome formation has been observed following transfection of the HCV replicon, making this an unlikely explanation (70). An alternative hypothesis is that typical autophagosome cargo, such as p62, is not incorporated into the specialized autophagosomes generated during HCV infection. If this is the case, assays measuring protein degradation levels would not be a reliable measure of autophagosome maturation.
HCV genome replication is attenuated following depletion of LAMP-2 or Rab7, both of which are essential for autolysosome formation (68). Treatment with either bafilomycin A1 or chloroquine, both of which inhibit autophagosome maturation, results in reduced viral RNA and protein expression (68, 71). Investigation of the retinoic acid-inducible gene I (RIG-I)-like receptor (RLR) signaling cascade following HCV infection has revealed a role for autophagic degradation during infection in suppressing the innate immune response to infection (68). Activation of the beta interferon (IFN-β) promoter by ectopically expressed RIG-I was measured in infected cells in both the presence and the absence of autophagic degradation. In control cells, HCV was able to inhibit RIG-I-mediated IFN-β activation. In the absence of autophagic degradation, infected cells showed a significant increase in IFN activation. The varied reports on the effects of autophagy in HCV production lead us to conclude that the process may play multiple roles in promoting viral replication.
POLIOVIRUS
Poliovirus (PV), the causative agent of poliomyelitis, is a member of the Picornaviridae family. It is one of the most well-characterized members of this family in terms of its molecular and cellular biology, biochemistry, structure, life cycle, and pathogenesis and therefore represents an important model virus (72). By 5 h postinfection, infected cells exhibit extensive accumulation of autophagic vacuoles throughout the cytoplasm (50, 73). Viral RNA replication proteins localize to the autophagosome membrane during infection (50, 74). LC3 and LAMP-1 also colocalize during infection, indicating autophagosome fusion with late endosomes and/or lysosomes (50). Staining infected cells with monodansylcadaverine (MDC), a lysosomotropic agent that is concentrated in acidic compartments by an ion-trapping mechanism, reveals that the lumen of autophagosomes acidifies relative to the cytosol (50, 75). Infection promotes autophagic protein degradation; however, unlike for dengue virus and HCV, degradation is not necessary for virus replication, since lysosomal protease inhibitors have no impact on the intracellular virus titer (76).
Our group recently showed that poliovirus utilizes both autophagosome formation and maturation of the autophagic vacuole to promote two separate and distinct steps in the virus life cycle. Inhibitors of autophagosome formation limit viral RNA replication (76). However, if autophagosome formation proceeds normally but vesicle acidification is inhibited, virus production remains attenuated. In the absence of acidic vesicles, viral entry, translation, RNA replication, and genome encapsidation all occur normally. Acidic vesicles are, however, required for the last step in the production of an infectious virion, marked by the internal cleavage of capsid protein VP0, which results in the maturation of a noninfectious provirion to an infectious virion (76). This cleavage step is attenuated in the absence of vesicle acidification, resulting in a decrease in the number of infectious virions produced. How an acidic vesicle can promote the maturation of a presumably cytosolic, nonenveloped virus is a current question of research focus.
POSSIBLE CONNECTIONS AMONG VIRUSES THAT BENEFIT FROM AUTOPHAGOSOME MATURATION
We have presented here three examples of how viruses benefit from autophagosome maturation. However, there are indications that some of these benefits may be conserved among the viruses. Of the viruses discussed, only HCV has been definitively shown to use autophagic degradation to downregulate immune signaling during infection (68). However, preliminary results indicate that the immune response to dengue virus infection may also be attenuated by autophagic degradation (68). Recently, Japanese encephalitis virus (JEV) has been shown to subvert autophagy as a viral immune evasion strategy. The mechanism may be similar to that used by HCV, since type I IFN activation is increased when JEV infects cells deficient in autophagy (77). Infection with JEV increases autolysosome formation in vivo, and this formation is essential for maximum virus production (77). Is it not yet known if autophagic degradation is responsible for restriction of the immune response during JEV infection. Interestingly, autophagy is essential for JEV production even in an IFN-defective background, indicating that the virus may have multiple uses for the autophagic pathway during infection.
