

Abstract
In natural infection, antibodies interact with HIV-1 primarily through nonfunctional forms of envelope glycoproteins (Env), including uncleaved (UNC) gp160 and gp41 stumps. These antigens are important to fully characterize, as they may be decoys that promote nonneutralizing responses and may also be targets for nonneutralizing effector responses. In this study, we compared the antigenic properties of Env expressed in situ on pseudovirion virus-like particle (VLP) surfaces and soluble gp120 using harmonized enzyme-linked immunosorbent assays (ELISAs) and a panel of 51 monoclonal antibodies (MAbs). Only 32 of 46 soluble gp120-reactive MAbs recognized the primary UNC gp160 antigen of VLPs. Indeed, many epitopes were poorly exposed (C1, V2, C1-C4, C4, C4-V3, CD4 induced [CD4i], and PGT group 3) or obscured (C2, C5, and C1-C5) on VLPs. In further studies, VLP Env exhibited an increased degree of inter-MAb competition, the epicenter of which was the base of the V3 loop, where PGT, 2G12, V3, and CD4 binding site specificities competed. UNC gp160 also underwent more drastic soluble CD4 (sCD4)-induced conformational changes than soluble gp120, exposing CD4i, C1-C4, and V2 epitopes. A greater propensity of UNC gp160 to undergo conformational changes was also suggested by the induction of CD4i MAb binding to VLPs by a V3 MAb as well as by soluble CD4. The same effect was not observed for soluble gp120. Taken together, our data suggest that membrane-expressed UNC gp160 exists in a less “triggered” conformational state than soluble gp120 and that MAb binding to UNC gp160 tends to have greater conformational consequences.
INTRODUCTION
Studies have shown that soluble HIV-1 envelope glycoprotein (Env) gp120 differs antigenically from the forms of Env that reside on virus or infected cell membranes (1–4). Since antibodies interact with HIV-1 particles via the latter in situ forms of Env, it is important to fully characterize these differences.
Over the last 30 years, substantial information has been gathered on the antigenic properties of both soluble (3, 5–19) and membrane-expressed (4, 16, 17, 20–44) forms of Env. However, few studies have reported direct comparisons (1, 3, 45). This is in part due to a lack of harmonized assays by which to make such comparisons, with soluble Env typically being analyzed by enzyme-linked immunosorbent assay (ELISA) and membrane Env usually being investigated by flow cytometry, immunoprecipitation, or virus capture (3, 34). One key early study compared the reactivities of a large set of gp120-directed monoclonal antibodies (MAbs) with Env expressed on the surfaces of HxB2-infected cells and monomeric gp120 (3), revealing generally reduced epitope exposure on membrane-expressed Env. Conversely, a few recently isolated MAbs, including PG9, PG16, CH01-04, PGT141-2, VRC03, and VRC06, can preferentially recognize native Env trimer expressed on membranes (9, 17).
Uncertainties regarding the exact nature of membrane Env rendered the significance of the above-mentioned comparative studies somewhat unclear. The observation that nonneutralizing MAbs (non-nAbs) can bind to the virus and infected cells conflicted with the previous widely held assumptions that virus particles express only native trimer and that MAb binding to trimers is the essence of the neutralization event (33–35, 40, 42, 46–48). This paradox was resolved by the finding of nonfunctional forms of Env on HIV-1 surfaces (33, 40). Thus, membrane Env generally is comprised of a mixture of Env isoforms that include the functional Env trimer, uncleaved (UNC) gp160, and gp41 stumps (33).
During natural infection, nonfunctional forms of Env are vastly preferred targets of antibodies, and as a consequence, serum responses are overwhelmingly nonneutralizing. Nonfunctional Env is important to understand in HIV-1 vaccine research for at least three reasons: (i) it may be involved in virus inhibition by other antibody mechanisms, such as antibody-dependent cell-mediated viral inhibition (ADCVI); (ii) it is immunodominant and therefore may act as an antigenic decoy that confers a valuable fitness advantage on the virus by allowing it to better evade nAbs; and (iii) it is possible that the non-nAb responses that rapidly develop against nonfunctional forms of Env during natural infection are not independent from the later development of nAbs. In fact, non-nAbs directed to nonfunctional Env may be stepping stones in nAb ontogeny. Thus, we envision a scenario in which nAbs may emerge from an early pool of non-nAbs that target UNC gp160 and later acquire mutations allowing them to cross-react with native trimers. For these reasons, to become better acquainted with our adversary and its evasion tactics, we decided to compare the antigenic topologies of membrane-expressed Env (principally UNC gp160) and soluble gp120 in detail.
One way to dissect Env topology is to examine MAb cross-competition relationships. Most work of this type has been done with soluble gp120 (5, 6, 11, 16, 19). However, limited competitions have been done on membrane Env by virus capture (22–25), by flow cytometry (2, 6, 9, 16–18), and by combinatorial neutralization assays designed to measure synergistic or antagonistic MAb binding to the native trimer (49–55). These studies shed some light on the conformational differences between soluble gp120 and membrane Env. For example, MAb VRC01 induces CD4-induced (CD4i) MAb binding to soluble gp120 (18) but not to the native trimer (2). In contrast, soluble CD4 (sCD4) induces exposure of the V3 loop on the native trimer but not on soluble gp120 (the latter is already fully exposed without added sCD4). Other competitive relationships have been reported for membrane Env that probably largely reflect MAb binding to UNC gp160 (22–25). The observation that point mutations often have disparate effects on MAb binding to the gp120 monomer and native trimer further highlights the marked conformational differences between these forms of Env (2, 4).
In this study, we built on the above-mentioned earlier studies in two major ways. First, we took advantage of our newly developed virus-like particle (VLP) ELISA (42) to facilitate harmonized comparisons between isolate-matched soluble and membrane-expressed forms of Env. Second, we investigated the binding patterns of several recently isolated novel MAbs in a comprehensive panel that was used to probe soluble gp120 and VLP Env epitope exposure and binding relationships. Our findings advance our understanding of the interactions between membrane-expressed Env and antibodies.
MATERIALS AND METHODS
Anti-HIV-1 Env monoclonal antibodies and gp120 C5-specific serum.
The MAbs used in this study were obtained from their producers, the AIDS reagent repositories of the Medical Research Council (United Kingdom) and the NIH, or were purchased from commercial suppliers. Detailed information on all these MAbs is provided in the HIV Molecular Immunology Database (http://www.hiv.lanl.gov/content/immunology/ab_search). Our MAb panel included the following (originators given in parentheses): EH21 (recognizes a peptide, TEKLWVTVYYGVPVWREATT, consisting of gp120 residues 31 to 50, according to the HxB2 reference strain [J. E. Robinson, unpublished data]), 133/290, 133/11, 133/237 (M. Niedrig), 522-149 (G. Robey), MAG45, MAG95, MAG97, and MAG104 (C. Y. Kang), all directed to the gp120 C1 segment (3, 11, 13, 56–60); A32, directed to a C1-C4 epitope (8, 11, 61); 212A, 3.12B, and 23B (J. Robinson), directed to C1-C5 discontinuous epitopes (11); B12 (G. Lewis), directed to a C2 epitope (3); M91 (F. di Marzo Veronese), 9301 (Dupont, Inc.), 8C6/1 (S. Ranjbar), 110.1 (Genetic Systems), 670-D (S. Zolla-Pazner), and sheep antiserum D7324 (Aalto Bio Reagents, Ireland), all directed to C5 epitopes (3, 11, 35); 8.22.2 (A. Pinter), G3-4 (M. Fung), 684-238, and SC258 (G. Robey), directed to the V2 loop (7, 10, 11); PG16 (D. Burton), directed to a quaternary V1/V2-dependent epitope (17); G3-42, G3-211, G3-299, G3-508, and G3-537 (M. Fung), directed to C4 and C4-V3 epitopes (3, 12, 62); E51 and 17b (J. Robinson), directed to CD4i epitopes (63); 2G12 (H. Katinger), directed to a unique glycan-dependent epitope on gp120 (64, 65); LA21 and CO11 (J. Robinson), directed to the gp120 V3 loop (38, 42); PGT121, PGT125, PGT130, PGT135, and PGT136 (D. Burton), directed to 3 epitope clusters involving the base of the V3 loop of gp120 and the glycan N332 (16); b6, b12 (D. Burton), 15e (J. Robinson), VRC01, and VRC03 (J. Mascola), directed to epitopes that overlap the CD4 binding site (CD4bs) (18, 42, 66); VRC06 (J. Mascola), directed to a hybrid CD4bs/CD4i epitope (9); 7B2 and 2.2B (J. Robinson), directed to the gp41 cluster I and II epitopes, respectively (33); and 10e8 (J. Mascola and M. Connors), 2F5, and 4E10 (H. Katinger), directed to the gp41 membrane-proximal ectodomain region (MPER) (67, 68). MAbs M91, B12 8C6.1, and 110.1 are ascites fluid antibodies, each with approximate IgG concentrations of 5 mg/ml.
Recombinant gp120 monomer, sCD4, and CD4-IgG2.
Recombinant monomeric JR-FL gp120 produced in Chinese hamster ovary (CHO) cells, CD4-IgG2, and sCD4 consisting of all the 4 outer domains were gifts from Progenics Pharmaceuticals (Tarrytown, NY).
Env plasmids.
The plasmid pCAGGS was used to express JR-FL Env on virus-like particles (VLPs) (33, 69). Envs were all truncated at amino acid 709 to produce a mutant termed gp160ΔCT, leaving a 3-amino-acid gp41 cytoplasmic tail. This truncation increases native trimer expression and produces pseudotyped virus that exhibits a neutralization sensitivity profile similar to those produced with a full-length version of JR-FL gp160, as we reported previously (70). Mutants were generated by QuikChange (Agilent Technologies) and numbered according to the HxB2 reference strain. The “SOS” mutations introduce an intermolecular disulfide bond between gp120 and gp41 (71). UNC SOS and UNC wild-type (WT) include K510S and R511S mutations at the primary gp160 cleavage site (42).
VLP production.
VLPs were produced by transiently transfecting 293T cells with a pCAGGS Env-expressing plasmid and the subgenomic plasmid pNL4-3.Luc.R-E-, using polyethyleneimine (42). Two days later, supernatants were collected, precleared by low-speed centrifugation, and pelleted at 50,000 × g in a Sorvall SS34 rotor. To remove residual medium, VLP pellets were diluted with 1 ml of phosphate-buffered saline (PBS), then recentrifuged in a microcentrifuge at 15,000 rpm, and resuspended in PBS at 1,000 times the original concentration. VLPs were referred to as WT-VLPs, UNC WT-VLPs, SOS-VLPs, or UNC SOS-VLPs, depending on the form of Env displayed on their surfaces (72). VLPs were inactivated using Aldrithiol (73), after which they were recentrifuged and washed with PBS.
Native PAGE.
VLP lysates treated with Triton X-100 and recombinant gp120 were resolved by blue native PAGE (BN-PAGE), followed by Western blotting to detect Env, as described previously (42, 46).
ELISAs using gp120 and VLPs.
ELISAs were performed as described previously (42). Briefly, Immulon II plates were coated overnight at 4°C with recombinant gp120 at 5 μg/ml or VLPs at 20 times their concentration in transfection supernatants. Following a PBS wash and blocking, MAbs were titrated against each antigen. Species-specific alkaline phosphatase anti-Fc conjugates (Accurate, Westbury, NY) and SigmaFAST p-nitrophenyl phosphate tablets (Sigma) were then used to detect binding. Plates were read at 405 nm. The MAb concentration resulting in an optical density (OD) of 0.5 (approximately 5 times background) was recorded as its titer.
In a competitive ELISA format, we used cold competitor MAbs at a fixed concentration of 10 μg/ml to inhibit binding of titrated biotinylated MAb to soluble gp120 or VLPs. MAbs were biotinylated using NHS-X-biotin reagent (Calbiochem) according to the manufacturer's instructions. Soluble CD4 was used at a concentration of 2 μg/ml. Biotinylated MAbs were detected using streptavidin-alkaline phosphatase (Vector, Burlingame, CA).
Neutralization assays.
Neutralization assays were performed in three different formats to determine the mechanism, as previously described (69, 70). Brief descriptions of each format follow.
(i) Standard format.
MAbs and VLPs were mixed for 1 h prior to addition to CF2Th.CD4.CCR5 target cells for 2 h at 37°C. SOS-VLPs were treated exactly as WT-VLPs, except that SOS-VLP infection was triggered at the end of virus-cell incubation by adding 5 mM dithiothreitol (DTT) for 10 min to break the gp120-gp41 disulfide, followed by a medium exchange (69, 70).