A recent publication investigated the role of autophagy in lipid metabolism during HCV infection. As with dengue virus, infection with HCV results in the appearance of autophagosomes filled with lipid cargo (78). Inhibition of autophagosome maturation through bafilomycin A1 treatment results in an accumulation of cholesterol in both HCV replicon cells and cells infected with HCV strain JFH1 (78). The purpose of increased autophagic breakdown of cholesterol during HCV infection remains elusive. One hypothesis is that the autophagic flux of cholesterol is needed for lipid droplet biogenesis during HCV infection. This would be very intriguing, as the lipid droplet area is decreased by autophagic degradation during dengue virus infection (78, 79). The differences in the proposed roles of autophagic degradation of lipids during infection may be a product of the different requirements each virus has for lipid droplets during infection. Unlike for dengue virus, lipid droplets are required for HCV virion assembly (79); therefore, increased surface area of these lipid droplets may aid HCV during replication. Conversely, dengue virus may promote destruction of these lipid pools to provide energy for virus replication occurring at an alternate site in the cell.
The data gathered thus far regarding the role of autophagy during poliovirus and dengue virus infections are almost exclusively from in vitro systems. Therefore, the effects of autophagic degradation on the host immune response to infection have not been assessed. If it is found that immune signaling during infection is attenuated through autophagic degradation, then autophagic degradation could play multiple proviral roles during infection. Recently, impairment of autophagosome formation has been shown to hamper formation of infectious dengue virus particles while having minimal effects on viral RNA replication (80). This suggests that both poliovirus and dengue virus may be utilizing the environment within a mature autophagosome to promote the final steps in production of viral progeny. It is not yet know if inhibitors of autophagosome maturation have an effect on dengue virus particle formation.
CONCLUSION
It is now appreciated that some viruses, such as poliovirus, dengue virus, and HCV, rely on the degradative activity of the autophagic pathway for efficient replication. While this review focused on the three viruses for which the role of autophagic maturation during infection has been elucidated, the story is far from complete. For example, both encephalomyocarditis virus (EMCV) and porcine reproductive and respiratory syndrome virus (PRRSV) subvert the autophagic pathway for optimal virus production while promoting autophagic protein degradation (81, 82). Replication of both PRRSV and EMCV has been shown to be sensitive to treatments known to restrict autophagosome maturation (83, 84). There is currently no model for the role that maturation of the autophagosome is playing during infection with either of these viruses. It will be interesting to learn if either virus shares a mechanism with one of the viruses presented in this review or if novel roles for autophagosome maturation are discovered. There is also recent evidence that treatment with the cathepsin inhibitor pepstatin A results in a decrease in influenza A virus production, which the authors of the study have linked to pepstatin A altering the regulation of autophagy (85). Together, these data indicate that multiple viruses may utilize autophagic degradation.
The question remains: how do these viruses thrive in a highly degradative environment? One possibility is that they have evolved to be resistant to degradation within the autolysosome. Alternatively, they may have developed a mechanism to avoid being trapped within the degradative vesicles. Finally, viral replication may occur outside the degradative autophagosome and thus be unaffected by the degradative environment within the vesicle.
It is often in the best interest of a virus to maintain the host cell's integrity until progeny virus production is complete. For example, many viruses have evolved mechanisms to prevent cellular apoptosis, a pathway that is linked to autophagy (86, 87). The way in which a virus interacts with the autophagic pathway may have important implications for cell viability throughout the infection. A number of studies have indicated that autophagy is induced by endoplasmic reticulum (ER) stress (88, 89). In both yeast and mammalian cells, autophagy has been shown to have prosurvival effects when the ER is overloaded with misfolded proteins (90–92). Inhibition of the autophagic pathway increases cell death following ER stress. This effect is due to the degradation of protein aggregates and misfolded proteins (90). Both HCV and EMCV have been shown to increase ER stress (68, 69, 93–95). By allowing autophagic degradation to proceed unperturbed, these viruses may be minimizing the risk of cell death prior to the completion of the replicative cycle.
Finally, a great deal has been learned about the functions of the host cell by studying the ways in which viruses regulate cellular pathways. A clearer picture of the mechanism by which viruses inhibit or promote autophagic degradation will not only improve our understanding of virus replication but also shed light on the ways in which the late stages of autophagy are regulated.
Footnotes
Published ahead of print 12 June 2013
REFERENCES
- 1. Randow F, Munz C. 2012. Autophagy in the regulation of pathogen replication and adaptive immunity. Trends Immunol. 33:475–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35 [DOI] [PubMed] [Google Scholar]
- 3. Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. 2006. CD40 induces macrophage anti–Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J. Clin. Invest. 116:2366–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Songane M, Kleinnijenhuis J, Netea MG, van Crevel R. 2012. The role of autophagy in host defence against Mycobacterium tuberculosis infection. Tuberculosis (Edinb.) 92:388–396 [DOI] [PubMed] [Google Scholar]
- 5. Kirkegaard K, Taylor MP, Jackson WT. 2004. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2:301–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kudchodkar SB, Levine B. 2009. Viruses and autophagy. Rev. Med. Virol. 19:359–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gaaloul I, Riabi S, Harrath R, Evans M, Salem NH, Mlayeh S, Huber S, Aouni M. 2012. Sudden unexpected death related to enterovirus myocarditis: histopathology, immunohistochemistry and molecular pathology diagnosis at post-mortem. BMC Infect. Dis. 12:212. 10.1186/1471-2334-12-212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li J, Lin C, Qu M, Li X, Gao Z, Zhang X, Liu Y, Huang Y, Wang X, Jia L, Li X, Liu G, Yan H, Chen L, Wang Q. 2013. Excretion of enterovirus 71 in persons infected with hand, foot and mouth disease. Virol. J. 10:31. 10.1186/1743-422X-10-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katoh H, Mori Y, Kambara H, Abe T, Fukuhara T, Morita E, Moriishi K, Kamitani W, Matsuura Y. 2011. Heterogeneous nuclear ribonucleoprotein A2 participates in the replication of Japanese encephalitis virus through an interaction with viral proteins and RNA. J. Virol. 85:10976–10988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Levine B, Klionsky DJ. 2004. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6:463–477 [DOI] [PubMed] [Google Scholar]
- 11. Xie Z, Klionsky DJ. 2007. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9:1102–1109 [DOI] [PubMed] [Google Scholar]
- 12. Pyo JO, Nah J, Jung YK. 2012. Molecules and their functions in autophagy. Exp. Mol. Med. 44:73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tovilovic G, Ristic B, Siljic M, Nikolic V, Kravic-Stevovic T, Dulovic M, Milenkovic M, Knezevic A, Bosnjak M, Bumbasirevic V, Stanojevic M, Trajkovic V. 2013. mTOR-independent autophagy counteracts apoptosis in herpes simplex virus type 1-infected U251 glioma cells. Microbes Infect. [Epub ahead of print.] 10.1016/j.micinf.2013.04.012 [DOI] [PubMed] [Google Scholar]
- 14. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xie Z, Nair U, Klionsky DJ. 2008. Dissecting autophagosome formation: the missing pieces. Autophagy 4:920–922 [DOI] [PubMed] [Google Scholar]
- 16. Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. 2001. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152:657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xie Z, Nair U, Klionsky DJ. 2008. Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 19:3290–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tanida I. 2011. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 14:2201–2214 [DOI] [PubMed] [Google Scholar]
- 19. Rubinsztein DC, Shpilka T, Elazar Z. 2012. Mechanisms of autophagosome biogenesis. Curr. Biol. 22:R29–R34 [DOI] [PubMed] [Google Scholar]
- 20. Weidberg H, Shvets E, Elazar Z. 2011. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 80:125–156 [DOI] [PubMed] [Google Scholar]
- 21. Eskelinen EL. 2005. Maturation of autophagic vacuoles in mammalian cells. Autophagy 1:1–10 [DOI] [PubMed] [Google Scholar]
- 22. Wang CW, Klionsky DJ. 2003. The molecular mechanism of autophagy. Mol. Med. 9:65–76 [PMC free article] [PubMed] [Google Scholar]