(ii) Post-CD4 format.
To measure neutralizing activity between the CD4 and CCR5 binding steps, VLPs were preincubated with 3 μg/ml sCD4 for 15 min at 37°C, followed by 1 h with titrated MAbs, before addition to target cells expressing only the CCR5 coreceptor (CF2Th.CCR5 cells) (74). As mentioned above, when SOS-VLPs were used, 5 mM DTT was added at the end of virus-cell incubation for 10 min, followed by a medium exchange.
(iii) Post-CD4/CCR5 format.
To measure neutralization after receptor binding, SOS-VLPs were allowed to attach to target cells (CF2Th.CD4.CCR5 cells) for 2 h. Unbound VLPs were washed away and titrated MAbs were added, followed by 1 h of incubation. As mentioned above, infection was then activated by a 10-min exposure to 5 mM DTT.
RESULTS
In addition to the native gp120/gp41 trimer, HIV-1 surfaces are populated by nonfunctional forms of Env that include UNC gp160 and gp41 stumps (33–35, 40, 42, 46–48). Relatively few MAbs are able to recognize the functional Env trimer and neutralize the virus. However, a wide range of MAb specificities can bind to HIV-1 virus particles via nonfunctional Env. In this analysis, we sought to improve our understanding of the antigenic properties of soluble gp120 and Env present in situ on VLP surfaces by investigating both antigens with a comprehensive panel of MAbs.
Native PAGE analysis of the Env compositions of soluble gp120 and VLPs.
To better define the Env components of our test antigens, we first examined WT-VLPs, SOS-VLPs, UNC SOS-VLPs and soluble gp120 by BN-PAGE-Western blotting (Fig. 1). JR-FL Env was chosen as our prototype because it is a tier 2 neutralization-resistant primary isolate that expresses efficiently and for which the gp160 precursor cleaves efficiently into gp120/gp41. This Env also provides useful continuity with our previous work (33, 42, 46, 70). Soluble gp120 was expressed in Chinese hamster ovary (CHO) cells, and VLPs were expressed in 293T cells. Conceivably, this difference in producer cells might cause host cell type-related glycan variations not encoded by the respective Env genes. However, there are in fact inherent differences in glycosylation patterns between particulate Env and soluble gp120, regardless of the producer cell. Soluble gp120 exhibits a greater proportion of mature, complex glycans, presumably due to the relative accessibility of its surface sugars to processing enzymes (75–77). Furthermore, VLPs are heavily populated by an early form of UNC gp160, termed gp160ER (ER stands for endoplasmic reticulum) that, in contrast to mature UNC gp160, exhibits largely untrimmed high-mannose glycans (46). Thus, the inherent differences in glycosylation patterns of membrane and soluble forms of Env probably supersede the effects of any differences arising from their expression in different mammalian cell lines.
Fig 1.
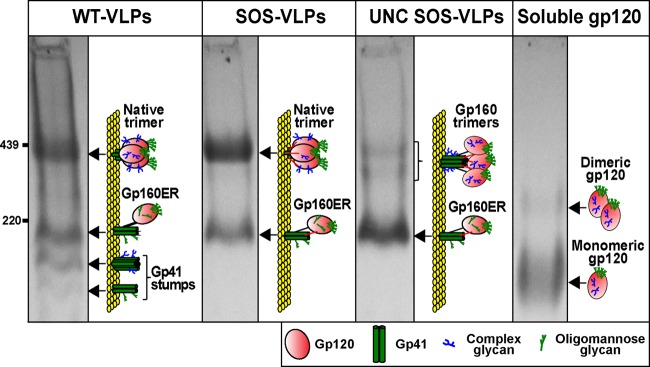
Comparison of VLP Env and soluble gp120 by BN-PAGE. The Env components of various VLPs and soluble gp120 were resolved by BN-PAGE-Western blotting and detected with anti-gp120 and anti-gp41 MAb cocktails. Ferritin was used as a size marker (GE Healthcare). Diagrams depict native trimer, UNC gp160 monomer (mostly gp160ER), soluble gp120, and gp41 stumps. Complex glycans are indicated by blue “tree” structures and oligomannose glycans by green “tree” structures. The SOS disulfide bond is depicted by red bars that link gp120 and gp41. Monomeric gp160ER, bearing largely only untrimmed high-mannose glycans, is a major form of Env on all VLPs. The faint doublet of oligomers on UNC SOS-VLPs (lane 3) is likely to include trimers of mature gp160 and gp160ER. Recombinant gp120 is largely monomeric, with a fraction of dimer.
Figure 1 shows the various forms of Env that decorate VLP surfaces. WT-VLPs exhibited a prominent native Env trimer, UNC gp160 monomer (consisting largely of gp160ER [46]), and gp41 stumps (Fig. 1, lane 1). SOS-VLPs exhibited only native Env trimer and UNC gp160 monomer (Fig. 1, lane 2). gp41 stumps were absent, because the SOS disulfide bond links gp120 and gp41 subunits and thereby prevents gp120 shedding. UNC SOS-VLP gp160 was largely monomeric, with faint traces of two oligomeric species (Fig. 1, lane 3; also see Fig. 3 in reference 46). The monomer consists largely of gp160ER, as evidenced by our prior observation that MAb 2G12 mediates a “supershift” of this monomer in BN-PAGE shift assays, commensurate with the binding of two 2G12 IgG molecules to twin binding sites found uniquely on gp160ER (33, 42, 46). The two oligomeric bands of UNC SOS-VLPs are likely to be trimers of gp160ER (lower oligomer band) and mature gp160, bearing some complex glycans (upper oligomer band). Soluble gp120 was also predominantly monomeric, with traces of a dimer that may arise through covalent (or possibly noncovalent) interactions between gp120 protomers (Fig. 1, lane 4) (78, 79).
Fig 3.

MAb neutralization of WT and SOS viruses in various assay formats. MAb neutralization of WT and SOS JR-FL viruses (an E168K N189A double mutant [E168K+N189A] was used to knock in the PG16 epitope) was evaluated in various neutralization assay formats. The neutralization sensitivity profile of the E168K+N189A mutant is identical to that of the parent virus.
Comparative antigenicities of Env expressed in situ on VLPs and soluble gp120.
Previous work showed that pseudovirion VLPs bear relatively high quantities of nonfunctional Env compared to live HIV-1 (20, 32, 33, 40, 80). However, nonfunctional Env is a universal contaminant of all HIV-1 preparations and dominates their antigenic landscape, regardless of its quantity. An antigenic analysis of VLPs should therefore provide information that is relevant to understanding the live, replicating virus. The observation that nonneutralizing MAbs efficiently capture infectious particles, even though they do not recognize the native trimer (33–35, 40), confirms that nonfunctional Env exists on intact virus particle surfaces and is not simply a derivative of noninfectious matter that copurifies with HIV-1 particles. This being the case, it is clearly of interest to investigate the antigenic properties of nonfunctional Env.
UNC gp160 is likely to be a major immunodominant antigen on viral membranes. This notion is supported by the observation that VLPs bearing both native trimer and UNC gp160 (e.g., WT-VLPs) exhibit antigenic properties similar to those of VLPs bearing only UNC gp160 (e.g., UNC WT-VLPs), both of which efficiently expose non-nAb epitopes (see Fig. 1 of reference 42). Given that the native trimer is highly compact, it is perhaps not surprising that the more conformationally “open” UNC gp160 is more immunogenic and may well be a driving force behind the largely nonneutralizing responses observed in natural HIV-1 infection. Since antibodies evidently interact with HIV-1 primarily through this nonfunctional UNC gp160, information on its antigenic topology is invaluable. Due to a lack of convenient assays to study membrane-presented forms of Env, however, there has been a relative shortfall of such information. To fill this knowledge gap, we developed a simple VLP ELISA (42) that facilitates direct, quantitative comparisons with soluble gp120 ELISAs.
Overview of MAb recognition of VLP Env and soluble gp120.
In a fresh attempt to identify conformational differences between membrane-expressed and soluble Env, we evaluated the epitope exposure patterns of all 4 antigens of Fig. 1 using a large panel of MAbs; results are shown in Fig. 2. We did not study MAbs PG9 and PG16, directed to quaternary V1/V2 epitopes, since we were primarily focused on binding to UNC gp160. In previous antigenicity studies, monomeric gp120 was usually captured by purified sheep serum, D7324, raised against the C5 region (3, 10–13), ostensibly to render its orientation consistent and to eliminate any possible conformational effects arising from direct gp120 adsorption. However, in the present study, we used monomeric gp120 to directly coat ELISA wells so that we could assay D7324 binding. To check what effect direct coating might have on epitope exposure or conformational flexibility, we examined the binding of MAbs 2G12 and 17b to directly coated gp120 and D7324-captured gp120 in the presence and absence of sCD4. 2G12 bound equivalently to both antigens, but 17b bound ∼5-fold more effectively to captured gp120 (data not shown). However, in the presence of sCD4, 17b binding to both gp120 presentations increased 50-fold (data not shown). Thus, while direct coating partially occludes 17b binding to gp120, it has little impact on its conformational state, as judged by the amplitude of sCD4-induced conformational changes.
Fig 2.

Epitope exposure of soluble gp120 and VLP Env. VLPs and soluble gp120 were probed with a panel of MAbs directed to various gp120 and gp41 epitopes by ELISA. aEpitope cluster recognized by each MAb: C represents conserved gp120 regions that are interspersed by variable loops (V). Letters in parentheses identify linear (L) and discontinuous (D) epitopes. The amino acid numbering is based on the HxB2 Env sequence. bBinding titers were calculated as the MAb concentration where its binding OD was 0.5 and are given in μg/ml. cThe mean VLP titer was calculated by averaging MAb titers of WT-VLPs, SOS-VLPs, and UNC SOS-VLPs. Progressively warmer-colored cells indicate tighter binding. dRatio of gp120 titer and mean VLP titer. Gray cells indicate a gp120 preference; cyan cells indicate a VLP preference. eMean titers of MAbs binding to each antigen. For soluble gp120, this titer excludes the gp41 MAbs. In all calculations, titers of >10 μg/ml are taken as 10 μg/ml.
In contrast to our previous study, we measured MAb endpoint titers at an OD at 405 nm of 0.5 rather than 50% effective concentrations (EC50s). This allowed weak binding to be recorded and factored into our calculations of relative antigenicity. The binding titers in Fig. 2 are color-coded to emphasize stronger binding with progressively warmer colors. The mean titers of each MAb against the 3 VLP antigens are given, as are soluble gp120/mean VLP titer ratios (Fig. 2, last two columns). Threefold and greater differences in titer ratio were considered significant.
Our data indicate a generalized preference for gp120, as illustrated by the ∼10-fold-stronger mean MAb titer against monomeric gp120 than VLP antigens (see mean titers at the bottom of Fig. 2). However, the individual MAb titers were not uniformly different by a 10-fold margin (Fig. 2). Thus, as noted in the last column of Fig. 2, while most MAbs preferentially recognized soluble gp120 (gray cells), some bound both antigens equivalently (white cells) and others bound better to VLPs (cyan cells). This implies a conformational distinction between the antigens, rather than a simple difference in their ELISA saturation. The ∼10-fold difference in mean epitope exposure may in fact be an underestimate considering that, as mentioned above, direct gp120 coating may decrease the exposure of certain epitopes like 17b to gp120.
(i) Soluble gp120-preferring MAbs.
Most MAbs, specifically those directed to the C1, C1-C4, C1-C5, C2, C5, V2, C4, C4-V3, and PGT group 3 epitopes, CD4i MAb E51, CD4-IgG2, and CD4bs MAb 15e, exhibited a preference for soluble gp120, as indicated by gray cells in the last column of Fig. 2.
MAbs targeting the extreme N terminus of the C1 region (residues 31 to 78) reacted weakly with VLPs. However, those that recognize C1 epitopes further downstream were nearly completely occluded, as were the C1-C5, C2, and C5 epitopes. Previously, Nyambi et al. showed that MAbs directed to the C-terminal part of the C5 region, specifically, 670-D (epitope, residues 498 to 504) and 1331A MAb (epitope, residues 503 to 511) efficiently capture HIV-1 (35). However, by ELISA, MAb 670-D did not recognize any of the VLPs. Similarly, MAb 110.1 and serum D7324, both directed to the extreme C terminus of the C5 region, also failed to react with VLPs. In a previous study, two other C5 MAbs, 858-D and 989-D, also exhibited weak VLP reactivity (35). Taken together, these data imply that like the C1 region, the C5 region is largely, if not completely, occluded on VLPs. In contrast, the epitopes of MAbs against C1-C4, V2, C4, and C4-V3, MAb E51 (in the absence of sCD4), PGT group 3 MAbs, and CD4-IgG2 were only partially occluded on VLPs.
(ii) Epitopes that are equally exposed on gp120 and VLP Env.
A second MAb group is indicated by white cells in the last column of Fig. 2 and includes several nAbs (2G12, PGT groups 1 and 2, b12, and VRC01) and nonneutralizing MAbs directed to the V3 loop (LA21 and CO11), CD4bs (b6), and CD4i (17b). In contrast to the other members of their respective epitope clusters, MAbs MAG104 (C1) and 8C6/1 (C5) also fell into this group. However, in our calculations, we took all titers of >10 μg/ml as 10 μg/ml. Therefore, the weak soluble gp120 binding and lack of VLP binding by these two MAbs could in fact indicate a preference for monomeric gp120 that is consistent with the other MAbs in their respective epitope groups.
(iii) VLP Env-preferring MAbs.
A third group consists of MAbs VRC03 and VRC06 (indicated by cyan cells in the last column of Fig. 2). Both MAbs preferentially bound to WT- and SOS-VLPs, in line with their known preference for native Env trimer (9). Unexpectedly, PGT130 also bound 3-fold more strongly to SOS-VLPs than UNC SOS-VLPs, suggesting a modest trimer preference.
Differential recognition of VLP Envs.
In addition to the mean VLP and soluble gp120 titer differences discussed above, there were some notable differences in MAb recognition of different VLP types (Fig. 2). These falls into 3 categories: preferential recognition of WT-VLPs, SOS-VLPs, or UNC gp160. As stated above, we took a >3-fold bias in titer as being significant.
(i) Preferential binding to WT-VLPs.
MAbs that recognize residues 61 to 78 of the gp120 C1 region preferentially bound WT-VLPs over SOS-VLPs. This may be because the SOS mutant involves a C5 mutation to create a novel gp120-gp41 disulfide, which may partially obscure the structurally apposing C1 region. Similarly, MAbs G3-4, 8.22.2, and SC258 all preferentially recognized WT-VLPs, suggesting that the SOS mutation leads to partial occlusion of the V2 loop. This was somewhat surprising, considering the disparate structural positions of the V2 and the C5 regions of gp120, and implies a transmitted effect. Nonneutralizing gp41 MAbs 7B2 and 2.2B recognized WT-VLPs but not SOS-VLPs (Fig. 2). Both epitopes were exposed on UNC WT gp160 (see Fig. 1 of reference 42) and gp41 stumps (see Fig. 6 of reference 42). The SOS mutation may directly impact the 7B2 epitope and could impose conformational constraints on the 2.2B epitope. In contrast, the gp41 MPER epitopes (2F5, 4E10, and 10e8) were equally exposed on WT-VLPs and SOS-VLPs.
Fig 6.
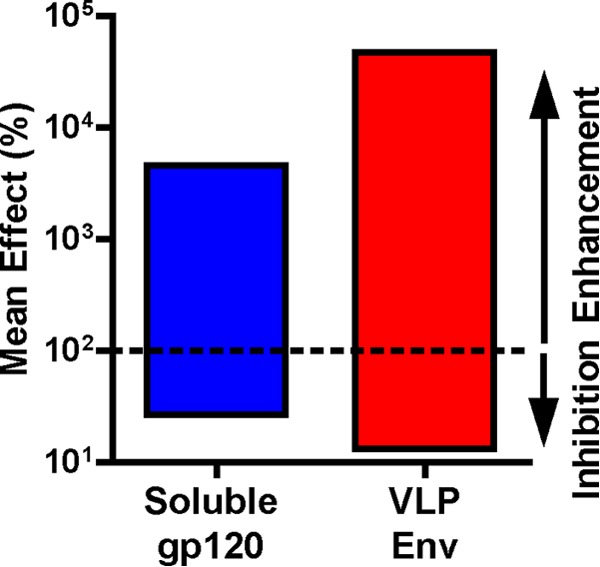
Conformational flexibility of soluble gp120 and VLPs. The overall mean inhibitions (red, orange, and yellow cells of Fig. 5; n = 53 data pairs) and mean enhancements (magenta and cyan cells of Fig. 5; n = 21 data pairs) for soluble gp120 and VLPs were calculated, where n is the total number of relevant data points for calculations. In many cases, competitions or enhancements were seen with only one antigen. However, data for both antigens were included in the calculation.
Although most of the binding patterns in Fig. 2 concern recognition of UNC gp160, it is possible that in some cases trimer binding is a contributing factor. Thus, the preferential binding of V2 MAbs to WT-VLPs could be related to differential V2 loop exposure on native Env WT and SOS trimers. To investigate this possibility, we tested the neutralizing activity of V2 MAb SC258 and the related quaternary V1/V2 loop MAb PG16 against SOS and WT-VLPs in the standard assay format (Fig. 3). Our previous work revealed that SOS-VLPs and WT-VLPs exhibit largely indistinguishable neutralization sensitivity profiles (70). Thus, MAbs b12, PGT121, and PGT125 all neutralized WT and SOS viruses equivalently. PG16 also neutralized both viruses equivalently (Fig. 3). Similar to the V3 MAb CO11, SC258 failed to neutralize either virus (Fig. 3) (69, 70). Only MAb 2F5 preferentially neutralized the SOS virus, as we reported previously (70). Together, these findings suggest that the preferential V2 MAb recognition of WT Env stems from increased binding to UNC gp160 and not to the native trimer.
(ii) Preferential binding to SOS-VLPs.
Several MAbs preferentially bound SOS-VLPs over WT-VLPs, namely, A32, G3-211, G3-299, 15e, and b12 (Fig. 2). Of these, cases with marginal differences could simply relate to the somewhat higher expression of SOS-VLP Env (Fig. 1, compare lanes 1 and 2; also see reference 42). The more significant differences (A32 plus sCD4, G3-299, 15e, and b12) probably reflect conformational distinctions between SOS and WT Env. It may not be a coincidence that all of these MAbs recognize epitopes involving the C4 region. Since b12 is the only member of this group that neutralizes JR-FL (70) (Fig. 3 and data not shown) and equally neutralizes SOS and WT viruses, this increased C4 exposure is likely to be a feature of UNC SOS gp160 rather than the native SOS trimer.
(iii) Preferential binding to UNC gp160.
The epitopes of MAbs MAG45, A32 plus sCD4, 8.22.2, G3-4, SC258, G3-211, G3-537, G3-508, G3-299, G3-42, PGT135, PGT136, CD4-IgG2, 15e, and 10e8 were all better exposed on UNC SOS-VLPs than SOS-VLPs (Fig. 2). This suggests a preference for the UNC gp160 monomer (Fig. 1). In the case of CD4-IgG2, this is consistent with its relatively weak affinity for the native trimer compared to other forms of Env (81).
Summary of epitope exposure analyses.
The antigenicity patterns of Fig. 2 are summarized in Fig. 4. V3 and CD4bs epitopes were exposed approximately equally on gp120 and VLPs. However, in all other cases, VLP epitope exposure was either reduced (part of the C1, V2, and C4 regions) or altogether eliminated (part of the C1, V5, and C5 regions). WT and SOS gp160ΔCT share similar topologies, with the biggest difference being the differential exposure of gp41 cluster I and II epitopes.
Fig 4.
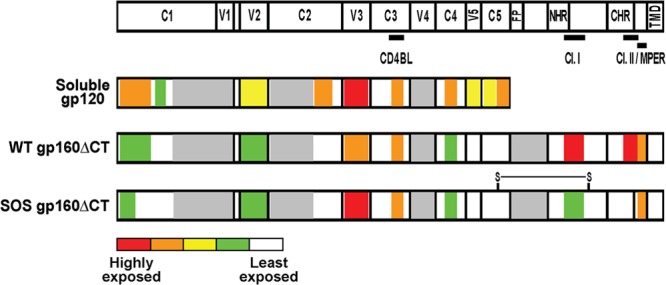
Summary of soluble gp120 and VLP Env epitope exposure. Schematic representations of soluble gp120 and VLP Env epitope exposure are shown, derived from the data of Fig. 2. The epitopes of the CD4 binding loop (CD4BL), gp41 cluster I (Cl. I) and cluster II (Cl. II), and MPER are indicated. The disulfide linkage on SOS gp160ΔCT is represented as “S-S.” Other abbreviations: FP, fusion peptide; NHR and CHR, N-heptad repeat and C-heptad repeat, respectively; TMD, transmembrane domain. The mean titer of MAb binding to each domain was calculated for each antigen. The relative exposure of Env domains is labeled according to the color scheme in Fig. 2. Warmer colors therefore indicate a greater epitope exposure; the most accessible regions are red, and the least accessible segments are white. Gray domains indicate unknown exposure due to a lack of MAbs.
Differential effects of sCD4 on MAb binding.
To identify any differences in receptor binding-induced conformational changes, we measured the effect of sCD4 binding on the binding of a subset of MAbs (Fig. 2). VLP binding of MAbs EH21, G3-42, and CO11 was marginally induced by sCD4. However, their binding to soluble gp120 was largely unaffected. This difference is evidenced by a 2-fold increase in binding ratio of all three MAbs in the last column of Fig. 2. In contrast, C1-C4-specific MAb A32 binding was induced by sCD4 on both VLPs and soluble gp120 to various extents. Soluble CD4 had a more dramatic effect on CD4i MAb (E51 and 17b) and V2 MAb G3-4 binding to VLPs than did soluble gp120, as evidenced by a 5- to 10-fold change in ratio in the last column of Fig. 2.
Although most VLP binding in Fig. 2 probably derives from recognition of UNC gp160, since sCD4 induces dramatic conformational changes in the trimer, nonneutralizing epitopes could be revealed and therefore could at least partly explain the sCD4-induced effects in Fig. 2. To investigate this possibility, we examined MAb activities in a previously published post-CD4 neutralization assay, using WT and SOS viruses (Fig. 3) (70). V3 MAb CO11 was highly potent in this format, consistent with our previous report (Fig. 3) (70). The extent of this neutralization enhancement was far more pronounced than the relatively modest effect of sCD4 on CO11 binding to VLPs by ELISA, suggesting that UNC gp160 still dominates the binding patterns in Fig. 2, even in the presence of sCD4. MAb 2F5 was also more potent in the post-CD4 format and was also the only MAb effective in the post-CD4/CCR5 neutralization format, consistent with the idea that only gp41-specific MAbs can neutralize HIV-1 after full receptor engagement (Fig. 3) (70). PGT MAb neutralization was unaffected by sCD4, consistent with the idea that these MAbs neutralize at a relatively late step, such as coreceptor binding. MAbs b12 and PG16 both exhibited lower activity in the post-CD4 format (Fig. 3), consistent with previous data (17, 70, 82, 83). SC258, G3-42, and A32 all remained nonneutralizing in the post-CD4 format, suggesting that they do not recognize the CD4-bound trimer. Taken together, these data suggest that the increased MAb binding to VLPs in the presence of sCD4 (Fig. 2) is largely governed by UNC gp160 and not the native trimer.
Cross-competition analysis of VLP and soluble Envs.
The decreased epitope exposure of VLP Env and the greater impact of sCD4 binding both hint at a more compact topology that could affect the binding relationships of ligands. To investigate, we cross-competed MAbs in ELISAs with both antigens (Fig. 5). To keep the resulting heat map sizes manageable, a subset of MAbs were selected from Fig. 2, generally those with the highest affinity of each epitope group. We omitted VRC03 and VRC06, as they are heavily trimer preferring. We also excluded 2F5 and 10e8, leaving 4E10 as the sole representative of MPER MAbs. To enhance binding of 17b and A32 MAbs to VLPs, competitions were selectively done in the presence of sCD4. In some cases, limited MAb availability prevented a full two-way competition analysis. Thus, for example, supplies of G3-42 and G3-4 were sufficient only for their use as biotinylated MAbs. For both MAbs, we confirmed that the respective biotinylated MAb binding was specific, by demonstrating that the cold self-competitor reduced their binding to <1% on both antigens (data not shown).
Fig 5.