- 23. Dunn WA., Jr 1990. Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J. Cell Biol. 110:1935–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mousavi SA, Kjeken R, Berg TO, Seglen PO, Berg T, Brech A. 2001. Effects of inhibitors of the vacuolar proton pump on hepatic heterophagy and autophagy. Biochim. Biophys. Acta 1510:243–257 [DOI] [PubMed] [Google Scholar]
- 25. Nakamura N, Matsuura A, Wada Y, Ohsumi Y. 1997. Acidification of vacuoles is required for autophagic degradation in the yeast, Saccharomyces cerevisiae. J. Biochem. 121:338–344 [DOI] [PubMed] [Google Scholar]
- 26. Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. 2006. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 281:36303–36316 [DOI] [PubMed] [Google Scholar]
- 27. Press B, Feng Y, Hoflack B, Wandinger-Ness A. 1998. Mutant Rab7 causes the accumulation of cathepsin D and cation-independent mannose 6-phosphate receptor in an early endocytic compartment. J. Cell Biol. 140:1075–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vitelli R, Santillo M, Lattero D, Chiariello M, Bifulco M, Bruni CB, Bucci C. 1997. Role of the small GTPase Rab7 in the late endocytic pathway. J. Biol. Chem. 272:4391–4397 [DOI] [PubMed] [Google Scholar]
- 29. Gutierrez MG, Munafó DB, Berón W, Colombo MI. 2004. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 117:2687–2697 [DOI] [PubMed] [Google Scholar]
- 30. Jäger S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. 2004. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 117:4837–4848 [DOI] [PubMed] [Google Scholar]
- 31. Eskelinen EL, Tanaka Y, Saftig P. 2003. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 13:137–145 [DOI] [PubMed] [Google Scholar]
- 32. Saftig P, Beertsen W, Eskelinen EL. 2008. LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy 4:510–512 [DOI] [PubMed] [Google Scholar]
- 33. Fortunato F, Burgers H, Bergmann F, Rieger P, Büchler MW, Kroemer G, Werner J. 2009. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology 137:350–360 [DOI] [PubMed] [Google Scholar]
- 34. Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. 2000. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406:902–906 [DOI] [PubMed] [Google Scholar]
- 35. Shi J, Luo H. 2012. Interplay between the cellular autophagy machinery and positive-stranded RNA viruses. Acta Biochim. Biophys. Sin. (Shanghai) 44:375–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taylor MP, Kirkegaard K. 2008. Potential subversion of autophagosomal pathway by picornaviruses. Autophagy 4:286–289 [DOI] [PubMed] [Google Scholar]
- 37. Klionsky DJ. 2005. The molecular machinery of autophagy: unanswered questions. J. Cell Sci. 118:7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Whitton JL, Cornell CT, Feuer R. 2005. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 3:765–776 [DOI] [PubMed] [Google Scholar]
- 39. Mackenzie J. 2005. Wrapping things up about virus RNA replication. Traffic 6:967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kemball CC, Alirezaei M, Flynn CT, Wood MR, Harkins S, Kiosses WB, Whitton JL. 2010. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 84:12110–12124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. 2008. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82:9143–9153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Crawford SE, Hyser JM, Utama B, Estes MK. 2012. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-β signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. U. S. A. 109:E3405–E3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Joshi AD, Swanson MS. 2011. Secrets of a successful pathogen: legionella resistance to progression along the autophagic pathway. Front. Microbiol. 2:138. 10.3389/fmicb.2011.00138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swanson MS, Fernandez-Moreira E. 2002. A microbial strategy to multiply in macrophages: the pregnant pause. Traffic 3:170–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Choy A, Dancourt J, Mugo B, O'Connor TJ, Isberg RR, Melia TJ, Roy CR. 2012. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338:1072–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deretic V, Levine B. 2009. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5:527–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K. 2009. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 5:937–945 [DOI] [PubMed] [Google Scholar]
- 48. Dreux M, Gastaminza P, Wieland SF, Chisari FV. 2009. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 106:14046–14051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dreux M, Chisari FV. 2009. Autophagy proteins promote hepatitis C virus replication. Autophagy 5:1224–1225 [DOI] [PubMed] [Google Scholar]
- 50. Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156. 10.1371/journal.pbio.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heaton NS, Randall G. 2010. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 8:422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khakpoor A, Panyasrivanit M, Wikan N, Smith DR. 2009. A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. J. Gen. Virol. 90:1093–1103 [DOI] [PubMed] [Google Scholar]
- 53. Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. 2005. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 1:84–91 [DOI] [PubMed] [Google Scholar]
- 54. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282:24131–24145 [DOI] [PubMed] [Google Scholar]
- 55. Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171:603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, Balduini W, Ballabio A, Bamber BA, Bampton ET, Banhegyi G, Bartholomew CR, Bassham DC, et al. 2012. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee YR, Lei HY, Liu MT, Wang JR, Chen SH, Jiang-Shieh YF, Lin YS, Yeh TM, Liu CC, Liu HS. 2008. Autophagic machinery activated by dengue virus enhances virus replication. Virology 374:240–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Halstead SB. 1988. Pathogenesis of dengue: challenges to molecular biology. Science 239:476–481 [DOI] [PubMed] [Google Scholar]
- 59. Holmes EC, Twiddy SS. 2003. The origin, emergence and evolutionary genetics of dengue virus. Infect. Genet. Evol. 3:19–28 [DOI] [PubMed] [Google Scholar]
- 60. Panyasrivanit M, Khakpoor A, Wikan N, Smith DR. 2009. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J. Gen. Virol. 90:448–456 [DOI] [PubMed] [Google Scholar]
- 61. Singh R, Cuervo AM. 2012. Lipophagy: connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012:282041. 10.1155/2012/282041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hoofnagle JH. 2002. Course and outcome of hepatitis C. Hepatology 36:S21–S29 [DOI] [PubMed] [Google Scholar]
- 63. Guévin C, Manna D, Belanger C, Konan KV, Mak P, Labonte P. 2010. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 405:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ferraris P, Blanchard E, Roingeard P. 2010. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 91:2230–2237 [DOI] [PubMed] [Google Scholar]
- 65. Su WC, Chao TC, Huang YL, Weng SC, Jeng KS, Lai MM. 2011. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 85:10561–10571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. 2011. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 53:406–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kimura S, Noda T, Yoshimori T. 2007. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3:452–460 [DOI] [PubMed] [Google Scholar]
- 68. Ke PY, Chen SS. 2011. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 121:37–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. 2008. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48:1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Taguwa S, Kambara H, Fujita N, Noda T, Yoshimori T, Koike K, Moriishi K, Matsuura Y. 2011. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 85:13185–13194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mizui T, Yamashina S, Tanida I, Takei Y, Ueno T, Sakamoto N, Ikejima K, Kitamura T, Enomoto N, Sakai T, Kominami E, Watanabe S. 2010. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J. Gastroenterol. 45:195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chase AJ, Semler BL. 2012. Viral subversion of host functions for picornavirus translation and RNA replication. Future Virol. 7:179–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dales S, Eggers HJ, Tamm I, Palade GE. 1965. Electron microscopic study of the formation of poliovirus. Virology 26:379–389 [DOI] [PubMed] [Google Scholar]