Ligand cross-competition on soluble gp120 and VLPs. MAb and sCD4 relationships were explored on soluble gp120 (A) and VLPs (B). Panels A and B are organized in identical fashions to facilitate comparisons. Data are expressed as percentages derived from the ratio of titers of biotinylated MAb (horizontal series) in the presence or absence of excess (10 μg/ml) unlabeled competitor (vertical series), wherein titers determined without competitor are taken as 100%. Colors are used to emphasize inhibition and enhancement. Thus, inhibitions are shown as warm colors and enhancements are shown in cyan and magenta. In cases where biotinylated MAbs were used in combination with sCD4 in a particular column, the reference control also included sCD4. Each datum is the average of at least two repeats and is presented as the nearest whole number. In most cases, SOS-VLPs were used for panel B. However, as indicated, other VLPs were used in some situations. For example, cells in which UNC VLPs were used are delimited by a blue box. Data that differ >3-fold between panels A and B, and in which one or the other datum point lacks competition or inhibition (i.e., is >30% and <200%), are bordered by black boxes. ND, not done. aThe mean effect was determined by averaging the horizontal numbers. bThe ratio of mean enhancements was determined by the following formula: mean percent effect on VLPs/mean percent effect on soluble gp120. The ratio of mean inhibitions was determined by the following formula: mean percent effect on soluble gp120/mean percent effect on VLPs. Ratios of <1 (dark blue shading) indicate a pronounced effect on soluble gp120, ratios of >1 (dark green) indicate a pronounced effect on VLPs, and ratios of 1.0 (white) indicate no significant competition differences between the two antigens.
The data in Fig. 5 were computed from competitions in which a fixed excess (10 μg/ml) of cold competitor MAb (in some instances supplemented with 2 μg/ml sCD4, as indicated) was added to antigen-coated ELISA wells that were later overlaid with a titrated biotinylated MAb. Each datum point is a function of biotinylated MAb titer in the presence or absence of competitor, taking the latter as the 100% binding reference point. In previous studies, biotinylated MAbs were used in competitions at fixed, subsaturating dilutions rather than titrated as we did here (5, 11, 19). Our relatively labor-intensive approach was used to help minimize possible errors relating to the often significantly different MAb affinities to gp120 and VLPs. Competitive effects are coded by progressively warmer coloring, commensurate with the strength of inhibition. Enhancements are colored in cyan and, in exceptional cases (>1,000%), magenta. The latter points are expressed to the nearest power of 10 to maintain a compactness of Fig. 5. Bidirectional MAb competitions may indicate epitope overlap, close proximity, or steric effects. For convenience, we describe such MAb epitopes to be “overlapping.” Unidirectional competitions indicate nonoverlapping, conformational effects. As mentioned above, effects are generally deemed significant when they differ from controls ∼3-fold. Thus, the warm-colored cells showing competitive effects begin at 30% of control binding. Some competitions were not done, largely to avoid redundancy, such as sCD4 competitions with CD4bs MAbs or gp41 MAb binding to soluble gp120. In other cases, data were omitted where binding was deemed too weak. For example, due to their weak binding EH21, 17b and A32 were not used as competitors in VLP ELISAs (Fig. 5).
To facilitate comparisons between soluble gp120 and VLP Env, the organizations of Fig. 5A and B are identical. Figure 5A and B are each divided into sections depending on the neutralizing activities of MAb competition pairs that in Fig. 5B are delimited by the use of distinct VLP antigens. Thus, SOS-VLPs were used for most gp120 MAbs, partly to take advantage of their high Env expression (Fig. 1, lane 2), which should ensure robust competitions. Furthermore, the SOS disulfide bond eliminates possible sCD4- or MAb-induced gp120 shedding that could complicate data interpretation if WT-VLPs are used (84). However, in all competitions involving MAbs 7B2 and 2.2B, WT-VLPs were used, as these MAbs do not efficiently recognize SOS-VLPs (Fig. 2).
Since the Env on VLPs is heterogeneous (Fig. 1), MAbs can possibly bind more than one form of Env, potentially complicating the measurement of their competitive relationships. This is a problem when non-nAbs (that bind only the UNC gp160 monomer) cannot compete with nAbs for binding to the native trimer, rendering competitions on SOS-VLPs futile. To address this problem, where necessary, we used UNC mutant VLPs, so that competitions occurred on a level “playing field,” essentially only on UNC gp160. Thus, in lower left section of Fig. 5B, bordered by a blue box, for gp120 MAb competitors, we used UNC SOS-VLPs and for 7B2 or 2.2B competitors, UNC WT-VLPs. Ultimately, the use of parent or UNC VLPs may not be a significant factor, as UNC gp160 probably dictates the outcome of most competitions in Fig. 5B, regardless of the UNC mutation.
It is worth noting that in Fig. 5, 17b plus sCD4 competitor is grouped with the neutralizing MAbs, because sCD4 induces the 17b epitope on native trimer, leading to potent neutralization (70). However, when used alone, 17b does not neutralize effectively and hence is grouped with the non-nAbs. Since neither EH21 nor A32 neutralizes in any format (Fig. 3 and data not shown) (70), these MAbs are grouped as non-nAbs even when used with sCD4. As a result of this strategy, in Fig. 5B, nAbs compete with other nAbs on both native trimer and UNC gp160 in the top left section. In all the remaining sections of Fig. 5B, competitions take place on UNC gp160 or, possibly, in the case of MAbs 7B2 and 2.2B, gp41 stumps.
As expected, competitor MAbs generally inhibited their biotinylated counterparts (Fig. 5). In cases where we analyzed more than one MAb directed to a single epitope cluster (CD4bs and PGT epitopes), intracluster MAb competitions were observed. However, VRC01 exhibited poor inhibition of b6 and b12 on soluble gp120, perhaps in part due to its relatively low soluble gp120 affinity (Fig. 2). Other MAbs whose epitopes are distinct but overlapping competed on both antigens. For example, MAbs CO11 (V3) and G3-508 (C4) both inhibited G3-42 (C4-V3) binding to both antigens, consistent with a previous study (11).
Tertiary ligand combinations involving sCD4 require special care to interpret. The patterns can be deconvoluted by following three guidelines. First, when sCD4 is included with a competitor MAb, the outcome is best judged in reference to the effects of the MAb and sCD4 alone. This helps determine the relative contribution(s) of the MAb or sCD4 to the observed effect(s). For example, on soluble gp120, 17b plus sCD4 mediated a net enhancement of biotinylated 17b. To explain this confusing result, the inductive effect of sCD4 engagement on the binding of biotinylated 17b may be more dominant than the inhibitory effect of also including the cold 17b competitor. Second, it is important to point out the distinction when using sCD4 with either competitor or biotinylated MAb. Specifically, when 17b competes against biotinylated A32 plus sCD4, control assays also use biotinylated A32 and sCD4, so any effect is solely mediated by 17b, not sCD4. In the reverse format, any effect on biotinylated A32 binding by 17b plus sCD4 could stem from the effects of 17b, sCD4, or both. Third, when sCD4 is added to both competitor and biotinylated MAbs, the final sCD4 concentration is twice that of competitions in which only one MAb has added sCD4 and therefore can lead to greater effects. Thus, when EH21 plus sCD4 enhances A32 plus sCD4 binding to soluble gp120 (Fig. 5A), the effect probably comes from a doubling of the sCD4 concentration and/or the effect of EH21. Similarly, EH21-plus-sCD4-mediated enhancement of 17b plus sCD4 on soluble gp120 is likely to be mediated by sCD4 rather than EH21 (Fig. 5A).
Overview of competitive effects on VLP Env and soluble gp120.
We first examined the general properties of each antigen (Fig. 5). To obtain an impression of the relative compactness and flexibility, we compared the mean amplitudes of inhibitions and enhancements. Thus, when significant enhancement or inhibition was noted for either antigen, data for both antigens were used to calculate overall mean enhancement and inhibitions (Fig. 6). This revealed that inhibitions were marginally stronger on VLPs than on soluble gp120 (mean values of 13.2% and 26.8%, respectively). Enhancements were also markedly stronger on VLPs than gp120 (mean values of 46,870% and 4,580%, respectively). Consistent with Fig. 2, sCD4 generally mediated more enhancement of MAb binding to VLPs than to soluble gp120 (Fig. 5). Thus, for example, sCD4 marginally enhanced CO11 binding to VLPs but had no effect on its binding to soluble gp120 (Fig. 5).
We next determined the mean competitive or enhancing effects of each MAb and sCD4, shown in columns on the right sides of Fig. 5A and B. The respective data from Fig. 5A and B were then calculated as a ratio (last two columns of Fig. 5A). Here, numbers greater than 1 (green cells) imply a stronger inhibition or enhancement on VLPs. Consistent with Fig. 6, inhibitions and enhancements were generally more effective on VLP Env. Thus, for example, sCD4 more effectively induced the 17b epitope on VLPs (compare Fig. 5A and B). Notably, although sCD4 was responsible for most of the enhancing effects, VRC01 and CO11 also surprisingly induced net enhancements. The only MAbs that did not show preferential effects on VLPs were 2G12 and EH21, which were virtually impartial to the context of their epitopes. Overall, this initial analysis implies that VLP Env exhibits increased epitope overlap, consistent with its relatively compact structure, and furthermore that VLP Env undergoes more dramatic conformational changes in instances of enhancement.
Conformational differences between gp120 and VLP Env.
Of particular interest in Fig. 5 are competitions by mismatched MAb pairs that reveal conformational relationships. Moreover, MAb-MAb combinations with different outcomes in Fig. 5A and B imply conformational differences between soluble gp120 and VLP Env. Since these are of special interest, we describe them first. For clarity, they are emphasized by bold black borders around individual cells in the respective locations of Fig. 5A and B. As mentioned above, differences of 3-fold or greater are considered significant. To emphasize qualitative differences, cells are boxed only when the respective datum in Fig. 5A or B shows competition or enhancement and the other shows little or no effect (i.e., the effect is >30 and <200%; white cells). This rules out cases of lesser interest, where a 3-fold difference occurs but where the inhibitory or enhancing effects are in the same direction.
Competitions between nAbs.
Focusing first on nAb-nAb competitions, we next discuss the differential effects in the top left quadrants of Fig. 5A and B (cells bordered by black boxes). Almost all cases involved the PGT MAbs, suggesting that these epitopes are at the nexus of competitive effects and impact many other epitopes. For convenience, below, bidirectional competitions are taken to imply epitope overlap, although they may also imply close proximity or steric relationships. Specific cases follow.
(i) PGT121 inhibited 2G12 binding to VLPs but not soluble gp120. In the reverse format, 2G12 strongly inhibited PGT121 binding to both antigens, consistent with a previous study (16). This suggests that PGT121 and 2G12 epitopes overlap on VLPs, but on soluble gp120, there is merely a nonoverlapping conformational relationship. Although 2G12 inhibited both PGT MAbs, PGT130 did not effectively inhibit 2G12, suggesting a conformational effect.
(ii) PGT130 inhibited PGT121 binding to VLPs to a 3-fold-greater extent than on gp120. In contrast, PGT121 strongly inhibited PGT130 binding to both antigens. Thus, there is a closer overlap between these epitopes on VLPs.
(iii) IgG1b12 enhanced PGT130 binding to soluble gp120 but not to VLPs. In contrast, PGT130 did not affect b12 binding to either antigen. Thus, there is a nonoverlapping conformational relationship on soluble gp120 but not on VLPs.
(iv) VRC01 inhibited PGT130 binding to VLPs but not soluble gp120. The reverse format was not tested. This implies a conformational relationship of VRC01 and PGT130 on VLPs only.
(v) 17b exacerbated sCD4-mediated inhibition of PGT130 binding to soluble gp120. However, on VLPs, the inhibition was weaker with 17b and sCD4 than with sCD4 alone, possibly due to conflicting enhancing and inhibiting effects. In the reverse format, PGT130 did not affect 17b plus sCD4 binding to either antigen. This suggests a nonoverlapping conformational relationship of PGT130 and 17b plus sCD4.
Competitions between nAbs and non-nAbs.
We next considered competitions between nAbs and non-nAbs. These competitions are in the top right and bottom left data quadrants in Fig. 5A and B. Specific cases of interest (shown in Fig. 5 by black boxes) were as follows.
(i) 2G12 inhibited G3-42 binding to VLPs but not to soluble gp120. The reverse combination was not tested. This suggests a conformational relationship on VLPs only.
(ii) VLPs exhibited greater CD4-mediated enhancement of G3-42 and CO11 binding. The same was observed for 17b and G3-4, but the datum was not boxed because enhancement was observed on both antigens. The reverse format was not tested. Thus, sCD4 has a more marked effect on several epitopes on VLPs than on soluble gp120. As mentioned above, most, if not all, of these effects are related to improved MAb binding to UNC gp160 in the presence of sCD4 (Fig. 2 and 3) (70).