- 74. Suhy DA, Giddings TH, Jr, Kirkegaard K. 2000. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 74:8953–8965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Niemann A, Baltes J, Elsässer H-P. 2001. Fluorescence properties and staining behavior of monodansylpentane, a structural homologue of the lysosomotropic agent monodansylcadaverine. J. Histochem. Cytochem. 49:177–185 [DOI] [PubMed] [Google Scholar]
- 76. Richards AL, Jackson WT. 2012. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathog. 8:e1003046. 10.1371/journal.ppat.1003046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jin R, Zhu W, Cao S, Chen R, Jin H, Liu Y, Wang S, Wang W, Xiao G. 2013. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS One 8:e52909. 10.1371/journal.pone.0052909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vescovo T, Romagnoli A, Perdomo AB, Corazzari M, Ciccosanti F, Alonzi T, Nardacci R, Ippolito G, Tripodi M, Garcia-Monzon C, Lo Iacono O, Piacentini M, Fimia GM. 2012. Autophagy protects cells from HCV-induced defects in lipid metabolism. Gastroenterology 142:644–653 [DOI] [PubMed] [Google Scholar]
- 79. Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097 [DOI] [PubMed] [Google Scholar]
- 80. Mateo R, Nagamine CM, Spagnolo J, Mendez E, Rahe M, Gale M, Jr, Yuan J, Kirkegaard K. 2013. Inhibition of cellular autophagy deranges dengue virion maturation. J. Virol. 87:1312–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang Y, Li Z, Ge X, Guo X, Yang H. 2011. Autophagy promotes the replication of encephalomyocarditis virus in host cells. Autophagy 7:613–628 [DOI] [PubMed] [Google Scholar]
- 82. Chen Q, Fang L, Wang D, Wang S, Li P, Li M, Luo R, Chen H, Xiao S. 2012. Induction of autophagy enhances porcine reproductive and respiratory syndrome virus replication. Virus Res. 163:650–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liu Q, Qin Y, Zhou L, Kou Q, Guo X, Ge X, Yang H, Hu H. 2012. Autophagy sustains the replication of porcine reproductive and respiratory virus in host cells. Virology 429:136–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Inglot AD. 1969. Comparison of the antiviral activity in vitro of some non-steroidal anti-inflammatory drugs. J. Gen. Virol. 4:203–214 [DOI] [PubMed] [Google Scholar]
- 85. Matarrese P, Nencioni L, Checconi P, Ciarlo L, Gambardella L, Ascione B, Sgarbanti R, Garaci E, Malorni W, Palamara AT. 2011. Pepstatin A alters host cell autophagic machinery and leads to a decrease in influenza A virus production. J. Cell. Physiol. 226:3368–3377 [DOI] [PubMed] [Google Scholar]
- 86. Lamkanfi M, Dixit VM. 2010. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8:44–54 [DOI] [PubMed] [Google Scholar]
- 87. Gordy C, He YW. 2012. The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell 3:17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yorimitsu T, Nair U, Yang Z, Klionsky DJ. 2006. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 281:30299–30304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Høyer-Hansen M, Jaattela M. 2007. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 14:1576–1582 [DOI] [PubMed] [Google Scholar]
- 90. Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T. 2007. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 16:618–629 [DOI] [PubMed] [Google Scholar]
- 91. Bernales S, McDonald KL, Walter P. 2006. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4:e423. 10.1371/journal.pbio.0040423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. 2006. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26:9220–9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ciccaglione AR, Costantino A, Tritarelli E, Marcantonio C, Equestre M, Marziliano N, Rapicetta M. 2005. Activation of endoplasmic reticulum stress response by hepatitis C virus proteins. Arch. Virol. 150:1339–1356 [DOI] [PubMed] [Google Scholar]
- 94. Joyce MA, Walters KA, Lamb SE, Yeh MM, Zhu LF, Kneteman N, Doyle JS, Katze MG, Tyrrell DL. 2009. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 5:e1000291. 10.1371/journal.ppat.1000291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rosebeck S, Sudini K, Chen T, Leaman DW. 2011. Involvement of Noxa in mediating cellular ER stress responses to lytic virus infection. Virology 417:293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]