(iii) PGT121 inhibited A32 plus sCD4 binding to VLPs but not to soluble gp120. In the reverse format, A32 plus sCD4 also inhibited PGT121 binding to VLPs more effectively. However, this is difficult to distinguish from inhibition of PGT121 by sCD4 alone. Overall, this suggests a conformational relationship between PGT121 and A32 in the presence of sCD4 on VLPs.
(iv) PGT121 also inhibited b6 more effectively on VLPs than on soluble gp120. In the reverse format, b6 inhibited PGT121 effectively on both antigens. This suggests a greater overlap between these epitopes on VLPs than on soluble gp120.
(v) In contrast, PGT121 inhibited 17b binding to soluble gp120 more effectively than it did on VLPs. In the reverse format, 17b had no effect on PGT121. In contrast, a previous study found modest X5 competition with PGT121 on soluble gp120 (17). Overall, there appears to be a nonoverlapping conformational relationship between PGT121 and 17b on gp120 but not on VLPs.
(vi) PGT121 inhibited CO11 on VLPs but not on soluble gp120. In the reverse format, CO11 inhibited PGT121 on both antigens. This indicates an overlap between these epitopes on VLPs but merely a conformational relationship on soluble gp120. CO11 also inhibited PGT130, but PGT130 was unable to compete with CO11 in the reverse format, suggesting a conformational effect. Overall, the competitions between PGT MAbs and CO11 were quite similar to those with PGT MAbs and 2G12 (Fig. 5). This is perhaps not surprising since they all target epitopes that involve parts of the V3 loop.
(vii) In contrast to PGT121, PGT130 did not exhibit any differences in its ability to compete with non-nAbs on the two antigens. In fact, PGT130 mediated very little competition in general. However, like PGT121, PGT130 showed more competition of b6 on VLPs. Overall, this suggests that there is a consistent an overlap between PGT and b6 epitopes on VLPs. The effects of b6 contrast with those of neutralizing CD4bs MAbs b12 and VRC01, which each exhibit distinct conformational relationships with PGT MAbs, as mentioned above.
(viii) The C4 MAb G3-508 inhibited 17b plus sCD4 on VLPs but not on soluble gp120. We did not test the reverse format. However, 17b plus sCD4 did not inhibit G3-42, a MAb with a related C4-V3 epitope. Thus, there appears to be a conformational relationship between G3-508 and 17b in the presence of sCD4 on VLPs but not on soluble gp120.
Competitions between non-nAbs.
As for those in “Competitions between nAbs and non-nAbs” above, competitions using only nonneutralizing MAbs (lower right quadrants of Fig. 5A and B) are all centered on UNC gp160. Specific cases of interest (shown in Fig. 5 by black boxes) were as follows.
(i) Assays using EH21 or A32 with sCD4 as inhibitors showed markedly more enhancement of MAbs 17b, G3-4, G3-42, and CO11 on VLPs than on soluble gp120. However, inspection of competitions using sCD4 alone reveals similar enhancements, suggesting that sCD4 alone largely mediates these effects.
(ii) MAb b6 inhibited 17b on soluble gp120 but not on VLPs. However, competition was not observed in the reverse format on soluble gp120 and was not tested on VLPs due to weak 17b binding. Taken together, this suggests a conformational relationship of 17b and b6 on soluble gp120.
(iii) The C4 MAb G3-508 inhibited CO11 on VLPs but not on soluble gp120. The reverse format was not tested. This is consistent with a relationship of the C4 and V3 regions, at least on VLPs, and also accounts for the existence of MAbs like G3-42 that recognize C4-V3 composite epitopes (12).
(iv) CO11 remarkably enhanced 17b binding on VLPs but not on soluble gp120, suggesting that CO11 triggers a conformational change in the bridging sheet of VLP Env that allows 17b to bind. Although this induction was potent, it fell short of the inductive effect of sCD4 on 17b binding to VLPs.
Other notable MAb-MAb relationships.
Notable MAb relationships that are not boxed in Fig. 5 are described below.
(i) Inhibitory effects.
(a) In contrast to VRC01, b6 and b12 inhibited 17b binding to soluble gp120, as reported previously (11). On VLPs, 17b binding was inhibited by b12 but not b6, as mentioned above.
(b) Unexpectedly, 2G12 inhibited A32 binding to soluble gp120. This is inconsistent with earlier reports (11, 19). Whether this discrepancy is related to differences in the gp120 isolate used, the producer cell line, or assay methodology is unclear. In the presence of sCD4, this competition was absent on both antigens. In the reverse format, A32 did not inhibit 2G12, suggesting a possible conformational relationship.
(c) Soluble CD4 inhibited PGT MAbs on both antigens but was more potent on VLPs (16).
(d) In gp41 competitions using WT-VLPs, 2.2B was partially inhibited by both 7B2 and 4E10. However, no competition was observed in the reciprocal format, suggesting a conformational relationship. There were no significant competitions between gp120 and gp41 MAbs.
(e) The C4 MAb G3-508 inhibited PGT121 on both antigens.
(ii) Enhancing effects.
(a) VRC01 enhanced 17b binding to soluble gp120, consistent with earlier reports (6, 18). Interestingly, this effect also occurred on VLPs. Since earlier work has shown that VRC01 binding does not induce 17b binding on the native trimer (18), this effect probably occurs on UNC gp160.
(b) A32 induced 17b binding to soluble gp120, as reported previously (11). However, in the reverse format, 17b also enhanced A32 binding to soluble gp120 (11, 19). 17b and A32 binding were too weak to make a similar experiment with VLPs feasible.
(c) EH21 enhanced A32 binding to soluble gp120. Previously, MAb M85, which recognizes an epitope similar to that recognized by EH21, had no effect on A32 binding to HxB2 gp120. This discrepancy may be due to the difference in either Env strains or MAbs (11).
(d) Both b6 and 17b marginally induced G3-4 binding to soluble gp120, consistent with a previous study (11). The effect of b6 was not observed on VLPs.
(e) A32 binding (in the presence or absence of sCD4) to soluble gp120, and in some cases to VLPs, was exacerbated in the presence of 17b, CO11, or EH21.
DISCUSSION
Due to methodological limitations, the antigenicity of Env expressed in situ on lipid membranes has not been comprehensively studied until now. In this study, we used a unified ELISA method to facilitate direct, harmonized comparisons of membrane-bound and soluble Envs over a range of MAb concentrations, with the intent to update and expand on the topological information gleaned from earlier studies (3, 6, 9, 10, 12, 22–24, 34, 35, 41). Our VLP ELISA was found to be sensitive and to exhibit a considerable dynamic range, as evidenced by dramatic (>105-fold) binding increases (Fig. 5), supporting the reliability of this approach.
Most of the binding patterns in our study concern the immunodominant form of membrane Env, namely, UNC gp160. Gathering more information on membrane UNC gp160 is important for several reasons. First, it is the major antigen through which antibodies bind free HIV-1 virus and infected cells, and therefore, it probably functions as a decoy antigen whose raison d'être may largely be to deflect antibody responses away from the native trimer. Indeed, we observed very strong binding of non-nAbs to VLPs, particularly those directed to V3 and CD4bs epitopes (Fig. 2). Second, based on the data of Fig. 2, it is possible that the germ line precursors of broad neutralizing antibodies (bnAbs) may initially interact with UNC gp160 rather than the native trimer, simply because it is an attractive and immunodominant antigen. Thus, UNC gp160 could be an integral part of nAb ontogeny, which might culminate in the appearance of mutations that allow cross-reactivity with the native trimer. If so, the competitive relationships on UNC gp160 in Fig. 5 could be important in dictating if and when nAbs develop. For example, b6-like nonneutralizing CD4bs antibodies might overlap the epitopes of PGT-like MAb precursors on UNC gp160 and therefore might interfere with the propagation and eventual development of mature PGT-like bnAbs. Third, UNC gp160 and gp41 stumps may impact virus opsonization, capture, and effector functions such as ADCVI, complement-mediated cellular inhibition effects, and/or transcytosis (61, 85–91).
Three lines of evidence suggest that membrane-expressed UNC gp160 is relatively compact. First, VLPs expose fewer epitopes than soluble gp120 (Fig. 4). There is also a clear trend of lower affinity for those MAbs that do bind. Thus, epitopes appear to be partially or fully hidden in a more compact conformation. Second, UNC gp160 exhibits increased competitive or enhancing MAb relationships, consistent with a generally greater proximity of epitopes (Fig. 6). Indeed, a previous study reported that soluble gp120 derived from a neutralization-resistant isolate exhibited more epitope proximities than soluble gp120 from a sensitive strain (19), showing that context can affect conformation. The greater proximities of multiple epitopes of UNC gp160 therefore suggest relatively compact conformation. Third, UNC gp160 undergoes more pronounced sCD4-induced conformational changes (Fig. 2). This suggests that gp120 can take on any of a range of conformational states (92), depending on its context, as shown in Fig. 7. Thus, if the native Env trimer is completely untriggered, UNC gp160 is somewhat more triggered, followed by soluble gp120, fully triggered sCD4-bound forms of Env, and gp120 cores (92). This model is perhaps best supported by considering CD4i epitope exposure as an arbiter of coreceptor exposure or “triggering.” Thus, the 17b (CD4i) epitope is constitutively exposed on the gp120 core, even in the absence of sCD4, consistent with full triggering (5, 92). Other forms of Env expose 17b less effectively but undergo conformational changes in the presence of sCD4 to expose this epitope. Thus, 17b is less well exposed on the full-length gp120 monomer and undergoes an ∼50-fold induction upon sCD4 binding (Fig. 2). VLPs expose the 17b epitope even less effectively and undergo an even more dramatic, 1,500-fold, induction upon the addition of sCD4 (Fig. 2). The unique induction of the 17b epitope by CO11 binding to VLPs suggests an inherent propensity for UNC gp160 conformational changes (Fig. 5). Finally, 17b fails to bind to the JR-FL native trimer (33) or neutralize infection (70), consistent with an untriggered resting state. The remarkable induction of 17b neutralization by adding sCD4 (>100,000-fold [70]) and the associated structural changes observed by cryo-electron microscopy (cryo-EM) (93) suggest that the native trimer undergoes a massive rearrangement as it achieves a fully triggered state. Assuming that after CD4 triggering, each form of Env reaches a similar state of induction regardless of its initial state, then the less triggered the ground state, the more profound are the changes that occur when sCD4 is added.
Fig 7.
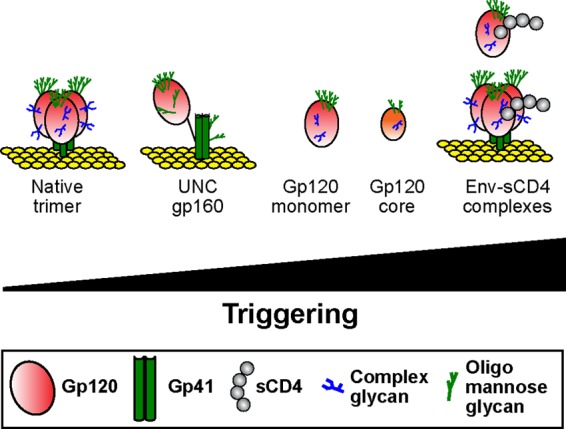
Spectrum of Env triggering states. Various forms of Env are aligned according to their inferred state of triggering, as judged by their relative exposure of CD4i epitopes, in the absence and presence of soluble CD4 ligand (gray circles). As in Fig. 1, complex glycans are indicated by blue tree structures and oligomannose glycans by green tree structures.
gp120 exists in a progressively less triggered state with the increasing complexity of its presentation. More specifically, progressive increases in compactness occur upon the addition of variable loops to the gp120 core and upon further addition of gp41, expression in membranes, and gp120/gp41 processing. Thus, the relatively untriggered state of UNC gp160 compared to soluble gp120 might be expected, considering that, like the native trimer, it resides in a plasma membrane, that it includes gp41, and also that it is a precursor of the native trimer.
An alternative explanation for the different antigenicities of monomeric gp120 and VLPs in Fig. 2 may relate to their glycosylation. gp160ER is a prominent form of Env species on VLP surfaces (46) and exhibits a relatively sparse coat of immature high-mannose glycans. For this reason, it may be particularly accessible to MAb and therefore immunodominant. In contrast, monomeric gp120 exhibits a substantial fraction of high-mass complex glycans (76). However, despite its thicker glycan blanket, soluble gp120 was more accessible to MAbs than VLP Env (Fig. 4), suggesting that another factor(s) accounts for the decreased MAb accessibility to VLP Env. As mentioned above, the context of the gp120 moiety in the form of UNC gp160 in membranes is probably more important in determining its antigenicity than its glycosylation pattern.
The epitope exposure patterns of VLP Env (summarized in Fig. 4) reveal that while CD4bs, V3, 2G12, PGT121, PGT125, PGT130, and all gp41 epitopes were well exposed, other epitopes either were poorly exposed (C4, CD4i, C1-C4, V2, PGT135, and PGT136) or were nearly completely occluded (C1, C2, C5, and C1-C5) (Fig. 2). The high exposure of nonneutralizing V3 and gp41 epitopes may explain their high immunogenicity (71) and their possible role as antigenic decoys.
The C1 and C5 epitopes are largely occluded on UNC gp160, presumably due to their interactions with gp41. The poor C5 exposure may explain the inefficient processing of gp160 into gp120 and gp41 by furin, due to limited accessibility. Our data may reflect the possible biological roles of UNC gp160 during infection and immune responses. Thus, for example, the lack of effective A32 or V2 loop MAb binding to surface UNC gp160 (Fig. 2) may limit the antibody-dependent cellular cytotoxicity (ADCC) activity (61) or α4β7 capture that has been reported to be associated with these determinants (94, 95). Thus, in light of the conformational differences observed in our study, we recommend that VLPs or similar membrane-expressed forms of Env be used in assays in place of soluble forms of Env for the routine assessment of relevant anti-Env responses and biological activities, so that we can assign proper weight to these effects.
Our antigenicity data (Fig. 2) largely agree with earlier studies (3, 6, 9, 10, 12, 41), although there were some discrepancies (22–24, 34, 35) that might be related to differences in the particular Env-virus isolate, producer cell lines, or assay methodology. In agreement, the V3 loop and gp41 cluster I epitopes were well exposed and the C2 and V2 domains were partially occluded. However, the CD4bs, 2G12, gp41 cluster II, and MPER epitopes were only modestly exposed by virus capture but were well exposed in our ELISAs. Furthermore, some C5 epitopes were well exposed by virus capture (35) but not in VLP ELISA (Fig. 2) or flow cytometry (3). The use of MAb 670-D in our study allowed us to bridge previous virus capture data with our ELISA data. Although 670-D captured HIV-1 (35), it failed to bind VLPs (Fig. 2). The basis for these discrepancies is more likely to be assay related. Having compared these assays, we suggest that VLP ELISA data are more reliable, due to this test's high interassay consistency and low nonspecific binding. The fact that our data are derived from titrations rather than single MAb dilutions adds to their reliability (20, 32, 33, 40, 80).
VLP ELISAs facilitated a detailed investigation of MAb binding relationships (Fig. 5), the findings of which were interpreted informatively by the use of Venn diagrams (Fig. 8). Bidirectional competitions are indicated as epitope overlaps, although they may also reflect close proximity or steric effects. Some MAb binding relationships were consistent on both soluble gp120 and VLP antigens (Fig. 8). For example, b12 was inhibited only by other CD4bs MAbs and sCD4. Others were specific to one or the other antigen. Some relationships were reported previously (6, 11, 17, 22–24, 44), while others are unique. The latter largely involve the PGT MAbs, which are the nexus of the increased epitope overlaps on VLP Env (Fig. 8). The differences between the antigens largely relate to changes in the conformation of surface loops. Thus, for example, on VLPs, PGT121 inhibited CO11 and 2G12 far more effectively. At the other end of the spectrum, the EH21 MAb did not have any notable effects on other epitopes, consistent with its epitope being largely isolated on both antigens (11, 19).
Fig 8.

Visual interpretation of ligand binding relationships on soluble gp120 and VLP Env. The ligand binding relationships from Fig. 5 are interpreted as Venn diagrams for gp120 (A) and VLPs (B). Each epitope footprint is depicted as a circle. Bidirectionally competing epitopes are shown as intersecting circles. Nonoverlapping, conformational effects are shown as contacting circles. MAbs with no effect on others are shown as isolated circles. For clarity, competitive effects are depicted in the central diagrams of panels A and B. Enhancements by sCD4 or MAbs (b12, VRC01, and CO11) are shown as additional diagrams emanating from the two central ones. For clarity, the color scheme of Fig. 5 is used. Thus, competitions are depicted by progressively warmer colors in the shared space of intersecting circles. Similarly, enhancements are depicted as cyan and magenta circles in the outer Venn diagrams. Black intersecting areas indicate untested relationships of tertiary ligand combinations that are expected to be competitive. Because a single circle did not provide sufficient space to depict all the binding relationships of MAbs b6, G3-42, VRC01, and sCD4, these ligands are in some cases depicted by two circles.
The PGT121 epitope overlaps with more MAbs than PGT130 and therefore is the overall focal point. Both MAbs exhibit relationships with 2G12, CO11, and CD4bs MAbs. Considering the N332-dependent epitopes of PGT MAbs at the base of the V3, the relationships with V3 MAbs and 2G12 are not surprising. The relationships with CD4bs MAbs are more revealing. The CD4bs-PGT MAb relationship was examined in more detail than previously (17), using different CD4bs MAbs and reciprocal competitions. This revealed complex and variable relationships, depending on the particular MAbs studied. While there are many similarities between PGT121 and PGT130 MAbs, there were a few interesting distinctions, some of which occurred on only one antigen. Perhaps most notable of these was the b12 enhancement of PGT130 but not PGT121 on soluble gp120 but not on VLPs. Together, these patterns imply a nonoverlapping conformational relationship of PGT and CD4bs epitopes. Possibly, the orientation of MAb binding to these epitopes can occasionally cause steric clashes, resulting in unpredictable, MAb-specific binding patterns. The generally increased competition around PGT epitopes reflects the closer spatial relationships on UNC gp160.
The relationships in Fig. 8 exhibit some general similarities with earlier data, but also some differences (6, 11, 16, 18, 19, 22, 24). One example is the effect of VRC01 on 17b. Consistent with earlier reports, VRC01 enhanced 17b binding to soluble gp120 (6, 18). Unexpectedly, and in contrast to previous flow cytometry data, this enhancement also occurred on VLPs (6, 18). Since VRC01 does not enhance 17b neutralization (see Fig. S7 in reference 18), it is likely that VRC01 induces 17b binding to UNC gp160, not the native trimer. The reasons for this discrepancy may be methodological and may relate to relative UNC gp160 expression. Interestingly, the VRC01-mediated enhancement of 17b binding to VLPs is similar to CO11-mediated induction of 17b on VLPs mentioned above, again showing the increased conformational flexibility of UNC gp160 upon MAb and sCD4 binding.
Recently, a high-resolution structure of membrane-extracted UNC gp160 was reported to be a trimer with antigenic properties similar to those of cell surface-expressed Env (96). Although only a small panel of nAbs and no nonneutralizing antibodies were used in the study of Mao et al. (96) considering the crucial role of gp120-gp41 processing in the assembly of native trimers (33), it is likely that nonneutralizing epitopes are exposed on these extracted UNC trimers, as they are on the membrane-associated UNC gp160 reported here. Considering that the study by Mao et al. used the same Env, JR-FL, as we used in this study, it is perhaps surprising that the extracted UNC gp160 is trimeric, whereas it is largely a monomer in membranes (Fig. 1). This apparent contradiction might be explained by our observation in BN-PAGE that UNC gp160 tends to multimerize or aggregate over time after it is extracted from membranes (data not shown).
In conclusion, our data provide new information on the major targets of the overwhelmingly dominant nonneutralizing response to natural HIV-1 infection. Despite this lack of neutralization, IgG engagement with these antigens is a key aspect of the interplay between HIV and antibodies during natural infection. Since these interactions may be associated with certain protective effects, our results may have broad implications for pathogenesis and vaccine design. A similar MAb cross-competition analysis is now under way using VLPs that have been protease digested to clear nonfunctional Env, leaving essentially only native Env trimer (42, 46). This should improve our understanding of functional Env topology and the interrelationships of neutralizing antibodies.
ACKNOWLEDGMENTS
This work was supported by grants RO1AI93278, RO1AI58763, and R21AI84714 (J.M.B.; NIH/NIAID/DAIDS) and the Torrey Pines Institute's AIDS and Infectious Disease Science Center (J.M.B.).
We thank IAVI, Progenics Pharmaceuticals, Inc., the MRC and NIH AIDS Reagent Programs, Quentin Sattentau, and the listed suppliers for providing MAbs, gp120, and sCD4. J.M.B. is grateful to J. P. Moore, whose early studies inspired this work.
Footnotes
Published ahead of print 5 June 2013
REFERENCES
- 1. Fouts TR, Binley JM, Trkola A, Robinson JE, Moore JP. 1997. Neutralization of the human immunodeficiency virus type 1 primary isolate JR-FL by human monoclonal antibodies correlates with antibody binding to the oligomeric form of the envelope glycoprotein complex. J. Virol. 71:2779–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li Y, O'Dell S, Walker LM, Wu X, Guenaga J, Feng Y, Schmidt SD, McKee K, Louder MK, Ledgerwood JE, Graham BS, Haynes BF, Burton DR, Wyatt RT, Mascola JR. 2011. Mechanism of neutralization by the broadly neutralizing HIV-1 monoclonal antibody VRC01. J. Virol. 85:8954–8967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moore JP, Sattentau QJ, Wyatt RT, Sodroski J. 1994. Probing the structure of the human immunodeficiency virus surface glycoprotein gp120 with a panel of monoclonal antibodies. J. Virol. 68:469–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pantophlet R, Saphire EO, Poignard P, Parren PW, Wilson IA, Burton DR. 2003. Fine mapping of the interaction of neutralizing and nonneutralizing monoclonal antibodies with the CD4 binding site of human immunodeficiency virus type 1 gp120. J. Virol. 77:642–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Binley JM, Wyatt RT, Desjardins E, Kwong PD, Hendrickson W, Moore JP, Sodroski J. 1998. Analysis of the interaction of antibodies with a conserved enzymatically deglycosylated core of the HIV type 1 envelope glycoprotein 120. AIDS Res. Hum. Retroviruses 14:191–198 [DOI] [PubMed] [Google Scholar]
- 6. Falkowska E, Ramos A, Feng Y, Zhou T, Moquin S, Walker LM, Wu X, Seaman MS, Wrin T, Kwong PD, Wyatt RT, Mascola JR, Poignard P, Burton DR. 2012. PGV04, an HIV-1 gp120 CD4 binding site antibody, is broad and potent in neutralization but does not induce conformational changes characteristic of CD4. J. Virol. 86:4394–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. He Y, Honnen WJ, Krachmarov CP, Burkhart M, Kayman SC, Corvalan J, Pinter A. 2002. Efficient isolation of novel human monoclonal antibodies with neutralizing activity against HIV-1 from transgenic mice expressing human Ig loci. J. Immunol. 169:595–605 [DOI] [PubMed] [Google Scholar]
- 8. Hioe CE, Visciano ML, Kumar R, Liu J, Mack EA, Simon RE, Levy DN, Tuen M. 2009. The use of immune complex vaccines to enhance antibody responses against neutralizing epitopes on HIV-1 envelope gp120. Vaccine 28:352–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y, O'Dell S, Wilson R, Wu X, Schmidt SD, Hogerkorp CM, Louder MK, Longo NS, Poulsen C, Guenaga J, Chakrabarti BK, Doria-Rose N, Roederer M, Connors M, Mascola JR, Wyatt RT. 2012. HIV-1 neutralizing antibodies display dual recognition of the primary and coreceptor binding sites and preferential binding to fully cleaved envelope glycoproteins. J. Virol. 86:11231–11241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moore JP, Sattentau QJ, Yoshiyama H, Thali M, Charles M, Sullivan N, Poon SW, Fung MS, Traincard F, Pinkus M, Robey G, Robinson JE, Ho DD, Sodroski J. 1993. Probing the structure of the V2 domain of human immunodeficiency virus type 1 surface glycoprotein gp120 with a panel of eight monoclonal antibodies: human immune response to the V1 and V2 domains. J. Virol. 67:6136–6151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moore JP, Sodroski J. 1996. Antibody cross-competition analysis of the human immunodeficiency virus type 1 gp120 exterior envelope glycoprotein. J. Virol. 70:1863–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moore JP, Thali M, Jameson BA, Vignaux F, Lewis GK, Poon SW, Charles M, Fung MS, Sun B, Durda PJ, Åkerblom L, Wahren B, Ho DD, Sattentau QJ, Sodroski J. 1993. Immunochemical analysis of the gp120 surface glycoprotein of human immunodeficiency virus type 1: probing the structure of the C4 and V4 domains and the interaction of the C4 domain with the V3 loop. J. Virol. 67:4785–4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moore JP, Willey RL, Lewis GK, Robinson J, Sodroski J. 1994. Immunological evidence for interactions between the first, second, and fifth conserved domains of the gp120 surface glycoprotein of human immunodeficiency virus type 1. J. Virol. 68:6836–6847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sattentau QJ, Moore JP. 1991. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J. Exp. Med. 174:407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Visciano ML, Tuen M, Gorny MK, Hioe CE. 2008. In vivo alteration of humoral responses to HIV-1 envelope glycoprotein gp120 by antibodies to the CD4-bionding site of gp120. Virology 372:409–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walker LM, Huber M, Doore KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Koff WC, Wilson IA, Burton DR, Poignard P. 2011. Broad neutralizing coverage of HIV by multiple highly potent antibodies. Nature 477:466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O'Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ye Y, Si ZH, Moore JP, Sodroski J. 2000. Association of structural changes in the V2 and V3 loops of the gp120 envelope glycoprotein with acquisition of neutralization resistance in a simian-human immunodeficiency virus passaged in vivo. J. Virol. 74:11955–11962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Agrawal N, Leaman DP, Rowcliffe E, Kinkead H, Nohria R, Akagi J, Bauer K, Du SX, Whalen RG, Burton DR, Zwick MB. 2011. Functional stability of unliganded envelope glycoprotein spikes among isolates of human immunodeficiency virus type 1 (HIV-1). PLoS One 6:e21339. 10.1371/journal.pone.0021339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bou-Habib DC, Roderiquez G, Oravecz T, Berman PW, Lusso P, Norcross MA. 1994. Cryptic nature of envelope V3 region epitopes protects primary monocytotropic human immunodeficiency virus type 1 from antibody neutralization. J. Virol. 68:6006–6013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cavacini L, Duval M, Song L, Sangster R, Xiang SH, Sodroski J, Posner M. 2003. Conformational changes in env oligomer induced by an antibody dependent on the V3 loop base. AIDS 17:658–689 [DOI] [PubMed] [Google Scholar]
- 23. Cavacini L, Posner M. 2004. Native HIV type 1 virion surface structures: relationships between antibody binding and neutralization or lessons from the viral capture assay. AIDS Res. Hum. Retroviruses 20:435–441 [DOI] [PubMed] [Google Scholar]
- 24. Cavacini LA, Duval M, Robinson J, Posner MR. 2002. Interactions of human antibodies, epitope exposure, antibody binding and neutralization of primary isolate HIV-1 virions. AIDS 16:2409–2417 [DOI] [PubMed] [Google Scholar]
- 25. Cavacini LA, Peterson JE, Nappi E, Duval M, Goldstein R, Mayer K, Posner MR. 1999. Minimal incidence of serum antibodies reactive with intact primary isolate virions in human immunodeficiency virus type 1-infected individuals. J. Virol. 73:9638–9641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chakrabarti BK, Walker LM, Guenaga JF, Ghobbeh A, Poignard P, Burton DR, Wyatt RT. 2011. Direct antibody access to the HIV-1 membrane-proximal external region positively correlates with neutralization sensitivity. J. Virol. 85:8217–8226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Finnegan CM, Berg W, Lewis GK, DeVico AL. 2001. Antigenic properties of the human immunodeficiency virus envelope during cell-cell fusion. J. Virol. 75:11096–11105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Finnegan CM, Berg W, Lewis GK, DeVico AL. 2002. Antigenic properties of the human immunodeficiency virus transmembrane glycoprotein during cell-cell fusion. J. Virol. 76:12123–12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gorny MK, Revesz K, Williams C, Volsky B, Louder MK, Anyangwe CA, Krachmarov C, Kayman SC, Pinter A, Nadas A, Nyambi PN, Mascola JR, Zolla-Pazner S. 2004. The V3 loop is accessible on the surface of most human immunodeficiency virus type 1 primary isolates and serves as a neutralization epitope. J. Virol. 78:2394–2404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Helseth E, Olshevsky U, Furman C, Sodroski S. 1991. Human immunodeficiency virus type 1 gp120 envelope glycoprotein regions important for association with the gp41 transmembrane glycoprotein. J. Virol. 65:2119–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Honnen WJ, Krachmarov C, Kayman SC, Gorny MK, Zolla-Pazner S, Pinter A. 2007. Type-specific epitopes targeted by monoclonal antibodies with exceptionally potent neutralizing activities for selected strains of human immunodeficiency virus type 1 map to a common region of the V2 domain of gp120 and differ only at single positions from the clade B consensus sequence. J. Virol. 81:1424–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leaman DP, Kinkead H, Zwick MB. 2010. In-solution virus capture assay helps deconstruct heterogenous antibody recognition of human immunodeficiency virus type 1. J. Virol. 84:3382–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moore PL, Crooks ET, Porter L, Zhu P, Cayanan CS, Grise H, Corcoran P, Zwick MB, Franti M, Morris L, Roux KH, Burton DR, Binley JM. 2006. Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J. Virol. 80:2515–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nyambi PN, Gorny MK, Bastiani L, van der Groen G, Williams C, Zolla-Pazner S. 1998. Mapping of epitopes exposed on intact human immunodeficiency virus type 1 (HIV-1) virions: a new strategy for studying the immunologic relatedness of HIV-1. J. Virol. 72:9384–9391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nyambi PN, Nadas A, Mbah A, Burda S, Williams C, Gorny MK, Zolla-Pazner S. 2000. Immunoreactivity of intact virions of human immunodeficiency virus type 1 (HIV-1) reveals the existence of fewer HIV-1 immunotypes than genotypes. J. Virol. 74:10670–10680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ota T, Doyle-Cooper C, Cooper AB, Huber M, Falkowska E, Doores KJ, Hangartner L, Le K, Sok D, Jardine J, Lifson J, Wu X, Mascola JR, Poignard P, Binley JM, Chakrabarti BK, Schief WR, Wyatt RT, Burton DR, Nemazee D. 2012. Anti-HIV B cell lines as candidate vaccine biosensors. J. Immunol. 189:4816–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pantophlet R. 2010. Antibody epitope exposure and neutralization of HIV-1. Curr. Pharm. Des. 16:3729–3743 [DOI] [PubMed] [Google Scholar]
- 38. Pantophlet R, Wrin T, Cavacini LA, Robinson JE, Burton DR. 2008. Neutralizing activity of antibodies to the V3 loop region of HIV-1 gp120 relative to their epitope fine specificity. Virology 381:251–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pinter A, Honnen WJ, He Y, Gorny MK, Zolla-Pazner S, Kayman SC. 2004. The V1/V2 domain of gp120 is a global regulator of the sensitivity of primary human immunodeficiency virus type 1 isolates to neutralization by antibodies commonly induced upon infection. J. Virol. 78:5205–5215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poignard P, Moulard M, Golez E, Vivona V, Franti M, Venturini S, Wang M, Parren PW, Burton DR. 2003. Heterogeneity of envelope molecules expressed on primary human immunodeficiency virus type 1 particles as probed by the binding of neutralizing and nonneutralizing antibodies. J. Virol. 77:353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sattentau QJ, Zolla-Pazner S, Poignard P. 1995. Epitope exposure on functional, oligomeric HIV-1 gp41 molecules. Virology 206:713–717 [DOI] [PubMed] [Google Scholar]
- 42. Tong T, Crooks ET, Osawa K, Binley JM. 2012. HIV-1 virus-like particles bearing pure env trimers expose neutralizing epitopes but occlude nonneutralizing epitopes. J. Virol. 86:3574–3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wyatt RT, Desjardin E, Olshevsky U, Nixon C, Binley J, Olshevksy V, Sodroski J. 1997. Analysis of the interaction of the human immunodeficiency virus type 1 gp120 envelope glycoprotein with the gp41 transmembrane glycoprotein. J. Virol. 71:9722–9731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wyatt RT, Moore J, Accola M, Desjardin E, Robinson J, Sodroski S. 1995. Involvement of the V1/V2 variable loop structure in the exposure of human immunodeficiency virus type 1 gp120 epitopes induced by receptor binding. J. Virol. 69:5723–5733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sattentau QJ, Moore JP. 1995. Human immunodeficiency virus type 1 neutralization is determined by epitope exposure on the gp120 oligomer. J. Exp. Med. 182:185–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Crooks ET, Tong T, Osawa K, Binley JM. 2011. Enzyme digests eliminate nonfunctional Env from HIV-1 particle surfaces, leaving native Env trimers intact and viral infectivity unaffected. J. Virol. 85:5825–5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gorny MY, Williams C, Volsky B, Revesz K, Cohen S, Polonis VR, Honnen WJ, Kayman SC, Krachmarov C, Pinter A, Zolla-Pazner S. 2002. Human monoclonal antibodies specific for conformation-sensitive epitopes of V3 neutralize human immunodeficiency virus type 1 primary isolates from various clades. J. Virol. 76:9035–9045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Herrera C, Spenlehauer C, Fung MS, Burton DR, Beddows S, Moore JP. 2003. Nonneutralizing antibodies to the CD4-binding site on the gp120 subunit of human immunodeficiency virus type 1 do not interfere with the activity of a neutralizing antibody against the same site. J. Virol. 77:1084–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Buchbinder A, Karwowska S, Gorny MK, Burda ST, Zolla-Pazner S. 1992. Synergy between human monoclonal antibodies to HIV extends their effective biologic activity against homologous and divergent strains. AIDS Res. Hum. Retroviruses 8:425–427 [DOI] [PubMed] [Google Scholar]
- 50. Doria-Rose NA, Louder MK, Yang Z, O'Dell S, Nason M, Schmidt SD, McKee K, Seaman MS, Bailer RT, Mascola JR. 2012. HIV-1 neutralization coverage is improved by combining monoclonal antibodies that target independent epitopes. J. Virol. 86:3393–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Laal S, Burda S, Gorny MK, Karwowska A, Buchbinder A, Zolla-Pazner S. 1994. Synergistic neutralization of human immunodeficiency virus type 1 by combinations of human monoclonal antibodies. J. Virol. 68:4001–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li A, Katinger H, Posner MR, Cavacini L, Zolla-Pazner S, Gorny MK, Sodroski J, Chou TC, Baba TW, Ruprecht R. 1998. Synergistic neutralization of simian-human immunodeficiency virus SHIV-vpu+ by triple and quadruple combinations of human monoclonal antibodies and high-titer anti-human immunodeficiency virus type 1 immunoglobulins. J. Virol. 72:3235–3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Verrier F, Nadas A, Gorny MK, Zolla-Pazner S. 2001. Additive effects characterize the interaction of antibodies involved in neutralization of the primary dualtropic human immunodeficiency virus type 1 isolate 89.6. J. Virol. 75:9177–9186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu W, Smith-Franklin BA, Li PL, Wood C, He J, Du Q, Bhat GJ, Kankasa C, Katinger H, Cavacini LA, Posner MR, Burton DR, Chou TC, Ruprecht R. 2001. Potent neutralization of primary human immunodeficiency virus clade C isolates with a synergistic combination of human monoclonal antibodies raised against clade B. J. Virol. 4:55–61 [PubMed] [Google Scholar]
- 55. Zwick MB, Wang M, Poignard P, Stiegler G, Katinger H, Burton DR, Parren PW. 2001. Neutralization synergy of human immunodeficiency virus type 1 primary isolates by cocktails of broadly neutralizing antibodies. J. Virol. 75:12198–12208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Decker JM, Bibollet-Ruche F, Wei X, Wang S, Levy DN, Wang W, Delaporte E, Peeters M, Derdeyn CA, Allen S, Hunter E, Saag MS, Hoxie JA, Hahn BH, Kwong PD, Robinson JE, Shaw GM. 2005. Antigenic conservation and immunogenicity of HIV coreceptor binding site. J. Exp. Med. 201:1407–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Haynes BF, Fleming J, St Clair EW, Katinger H, Stiegler G, Kunert R, Robinson J, Scearce RM, Plonk K, Staats HF, Ortel TL, Liao HX, Alam SM. 2005. Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 308:1906–1908 [DOI] [PubMed] [Google Scholar]
- 58. Kang CY, Hariharan K, Nara PL, Sodroski J, Moore JP. 1994. Immunization with a soluble CD4-gp120 complex preferentially induces neutralizing anti-human immunodeficiency virus type 1 antibodies directed to conformational-dependent epitopes of gp120. J. Virol. 68:5854–5862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Niedrig M, Harthus HP, Hinkula J, Broker M, Bickhard H, Pauli G, Gelderblom HR, Wahren B. 1992. Inhibition of viral replication by monoclonal antibodies directed against human immunodeficiency virus gp120. J. Gen. Virol. 73:2451–2455 [DOI] [PubMed] [Google Scholar]
- 60. Tuen M, Visciano ML, Chien PC, Jr, Cohen S, Chen PD, Robinson J, He Y, Pinter A, Gorny MK, Hioe CE. 2005. Characterization of antibodies that inhibit HIV gp120 antigen processing and presentation. Eur. J. Immunol. 35:2541–2551 [DOI] [PubMed] [Google Scholar]
- 61. Ferrari G, Pollara J, Kozink D, Harms T, Drinker M, Freel S, Moody MA, Alam SM, Tomaras GD, Ochsenbauer C, Kappes JC, Shaw GM, Hoxie JA, Robinson JE, Haynes BF. 2011. An HIV-1 gp120 envelope human monoclonal antibody that recognizes a C1 conformational epitope mediates potent antibody-dependent cellular cytotoxicity (ADCC) activity and defines a common ADCC epitope in human HIV-1 serum. J. Virol. 85:7029–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sun NC, Ho DD, Sun CR, Liou RS, Gordon W, Fung MS, Li XL, Ting RC, Lee TH, Chang NT, Chang TW. 1989. Generation and characterization of monoclonal antibodies to the putative CD4-binding domain of human immunodeficiency virus type 1 gp120. J. Virol. 63:3579–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Labrijn AF, Poignard P, Raja A, Zwick MB, Delgado K, Franti M, Binley J, Vivona V, Grundner C, Huang CC, Venturi M, Petropoulos CJ, Wrin T, Dimitrov DS, Robinson J, Kwong PD, Wyatt RT, Sodroski J, Burton DR. 2003. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J. Virol. 77:10557–10565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sanders RW, Venturi M, Schiffner L, Kalyanaraman R, Katinger H, Lloyd KO, Kwong PD, Moore JP. 2002. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J. Virol. 76:7293–7305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Scanlan CN, Panthophlet R, Wormald MR, Ollmann Saphire E, Stanfield R, Wilson IA, Katinger H, Dwek RA, Rudd PM, Burton DR. 2002. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of α1→2 mannose residues on the outer face of gp120. J. Virol. 76:7306–7321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Barbas CF, Collet TA, III, Amberg W, Roben P, Binley JM, Hoekstra D, Cababa D, Jones TM, Williamson RA, Pilkington GR, Haigwood NL, Cabezas E, Satterthwait AC, Sanz I, Burton DR. 1993. Molecular profile of an antibody response to HIV-1 as probed by combinatorial libraries. J. Mol. Biol. 230:812–823 [DOI] [PubMed] [Google Scholar]
- 67. Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. 2012. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491:406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zwick MB, Saphire EO, Burton DR. 2004. gp41: HIV's shy protein. Nat. Med. 10:133–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Binley JM, Cayanan CS, Wiley C, Schulke N, Olson WC, Burton DR. 2003. Redox-triggered infection by disulfide-shackled human immunodeficiency virus type 1 pseudovirions. J. Virol. 77:5678–5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Crooks ET, Moore PL, Richman D, Robinson J, Crooks JA, Franti M, Schulke N, Binley JM. 2005. Characterizing anti-HIV monoclonal antibodies and immune sera by defining the mechanism of neutralization. Hum. Antibodies 14:101–113 [PMC free article] [PubMed] [Google Scholar]
- 71. Binley JM, Sander RW, Clas B, Schulke N, Master A, Guo Y, Kajumo F, Anselma DJ, Maddon PJ, Olson WC, Moore JP. 2000. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J. Virol. 74:627–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Crooks ET, Moore PL, Franti M, Cayanan SC, Zhu P, Jiang P, de Vries RP, Wiley C, Zharkikh I, Schulke N, Roux KH, Montefiori DC, Burton DR, Binley JM. 2007. A comparative immunogenicity study of HIV-1 virus-like particles bearing various forms of envelope proteins, particles bearing no envelope and soluble monomeric gp120. Virology 366:245–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rossio JL, Esser MT, Suryanarayana K, Schneider DK, Bess JW, Jr, Vasquez GM, Wiltrout TA, Chertova E, Grimes MK, Sattentau Q, Arthur LO, Henderson LE, Lifson JD. 1998. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J. Virol. 72:7992–8001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kolchinsky P, Kiprilov E, Sodroski J. 2001. Increased neutralization sensitivity of CD4-independent human immunodeficiency virus variants. J. Virol. 75:2041–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bonomelli C, Doores KJ, Dunlop DC, Thaney V, Dwek RA, Burton DR, Crispin M, Scanlan CN. 2011. The glycan shield of HIV is predominantly oligomannose independently of production system or viral clade. PLoS One 6:e23521. 10.1371/journal.pone.0023521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Doores KJ, Bonomelli C, Harvey DJ, Vasiljevic S, Dwek RA, Burton DR, Crispin M, Scanlan CN. 2010. Envelope glycans of immunodeficiency virions are almost entirely oligomannose antigens. Proc. Natl. Acad. Sci. U. S. A. 107:13800–13805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Raska M, Takahashi K, Czernekova L, Zachova K, Hall S, Moldoveanu Z, Elliott MC, Wilson L, Brown R, Jancova D, Barnes S, Vrbkova J, Tomana M, Smith PD, Mestecky J, Renfrow MB, Noak J. 2010. Glycosylation patterns of HIV-1 gp120 depend on the type of expressing cells and affect antibody recognition. J. Biol. Chem. 285:20860–20869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Center RJ, Earl PL, Lebowitz J, Schuck P, Moss B. 2000. The human immunodeficiency virus type 1 gp120 V2 domain mediates gp41-independent intersubunit contacts. J. Virol. 74:4448–4455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Finzi A, Pacheco B, Zeng X, Kwon YD, Kwong PD, Sodroski J. 2010. Conformational characterization of aberrant disulfide-linked HIV-1 gp120 dimers secreted from overexpressing cells. J. Virol. Methods 168:155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Leaman DP, Zwick MB. 2013. Increased functional stability and homogeneity of viral envelope spikes through directed evolution. PLoS Pathog. 9:e1003184. 10.1371/journal.ppat.1003184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pancera M, Wyatt R. 2005. Selective recognition of oligomeric HIV-1 primary isolate envelope glycoproteins by potently neutralizing ligands requires efficiency precursor cleavage. Virology 332:145–156 [DOI] [PubMed] [Google Scholar]
- 82. Julien JP, Lee JH, Cupo A, Murin CD, Berking R, Hoffenberg S, Caufield MJ, King CR, Marozsan AJ, Klasse PJ, Sanders RW, Moore JP, Wilson IA, Ward AB. 2013. Asymmetric recognition of the HIV-1 trimer by broadly neutralizing antibody PG9. Proc. Natl. Acad. Sci. U. S. A. 110:4351–4356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Loving R, Sjoberg M, Wu SR, Binley JM, Garoff H. 2013. Inhibition of the HIV-1 spike by single PG9/16 antibody binding suggests a coordinated activation model for its three protomeric units. J. Virol. 10.1128/JVI.00530-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ruprecht CR, Krarup A, Reynell L, Mann AM, Brandenberg OF, Berlinger L, Abela IA, Regoes RR, Gunthard HF, Rusert P, Trkola A. 2011. MPER-specific antibodies induce gp120 shedding and irreversibly neutralize HIV-1. J. Exp. Med. 208:439–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Burton DR, Hessell AJ, Keele BF, Klasse PJ, Ketas TA, Moldt B, Dunlop DC, Poignard P, Doyle LA, Cavacini L, Veazey RS, Moore JP. 2011. Limited or no protection by weakly or nonneutralizing antibodies against vaginal SHIV challenge of macaques compared with a strongly neutralizing antibody. Proc. Natl. Acad. Sci. U. S. A. 108:11181–11186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Florese RH, van Rompay KK, Aldrich K, Forthal DN, Landucci G, Mahalanabis N, Haigwood N, Venzon D, Kalyanaraman VS, Marthas ML, Robert-Guroff M. 2006. Evaluation of passively transferred, nonneutralizing antibody-dependent cellular cytotoxicity-mediating IgG in protection of neonatal rhesus macaques against oral SIVmac251 challenge. J. Immunol. 177:4028–4036 [DOI] [PubMed] [Google Scholar]
- 87. Forthal DN, Landucci G, Gorny MK, Zolla-Pazner S, Robinson WE., Jr 1995. Functional activities of 20 human immunodeficiency virus type 1 (HIV-1)-specific human monoclonal antibodies. AIDS Res. Hum. Retroviruses 11:1095–1099 [DOI] [PubMed] [Google Scholar]
- 88. Girard MP, Plotkin SA. 2012. HIV vaccine development at the turn of the 21st century. Curr. Opin. HIV AIDS 7:4–9 [DOI] [PubMed] [Google Scholar]
- 89. Moore JP, Cao Y, Ho DD, Koup RA. 1994. Development of the anti-gp120 antibody response during seroconversion to human immunodeficiency virus type 1. J. Virol. 68:5142–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shen R, Drelichman ER, Bimczok D, Ochsenbauer C, Kappes JC, Cannon JA, Tudor D, Bomsel M, Smythies LE, Smith PD. 2010. gp41-specific antibody blocks cell-free HIV type 1 transcytosis through human rectal mucosa and model colonic epithelium. J. Immunol. 184:3648–3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tomaras GD, Yater NL, Liu P, Qin L, Fouda GG, Chavez LL, Decamp AC, Parks RJ, Ashley VC, Lucas JT, Cohen M, Eron J, Hicks CB, Liao HX, Self SG, Landucci G, Forthal DN, Weinhold KJ, Keele BF, Hahn BH, Greenberg ML, Morris L, Karim SS, Blattner WA, Montefiori DC, Shaw GM, Perelson AS, Haynes BF. 2008. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J. Virol. 82:12449–12463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kwon YD, Finzi A, Wu X, Dogo-Isonagie C, Lee LK, Moore LR, Schmidt SD, Stuckey J, Yang Y, Zhou T, Zhu J, Vicic DA, Debnath AK, Shapiro L, Bewley CA, Mascola JR, Sodroski JG, Kwong PD. 2012. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl. Acad. Sci. U. S. A. 109:5663–5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tran EE, Borgnia MJ, Kuybeda O, Schauder DM, Bartesaghi A, Fank GA, Sapiro G, Milne JL, Subramaniam S. 2012. Structural mechanism of trimeric HIV-1 envelope glycoprotein activation. PLoS Pathog. 8:e1002797. 10.1371/journal.ppat.1002797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Arthos J, Cicala C, Martinelli E, Macleod K, van Ryk D, Wei D, Xiao Z, Veenstra TD, Conrad TP, Lempicki RA, McLaughlin S, Pascuccio M, Gopaul R, McNally J, Cruz CC, Censoplano N, Chung E, Reitano KN, Kottilil S, Goode DJ, Fauci AS. 2008. HIV-1 envelope protein binds to and signals through integrin alpha4beta7, the gut mucosal homing receptor for peripheral T cells. Nat. Immunol. 9:301–309 [DOI] [PubMed] [Google Scholar]
- 95. Parrish NF, Wilen CB, Banks LB, Iyer SS, Pfaff JM, Salazar-Gonzales JF, Salazar MG, Decker JM, Parrish EH, Berg A, Hopper J, Hora B, Kumar A, Mahlokozera T, Yuan S, Coleman C, Vermeulen M, Ding H, Ochsenbauer C, Tilton JC, Permar SR, Kappes JC, Betts MR, Busch MP, Gao F, Montefiori D, Haynes BF, Shaw GM, Hahn BH, Doms RW. 2012. Transmitted/founder and chronic subtype C HIV-1 use CD4 and CCR5 receptors with equal efficiency and are not inhibited by blocking the α4β7. PLoS Pathog. 8:e1002686. 10.1371/journal.ppat.1002686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mao Y, Wang L, Gu C, Herschhorn A, Xiang SH, Haim H, Yang X, Sodroski J. 2012. Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat. Struct. Mol. Biol. 19:893–899 [DOI] [PMC free article] [PubMed] [Google Scholar]